

Abstract
Phosphocreatine is a major cellular source of high energy phosphates, which is crucial to maintain cell viability under conditions of impaired metabolic states, such as decreased oxygen and energy availability (i.e., ischemia). Many methods exist for the bulk analysis of phosphocreatine and its dephosphorylated product creatine; however, no method exists to image the distribution of creatine or phosphocreatine at the cellular level. In this study, Fourier transform infrared (FTIR) spectroscopic imaging has revealed the ex vivo development of creatine microdeposits in situ in the brain region most affected by the disease, the cerebellum of cerebral malaria (CM) diseased mice; however, such deposits were also observed at significantly lower levels in the brains of control mice and mice with severe malaria. In addition, the number of deposits was observed to increase in a time-dependent manner during dehydration post tissue cutting. This challenges the hypotheses in recent reports of FTIR spectroscopic imaging where creatine microdeposits found in situ within thin sections from epileptic, Alzheimer’s (AD), and amlyoid lateral sclerosis (ALS) diseased brains were proposed to be disease specific markers and/or postulated to contribute to the brain pathogenesis. As such, a detailed investigation was undertaken, which has established that the creatine microdeposits exist as the highly soluble HCl salt or zwitterion and are an ex-vivo tissue processing artifact and, hence, have no effect on disease pathogenesis. They occur as a result of creatine crystallization during dehydration (i.e., air-drying) of thin sections of brain tissue. As ischemia and decreased aerobic (oxidative metabolism) are common to many brain disorders, regions of elevated creatine-to-phosphocreatine ratio are likely to promote crystal formation during tissue dehydration (due to the lower water solubility of creatine relative to phosphocreatine). The results of this study have demonstrated that although the deposits do not occur in vivo, and do not directly play any role in disease pathogenesis, increased levels of creatine deposits within air-dried tissue sections serve as a highly valuable marker for the identification of tissue regions with an altered metabolic status. In this study, the location of crystalline creatine deposits were used to identify whether an altered metabolic state exists within the molecular and granular layers of the cerebellum during CM, which complements the recent discovery of decreased oxygen availability in the brain during this disease.
Keywords: FTIR imaging, cerebral malaria, metabolism, creatine, ischemia, neurodegeneration
The total brain creatine pool (CrT) is the sum of the phosphorylated (CrP) and dephosphorylated (Cr) forms, and serves as an immediate supply of high energy phosphates under ATP depleting conditions.1,2 A decrease in the CrP level, and a corresponding increase in the dephosphorylated product (Cr), is observed in many neurodegenerative disorders in which ischemic conditions (reduced energy and O2 supply) induce cellular energy failure and ATP depletion.1−3 Despite the increase in the Cr:CrP ratio, the CrT content (Cr + CrP) remains constant.2,4 Indeed, it is the stability of the CrT levels under a wide range of neurological conditions that enable this metabolite to be used for normalization of N-acetyl aspartate (NAA) signals in magnetic resonance imaging (MRI) studies.4 Therefore, a method capable of identifying/investigating the Cr to CrP equilibrium, as opposed to total creatine levels, would be highly valuable in studies of changes in this equilibrium as a result of neurodegenerative disorders.
Fourier transform infrared (FTIR) spectroscopic mapping and/or imaging are invaluable methods for studying basic physiology and disease processes, as they measure changes in the distribution and concentration of biological molecules.5−15 Recently, FTIR spectroscopic imaging and mapping has been used to detect microcrystalline deposits of creatine within the hippocampus of mice in rodent models of Alzheimer's disease and epilepsy.5,8,16 Likewise, studies by the same authors have verified the existence of similar deposits postmortem in the brain tissue of human patients who suffered from Alzheimer's disease (AD) or amyotrophic lateral sclerosis (ALS).6,7 In these studies, it was hypothesized that the observations are disease specific and may reflect an increase in the Cr/CrP ratio due to a decrease in tricarboxylic acid (TCA) cycle activity and decreased oxidative metabolism, or a decrease in creatine kinase function during epilepsy, AD, and ALS.5−8,16
Evidence for decreases in both TCA activity and oxidative metabolism are observed in AD and ALS, and in a wide range of other neurological disorders with distinctly different pathologies.17−31 For example, decreased TCA cycle activity and decreased oxidative metabolism is observed in murine and human cerebral malaria (CM) as a consequence of brain ischemia.19−24 In direct contrast to AD pathology, CM predominantly affects children under the age of five in sub-Saharan Africa (approximately one million deaths each year), and the disease has a rapid time-course from onset of clinical symptoms to death (less than 1 week, in both humans and animal models).22,32−37 Previously, there was no evidence for the involvement of altered brain creatine kinase function during CM pathogenesis.22,37−41 Despite these significant differences in pathology, the following results, which employ FTIR spectroscopic imaging for the analysis of cryofixed air-dried tissue sections, have been used to ascertain whether changes in the Cr/CrP ratio is also common to CM.
Results and Discussion
Morphology and Distribution of Creatine Deposits in Cerebellar Tissue
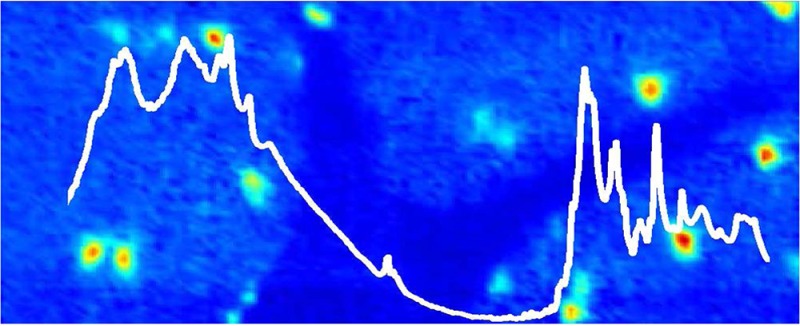
As described by the current authors and other researchers, FTIR spectroscopic functional group images generated from band intensities in the second-derivatives of the spectra reveal chemical contrast characteristic of specific tissue layers.10,11,42,43 In this study, false color functional group images were generated from the second-derivative intensity of the lipid carbonyl ν(C=O) band centered at 1742 cm–1, and the organophosphate νs(PO2–) band centered at 1087 cm–1, and were used to visualize the distribution of the three major tissue layers of the cerebellum (inner white matter, granular layer, and molecular layer). Functional group images generated from the second-derivative intensity of the νs(COO–) band centered at 1402 cm–1, which were attributable to creatine, were used to visualize the location of crystalline creatine microdeposits, as described by other researchers.6−8,16 In the results reported herein, the creatine deposits were observed only in the molecular and granular layers of the cerebellum, and no deposits were observed within the inner white matter, which is consistent with the distribution of cellular creatine receptors and, therefore, the likely distribution of creatine.44 A representative example of images of a crystalline creatine microdeposit observed at the boundary of the granular and molecular tissue layers of a cerebellar tissue section from a control mouse is presented in Figure 1. Although the creatine deposit is clearly evident in both the optical image (Figure 1A) of the unstained section and the false color functional image generated from the second-derivative intensity at 1402 cm–1 (Figure 1E), no visible evidence of the creatine deposit was observed after the tissue section was stained (H&E) for routine histology. In addition, it was noted that spectra collected from the center of the deposit showed a strong additional peak centered at 2780 cm–1, while spectra collected from the periphery of the deposit displayed a distinct shoulder at 1395 cm–1, in addition to the main band at 1402 cm–1.
Figure 1.
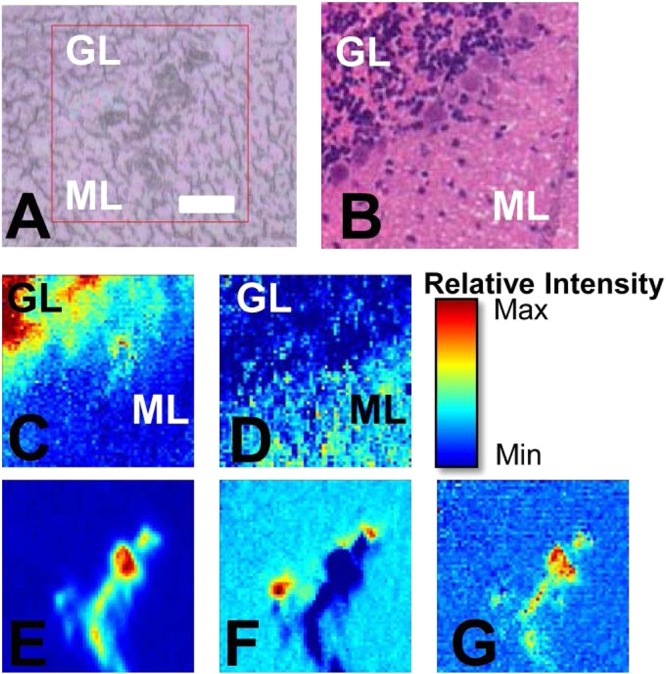
Representative example of a creatine microdeposit in the molecular layer (ML) adjacent to the granular layer (GL) of the murine cerebellum. (A) Optical image of the unstained tissue section and (B) optical image of the H&E stained tissue section (stained after FTIR imaging). (C–G) FTIR functional group images: (C) the νs(PO2–) band centered at 1087 cm–1 shows the distribution of the GL (highest intensity, red) and ML (lowest intensity, blue); (D) the ν(C=O) band centered at 1742 cm–1 shows the distribution of GL (lowest intensity, blue) and ML (highest intensity, red); (E) the νs(COO–) band at 1402 cm–1 shows the crystalline creatine microdeposits; (F) the νs(COO–) band at 1395 cm–1 shows the periphery of the creatine microdeposits; and (G) the band centered at 2780 cm–1. Scale bar = 50 μm.
The creatine microdeposits observed in thin air-dried mouse cerebellum sections in this study (Figure 1) bear a striking resemblance to the deposits previously reported in the hippocampus from animal models of AD and epilepsy, and from postmortem human AD and ALS tissue.6−8,14 Spectra recorded from the deposits observed in this study (Figure 2) show the typical bands of creatine, as previously characterized (with bands centered at 1402 and 1305 cm–1 the most striking), and the relative intensity of the bands compared to surrounding tissue supports a crystalline state.6−8 Following the observation of the additional band centered at 2878 cm–1 in the center of the deposit, and the shoulder centered at 1395 cm–1 in the periphery of the deposit, multivariate analysis (K-means clustering) was performed (Figure 2). The results of a five-group cluster clearly revealed the distribution of the granular and molecular layers, as well as separated the periphery from the center of the creatine deposit (Figure 2B). A thorough investigation of the spectroscopic differences within the cerebellum has already been presented in the literature, and the results of this study support the previous work (Figure 2C, D, and see Supporting Information Figure 1).10,42,43 The average spectra of the molecular layer displayed increased band area and second-derivative intensity for the lipid carbonyl band (1742 cm–1) and decreased band area and second-derivative intensity for the organophosphate band (1087 cm–1) relative to those in the spectra from the granular layer. Visual inspection of the average spectrum for each cluster assigned to the creatine deposit (Figure 2C–H) confirmed that there is a general trend of decreasing second-derivative intensity at 2787 cm–1, and increasing intensity at 1395 cm–1 traveling from the center to the periphery of the creatine deposit. Possible assignments for this band include N–H stretching of the HCl salt, NH3+ stretching of the zwitterion, or a Fermi resonance of the NH3+ group.45,46 Although the exact assignment remains unknown, the appearance of this band indicates the presence of either the creatine zwitterion or HCl salt, both of which are highly soluble. Likewise, the occurrence of an additional band at 1395 cm–1, red-shifted from the main band centered at 1402 cm–1, may be indicative of increased hydration and hydrogen bonding of the creatine (hence, a red-shift to a longer wavelength) around the crystal periphery. Consistent with this observation, the crystals readily dissolved in aqueous PBS washing or fixation media during routine histology (Figure 1B), as has previously been demonstrated.6−8
Figure 2.
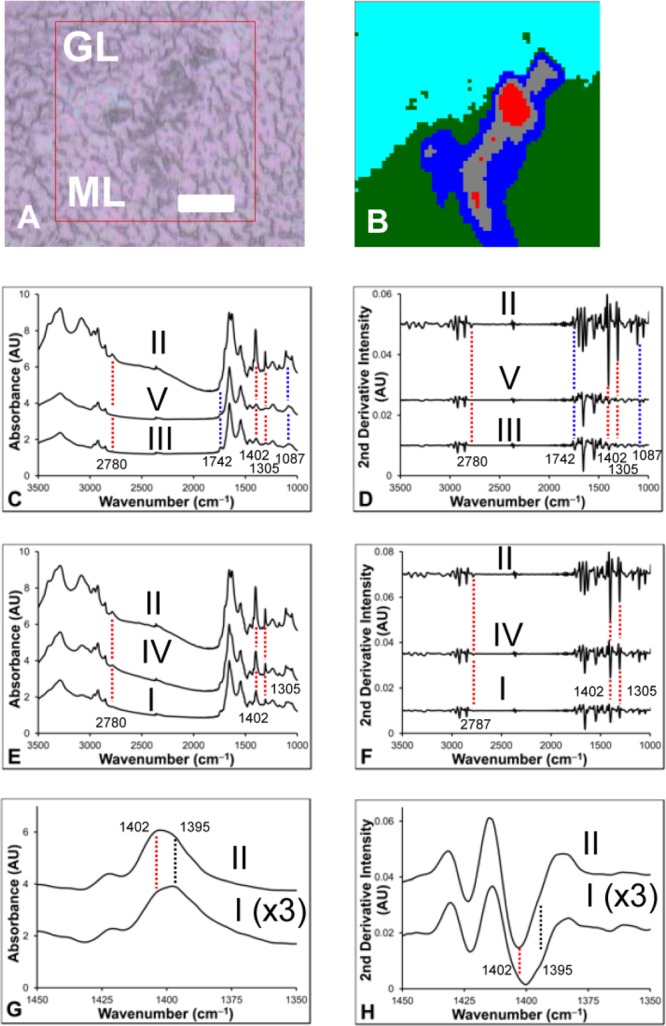
Multivariate analysis (five group K-means cluster) of an FTIR spectroscopic image of a creatine deposit. (A) Optical image. (B) Five-cluster image showing creatine deposit periphery (cluster I, dark blue), creatine deposit center (cluster II, red), molecular layer (cluster III, green), region between center and periphery of the creatine deposit (cluter IV, gray), and granular layer (cluster V, light blue). (C–H) The average spectra (raw, and second-derivative) for each cluster are presented to highlight the spectroscopic differences between the molecular and granular layers of the cerebellum and the creatine deposit (C, D) and within the creatine deposit itself (E–H). Spectra have been offset to clarify the location of spectroscopic differences. The average spectra of cluster I in (G) and (H) has been scaled by a factor of ×3 to highlight the appearance of the shoulder at 1395 cm–1. Blue dashed lines highlight differences between molecular and granular layers. Red and black dashed lines highlight differences within the creatine deposit. Scale bar = 50 μm.
Previous reports of the occurrence of such microdeposits in the hippocampus during epilepsy or AD did not determine the location of the deposits as either intra- or extracellular.6−8,16 The creatine deposits reported herein did not show any correlation to cellular morphology across the molecular or granular layers, as observed by examination of the optical visible light image, FTIR functional group images, K-means cluster images, or histological (H&E) optical images. However, due to the significant difference in cellular layout and density within the molecular and granular layers of the cerebellum, a difference in the morphology of the deposits would be expected if they were intracellular. Therefore, the random distribution and morphology of the deposits across both the molecular and granular layers of the cerebellum can only be produced by an extracellular localization. Interestingly, while focusing on to the deposits with visible light prior to FTIR spectroscopic imaging, it was observed that the focal plane of the deposits was above the focal plane of the tissue section. Again, this suggests that the deposits are not localized within cells, but rather are on the surface of the cut tissue section.
Ex Vivo Formation of Crystalline Creatine Deposits
It has previously been suggested that creatine deposits observed in situ, exist in vivo, and may play a role in disease pathogenesis.6 While the possibility of Cr pooling and crystallization ex vivo has previously been mentioned,8 no previous study has conclusively demonstrated if creatine deposits observed in situ exist in vivo, or if they are an ex vivo artifact of tissue processing and dehydration. To investigate the possibility that creatine deposits are ex vivo, images were collected immediately following cutting of the tissue section (<2 min air-drying) and then again 2 and 4 h later (air-dried 4 h), or after complete dehydration (freeze-dried). In all cases, the appearance of creatine microdesposits was observed to increase with tissue dehydration. A respresentative example is presented in Figure 3, where no creatine deposits were observed in the fresh cut tissue section (<2 min air-drying), but three deposits were apparent after the tissue section was air-dried for 4 h. Spectra collected from the fresh cut tissue (<2 min air-dried) did not display characteristic crystalline creatine bands (i.e., no sharp bands at 1402 or 1305 cm–1); however, spectra collected from the same location after the tissue section was air-dried (4 h) contained these obvious spectroscopic features of crystalline creatine (Figure 4). The increase in size and number of the creatine deposits in a time-course fashion during tissue dehydration provided the first conclusive evidence of ex vivo crystallization of creatine within tissue sections, and proves conclusively that observation of crystalline creatine deposits in situ does not necessarily correlate to their existence in vivo.
Figure 3.
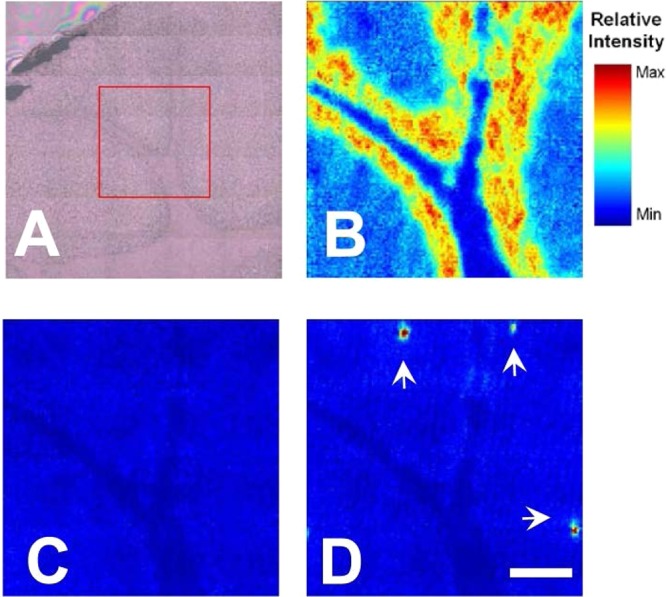
Time-course formation of creatine crystals during tissue dehydration. (A) Optical image of tissue section prior to data collection (no creatine deposits are visible). (B) FTIR functional group image of the νs(PO2–) band at 1087 cm–1 (2 min air-dried), which shows the distribution of the white matter (very dark blue), granular layer (yellow/red), and molecular tissue layers (blue). (C,D) False color FTIR functional group images of the νs(COO–) band at 1402 cm–1, which show the absence of creatine deposits in the 2-min air-dried tissue section (C), and the appearance of creatine deposits at the same location after the tissue section was air-dried for 4 h (D). Images (C) and (D) have been set to the same intensity scale to highlight the intensity increase upon formation of crystalline creatine deposits. White arrows indicate the position of crystalline creatine deposits. Scale bar = 100 μm.
Figure 4.

Representative FTIR spectra (A) raw spectra (normalized) and (B) second-derivative, taken from the same location in a tissue section air-dried for 2 min or 4 h. The significant increase in band intensity at 1402 and 1305 cm–1, characteristic of crystalline creatine, is clearly evident in the tissue section air-dried for 4 h.
In order to further address the possibility that the creatine deposits do not exist in vivo, serial sections of tissue were imaged and the locations of creatine deposits were observed. Despite repeating the experiment for tissue sections of various thicknesses (5, 7, 10, 14, and 20 μm) creatine deposits were never localized in the same region of consecutive tissue sections. If creatine deposits were observed in two adjacent sections, their location varied by 100–1000 μm (data not shown). Therefore, if the deposits were to exist in vivo, they must have a thickness of less than one tissue section (i.e., 5 μm). This seems unlikely considering the X and Y dimensions of the deposits in the plane of the tissue section ranged from 20 to 100 μm. Therefore, the failure to observe the creatine deposits in the same location in adjacent sections, a deposit morphology which reflected an extracellular localization, a different focal plane between the tissue surface, and the surface of the deposits, and the observation of an increase in the size and number of deposits during tissue dehydration, all provided conclusive evidence that the creatine deposits were an ex vivo artifact of tissue dehydration. Further, observation of the band at 2780 cm–1, and evidence for the existence of a hydrophilic zwitterion or HCl salt (both highly hydrophilic), highlights the unlikelihood that such crystals could exist in vivo under hydrated conditions.
In addition, previous studies have reported that the creatine deposits are pigmented.7 Although crystalline creatine is colorless, various impurities within the crystal could account for pigmentation, as proposed previously.7 However, based on the results of this study, an alternative theory is proposed: the dark appearance of the deposits may be simply due to a “shadowing” effect of light reflecting off the numerous irregular edges of the creatine crystals on the surface of the tissue section. Consistent with this theory, the darkest color within the deposits was observed around the periphery of the crystal in this study, and in the previous reports.7
As presented in Figures 1 and 2, although all creatine deposits displayed strong bands that were characteristic of crystalline creatine, closer examination of the second derivatives of the FTIR spectra and functional group images revealed a red-shift of the position of the creatine band centered at 1402–1395 cm–1 in spectra collected from the center relative to the periphery of the microdeposits. As already discussed, it is proposed that the red-shift in band position was a result of increased hydrogen bonding on the periphery of the crystalline deposits, which is consistent with pooling of soluble creatine in tissues postsectioning, followed by a substantial increase in creatine concentration and subsequent crystallization during dehydration. The variations in the crystal morphology and structure (as shown by changes in the position and intensity of the FTIR bands) of creatine due to crystallization across a concentration gradient supported this hypothesis. If so, such a pooling effect must be taken into account when interpreting the location and morphology of crystalline creatine deposits. As the creatine crystals display numerous characteristic bands across the entire mid infrared range, they may be resolved with a spatial resolution of several micrometers. However, due to the pooling effect, the exact location and morphology of the creatine deposits at the micrometer level is most likely effected to a large extent by the morphology of surface of the cut tissue section.
While the creatine deposits may be resolved with micrometer spatial resolution, they most likely did not provide physiological information at this level. More realistically, they revealed the approximate tissue location that contained a pool of elevated Cr to CrP ratio in vivo. Therefore, based on the results of this study, it is recommended that conclusions drawn from the location of crystalline deposits be limited to identify tissue regions (but not cellular or subcellular location) that contained an elevated Cr to CrP ratio in vivo.
CM diseased mice displayed a significant (P < 0.05) increase in the occurrence of deposits in both the granular (P < 0.005) and molecular layers (P < 0.001) relative to control mice (Student's t test). A similar significant increase was observed in the granular (P < 0.005) and molecular (P < 0.001) layers of CM diseased mice relative to SM diseased mice. No significant difference was observed between control and SM diseased mice. No significant difference in the occurrence of deposits between the granular and molecular layers was observed for any of the animal groups.
Occurrence of Creatine Deposits in Control and Malarial Diseased Mice Is Not Disease Specific
In previous reports where creatine deposits were observed during epilepsy, AD, and ALS diseased brain samples, the authors stated that the deposits are most likely disease specific markers of the biochemical pathways contributing to the pathology of the respective diseases.5−8,16 Indeed, in the results reported herein and in the previous work of others, the deposits were observed in the tissue region most affected by the disease (i.e., cerebellum during CM, hippocampus during epilepsy and AD, and brain stem during ALS).6−8,16
Although the formation of creatine deposits may occur through disease-specific chemical pathways, there is no known involvement of creatine kinase during CM; hence, the creatine deposits can obviously be produced by processes that are independent of this pathway.22,37−41
It is crucial to note that the creatine deposits were observed in healthy control tissue in the current study and in studies by other groups.6−8,16 As the deposits have now been observed in samples collected from four different neurodegenerative diseases, and also in healthy control tissue for each of these studies, it is clear that creatine deposits are not unique markers of a disease and it would seem unlikely that a specific chemical pathway is responsible for the formation of the deposits. A more likely cause of the deposits is a generic biochemical event, common to neurodegeneration. In addition, it is known that cerebral ischemia and altered cerebral metabolism occur rapidly (within minutes) of animal sacrifice.48 Therefore, the occurrence of the creatine deposits in healthy control tissue is most likely due to ischemic conditions artificially induced during sacrifice of the animal and removal of the brain.
In this study, creatine deposits were distributed throughout the molecular and granular layers of the cerebellum in control mice, mice suffering noncerebral malaria (severe malarial anemia, SM), and CM-diseased mice.
There was a significant increase in the occurrence of the deposits in the molecular and granular layers of the cerebellum in CM diseased mice relative to control mice (Table 1). There was no increase in the incidence of deposits in the cerebellum of mice suffering SM relative to control mice (Table 1).
Table 1. Occurrence of Crystalline Creatine Microdeposits in Cerebellum Sections Cut from Control Mice, Mice Suffering SM, and Mice Suffering CMa.
| GL,
ML |
|||||||
|---|---|---|---|---|---|---|---|
| animal type | animal 1 | animal 2 | animal 3 | animal 4 | animal 5 | animal 6 | mean |
| control | 0, 1 | 2, 3 | 7, 2 | 1, 0 | 0, 0 | 0, 0 | 2, 1 |
| SM | 0, 1 | 1, 0 | 0, 0 | 0, 0 | 2, 2 | 0, 0 | 1, 1 |
| CM | 19, 11 | 9, 20 | 12, 13 | 4, 11 | 16, 16 | 4, 11 | 11,b 14c |
For each brain, 10 replicate sections were analyzed. The numbers reported (GL = granular layer, ML = molecular layer) are the sum of the creatine deposits observed within the granular and molecular layers for the 10 tissue sections from each animal. No deposits were observed in the inner white matter. The reported mean is rounded to the nearest whole number.
p < 0.005.
p < 0.001.
In CM-diseased mice, there was no correlation between the location or morphology of the deposits and the characteristic histological features of CM (i.e., sequestered cells within blood microvessels, vascular hemorrhage).49,50 However, the aforementioned characteristic features of CM were present in all the analyzed CM diseased tissue sections. Previous analyses of bulk brain homogenates have demonstrated that a metabolic profile characteristic of reduced blood supply to the brain (ischemia) exists during murine CM.19,22−24,37,39−41 Moreover, a recent study provided clear evidence that decreased oxygen availability (hypoxia) is present in the brain during murine CM.47 Therefore, through the use of creatine microdeposits as markers of altered brain metabolism, this study has provided the first demonstration that the characteristic pathological hallmarks of CM (sequestration of immune cells within blood microvessels and microvascular hemorrhage) occur concomitant with an altered brain metabolic profile within the molecular and granular layers of the cerebellum. Due to the importance of these tissue layers for both the efferent and afferent pathways of the granule and purkinje neurons of the cerebellum, altered metabolism in these regions may, in part, account for some of the neurological deficits observed during CM.
Conclusions
In summary, this study has demonstrated that crystalline creatine microdeposits do not exist in vivo, and are an ex vivo postprocessing artifact caused by dehydration of tissue sections. This is a crucial finding and an important consideration for all future studies and the interpretation of crystalline creatine deposits in air-dried cryofixed sections of brain tissue. Despite establishing that the creatine deposits do not exist in vivo, this study has confirmed that the observation of crystalline creatine deposits does reflect an in vivo state of elevated aqueous Cr to CrP ratio. As such, this study, in combination with the previous work of others,6−8,16 validates the use of FTIR spectroscopic mapping and or imaging to screen sections of air-dried, cryofixed brain tissue to identify tissue regions in which altered metabolic status was present in vivo. In this instance, the increased presence of creatine deposits in the cerebellum of CM-diseased mice has been used to provide further evidence that cerebral ischemia is a component of disease pathogenesis, and results in altered brain metabolism within the molecular and granular layers of the cerebellum. To the best of the authors’ knowledge, this is the first such study to successfully image direct biochemical markers of altered metabolism during CM at this level of spatial resolution. In future studies, this approach, in combination with information on lipid and protein oxidation provided by infrared spectra collected from nearby tissue regions, or elemental distributions provided by elemental mapping of the same or adjacent tissue sections, may be a useful tool to identify and study the biochemical alterations that occur in tissue regions most vulnerable to ischemia and altered metabolic status during neurodegeneration.
Methods
Animal Models
In this study, a Plasmodium berghei ANKA (PbA) model of CM was used, with the same experimental parameters that have been described elsewhere.21,34−36,40 The Plasmodium berghei K173 (PbK) model of SM was used as a malaria control, in which malaria parasites are present, but no cerebral complications are observed.21,34−36,40 For each model, a total of 6 animals and 10 tissue sections from each brain were studied. The studies were undertaken with the approval of the University of Sydney Animal Ethics Committee.
Sample Preparation
Blood was drained from the brains of control and CM-diseased mice, and the brains were embedded in optimal cutting temperature (OCT) medium and snap frozen in liquid-nitrogen-cooled hexane, as previously described.42,43 Thin sections (5, 7, 10, 14, and 20 μm thick) of the cerebellum were cut and mounted on low E Kevley slides, and FTIR spectroscopic images were collected after one of the following drying conditions: air-dried and stored on desiccant prior to FTIR spectroscopic imaging analyses; freeze-dried at −80 °C under vacuum conditions following initial FTIR spectroscopic analysis of the air-dried tissue section; or analyzed immediately after tissue sectioning (“fresh” 2 min air-drying) and then reimaged at 2 and 4 h time points postsectioning. Following FTIR imaging, tissue sections were fixed in 4% phosphate buffered formalin solution and stained with hemotxylin and eosin for routine histological analysis as described previously.42,43
FTIR Spectroscopic Imaging
All tissue sections were kept ice cold (4 °C) prior to analysis. FTIR spectroscopic images were collected using a Bruker Vertex 80v FTIR spectrometer, coupled to a Hyperion 3000 microscope. The microscope is equipped with a liquid-nitrogen-cooled 64 × 64 focal plane array (FPA) detector. Opus software (version 6.5, Bruker, Ettlingen, Germany) was used for instrument control and data collection. Spectra were acquired with the coaddition of 64 scans at 4 cm–1 over the range 3600–900 cm–1. Images were collected using a ×15 microscope objective and 2 × 2 pixel binning. A background image was collected from a blank substrate before the collection of each sample image. FTIR spectroscopic images of samples mounted on Kevley slides were collected in reflection (transflection) mode.
Choice of Substrate
It has recently been reported that “transflection” measurements made on reflective substrates, suffer from an electric field standing wave artifact (EFSW).49 It has been demonstrated that this artifact, which was previously assumed to be insignificant is, in fact, a major contributor to the variance observed on FTIR transflection measurements of biological samples.51,52 In this study, it is expected that spectra suffer from this artifact. The choice of low-e Kevley slides and transflection measurements was made to maintain consistency with the previous published research on imaging creatine in brain tissue.6−8,16 Although the spectra may contain the transflection and EFSW artifact, in principle, this should not affect the number of creatine deposits observed in the tissue section through both FTIR and optical microscopy. This study also demonstrated that the spectroscopic features reported in this paper are also observed in FTIR spectroscopic images collected in transmission mode from thin sections mounted on Si3N4 windows (see the Supporting Information), which do not suffer from this artifact and also have the advantage of being able to be used with a large range of microscopies and microspectroscopies.53
Data Analyses
FTIR spectroscopic data were processed (normalized to the area of the amide I band at 1690–1610 cm–1) and functional group images were generated using Cytospec software (Cytospec, version 1.2.04). The time-course FTIR functional group images were adjusted to the same intensity scale, to better highlight the appearance of creatine deposits. Second-derivative spectra were generated in Cytospec and or Opus, with a nine-point Savitsky-Golay smoothing function, and functional group images were generated from the intensity of selected bands within the second-derivatives of the spectra. K-means clusters (5 clusters) were generated from the second-derivatives of the spectra across the amide and fingerprint region (1760–1000 cm–1). A limitation of generating functional group maps is that second-derivative intensity is only proportional to band area when a constant band width is assumed. However, due to the significant effect of scattering artifacts, which predominantly affect raw spectra, second-derivatives of spectra have been utilized in order to minimize the effects of baseline artifacts due to scattering. Although differences in the raw spectra and their second-derivatives were present due to scattering artifacts and variation in band width, due to the drastic chemical difference between brain tissue and crystalline creatine, this did not confound the results for this study. In fact, nearly identical results and identical conclusions were obtained between simple visual observations of the creatine deposits in optical images, the FTIR functional group images generated from raw data, or the FTIR functional group images generated from second-derivative intensity (see Supporting Information Figure 2).
Acknowledgments
The authors also acknowledge the expert assistance of Dr. Jane Radford and the Laboratory of Histopathology at The University of Sydney in sample preparation.
Supporting Information Available
Figures of the average spectra of the molecular and granular layers calculated from the K means cluster; optical and FTIR functional group image of a creatine microdeposit; and the effect of data pretreatment on the visualization of creatine deposits in FTIR functional group images. This material is available free of charge via the Internet at http://pubs.acs.org.
M.J.H. gratefully acknowledges funding from the Australian Government through an Australian Postgraduate Award scholarship and a John A. Lamberton Scholarship from The University of Sydney. The authors gratefully acknowledge funding support from the Australian Research Council, ARC (PAL DP0664706 and DP0984722, including an ARC Professorial Fellowship, GEG Discovery DP0774425, NHH Discovery DP0987074, PAL and EAC LIEF LE0883036), NHMRC (NHH 512469, GEG 464893, and 512101).
The authors declare no competing financial interest.
Supplementary Material
References
- van der Toorn A.; Syková E.; Dijkhuizen R. M.; Vořišek I.; Vargová L.; Škobisová E.; van Lookeren Campagne M.; Reese T.; Nicolay K. (1996) Dynamic changes in water ADC, energy metabolism, extracellular space volume, and tortuosity in neonatal rat brain during global ischemia. Magn. Reson. Med. 36, 52–60. [DOI] [PubMed] [Google Scholar]
- Ames A. (2000) CNS energy metabolism as related to function. Brain Res. Rev. 34, 42–68. [DOI] [PubMed] [Google Scholar]
- Shimizu H.; Graham S. H.; Chang L. H.; Mintorovitch J.; James T. L.; Faden A. I.; Weinstein P. R. (1993) Relationship between extracellular neurotransmitter amino acids and energy metabolism during cerebral ischemia in rats monitored by microdialysis and in vivo magnetic resonance spectroscopy. Brain Res. 605, 33–42. [DOI] [PubMed] [Google Scholar]
- Petroff O. A. C.; Ogino T.; Alger J. R. (1988) High-Resolution Proton Magnetic Resonance Spectroscopy of Rabbit Brain: Regional Metabolite Levels and Postmortem Changes. J. Neurochem. 51, 163–171. [DOI] [PubMed] [Google Scholar]
- Burklen T. S.; Schlattner U.; Homayouni R.; Gough K.; Rak M.; Szeghalmi A.; Wallimann T. (2006) The creatine kinase/creatine connection to Alzheimer’s disease: CK inactivation, APP-CK complexes, and focal creatine deposits. J. Biomed. Biotechnol. 2006, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallant M.; Rak M.; Szeghalmi A.; Del Bigio M. R.; Westaway D.; Yang J.; Julian R.; Gough K. M. (2006) Focally elevated creatine detected in amyloid precursor protein (APP) transgenic mice and alzheimer disease brain tissue. J. Biol. Chem. 281, 5–8. [DOI] [PubMed] [Google Scholar]
- Kastyak M. Z.; Szczerbowska-Boruchowska M.; Adamek D.; Tomik B.; Lankosz M.; Gough K. M. (2010) Pigmented creatine deposits in Amyotrophic Lateral Sclerosis central nervous system tissues identified by synchrotron Fourier Transform Infrared microspectroscopy and X-ray fluorescence spectromicroscopy. Neuroscience 166, 1119–1128. [DOI] [PubMed] [Google Scholar]
- Kuzyk A.; Kastyak M.; Agrawal V.; Gallant M.; Sivakumar G.; Rak M.; Del Bigio M. R.; Westaway D.; Julian R.; Gough K. M. (2010) Association among amyloid plaque, lipid, and creatine in hippocampus of TgCRND8 mouse model for Alzheimer disease. J. Biol. Chem. 285, 31202–31207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chwiej J.; Dulinska J.; Janeczko K.; Dumas P.; Eichert D.; Dudala J.; Setkowicz Z. (2010) Synchrotron FTIR micro-spectroscopy study of the rat hippocampal formation after pilocarpine-evoked seizures. J. Chem. Neuroanat. 40, 140–147. [DOI] [PubMed] [Google Scholar]
- Heraud P.; Caine S.; Campanale N.; Karnezis T.; McNaughton D.; Wood B. R.; Tobin M. J.; Bernard C. C. A. (2010) Early Dectection of the Chemical Changes Occurring During the Induction and Prevention of Autoimmune-mediated Demyelination Detected by FT-IR Imaging. NeuroImage 49, 1180–1189. [DOI] [PubMed] [Google Scholar]
- Kneipp J.; Miller L. M.; Joncic M.; Kittel M.; Lasch P.; Beekes M.; Naumann D. (2003) In situ identification of protein structural changes in prion-infected tissue. Biochim. Biophys. Acta, Mol. Basis Dis. 1639, 152–158. [DOI] [PubMed] [Google Scholar]
- Miller L. M.; Wang Q.; Telivala T. P.; Smith R. J.; Lanzirotti A.; Miklossy J. (2006) Synchrotron-based infrared and X-ray imaging shows focalized accumulation of Cu and Zn co-localized with [beta]-amyloid deposits in Alzheimer’s disease. J. Struct. Biol. 155, 30–37. [DOI] [PubMed] [Google Scholar]
- Leskovjan A. C.; Kretlow A.; Miller L. M. (2010) Fourier Transform Infrared Imaging Showing Reduced Unsaturated Lipid Content in the Hippocampus of a Mouse Model of Alzheimer’s Disease. Anal. Chem. 82, 2711–2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter E. A.; Tam K. K.; Armstrong R.; Lay P. A. (2009) Vibrational Spectroscopic Mapping and Imaging of Tissues and Cells. Biophys. Rev. 1, 95–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aitken J. B.; Carter E. A.; Eastgate H.; Hackett M. J.; Harris H. H.; Levina A.; Lee Y.-C.; Chen C.-I.; Lai B.; Vogt S.; Lay P. A. (2010) Biomedical Applications of X-Ray Absorption and Vibrational Spectroscopic Microscopies in Obtaining Structural Information from Complex Systems. Radiat. Phys. Chem. 79, 176–184. [Google Scholar]
- Dulinska J.; Setkowicz Z.; Janeczko K.; Sandt C.; Dumas P.; Uram L.; Gzielo-Jurek K.; Chwiej J. (2012) Synchrotron radiation Fourier-transform infrared and Raman microspectroscopy study showing an increased frequency of creatine inclusions in the rat hippocampal formation following pilocarpine-induced seizures. Anal. Bioanal. Chem. 402, 2267–2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy A. M.; Frackowiak R. S. J.; Newman S. K.; Bloomfield P. M.; Seaward J.; Roques P.; Lewington G.; Cunningham V. J.; Rossor M. N. (1995) Deficits in cerebral glucose metabolism demonstrated by positron emission tomography in individuals at risk of familial Alzheimer’s disease. Neurosci. Lett. 186, 17–20. [DOI] [PubMed] [Google Scholar]
- Schubert D. (2005) Glucose metabolism and Alzheimer’s disease. Ageing Res. Rev. 4, 240–257. [DOI] [PubMed] [Google Scholar]
- Grau G. E.; de Kossodo S. (1994) Cerebral Malaria: mediators, mechanical obstruction or more?. Parasitol. Today 10, 408–409. [DOI] [PubMed] [Google Scholar]
- Hunt N. H.; Golenser J.; Chan-Ling T.; Parekh S.; Rae C.; Potter S.; Medana I. M.; Miu J.; Ball H. J. (2006) Immunopathogenesis of cerebral malaria. Int. J. Parasitol. 36, 569–582. [DOI] [PubMed] [Google Scholar]
- Hunt N. H.; Grau G. E. (2003) Cytokines: accelerators and brakes in the pathogenesis of cerebral malaria. Trends Immunol. 24, 491–499. [DOI] [PubMed] [Google Scholar]
- Sanni L. A.; Rae C.; Maitland A.; Stocker R.; Hunt N. H. (2001) Is Ischemia Involved in the Pathogenesis of Murine Cerebral Malaria. Am. J. Pathol. 159, 1105–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrel D. A.; White N. J.; Veall N.; Looareesuwan S.; Chanthavanich P.; Phillips R. E.; Karbwang J.; Pongpaew P.; Krishna S. (1988) Cerebral anaerobic glycolysis and reduced cerebral oxygen transport in human cerebral malaria. Lancet 2, 534–538. [DOI] [PubMed] [Google Scholar]
- White N. J.; Looareesuwan S.; Phillips R. E.; Warrell D. A.; Chanthavanich P.; Pongpaew P. (1985) Pathophysiological and prognostic significance of cerebrospinal-fluid lactate in cerebral malaria. Lancet 1, 776–778. [DOI] [PubMed] [Google Scholar]
- Berkley J. A.; Mwangi I.; Mellington F.; Mwarumba S.; Marsh K. (1999) Cerebral malaria versus bacterial meningitis in children with impaired consciousness. Q. J. Med. 92, 151–157. [DOI] [PubMed] [Google Scholar]
- Weber J. R. (2007) Cellular damage in bacterial meningitis: An interplay of bacterial and host driven toxicity. J. Neuroimmunol. 184, 45–52. [DOI] [PubMed] [Google Scholar]
- Quagliarello V.; Scheld W. M. (1992) Bacterial Meningitis: pathogensis, pathophysiology and progress. N. Engl. J. Med. 327, 864–872. [DOI] [PubMed] [Google Scholar]
- Nau R.; Bruck W. (2002) Neuronal injury in bacterial meningitis: mechanisms and implications for therapy. Trends Neurosci. 25, 38–45. [DOI] [PubMed] [Google Scholar]
- DiMauro J.-P. P.; Yang L.; Fuller S. W.; Browne S. E. (2005) Metabolic abnormalities precede pathologic changes in the G93A SOD1 mouse model of ALS. J. Cereb. Blood Flow Metab. S64–S64. [Google Scholar]
- Beal M. F.; Hyman B. T.; Koroshetz W. (1993) Do defects in mitochondrial energy metabolism underlie the pathology of neurodegenerative diseases?. Trends Neurosci. 16, 125–131. [DOI] [PubMed] [Google Scholar]
- Briellmann R. S.; Wellard R. M.; Jackson G. D. (2005) Seizure-associated Abnormalities in Epilepsy: Evidence from MR Imaging. Epilepsia 46, 760–766. [DOI] [PubMed] [Google Scholar]
- WHO. (2009) World Malaria Report 2009, World Health Organisation, Geneva. [Google Scholar]
- Luhan M. (1996) Malaria. WHO Press Office Fact Sheet 94, World Health Organisation, Geneva. [Google Scholar]
- de Souza J. B.; Riley E. M. (2002) Cerebral Malaria: the contribution of studies in animal models to our understanding of immunopathogenesis. Microbes Infect. 4, 291–300. [DOI] [PubMed] [Google Scholar]
- Combes V.; De Souza J. B.; Renie L.; Hunt N. H.; Grau G. E. (2005) Cerebral Malaria: Which parasite? Which model?. Drug Discovery Today: Dis. Models 2, 141–147. [Google Scholar]
- Lou J.; Lucas R.; Grau G. E. (2001) Pathogenesis of cerebral malaria: Recent experimental data and possible applications for humans. Clin. Microbiol. Rev. 14, 810–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh S. B.; Bubb W. A.; Hunt N. H.; Rae C. (2006) Brain metabolic markers reflect susceptibility status in cytokine gene knockout mice with murine cerebral malaria. Int. J. Parasitol. 36, 1409–1418. [DOI] [PubMed] [Google Scholar]
- Miller K. D.; White N. J.; Lott J. A.; Roberts J. M.; Greenwood B. M. (1989) Biochemical Evidence of Muscle Injury in African Children with Severe Malaria. J. Inf. Dis. 159, 139–142. [DOI] [PubMed] [Google Scholar]
- Penet M.; Kober F.; Gouny-Confort S.; Le Fur Y.; Dalmasso C.; Coltel N.; Liprandi A.; Gulian J.; Grau G. E.; Cozzone P. J.; Viola A. (2007) Magnetic Resonance Spectroscopy Reveals an Impaired Brain Metabolic Profile in Mice Resistant to Cerebral Malaria Infected with Plasmodium berghei ANKA. J. Biol. Chem. 282, 14505–14514. [DOI] [PubMed] [Google Scholar]
- Penet M.; Viola A.; Confort-Gouny S.; Le Fur Y.; Duhamel G.; Kober F.; Ibarrola D.; Izquierdo M.; Coltel N.; Gharib B.; Grau G. E.; Cozzone P. J. (2005) Imaging Experimental Cerebral Malaria In Vivo: Significant Role of Ischemic Brain Edema. J. Neurosci. 25, 7352–7358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rae C.; McQuillan J. A.; Parekh S. B.; Bubb W. A.; Weiser S.; Balcar V. J.; Hansen A. M.; Ball H. J.; Hunt N. H. (2004) Brain Gene Expression, Metabolism, and Bioenergetics: Inter-relationships in Murine Models of Cerebral and Noncerebral Malaria. FASEB J. 18, 499–510. [DOI] [PubMed] [Google Scholar]
- Hackett M. J.; Siegele R.; El-Assaad F.; McQuillan J. A.; Aitken J. B.; Carter E. A.; Grau G. E.; Hunt N. H.; Cohen D.; Lay P. A. (2011) Investigation of the mouse cerebellum using STIM and μ-PIXE spectrometric and FTIR spectroscopic mapping and imaging. Nucl. Instrum. Methods Phys. Res., Sect. B 269, 2260–2263. [Google Scholar]
- Hackett M. J.; McQuillan J. A.; El-Assaad F.; Aitkin J. B.; Levina A.; Cohen D.; Siegele R.; Carter E. A.; Grau G. E.; Hunt N. H.; Lay P. A. (2011) Chemical alterations to murine brain tissue induced by formalin fixation: Implications for biospectroscopic imaging and mapping studies of disease pathogenesis. Analyst 136, 2941–2952. [DOI] [PubMed] [Google Scholar]
- Braissant O.; Henry H.; Loup M.; Eilers B.; Bachmann C. (2001) Endogenous synthesis and transport of creatine in the rat brain: an in situ hybridization study. Mol. Brain Res. 86, 193–201. [DOI] [PubMed] [Google Scholar]
- Rozenberg M.; Shoham G.; Reva I.; Fausto R. (2005) A Correlation between the Proton Stretching Vibration Red Shift and the Hydrogen Bond Length in Polycrystalline Amino Acids and Peptides. Phys. Chem. Chem. Phys. 7, 2376–2383. [DOI] [PubMed] [Google Scholar]
- Leifer A.; Lippincott E. R. (1956) The Infrared Spectra of Some Amino Acids. J. Am. Chem. Soc. 79, 5098–5101. [Google Scholar]
- Hempel C.; Combes V.; Hunt N. H.; Kurtzhals J. A. L.; Grau G. E. R. (2011) CNS Hypoxia Is More Pronounced in Murine Cerebral than Noncerebral Malaria and Is Reversed by Erythropoietin. Am. J. Pathol. 179, 1939–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shank R. P.; Aprison M. H. (1971) Post mortem changes in the content and specific radioactivity of several amino acids in four areas of the rat brain. J. Neurobiol. 2, 145–151. [DOI] [PubMed] [Google Scholar]
- Chan-Ling T.; Neill A. L.; Hunt N. H. (1992) Early microvascular changes in murine cerebral malaria detected in retinal wholemounts. Am. J. Pathol. 140, 1121–1130. [PMC free article] [PubMed] [Google Scholar]
- Sein K. K.; Maeno Y.; Thuc H. V.; Anh T. K.; Aikawa M. (1993) Differential sequestration of parasitised erythrocytes in the cerebrum and cerebellum in human cerebral malaria. Am. J. Trop. Med. Hyg. 48, 504–511. [DOI] [PubMed] [Google Scholar]
- Filik J.; Frogley M. D.; Pijanka J. K.; Wehbe K.; Cinque G. (2012) Electric field standing wave artefacts in FTIR micro-spectroscopy of biological materials. Analyst 137, 853–861. [DOI] [PubMed] [Google Scholar]
- Bassan P.; Byrne H. J.; Lee J.; Bonnier F.; Clarke C.; Dumas P.; Gazi E.; Brown M. D.; Clarke N. W.; Gardner P. (2009) Reflection contributions to the dispersion artefact in FTIR spectra of single biological cells. Analyst 134, 1370–1377. [DOI] [PubMed] [Google Scholar]
- Carter E. A.; Rayner B. S.; McLeod A. I.; Wu L. E.; Marshall C. P.; Levina A.; Aitken J. B.; Witting P. K.; Lai B.; Cai Z.; Vogt S.; Lee Y.-C.; Chen C.-I.; Tobin M. J.; Harris H. H.; Lay P. A. (2010) Silicon nitride as a versatile growth substrate for microspectroscopic imaging and mapping of individual cells. Mol. BioSyst. 6, 1316–1322. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.