

Abstract
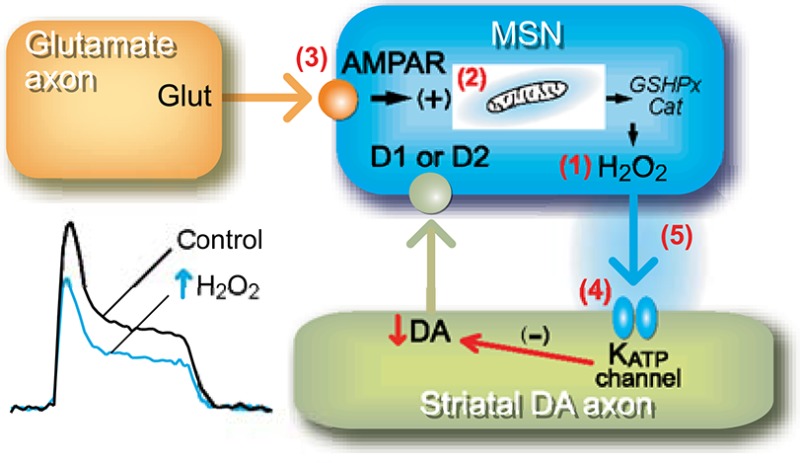
Here we review evidence that the reactive oxygen species, hydrogen peroxide (H2O2), meets the criteria for classification as a neuromodulator through its effects on striatal dopamine (DA) release. This evidence was obtained using fast-scan cyclic voltammetry to detect evoked DA release in striatal slices, along with whole-cell and fluorescence imaging to monitor cellular activity and H2O2 generation in striatal medium spiny neurons (MSNs). The data show that (1) exogenous H2O2 suppresses DA release in dorsal striatum and nucleus accumbens shell and the same effect is seen with elevation of endogenous H2O2 levels; (2) H2O2 is generated downstream from glutamatergic AMPA receptor activation in MSNs, but not DA axons; (3) generation of modulatory H2O2 is activity dependent; (4) H2O2 generated in MSNs diffuses to DA axons to cause transient DA release suppression by activating ATP-sensitive K+ (KATP) channels on DA axons; and (5) the amplitude of H2O2-dependent inhibition of DA release is attenuated by enzymatic degradation of H2O2, but the subsecond time course is determined by H2O2 diffusion rate and/or KATP-channel kinetics. In the dorsal striatum, neuromodulatory H2O2 is an intermediate in the regulation of DA release by the classical neurotransmitters glutamate and GABA, as well as other neuromodulators, including cannabinoids. However, modulatory actions of H2O2 occur in other regions and cell types, as well, consistent with the widespread expression of KATP and other H2O2-sensitive channels throughout the CNS.
Keywords: Brain slices, dorsal striatum, fast-scan cyclic voltammetry, fluorescence imaging, transmitter release, review
The striatum is the largest component of the basal ganglia and receives dense dopamine (DA) innervation from midbrain DA neurons in the substantia nigra pars compacta (SNc) and ventral tegmental area (VTA).1 The primary targets of these DA afferents are medium spiny neurons (MSNs) that constitute over 90% of the striatal neuronal population and are the main output neurons of this region. Axons arising from midbrain DA neurons form symmetric synapses on MSNs, primarily on spine necks, adjacent to glutamatergic input to spine heads.2,3 This architectural arrangement enables DA to modulate glutamate-induced MSN excitability with differential outcomes according to whether the target MSN also expresses DA D1 receptors (D1Rs) or DA D2 receptors (D2Rs); DA enhances MSN excitability and up-state MSN spiking via D1Rs and decreases these via D2Rs.4 Thus, converging glutamatergic and dopaminergic afferents control striatal output at the level of individual spines to regulate motor and cognitive function.4−6 It is relevant to note that D1Rs and D2Rs are predominantly extrasynaptic,7 so that rapid electrochemical measurements of extracellular DA concentration ([DA]o) can provide a direct index of DA transmission (e.g., 8–10).
The crucial role that DA plays in normal motor behavior has been illuminated by the findings that disturbances in DA function contribute to basal-ganglia circuit dysfunction and that DA depletion leads to the motor deficits of Parkinson’s disease.11−13 Thus, elucidating factors that regulate DA release in the striatum is of relevance for understanding the role of DA in normal brain function, as well as for providing therapeutic insight for neuropathological conditions like Parkinson’s disease.
Our laboratory examines the regulation of striatal DA release using fast-scan cyclic voltammetry (FCV) with carbon-fiber microelectrodes. Because FCV detects dynamic changes in [DA]o with high temporal and spatial resolution, this technique is ideally suited to determine factors that influence the amplitude, duration, and sphere of influence of DA signaling.14−17 Basic factors that we have examined include the pattern of stimulation, Ca2+-dependence of DA release, inhibition of DA release by presynaptic DA autoreceptors, and DA clearance by the DA transporter (DAT); we have also examined the effect of a number of local neurotransmitters and neuromodulators that can act on DA axons either directly or indirectly to alter evoked [DA]o (for review, see ref (10)).
One unconventional and under-appreciated modulator of striatal DA release is hydrogen peroxide (H2O2). H2O2 is most commonly known as a reactive oxygen species (ROS) that is formed from cellular oxidative metabolism and is often viewed as being toxic. However, H2O2 is not a free radical and, consequently, is not as reactive as other ROS.18 In fact, increasing evidence indicates that H2O2 plays an important role in normal cellular signaling processes, as well as in the modulation of transmitter release.19
Here we review the evidence that endogenous H2O2 fulfills criteria for classification as a neuromodulator through its actions on striatal DA release. We would note, however, that actions of H2O2 are not limited to DA axons, and possibly not even to the brain, as summarized in Concluding Remarks. Criteria often used to classify a molecule as a transmitter or modulator include the following: (1) same response elicited by the exogenous and endogenous substance; (2) synthesis of the substance within a releasing cell; (3) release in response to depolarization or receptor activation; (4) receptor/sensor for the substance at a target site; and (5) mechanism for deactivation. Using a combination of FCV to detect evoked [DA]o in striatal slices, whole-cell recording of striatal MSN activity, and fluorescence imaging to monitor H2O2 generation, we have shown that H2O2 is generated downstream from glutamatergic AMPA receptor (AMPAR) activation in MSNs, and then diffuses to DA axons where it causes transient suppression of striatal DA release through activation of ATP-sensitive K+ (KATP) channels on DA axons. Through this regulatory pathway, H2O2 acts as a key intermediate for DA modulation by glutamate and GABA, as well as other neuromodulators including the cannabinoids.
Criterion 1: Both Exogenous and Endogenous H2O2 Suppress DA Release
The first criterion for classification of a substance as a neuromodulator is that the same response must be elicited by the exogenous and endogenous substance. Our initial studies of H2O2-dependent regulation of striatal DA release revealed that brief exposure of guinea-pig striatal slices to exogenous H2O2 (1.5 mM for 15 min) suppresses [DA]o, evoked by pulse-train stimulation (30–50 pulses at 10 Hz, 0.4–0.8 mA pulse amplitude) in the dorsal striatum20 (Figure 1A). This effect is not accompanied by a decrease in striatal DA content, indicating that it is not a consequence of loss of the releasable pool of DA.20 Moreover, the effect is reversible and occurs without causing lipid peroxidation, demonstrating that H2O2 exposure under these conditions does not cause oxidative damage.20 Subsequently, we found that elevation of endogenous H2O2 has a similar effect on the peak amplitude of evoked [DA]o. Amplification of endogenous H2O2 levels following inhibition of the H2O2-metabolizing enzyme glutathione (GSH) peroxidase with mercaptosuccinate (MCS) also causes a reversible decrease in pulse-train evoked [DA]o21 (Figure 1B), which is also not accompanied by a change in DA content.22 Similar suppression of pulse-train evoked [DA]o by exogenous and endogenous H2O2 is also seen in the ventral striatum, including the nucleus accumbens (NAc) shell23 (Figure 1C). The inhibitory effect of MCS on evoked [DA]o persists when the DAT is inhibited, indicating that endogenous H2O2 decreases evoked [DA]o by inhibiting release rather than by enhancing DA uptake.21
Figure 1.

Both exogenous and endogenous H2O2 suppress striatal DA release. (A) Left: Exogenous H2O2 (1.5 mM, 15 min) inhibits pulse-train evoked extracellular DA concentration ([DA]o) in the dorsal striatum (caudate putamen, CPu). Right: DA release-suppression is fully reversibly upon washout (wash). (B) Amplification of endogenous H2O2 levels by inhibition of the H2O2 metabolizing enzyme GSH peroxidase with mecaptosuccinate (MCS, 1 mM), also reversibly inhibits pulse-train evoked [DA]o in the CPu. (C) Both exogenous H2O2 (left) and MCS (right) reversibly decrease evoked [DA]o in the nucleus accumbens (NAc) shell, demonstrating sensitivity of DA release regulation by H2O2 throughout the striatal complex. Data are means ± SEM (A is modified from ref (20), B is modified from ref (21), and C is modified from ref (23)).
It is important to emphasize, however, that DA release inhibition by endogenous H2O2 occurs under physiological conditions, that is, in the absence of GSH peroxidase inhibition. As discussed in the following sections, generation of inhibitory H2O2 occurs downstream from AMPAR activation in dorsal striatum during local pulse-train stimulation under control conditions.21,22 A consequence of this is an increase in pulse-train evoked [DA]o when AMPARs are blocked, indicating normal glutamatergic inhibition of DA release that is completely prevented in the presence of exogenous GSH peroxidase or catalase.21
Although exogenous H2O2 also suppresses [DA]o evoked by a single stimulus pulse, inhibition of GSH peroxidase by MCS does not.21 This makes two points. First, basal levels of H2O2 in the striatum are insufficient to modulate axonal DA tonically release, even with amplification by GSH peroxidase inhibition; this contrasts with the more significant role of basal H2O2 levels in regulating the firing rate of DA neurons in the SNc.24 The second point is that AMPAR-dependent modulatory H2O2 is generated dynamically during the first few pulses of a stimulus train, as seen in our H2O2 imaging studies (Figure 2B), with consequent, H2O2-dependent inhibition of DA released by subsequent pulses, even under control conditions. Overall, these findings demonstrate that endogenous H2O2 has a similar effect to that of exogenous H2O2 in suppressing axonal DA release in the striatum, thereby fulfilling the first criterion for classification as a neuromodulator.
Figure 2.

Synthesis and regulation of H2O2 in striatal medium spiny neurons, not DA axons. (A) Generation of H2O2 by the mitochondrial respiratory chain in which reduction of O2 forms the superoxide radical (•O2–), which then produces H2O2 either through catalysis by superoxide dismutase (SOD) or by spontaneous dismutation. The antioxidant enzymes catalase and GSH peroxidase metabolize H2O2 thereby regulating endogenous H2O2 levels. (B) Upper: Basal and activity-dependent H2O2 generation in a striatal MSN indicated by a 30% increase in DCF fluorescence intensity (FI) with local pulse-train stimulation (30 pulses, 10 Hz) under control conditions (black trace). Lower: Simultaneous whole-cell recording of membrane potential (Vmemb) shows that a single action potential is generated for each stimulus pulse within the pulse train. Blockade of AMPARs with GYKI-52466 (GYKI, 50 μM; red traces) prevents both stimulus-induced MSN firing and H2O2 generation. When H2O2 metabolism is compromised by inhibiting GSH peroxidase activity with MCS (1 mM; blue trace), stimulus-induced H2O2 is enhanced. (C) The average increase in stimulus-induced H2O2, indicated by DCF FI, is eliminated by GYKI and catalase, and doubled by MCS, demonstrating the H2O2 dependence of the DCF response and our ability to alter H2O2 levels in MSNs by manipulating peroxidase activity (**p < 0.01; ***p < 0.001 vs each respective basal DCF-FI, ###p < 0.001 vs control DCF-FI). (D, E) Differential regulation by GYKI and MCS on striatal DA release evoked at a single site by alternating local stimulation and selective stimulation of DA pathways demonstrates that modulatory H2O2 is not generated by DA axons. (D) Blockade of AMPARs with GYKI (50–100 μM) increases [DA]o evoked by local pulse-train stimulation indicating that endogenous glutamate normally enhances DA release (***p < 0.001 vs local control). By contrast, GYKI has no effect on [DA]o evoked by selective DA pathway stimulation, thereby confirming the absence of local glutamate release with this stimulus paradigm. (E) Inhibition of GSH peroxidase by MCS (1 mM) suppresses [DA]o evoked by local stimulation (***p < 0.001 vs local control) but has no effect on DA release evoked by pathway stimulation, demonstrating a lack of modulatory H2O2 generation by DA axons. Data are means ± SEM (B–E are modified from ref (22)).
Criterion 2: Synthesis of H2O2 in Striatal Medium Spiny Neurons
The second criterion that a neuromodulator must be synthesized in neurons is easily met by H2O2. Indeed, H2O2 is generated in all cells during mitochondrial respiration.25−27 This process generates H2O2 by mitochondrial electron transport; O2 is reduced to form the superoxide radical (•O2–), which is then converted to H2O2 either through catalysis by superoxide dismutase (SOD) or by spontaneous dismutation (Figure 2A). Other less ubiquitous subcellular sources of •O2–, and thus H2O2 generation, include the enzymes monoamine oxidase (MAO)28,29 and NADPH oxidase.30−34 It has been reported that the amount of H2O2 produced by brain mitochondria can reach 5% of the total O2 metabolized.35
Absolute levels of H2O2 within a given cell depend on the dynamic balance between the rate of H2O2 production and the rate of H2O2 metabolism by antioxidant enzymes, as well as diffusion away from the site of generation. The main antioxidant enzymes that regulate cellular H2O2 levels are catalase, which is located in intracellular peroxisomes of neurons and glia,18,36,37 and GSH peroxidase, which is present in mitochondria and in the cytosol, especially in glia.38 These enzymes catalyze the decomposition of H2O2 to water and O2. H2O2 levels are also regulated, albeit to a lesser extent, by peroxiredoxins and cellular thiols.39−43
Given that striatal MSNs, which express AMPARs, constitute >90% of striatal neurons and are a prime target of DA axons, we hypothesized that these cells could be an important source of modulatory H2O2. To visualize H2O2 generation in individual MSNs, we used real-time fluorescence imaging of H2O2-activated dichlorofluorescein (DCF), coupled with whole-cell recording for simultaneous monitoring of neuronal activation in guinea-pig striatal slices. Although DCF fluorescence imaging is not suitable for quantitative evaluation of absolute H2O2 concentration, this dye can be used to monitor relative differences in basal and stimulated levels of H2O2.19,44 Despite the fact that MSNs are electrically silent in brain slices, basal DCF fluorescence is detected in all cells tested, presumably reflecting tonic H2O2 production through mitochondrial respiration required to maintain ion gradients.22 During local electrical stimulation of dorsal striatum in ex vivo brain slices, each pulse of a stimulus train (e.g., 30 pulses, 10 Hz) elicits a single action potential in recorded MSNs (Figure 2B); these spikes are AMPAR-dependent, as discussed further below. This stimulus paradigm simultaneously produces a ∼30% increase in DCF fluorescence in a majority of MSNs22 (Figure 2B). The observations that stimulus-induced increases in DCF fluorescence are absent in the presence of the H2O2 scavenging enzyme catalase and are doubled when the H2O2 metabolizing enzyme GSH peroxidase is inhibited by MCS confirmed H2O2 detection22 (Figure 2C). Thus, H2O2 is synthesized in striatal MSNs, with regulation of H2O2 levels by cellular peroxidases, thereby fulfilling the second criterion for classification as a neuromodulator.
DA Axons Are Not a Source of Modulatory H2O2
Because all cells are capable of producing H2O2, an additional source of H2O2 for DA modulation could be DA axons themselves. However, several observations prove that this is not the case. First, DA release modulation by endogenous H2O2 requires AMPAR activation; the effect of MCS on pulse-train evoked [DA]o is lost when AMPARs are blocked, indicating that there is no remaining H2O2 signal to amplify and thus the source of modulatory H2O2 is AMPAR-dependent.21 Second, blockade of AMPARs with an antagonist, GYKI-52466, prevents H2O2 generation in MSNs during local pulse-train stimulation (Figure 2B), which leads to a 2-fold increase in local pulse-train evoked [DA]o (Figure 2D). Because DA axons in dorsal striatum lack AMPARs,45−47 it is unlikely that modulation of DA release by H2O2 is a self-regulatory process.
The third and most direct observation for lack of DA axon involvement comes from experiments using parasagittal slices; this preparation allows the use of distal stimulation of nigrostriatal DA axons to evoke DA release and thereby avoid the concurrent glutamate release that accompanies local stimulation.22 The absence of local glutamate release was shown by the lack of effect of AMPAR antagonism by GYKI-52466 on pathway-evoked [DA]o, but the usual enhancement of locally evoked [DA]o at the same recording site (Figure 2D). We also found no increase in DCF fluorescence in MSNs or induction of MSN spiking with DA pathway stimulation;22 the absence of co-released glutamate from DA axons in dorsal striatum was later confirmed in optogenetic studies.48 Importantly, we found no evidence for an AMPAR-independent contribution from DA axons to the generation of modulatory H2O2: inhibition of GSH peroxidase by MCS also has no effect on pathway-evoked DA release22 (Figure 2E). Thus, these data not only show that DA axons are not the primary source of neuromodulatory H2O2, but also confirm that H2O2 is necessarily a diffusible messenger in the striatum.
What Subcellular Processes Generate Modulatory H2O2?
As noted above, there are at least three subcellular sources of H2O2: mitochondrial respiration; MAO; and NADPH oxidase. We have shown that the usual effects of GSH peroxidase inhibition or AMPAR blockade on evoked [DA]o are completely lost in the presence of rotenone, a complex I inhibitor, and succinate, a mitochondrial complex II substrate, which together limit H2O2 production, but maintain tissue ATP content.49 These data implicate mitochondrial complex I as the primary source of rapid neuronal signaling by H2O2 in the striatum. Another source with particular relevance in monoaminergic regions is MAO, which catalyzes deamination of DA and other biogenic amines via a two-electron reduction of O2 to H2O2, with one molecule of H2O2 produced for each molecule of DA metabolized.50,51 The isoforms of MAO are type A (MAO-A) and type B (MAO-B), with MAO-A primarily in neurons and MAO-B primarily in glia.29,52 However, the ability of H2O2 amplification by MCS to suppress pulse-train evoked [DA]o is unaltered by a cocktail of MAO-A and MAO-B inhibitors, as is the effect of the AMPAR antagonist GYKI-52466.49 NADPH oxidase belongs to a family of membrane-associated, multisubunit enzymes that catalyze the one-electron reduction of O2 to form •O2– and subsequently H2O2.30−34 However, inhibition of NADPH oxidase by phenylarsine oxide also fails to prevent the effect of MCS or GYKI-5466 on evoked [DA]o.49 Thus, dynamic, glutamate-dependent modulation of striatal DA release requires H2O2 that originates from mitochondria, rather than MAO or NADPH oxidase. Of course, this does not exclude roles of H2O2 derived from MAO-dependent DA metabolism or NADPH oxidase in other aspects of neuronal regulation on longer time scales.18,32,53−56
Criterion 3: H2O2 Generation in MSNs Requires Synaptic Glutamatergic Depolarization
Activity-dependent H2O2 generation in striatal MSNs not only implicates these neurons as a source of modulatory H2O2, but also implies that H2O2 is produced in response to neuronal activation, meeting the third criterion for classification as a neuromodulator. Indeed, stimulus-induced H2O2 generation in striatal MSNs does not occur when Na+ channels are blocked by TTX. However, this process also requires glutamatergic activation of AMPARs on these neurons. Antagonism of AMPARs with GYKI-52466 during local electrical stimulation in dorsal striatum prevents generation of action potentials, as well as H2O2 elevation (Figure 2B). By contrast, neither of these parameters is altered by antagonism of NMDA receptors (NMDARs) under the same experimental conditions.22 Notably, we found no detectable H2O2 elevation when action potentials are generated in single MSNs by current injection pulses, implying that depolarization alone may not be sufficient to generate modulatory H2O2 in these cells.22 This result suggests a key role for glutamate in striatal H2O2 generation, and is consistent with previous studies in cultured cells showing glutamate-dependent increases in intracellular ROS production.26,57−60
How is H2O2 “released”? Once generated, H2O2 apparently quickly diffuses away from a site of generation; as a neutral molecule, it can be relatively membrane permeable, although recent studies suggest that it may escape the intracellular compartment via aquaporin channels.61,62 However, the fact that H2O2 generated in MSNs can inhibit DA release from striatal DA axons provides functional evidence that modulatory H2O2 is released from MSNs. Thus, H2O2 “release” in response to transmitter action and consequent depolarization addresses the third criteria for H2O2 as a striatal neuromodulator.
Criterion 4: Endogenous H2O2 Suppresses DA Release via KATP Channels on DA Axons
The main targets of H2O2 for DA release regulation have been identified as KATP channels,63 which fulfills the fourth criterion for classification of H2O2 as a neuromodulator. Binding and immunohistochemical studies show that KATP channels are highly expressed in the striatum.64−70 The role of H2O2-sensitive KATP channels in axonal DA release regulation in dorsal striatum was first demonstrated by prevention of the usual changes in pulse-train evoked [DA]o with GSH peroxidase inhibition by MCS or AMPAR antagonism by GYKI-52466 in the presence of a KATP channel blocker, like tolbutamide orglibenclamide21,63 (Figure 3A). These and other studies show that activity-dependent H2O2 causes KATP-channel opening, with elevation of H2O2 leading to KATP-channel-dependent suppression of axonal DA release or DA neuron firing.21,24,63
Figure 3.

Endogenous H2O2 suppresses DA release via KATP channels on DA axons. (A) The KATP-channel blocker, glibenclamide (3 μM), increases pulse-train evoked [DA]o in the dorsal striatum and prevents the usual suppression of DA release by the GSH peroxidase inhibitor MCS or enhancement of release by the AMPAR antagonist GYKI-52466 (GYKI). (B) The SUR1 subunit sensitive KATP-channel opener diazoxide (30 μM) decreases pulse-train evoked [DA]o and prevents the usual effects of MCS and GYKI. (C) The SUR2 subunit sensitive KATP-channel opener cromakalim (30 μM) also suppresses pulse-train evoked [DA]o but in contrast to diazoxide does not alter the effects of MCS and GYKI. (D) Immunohistochemical staining of striatum with an anti-tyrosine hydroxylase (TH) antibody for DA axons (green) and an anti-Kir6.2 antibody (red) for KATP channels. The upper panel shows colocalization of KATP channels in DA axons, as well as other cells and processes. The lower panel shows KATP-channel labeling along the length of an extended DA axon. (E) Independent suppression of single pulse evoked DA release by diazoxide and cromakalim suggest that both SUR1 and SUR2 KATP channels are functionally present on DA axons. Data are means ± SEM (A–C are modified from ref (63); D and E are modified from ref (70)).
Many neuromodulators have receptors that are activated selectively, if not specifically, by a given modulatory agent; for example, the D1 and D2 family receptors are activated selectively by DA. However, KATP channels have other known regulators besides H2O2, the most obvious of which are low ATP and/or elevated ADP; through these, KATP channels act to couple the metabolic state of a cell to membrane excitability.71 Previous studies using inside-out membrane patches from cardiac cells have shown a direct, concentration-dependent effect of H2O2 on KATP-channel opening by decreasing channel sensitivity to ATP.72,73 However, whether H2O2 regulation of KATP channels in intact DA neurons is direct or indirect has not yet been established. KATP channels are octameric proteins74,75 composed of four inward rectifier K+ channel subunits that form a central pore, typically Kir6.2 in neurons and Kir6.1 in glia,69,76−78 and four surrounding sulfonylurea-binding subunits, either SUR1 or SUR2.71,79,80 Channels based on SUR1 or SUR2 subunits can be distinguished by their differential sensitivity to KATP-channel openers.81,82 Both SUR1-acting diazoxide and SUR2-acting cromakalim decrease pulse-train evoked [DA]o by roughly 30%63 (Figure 3B,C). However, suppression of evoked [DA]o by MCS is occluded only by diazoxide and not cromakalim, indicating that SUR1-based KATP channels are the primary target of neuromodulatory H2O263 (Figure 3B,C).
Identification of KATP channels as the targets through which H2O2 inhibits DA release fulfills the fourth criterion of having a “receptor” that mediates the action of a putative neuromodulator. However, the use of pulse-train stimulation alone cannot indicate the location at which these H2O2-sensitive KATP channels alter DA release; that is, whether responses are mediated by KATP channels on DA axons or elsewhere in the striatal microcircuitry. The most straightforward explanation for rapid, reversible inhibition of striatal DA release by H2O2 is that KATP channels are located directly on DA axons. We tested this hypothesis using immunohistochemical methods, which confirmed that at least 30% of KATP channels containing the Kir6.2 subunit in dorsal striatum are expressed by DA axons70 (Figure 3D). Functional studies examining the effect of SUR-subunit selective openers on [DA]o evoked by single-pulse stimulation, that is not modulated by glutamate, GABA, or D2 autoreceptors,21,−86 show that either SUR1-sensitive diazoxide or SUR2-sensitive cromakalim can independently decrease DA release70 (Figure 3E).
Although DA release elicited by a single pulse is independent from regulation by glutamate and GABA,21,86 acetylcholine (ACh) release from striatal cholinergic interneurons exerts a strong modulatory effect on single-pulse evoked release through nicotinic ACh receptors (nAChRs) on DA axons: when nAChRs are blocked, single pulse evoked [DA]o is suppressed, but the frequency-responsiveness of brief pulse trains is enhanced.87−90 Given that cholinergic interneurons also express SUR1-based KATP channels that can hyperpolarize these cells,91 one could speculate that H2O2 regulation of DA release might occur indirectly by inhibiting ACh release. However, this is not the case. First, activation of KATP channels by diazoxide suppresses DA release evoked by 5-pulse trains to a similar degree across all frequencies tested, with no change in the frequency-dependence of evoked [DA]o.70 Thus, activation of KATP channels fails to recreate the dynamic pattern of striatal DA release with frequency that is characteristic of decreased ACh release. Second, the effect of MCS on evoked [DA]o is unaltered when nAChRs are blocked by mecamylamine, indicating that inhibition of axonal DA release by endogenous H2O2 is also independent of H2O2 effects on cholinergic interneurons.70
Together, these anatomical and functional observations confirm the presence of striatal “receptors” in the form of KATP channels for neuromodulatory H2O2. Moreover, they show that H2O2 regulates DA release by activation of KATP channels directly on DA axons.
Criterion 5: the Roles of Diffusion and Enzymatic Degradation in Termination of H2O2 Effects
The final criterion for a substance being a neuromodulator is that there must be a mechanism for inactivation. As already noted, H2O2 is metabolized primarily by the antioxidant enzymes GSH peroxidase and catalase (Figure 2A); inhibition of GSH peroxidase leading to amplification of endogenous H2O2 levels in cells in which it is generated (e.g., MSNs) (Figure 2C), with subsequent enhancement of H2O2-dependent suppression of DA release (Figures 1B, 2E). However, our studies have also shown that exogenous application of GSH peroxidase or catalase completely prevents the increase in striatal DA release normally seen with the AMPAR antagonist GYKI-52466 (21) (Figure 4A,B). Moreover, exogenous catalase reverses the decrease in DA release usually seen when GSH peroxidase is inhibited with MCS21 (Figure 4C). These data not only demonstrate that the actions of endogenous H2O2 on striatal DA release are regulated by enzymatic degradation but they also confirm the role of H2O2 in glutamate-dependent regulation of DA release. Given the ability of H2O2 to cross plasma membranes, inactivation of dynamically generated H2O2 is likely to occur via diffusion and enzymatic inactivation in neighboring cells, as well as by regulation in the generating cell.
Figure 4.

Enzymatic degradation in termination of H2O2 DA release regulation. (A, B) Exogenous application of the H2O2 metabolizing enzymes glutathione peroxidase (GSHPx) or catalase (Cat) completely prevents the increase in pulse-train evoked striatal [DA]o normally seen with the AMPAR antagonist GYKI-52466 in dorsal striatum. By contrast, heat-inactivated forms of these enzymes (I-GSHPx or I-Cat) are without effect. (C) Exogenous catalase also reverses the decrease in DA release usually seen when GSH peroxidase is inhibited with MCS. These data demonstrate the scavenging nature of these antioxidant enzymes in regulating and terminating the DA modulatory effects of endogenous H2O2. Data are means ± SEM (modified from ref (21)).
DA Release Suppression by Endogenous H2O2 and KATP Channels Is Rapid Yet Transient
What is the time frame for H2O2 induced suppression of DA release? Our studies of monitoring H2O2 in striatal MSNs with DCF suggest that H2O2 generation is rapid, occurring within a few hundred milliseconds following initiation of a 10 Hz stimulus burst22 (Figure 2B). In addition, suppression of pulse-train evoked [DA]o during GSH peroxidase inhibition or enhancement of pulse-train evoked [DA]o by AMPAR antagonism or KATP-channel blockade follows a similar time frame and is maintained throughout the stimulus.21,22,63 However, these observations provide little information about the dynamic window for H2O2/KATP-channel-dependent modulation of striatal DA release.
We determined this regulatory time window using a paired pulse-paradigm in which the influence of endogenous H2O2 generated during an initial stimulus is assessed on [DA]o evoked by a subsequent test pulse applied at varying interpulse intervals70 (Figure 5A). These studies revealed that when KATP channels are blocked by glibenclamide, DA release evoked by a subsequent test pulse at 500 or 1000 ms after the initial stimulus is enhanced, with loss of the effect by 1500 ms. These data indicate a 1 s time window for KATP-channel activation and DA release inhibition after the stimulus70 (Figure 5A,B). The delay in onset of striatal DA release regulation by H2O2 at intervals of less than 500 ms could reflect a number of factors, including the time required for glutamate-AMPAR dependent H2O2 generation and diffusion of modulatory H2O2 from the site of generation (e.g., MSNs), to DA axons,22 as well as the time required for KATP-channel activation. In parallel experiments, we found that amplification of endogenous H2O2 levels by GSH peroxidase inhibition enhances DA release suppression at the same times that KATP-channel blockade led to enhanced evoked [DA]o after the initial stimulus70 (Figure 5B). This suggests that H2O2 metabolism, at least by GSH peroxidase, plays a greater role in regulating the amplitude of H2O2 signaling than in limiting its time course and, therefore, implicates diffusion and KATP-channel kinetics in the termination of action. Overall, these data show that the time window for H2O2/KATP-channel-dependent suppression of DA release is 500–1000 ms after a burst of activity.
Figure 5.

Time course of DA release suppression by endogenous H2O2 and KATP channels. (A) Representative [DA]o versus time records from the dorsal striatum using a paired-pulse stimulation paradigm in which a pseudo-one pulse stimulus (10 pulses, 100 Hz; P1) is followed by a single test pulse (P2) at varying time intervals (P1 + P2, black traces), with subtracted P1 (red traces). Note the recovery of P2 amplitude with time in the absence and presence of the KATP-channel blocker glibenclamide (3 μM). (B) Mean data showing the recovery in amplitude of [DA]o relative to P1 under control conditions, in the presence of glibenclamide (Gliben) or in MCS, a GSH peroxidase inhibitor. Glibenclamide significantly increased [DA]o evoked by P2 at 500 and 1000 ms, whereas MCS significantly decreased P2 at these time points, thereby revealing a role for endogenous H2O2 generation by P1 stimulation in DA release suppression; P2 values returned to control levels by 1500 ms (*p < 0.05; **p < 0.01 vs control). These data indicate rapid yet transient suppression of axonal DA release by endogenous H2O2 and KATP channels. Data are means ± SEM (modified from ref (70)).
H2O2 Mediates DA Release Regulation by Glutamate, GABA, and Cannabinoids
Glutamate
Given the close apposition of dopaminergic and glutamatergic inputs on MSN spines,2,3 it is not surprising that glutamate regulates striatal DA release. However, the mechanism by which glutamate regulates DA release has been controversial. This may be in part due to the use of glutamate agonists that can induce widespread depolarization, including pathophysiological spreading depression.92,93 Glutamatergic transmission is generally regarded as being “hard-wired”, with spillover limited by avid uptake.94−97 When glutamate spillover does occur, for example, when uptake is compromised, DA release is inhibited via metabotropic glutamate receptors on DA axons.98 However, because DA axons in the dorsal striatum lack both AMPARs and NMDARs,45−47 any effect of glutamate on DA release via these ionotropic receptors must be indirect. Our studies have revealed that glutamatergic activation of AMPARs inhibits striatal DA via H2O2 and KATP channels on DA axons, as outlined above. Glutamatergic excitation increases H2O2 generation in a large population of striatal MSNs via activation of AMPARs. When AMPARs are blocked in dorsal striatum, dynamic H2O2 generation in MSNs is prevented, leading to an increase in pulse-train evoked DA release21,22 (Figure 2B,C). Blocking NMDARs under the same conditions has no effect on H2O2 generation in MSNs or evoked [DA]o.21,22 The observations that the effect of GYKI-52466 is absent when the H2O2 scavenging enzymes catalase and GSH peroxidase are present21 (Figure 4A,B), when mitochondrial H2O2 generation is prevented,49 or when H2O2-sensitive KATP channels are blocked (Figure 3A) confirm that glutamatergic AMPAR-modulation of DA release occurs exclusively through mitochondrial H2O2 and KATP-channel activation.
GABA
Regulation of axonal DA release in dorsal striatum by GABA is also unconventional. Blockade of GABAA receptors (GABAARs) with picrotoxin causes a roughly 50% decrease in pulse-train evoked [DA]o in the dorsal striatum, showing that GABA enhances DA release whereas blockade of GABABRs is without effect.21 The influence of GABA on DA release, like that of glutamate, must be indirect, because DA axons in dorsal striatum do not appear to express ionotropic GABARs.99 Our studies have revealed that GABAergic modulation of DA release also occurs via H2O2, with complete prevention of the effect of GABAAR blockade by catalase21 and by the KATP-channel blocker glibenclamide.63 Moreover, picrotoxin has no effect when AMPARs are blocked,21 indicating that the actions of GABA at GABAARs must be mediated by the pool of H2O2 generated downstream from glutamate acting at AMPARs. The localization of GABAARs near spines on the dendrites of MSNs99 provides ideal placement for the established role of GABA in attenuating glutamatergic excitation of MSNs, as well as for attenuation of glutamate-dependent H2O2 generation and DA release suppression.
Glutamate input to MSNs generates modulatory H2O2 that diffuses to adjacent DA axons, opens SUR1-based KATP channels that hyperpolarize DA axons, and thereby inhibits DA release. GABAergic activation of GABAARs curtails AMPAR-dependent MSN excitation and consequent H2O2 generation from mitochondria.19 In the absence of glutamatergic input, or when AMPARs are blocked, H2O2 generation is minimized, GABAAR-dependent regulation is lost, and DA release is no longer inhibited by H2O2 acting at KATP channels. Thus, GABA has no direct influence on DA release when MSNs are not activated by glutamate.
Cannabinoids
Given the critical role that H2O2 plays in enabling the effects of glutamate and GABA on DA transmission in the dorsal striatum, it is not surprising to find that H2O2 is also involved in mediating the effect of other modulators on DA release that act by altering either glutamate or GABA transmission. One example is the regulation of DA release by cannabinoid activation of CB1 receptors (CB1Rs). Consistent with in vivo studies, pulse-train evoked [DA]o in the dorsal striatum in striatal slices is not altered by CB1R antagonists, indicating the absence of DA release regulation by endocannabinoids with brief, mild stimulation. However, CB1R agonists cause a decrease in pulse-train evoked [DA]o whereas single-pulse evoked [DA]o is unaffected, which implicates the involvement of local striatal circuitry in consequences of CB1R activation on DA release, rather than direct effects on DA axons.100,101 The effect of CB1R activation on pulse-train evoked [DA]o is prevented by GABAAR blockade, by catalase, and by blockade of KATP channels.101 Thus, these data implicate presynaptic inhibition of GABA release via presynaptic CB1Rs, with consequent increase in MSN activation and H2O2 generation. Consistent with this notion, the effect of the CB1R agonist WIN55,212-2 in dorsal striatum is also lost with AMPAR antagonism (Sidló and Rice, unpublished). Local inhibition of DA release consequent to GABA release inhibition might explain CB1R-agonist induced catalepsy, despite evidence for increased phasic DA-neuron activity.102
Concluding Remarks
The mechanistic studies reviewed here demonstrate that H2O2 meets the criteria for classification as a neuromodulator: both exogenous and endogenous H2O2 suppress DA release; endogenous H2O2 is generated by mitochondria in striatal MSNs in a manner that requires glutamatergic depolarization via AMPARs; dynamically generated H2O2 leaves the cell of generation and diffuses through the extracellular compartment to activate KATP channels located directly on DA axons, thereby inhibiting exocytotic DA release; finally, the action of modulatory H2O2 is regulated by the metabolizing enzymes GSH peroxidase and catalase. Notably, findings from the dorsal striatum indicate that regulation of axonal DA release by glutamate and GABA acting via ionotropic receptors in that region occurs exclusively through modulatory H2O2. As a result, other on-demand modulators such as the cannabinoids that can alter glutamate or GABA transmission also rely on H2O2 to deliver the final message to DA axons.
Although the evidence summarized here is focused on H2O2-dependent modulation of axonal DA release in the striatum, the ubiquitous presence of mitochondria as sources of H2O2 in all cells and the widespread distribution of KATP channels throughout the CNS64−70,76,89,90,103 imply that neuromodulation by H2O2 could be equally widespread. Indeed, endogenous H2O2 acts via KATP channels to regulate the activity of SNc DA neurons,24 striatal GABAergic MSNs,104 and substantia nigra pars reticulata GABAergic neurons.105,106 In this light, it is possible that H2O2-dependent modulation could occur in other KATP-channel expressing cells elsewhere in the body, including pancreatic β-cells and cardiac myocytes.71−73,107,108
Lastly, it should be noted that KATP-channels are not the only ion-channel target of endogenous H2O2. Among the most intriguing of these is a subtype of transient receptor potential (TRP) channels, the TRP melastatin 2 (TRPM2) channel, which is uniquely sensitive to activation by H2O2.109,110 In contrast to KATP channels, which hyperpolarize neuronal membranes and decrease cell excitability, TRP channels are nonselective cation channels that cause membrane depolarization. In guinea-pig striatal MSNs and SNr GABAergic neurons, the primary effect of elevation of endogenous H2O2 is TRP-channel mediated depolarization and increased activity.104−106 The specific channel subtype underlying these effects in SNr neurons has been identified as TRPM2.106 Although SNr GABAergic neurons also express KATP channels, the predominant effect of H2O2 elevation on these cells in guinea-pig SNr (albeit not in mouse SNr) is depolarization and increased firing rate.105 In contrast, although DA neurons in guinea-pig SNc express TRPM2,106 as well as KATP channels, the predominant effect of H2O2 elevation on SNc neurons is hyperpolarization.24 These data indicate that the net effect of H2O2 on a given cell or transmitter release site will reflect the balance between H2O2-sensitive target channels expressed and thereby provide cell-type-specific patterns of modulation.
Acknowledgments
We would like to thank past and present members of the Rice Laboratory for their contributions to the original studies reviewed in this article.
Author Contributions
Both authors contributed to the writing of this review and to the preparation of the figures.
This work was supported by NIH/NINDS grant NS036362 and the Olympia and Attilio Ricciardi Research Fund.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Dahlström A.; Fuxe K. (1964) Evidence of the existence of monoamine-containing neurons in the central nervous system. I: Demonstration of monoamines in the cell bodies of brain stem neurons. Acta. Physiol. Scand. 62, 1–55. [PubMed] [Google Scholar]
- Freund T. F.; Powell J. F.; Smith A. D. (1984) Tyrosine hydroxylase-immunoreactive boutons in synaptic contact with identified striatonigral neurons, with particular reference to dendritic spines. Neuroscience 13, 1189–1215. [DOI] [PubMed] [Google Scholar]
- Smith A. D.; Bolam J. P. (1990) The neural artwork of the basal ganglia as revealed by the study of synaptic connections of identified neurons. Trends Neurosci. 13, 259–265. [DOI] [PubMed] [Google Scholar]
- Surmeier D. J.; Carrillo-Reid L.; Bargas J. (2011) Dopaminergic modulation of striatal neurons, circuits, and assemblies. Neuroscience 198, 3–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cagniard B.; Beeler J. A.; Britt J. P.; McGehee D. S.; Marinelli M.; Zhuang X. (2006) Dopamine scales performance in the absence of new learning. Neuron 51, 541–547. [DOI] [PubMed] [Google Scholar]
- Gerfen C. R.; Surmeier D. J. (2011) Modulation of striatal projection systems by dopamine. Annu. Rev. Neurosci. 34, 441–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung K. K.; Bolam J. P.; Smith A. D.; Hersch S. M.; Ciliax B. J.; Levey A. I. (1995) Immunocytochemical localization of D1 and D2 dopamine receptors in the basal ganglia of the rat: light and electron microscopy. Neuroscience 65, 709–730. [DOI] [PubMed] [Google Scholar]
- Cragg S. J.; Rice M. E. (2004) DAncing past the DAT at a DA synapse. Trends Neurosci. 27, 270–277. [DOI] [PubMed] [Google Scholar]
- Rice M. E.; Cragg S. J. (2008) Dopamine spillover after quantal release: rethinking dopamine transmission in the nigrostriatal pathway. Brain Res. Rev. 58, 303–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice M. E.; Patel J. C.; Cragg S. J. (2011) Dopamine release in the basal ganglia. Neuroscience 198, 112–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson A. (2002) Treatment of Parkinson’s with L-DOPA. The early discovery phase, and a comment on current problems. J. Neural Transm. 109, 777–787. [DOI] [PubMed] [Google Scholar]
- Mallet N.; Pogosyan A.; Sharott A.; Csicsvari J.; Bolam J. P.; Brown P.; Magill P. J. (2008) Disrupted dopamine transmission and the emergence of exaggerated beta oscillations in subthalamic nucleus and cerebral cortex. J. Neurosci. 28, 4795–4806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichmann T.; Dostrovsky J. O. (2011) Pathological basal ganglia activity in movement disorders. Neuroscience 198, 232–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar J.; Stamford J. A.; Kruk Z. L.; Wightman R. M. (1985) Electrochemical, pharmacological and electrophysiological evidence of rapid dopamine release and removal in the rat caudate nucleus following electrical stimulation of the median forebrain bundle. Eur. J. Pharmacol. 109, 341–348. [DOI] [PubMed] [Google Scholar]
- Bull D. R.; Palij P.; Sheehan M. J.; Millar J.; Stamford J. A.; Kruk Z. L.; Humphrey P. P. (1990) Application of fast cyclic voltammetry to measurement of electrically evoked dopamine overflow from brain slices in vitro. J. Neurosci. Methods 32, 37–44. [DOI] [PubMed] [Google Scholar]
- Patel J. C., and Rice M. E. (2006) Monitoring dopamine release in brain slices. In Encyclopedia of Sensors, Vol. 6 (Grimes C. A., Dickey E. C., Pishko M. V., Eds.), pp 313–334, American Scientific Publishers, Stevenson Ranch, CA. [Google Scholar]
- Wightman R. M. (2006) Detection technologies. Probing cellular chemistry in biological systems with microelectrodes. Science 311, 1570–1574. [DOI] [PubMed] [Google Scholar]
- Cohen G. (1994) Enzymatic/nonenzymatic sources of oxyradicals and regulation of antioxidant defenses. Ann. N.Y. Acad. Sci. 73, 8–14. [DOI] [PubMed] [Google Scholar]
- Rice M. E. (2011) H2O2: a dynamic neuromodulator. The Neuroscientist 17, 389–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B. T.; Avshalumov M. V.; Rice M. E. (2001) H2O2 is a novel, endogenous modulator of synaptic dopamine release. J. Neurophysiol. 85, 2468–2476. [DOI] [PubMed] [Google Scholar]
- Avshalumov M. V.; Chen B. T.; Marshall S. P.; Peña D. M.; Rice M. E. (2003) Glutamate-dependent inhibition of dopamine release in striatum is mediated by a new diffusible messenger, H2O2. J. Neurosci. 23, 2744–2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avshalumov M. V.; Patel J. C.; Rice M. E. (2008) AMPA receptor-dependent H2O2 generation in striatal medium spiny neurons, but not dopamine axons: one source of a retrograde signal that can inhibit dopamine release. J. Neurophysiol. 100, 1590–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B. T.; Avshalumov M. V.; Rice M. E. (2002) Modulation of somatodendritic dopamine release by endogenous H2O2: susceptibility in substantia nigra but resistance in VTA. J. Neurophysiol. 87, 1155–1158. [DOI] [PubMed] [Google Scholar]
- Avshalumov M. V.; Chen B. T.; Koós T.; Tepper J. M.; Rice M. E. (2005) Endogenous hydrogen peroxide regulates the excitability of midbrain dopamine neurons via ATP-sensitive potassium channels. J. Neurosci. 25, 4222–4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boveris A.; Chance B. (1973) The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem. J. 134, 707–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugan L. L.; Sensi S. L.; Canzoniero L. M.; Handran S. D.; Rothman S. M.; Lin T. S.; Goldberg M. P.; Choi D. W. (1995) Mitochondrial production of reactive oxygen species in cortical neurons following exposure to N-methyl-d-aspartate. J. Neurosci. 15, 6377–6388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Fiskum G.; Schubert D. (2002) Generation of reactive oxygen species by the mitochondrial electron transport chain. J. Neurochem. 80, 780–787. [DOI] [PubMed] [Google Scholar]
- Maker H. S.; Weiss C.; Silides D. J.; Cohen G. (1981) Coupling of dopamine oxidation (monoamine oxidase activity) to glutathione oxidation via the generation of hydrogen peroxide in rat brain homogenates. J. Neurochem. 36, 589–593. [DOI] [PubMed] [Google Scholar]
- Azzaro A. J.; King J.; Kotzuk J.; Schoepp D. D.; Frost J.; Schochet S. (1985) Guinea pig striatum as a model of human dopamine deamination: the role of monoamine oxidase isozyme ratio, localization, and affinity for substrate in synaptic dopamine metabolism. J. Neurochem. 45, 949–956. [DOI] [PubMed] [Google Scholar]
- Babior B. M. (1984) Oxidants from phagocytes: agents of defense and destruction. Blood 64, 959–966. [PubMed] [Google Scholar]
- Lambeth J. D. (2004) NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 4, 181–189. [DOI] [PubMed] [Google Scholar]
- Infanger D. W.; Sharma R. V.; Davisson R. L. (2006) NADPH oxidases of the brain: distribution, regulation, and function. Antioxid. Redox Signal. 8, 1583–1596. [DOI] [PubMed] [Google Scholar]
- Rhee S. G. (2006) H2O2, a necessary evil for cell signaling. Science 312, 1882–1883. [DOI] [PubMed] [Google Scholar]
- Bedard K.; Krause K. H. (2007) The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 87, 245–313. [DOI] [PubMed] [Google Scholar]
- Arnaiz S. L.; Coronel M. F.; Boveris A. (1999) Nitric oxide, superoxide, and hydrogen peroxide production in brain mitochondria after haloperidol treatment. Nitric Oxide 3, 235–243. [DOI] [PubMed] [Google Scholar]
- Peuchen S.; Bolanos J. P.; Heales S. J.; Almeida A.; Duchen M. R.; Clark J. B. (1997) Interrelationships between astrocyte function, oxidative stress and antioxidant status within the central nervous system. Prog. Neurobiol. 52, 261–281. [DOI] [PubMed] [Google Scholar]
- Dringen R.; Pawlowski P. G.; Hirrlinger J. (2005) Peroxide detoxification by brain cells. J. Neurosci. Res. 79, 157–165. [DOI] [PubMed] [Google Scholar]
- Stults F. H.; Forstrom J. W.; Chiu D. T. Y.; Tappel A. L. (1977) Rat liver glutathione peroxidase: purification and study of multiple forms. Arch. Biochem. Biophys. 183, 490–497. [DOI] [PubMed] [Google Scholar]
- Rhee S. G.; Kang S. W.; Chang T. S.; Jeong W.; Kim K. (2001) Peroxiredoxins: a novel family of peroxidases. IUBMB Life 52, 35–41. [DOI] [PubMed] [Google Scholar]
- Rhee S. G.; Kang S. W.; Jeong W.; Chang T. S.; Yang K. S.; Woo. H. A. (2005) Intracellular messenger function of hydrogen peroxide and its regulation by peroxiredoxins. Curr. Opin. Cell Biol. 17, 183–189. [DOI] [PubMed] [Google Scholar]
- Hofmann B.; Hecht H. J.; Flohe L. (2002) Peroxiredoxins. Biol. Chem. 383, 347–364. [DOI] [PubMed] [Google Scholar]
- Adimora N. J.; Jones D. P.; Kemp M. L. (2010) A model of redox kinetics implicates the thiol proteome in cellular hydrogen peroxide responses. Antioxid. Redox Signaling 13, 731–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishina N. M.; Tyurin-Kuzmin P. A.; Markvicheva K. N.; Vorotnikov A. V.; Tkachuk V. A.; Laketa V.; Schultz C.; Lukyanov S.; Belousov V. V. (2011) Does cellular hydrogen peroxide diffuse or act locally?. Antioxid. Redox Signaling 14, 1–7. [DOI] [PubMed] [Google Scholar]
- Avshalumov M. V.; Bao L.; Patel J. C.; Rice M. E. (2007) H2O2 signaling in the nigrostriatal dopamine pathway via ATP-sensitive potassium channels: issues and answers. Antioxid. Redox Signaling 9, 219–231. [DOI] [PubMed] [Google Scholar]
- Bernard V.; Bolam. J. P. (1998) Subcellular and subsynaptic distribution of the NR1 subunit of the NMDA receptor in the neostriatum and globus pallidus of the rat: colocalization at synapses with the GluR2/3 subunit of the AMPA receptor. Eur. J. Neurosci. 10, 3721–3738. [DOI] [PubMed] [Google Scholar]
- Bernard V.; Somogyi P.; Bolam J. P. (1997) Cellular, subcellular, and subsynaptic distribution of AMPA-type glutamate receptor subunits in the neostriatum of the rat. J. Neurosci. 17, 819–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q.; Veenman L.; Knopp K.; Yan Z.; Medina L.; Song W. J.; Surmeier D. J.; Reiner A. (1998) Evidence for the preferential localization of glutamate receptor-1 subunits of AMPA receptors to the dendritic spines of medium spiny neurons in rat striatum. Neuroscience 83, 749–761. [DOI] [PubMed] [Google Scholar]
- Stuber G. D.; Hnasko T. S.; Britt J. P.; Edwards R. H.; Bonci A. (2010) Dopaminergic terminals in the nucleus accumbens but not the dorsal striatum corelease glutamate. J. Neurosci. 30, 8229–8233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao L.; Avshalumov M. V.; Patel J. C.; Lee C. R.; Miller E. W.; Chang C. J.; Rice M. E. (2009) Mitochondria are the source of hydrogen peroxide for dynamic brain-cell signaling. J. Neurosci. 29, 9002–9010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandri G.; Panfili E.; Ernster L. (1990) Hydrogen peroxide production by monoamine oxidase in isolated rat-brain mitochondria: its effect on glutathione levels and Ca2+ efflux. Biochim. Biophys. Acta 1035, 300–305. [DOI] [PubMed] [Google Scholar]
- Cohen G.; Farooqui R.; Kesler N. (1997) Parkinson disease: a new link between monoamine oxidase and mitochondrial electron flow. Proc. Natl. Acad. Sci. U.S.A. 94, 4890–4894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt P.; Maxwell G. D.; Pintar J. E. (1985) Specific cellular expression of monoamine oxidase B during early stages of quail embryogenesis. Dev. Biol. 110, 346–361. [DOI] [PubMed] [Google Scholar]
- Zekry D.; Epperson T. K.; Krause K. H. (2003) A role for NOX NADPH oxidases in Alzheimer’s disease and other types of dementia?. IUBMB Life 55, 307–313. [DOI] [PubMed] [Google Scholar]
- Kishida K. T.; Klann E. (2007) Sources and targets of reactive oxygen species in synaptic plasticity and memory. Antioxid. Redox Signaling 9, 233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller E. W.; Tulyathan O.; Isacoff E. Y.; Chang C. J. (2007) Molecular imaging of hydrogen peroxide produced for cell signaling. Nat. Chem. Biol. 3, 263–267. [DOI] [PubMed] [Google Scholar]
- Brennan A. M.; Suh S. W.; Won S. J.; Narasimhan P.; Kauppinen T. M.; Lee H.; Edling Y.; Chan P. H.; Swanson R. A. (2009) NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat. Neurosci. 12, 857–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindokas V. P.; Jordan J.; Lee C. C.; Miller R. J. (1996) Superoxide production in rat hippocampal neurons: selective imaging with hydroethidine. J. Neurosci. 16, 1324–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carriedo S. G.; Sensi S. L.; Yin H. Z.; Weiss J. H. (2000) AMPA exposures induce mitochondrial Ca2+ overload and ROS generation in spinal motor neurons in vitro. J. Neurosci. 20, 240–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafon-Cazal M.; Pietri S.; Culcasi M.; Bockaert J. (1993) NMDA-dependent superoxide production and neurotoxicity. Nature 364, 535–537. [DOI] [PubMed] [Google Scholar]
- Reynolds I. J.; Hastings T. G. (1995) Glutamate induces the production of reactive oxygen species in cultured forebrain neurons following NMDA receptor activation. J. Neurosci. 15, 3318–3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienert G. P.; Schjoerring J. K.; Jahn T. P. (2006) Membrane transport of hydrogen peroxide. Biochim. Biophys. Acta 1758, 994–1003. [DOI] [PubMed] [Google Scholar]
- Bienert G. P.; Møller A. L.; Kristiansen K. A.; Schulz A.; Møller I. M.; Schjoerring J. K.; Jahn T. P. (2007) Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J. Biol. Chem. 282, 1183–1192. [DOI] [PubMed] [Google Scholar]
- Avshalumov M. V.; Rice M. E. (2003) Activation of ATP-sensitive K+ (KATP) channels by H2O2 underlies glutamate-dependent inhibition of striatal dopamine release. Proc. Natl. Acad. Sci. U.S.A. 100, 11729–11734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourre C.; Ben Ari Y.; Bernardi H.; Fosset M.; Lazdunski M. (1989) Antidiabetic sulfonylureas: localization of binding sites in the brain and effects on the hyperpolarization induced by anoxia in hippocampal slices. Brain Res. 486, 159–164. [DOI] [PubMed] [Google Scholar]
- Treherne J. M.; Ashford M. L. (1991) The regional distribution of sulphonylurea binding sites in rat brain. Neuroscience 40, 523–531. [DOI] [PubMed] [Google Scholar]
- Zini S.; Tremblay E.; Pollard H.; Moreau J.; Ben-Ari Y. (1993) Regional distribution of sulfonylurea receptors in the brain of rodent and primate. Neuroscience 55, 1085–1091. [DOI] [PubMed] [Google Scholar]
- Schwanstecher C.; Panten U. (1994) Identification of an ATP-sensitive K+ channel in spiny neurons of rat caudate nucleus. Pflugers Arch. 427, 187–199. [DOI] [PubMed] [Google Scholar]
- Dunn-Meynell A. A.; Routh V. H.; McArdle J. J.; Levin B. E. (1997) Low-affinity sulfonylurea binding sites reside on neuronal cell bodies in the brain. Brain Res. 745, 1–9. [DOI] [PubMed] [Google Scholar]
- Dunn-Meynell A. A.; Rawson N. E.; Levin B. E. (1998) Distribution and phenotype of neurons containing the ATP-sensitive K+ channel in rat brain. Brain Res. 814, 41–54. [DOI] [PubMed] [Google Scholar]
- Patel J. C.; Witkovsky P.; Coetzee W. A.; Rice M. E. (2011) Subsecond regulation of striatal dopamine release by presynaptic KATP channels. J. Neurochem. 118, 721–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols C. G. (2006) KATP channels as molecular sensors of cellular metabolism. Nature 440, 470–476. [DOI] [PubMed] [Google Scholar]
- Ichinari K.; Kakei M.; Matsuoka T.; Nakashima H.; Tanaka H. (1996) Direct activation of the ATP-sensitive potassium channel by oxygen free radicals in guinea-pig ventricular cells: its potentiation by MgADP. J. Mol. Cell. Cardiol. 28, 1867–1877. [DOI] [PubMed] [Google Scholar]
- Tokube K.; Kiyosue T.; Arita M. (1998) Effects of hydroxyl radicals on KATP channels in guinea-pig ventricular myocytes. Pflugers Arch. 437, 155–157. [DOI] [PubMed] [Google Scholar]
- Clement J. P.; Kunjilwar K.; Gonzalez G.; Schwanstecher M.; Panten U.; Aguilar-Bryan L.; Bryan J. (1997) Association and stoichiometry of KATP channel subunits. Neuron 18, 827–838. [DOI] [PubMed] [Google Scholar]
- Shyng S.; Nichols C. G. (1997) Octameric stoichiometry of the KATP channel complex. J. Gen. Physiol. 110, 655–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karschin C.; Ecke C.; Ashcroft F. M.; Karschin A. (1997) Overlapping distribution of K-ATP channel-forming Kir6.2 subunit and the sulfonylurea receptor SUR1 in rodent brain. FEBS Lett. 401, 59–64. [DOI] [PubMed] [Google Scholar]
- Ashcroft F. M.; Gribble F. M. (1998) Correlating structure and function in ATP-sensitive K+ channels. Trends Neurosci. 21, 288–294. [DOI] [PubMed] [Google Scholar]
- Thomzig A.; Wenzel M.; Karschin C.; Eaton M. J.; Skatchkov S. N.; Karschin A.; Veh R. W. (2001) Kir6.1 is the principal pore-forming subunit of astrocyte but not neuronal plasma membrane K-ATP channels. Mol. Cell. Neurosci. 18, 671–690. [DOI] [PubMed] [Google Scholar]
- Babenko A. P.; Aguilar-Bryan L.; Bryan J. (1998) A view of SUR/KIR6.X, KATP channels. Annu. Rev. Physiol. 60, 667–687. [DOI] [PubMed] [Google Scholar]
- Aguilar-Bryan L.; Clement J. P. T.; Gonzalez G.; Kunjilwar K.; Babenko A.; Bryan J. (1998) Toward understanding the assembly and structure of KATP channels. Physiol. Rev. 78, 227–245. [DOI] [PubMed] [Google Scholar]
- Inagaki N.; Gonoi T.; Clement J. P.; Wang C. Z.; Aguilar-Bryan L.; Bryan J.; Seino S. (1996) A family of sulfonylurea receptors determines the pharmacological properties of ATP-sensitive K+ channels. Neuron 16, 1011–1017. [DOI] [PubMed] [Google Scholar]
- Babenko A. P.; Gonzalez G.; Bryan J. (2000) Pharmaco-topology of sulfonylurea receptors. Separate domains of the regulatory subunits of KATP channel isoforms are required for selective interaction with K+ channel openers. J. Biol. Chem. 275, 717–720. [DOI] [PubMed] [Google Scholar]
- Limberger N.; Trout S. J.; Kruk Z. L.; Starke K. (1991) Real time” measurement of endogenous dopamine release during short trains of pulses in slices of rat neostriatum and nucleus accumbens: role of autoinhibition. Naunyn-Schmiedeberg's Arch. Pharmacol. 344, 623–629. [DOI] [PubMed] [Google Scholar]
- Patel J.; Trout S. J.; Kruk Z. L. (1992) Regional differences in evoked dopamine efflux in brain slices of rat anterior and posterior caudate putamen. Naunyn-Schmiedeberg's Arch. Pharmacol. 346, 267–276. [DOI] [PubMed] [Google Scholar]
- Phillips P. E. M.; Hancock P. J.; Stamford J. A. (2002) Time window of autoreceptor-mediated inhibition of limbic and striatal dopamine release. Synapse 44, 15–22. [DOI] [PubMed] [Google Scholar]
- Chen B. T.; Moran K. A.; Avshalumov M. V.; Rice M. E. (2006) Limited regulation of somatodendritic dopamine release by voltage-sensitive Ca2+ channels contrasted with strong regulation of axonal dopamine release. J. Neurochem. 96, 645–655. [DOI] [PubMed] [Google Scholar]
- Zhou F. M.; Liang Y.; Dani J. A. (2001) Endogenous nicotinic cholinergic activity regulates dopamine release in the striatum. Nat. Neurosci. 4, 1224–1229. [DOI] [PubMed] [Google Scholar]
- Rice M. E.; Cragg S. J. (2004) Nicotine amplifies reward-related dopamine signals in striatum. Nat. Neurosci. 7, 583–584. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Sulzer D. (2004) Frequency-dependent modulation of dopamine release by nicotine. Nat. Neurosci. 7, 581–582. [DOI] [PubMed] [Google Scholar]
- Patel J. C.; Rossignol E.; Rice M. E.; Machold R. P. (2012) Opposing regulation of dopaminergic activity and exploratory motor behavior by forebrain and brainstem cholinergic circuits.. Nature Commun. 10.108/ncomms2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomzig A.; Prüss H.; Veh R. W. (2003) The Kir6.1-protein, a pore-forming subunit of ATP-sensitive potassium channels, is prominently expressed by giant cholinergic interneuron in the striatum of the rat brain. Brain Res. 986, 132–138. [DOI] [PubMed] [Google Scholar]
- Moghaddam B.; Gruen R. J.; Roth R. H.; Bunney B. S.; Adams R. N. (1990) Effect of L-glutamate on the release of striatal dopamine: in vivo dialysis and electrochemical studies. Brain Res. 518, 55–60. [DOI] [PubMed] [Google Scholar]
- Westerink B. H.; Santiago M.; De Vries J. B. (1992) In vivo evidence for a concordant response of terminal and dendritic dopamine release during intranigral infusion of drugs. Naunyn Schmied. Arch. Pharmacol. 345, 523–529. [DOI] [PubMed] [Google Scholar]
- Barbour B. (2001) An evaluation of synapse independence. J. Neurosci. 21, 7969–7984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergles D. E.; Diamond J. S.; Jahr C. E. (1999) Clearance of glutamate inside the synapse and beyond. Curr. Opin. Neurobiol. 9, 293–298. [DOI] [PubMed] [Google Scholar]
- Galvan A.; Kuwajima M.; Smith Y. (2006) Glutamate and GABA receptors and transporters in the basal ganglia: what does their subsynaptic localization reveal about their function?. Neuroscience 143, 351–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusakov D. A.; Kullmann D. M.; Stewart M. G. (1999) Hippocampal synapses: do they talk to their neighbours?. Trends Neurosci. 22, 382–388. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Sulzer D. (2003) Glutamate spillover in the striatum depresses dopaminergic transmission by activating group I metabotropic glutamate receptors. J. Neurosci. 23, 10585–10592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiyama F.; Fritschy J. M.; Stephenson F. A.; Bolam J. P. (2000) Synaptic localization of GABAA receptor subunits in the striatum of the rat. J. Comp. Neurol. 416, 158–172. [PubMed] [Google Scholar]
- Szabo B.; Muller T.; Koch H. (1999) Effects of cannabinoids on dopamine release in the corpus striatum and the nucleus accumbens in vitro. J. Neurochem. 73, 1084–1089. [DOI] [PubMed] [Google Scholar]
- Sidló Z.; Reggio P. H.; Rice M. E. (2008) Inhibition of striatal dopamine release by CB1 receptor activation requires nonsynaptic communication via GABA, H2O2, and KATP channels. Neurochem. Int. 52, 80–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheer J. F.; Wassum K. M.; Heien M. L.; Phillips P. E. M.; Wightman R. M. (2004) Cannabinoids enhance subsecond dopamine release in the nucleus accumbens of awake rats. J. Neurosci. 24, 4393–4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiemann J.; Schlaudraff F.; Klose V.; Bingmer M.; Seino S.; Magill P. J.; Zaghloul K. A.; Schneider G.; Liss B.; Roeper J. (2012) K-ATP channels in dopamine substantia nigra neurons control bursting and novelty-induced exploration. Nat. Neurosci. 15, 1272–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao L.; Avshalumov M. V.; Rice M. E. (2005) Partial mitochondrial inhibition causes suppression of striatal dopamine release and depolarization of medium spiny neuron via H2O2 elevation in the absence of ATP depletion. J. Neurosci. 25, 10029–10040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C. R.; Witkovsky P.; Rice M. E. (2011) Regulation of substantia nigra pars reticulata GABAergic neuron activity by H2O2 via flufenamic acid-sensitive channels and K-ATP channels. Front. Syst. Neurosci. 5, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C. R., Machold R. P., Witkovsky P., and Rice. M. E.. TRPM2 channels are required for NMDA-induced burst firing and contribute to H2O2-dependent modulation in substantia nigra pars reticulata GABAergic neurons. J. Neurosci. (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- McTaggart J. S.; Clark R. H.; Ashcroft F. M. (2010) The role of the KATP channel in glucose homeostasis in health and disease: more than meets the islet. J. Physiol. 588, 3201–3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flagg T. P.; Enkvetchakul D.; Koster J. C.; Nichols C. G. (2010) Muscle KATP channels: recent insights to energy sensing and myoprotection. Physiol. Rev. 90, 799–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehage E.; Eisfeld J.; Heiner I.; Jüngling E.; Zitt C.; Lückhoff A. (2002) Activation of the cation channel long transient receptor potential channel 2 (LTRPC2) by hydrogen peroxide. A splice variant reveals a mode of activation independent of ADP-ribose. J. Biol. Chem. 277, 23150–23156. [DOI] [PubMed] [Google Scholar]
- Fleig A.; Penner R. (2004) The TRPM ion channel subfamily: molecular, biophysical and functional features. Trends Pharmacol. Sci. 25, 633–639. [DOI] [PubMed] [Google Scholar]