

Abstract
We previously reported on the synthesis of substituted phenyl-4-hydroxy-1-piperidyl indole analogues with nanomolar affinity at D2 dopamine receptors, ranging from 10- to 100-fold selective for D2 compared to the D3 dopamine receptor subtype. More recently, we evaluated a panel of aripiprazole analogues, identifying several analogues that also exhibit D2 vs D3 dopamine receptor binding selectivity. These studies further characterize the intrinsic efficacy of the compound with the greatest binding selectivity from each chemical class, 1-((5-methoxy-1H-indol-3-yl)methyl)-4-(4-(methylthio)phenyl)piperidin-4-ol (SV 293) and 7-(4-(4-(2-methoxyphenyl)piperazin-1-yl)butoxy)-3,4-dihydroquinolin-2(1H)-one (SV-III-130s), using an adenylyl cyclase inhibition assay, a G-protein-coupled inward-rectifying potassium (GIRK) channel activation assay, and a cell based phospho-MAPK (pERK1/2) assay. SV 293 was found to be a neutral antagonist at D2 dopamine receptors using all three assays. SV-III-130s is a partial agonist using an adenylyl cyclase inhibition assay but an antagonist in the GIRK and phospho ERK1/2 assays. To define the molecular basis for the binding selectivity, the affinity of these two compounds was evaluated using (a) wild type human D2 and D3 receptors and (b) a panel of chimeric D2/D3 dopamine receptors. Computer-assisted modeling techniques were used to dock these compounds to the human D2 and D3 dopamine receptor subtypes. It is hoped that these studies on D2 receptor selective ligands will be useful in the future design of (a) receptor selective ligands used to define the function of D2-like receptor subtypes, (b) novel pharmacotherapeutic agents, and/or (c) in vitro and in vivo imaging agents.
Keywords: Dopamine receptors, binding selectivity, functional selectivity, GPCR structure, D2-like dopamine receptors, GPCR model building, ligand−receptor docking
There are three dopaminergic pathways in the brain: the nigrostriatal pathway, the mesocorticolimbic pathway, and the tuberoinfundibular pathway. These pathways are involved in movement coordination, cognition, emotion, memory, reward, and regulation of prolactin secretion. Alterations in the dopaminergic pathways are thought to be involved in the pathogenesis of neurological, neuropsychiatric, and hormonal disorders.1−6 Modulation of the dopaminergic pathways is also thought to occur as a consequence of acute or chronic abuse of pyschostimulants.7,8
Previous studies have defined two types of dopamine receptors, the D1-like (D1 and D5 subtypes) and D2-like (D2, D3, and D4 subtypes) receptors. D1-like receptors are linked to the activation of adenylyl cyclase via coupling to the Gs/Golf class of G proteins.9 Stimulation of the D2-like receptors results in coupling with the Gi/Go class of G proteins, leading to the inhibition of adenylyl cyclase activity.10,11 Agonist activation of D2-like receptors can also lead to (a) activation of G-protein-coupled inward rectifying potassium (GIRK) channels, (b) stimulation of mitogenesis, (c) an increase in phospholipase D activity, and (d) phosphorylation of ERK1/2.12−16
D2 and D3 dopamine receptors share approximately 46% amino acid homology. However, the transmembrane spanning (TMS) regions, which play a major role in the construction of the orthosteric binding site, share 78% homology.17 Despite similarities in the structural and pharmacological properties of the D2 and D3 receptors, D2 and D3 receptors differ in their (a) neuroanatomical localization,18 (b) trafficking and signal activation,19 and (c) regulation and desensitization.12 D2 or D3 dopamine receptor selective ligands would be useful pharmacologic tools to precisely define the role of these two receptor subtypes in a variety of physiological and behavioral situations. However, due to the high degree of homology, it has been difficult to obtain compounds that can bind selectively to either the D2 or the D3 dopamine receptor subtype.20−23
We recently reported the results of homology model building studies for the human D2 and D3 dopamine receptors.24 Shortly thereafter, X-ray diffraction studies for the human D3 dopamine receptor were reported. Our model for the D3 receptor was found to be in good agreement with the coordinates reported from the X-ray diffraction studies.25 A comparison of the D3 homology model to the crystal structure had a heavy atom RMSC of 2.88 Å comparable to the cystal structure’s reported resolution of 2.89 Å. In our study,24 examples of substituted phenylpiperazines were aligned against the receptor binding conformations, refined using the antagonist haloperidol and three-dimensional quantitative structure activity relationship (3D-QSAR) analyses. These studies indicated that although D2 and D3 receptors share similar folding patterns and 3-D conformations, the slight sequence differences result in the binding cavity of the D2 receptor being comparably shallower than for the D3 receptor. This finding explains, in part, why these extended ligands bind with greater affinity at the D3 receptor compared to the D2 receptor subtype. We concluded that ligands capable of exploiting differences in the contour or topography of the two binding sites were required to obtain D3 versus D2 receptor subtype affinity selective compounds.
Concurrent with those studies, we reported on a series of indoles structurally which (a) have nanomolar affinity at D2 receptors and (b) range from 10- to 100-fold selectivity for D2 compared to the D3 receptor subtype.26 More recently, we synthesized a panel of aripiprazole analogues and identified several compounds with D2 versus D3 dopamine receptor binding selectivity.27
In this report, we investigated the molecular basis for the binding selectivity of two D2 dopamine receptor selective compounds, 1-((5-methoxy-1H-indol-3-yl)methyl)-4-(4-(methylthio)phenyl)piperidin-4-ol (SV 293) and 7-(4-(4-(2-methoxyphenyl)piperazin-1-yl)butoxy)-3,4-dihydroquinolin-2(1H)-one (SV-III-130s). We have (a) further investigated the intrinsic efficacy of these two compounds, (b) compared the affinity of these compounds for both wild type D2 and D3 receptors and a panel of chimeric D2/D3 dopamine receptors, and (c) used computer-assisted modeling techniques to dock these compounds to our models of the D2 and D3 dopamine receptor subtypes24 to further define the molecular basis for the observed receptor subtype binding selectivity.
Results
Background
We previously reported that the radiolabeled benzamide 125I-IABN is a D2-like (D2, D3, and D4) dopamine receptor subtype selective antagonist, although it has similar affinity at the D2 and D3 receptor subtypes.28 We have used this radioligand extensively in our studies to identify D2 and D3 dopamine receptor selective compounds.22,26,27,29 Over the last several years, we identified two different classes of compounds that exhibit D2 versus D3 dopamine receptor subtype binding selectivity. First, SV 293 is a member of a panel of indole substituted phenylpiperadines, structurally related to the butyrophenone haloperidol, that exhibits >100-fold binding selectivity for the D2 receptor compared to the D3 dopamine receptor subtype. We previously reported that two compounds structurally related to SV 293 were antagonists at human D2 dopamine receptors expressed in HEK-293 cells.26 Second, we reported on the synthesis and characterization of a series of derivatives of aripiprazole and showed that several of those compounds also exhibit D2 versus D3 dopamine receptor binding selectivity.27 The most selective compound, designated SV-III-130s, (a) exhibited sub-nanomolar binding affinity at D2 receptors, (b) exhibited 60-fold D2 versus D3 receptor binding selectivity, and (c) was found to be a partial agonist at human D2 dopamine receptors when evaluated for intrinsic efficacy using the adenylyl cyclase inhibition assay.
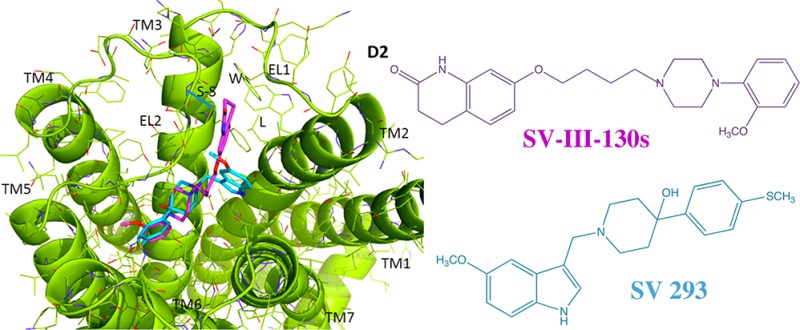
The structures of SV 293 and SV-III-130s are shown in Figure 1. The affinity of these compounds at human D2 and D3 dopamine receptors is presented in Table 1. Table 1 also shows the intrinsic efficacy of these compounds at human D2 receptors using a forskolin-dependent adenylyl cyclase inhibition assay in stably transfected HEK cells. In this assay, the intrinsic activity of our compounds was normalized to the efficacy observed for the full D2 receptor agonist quinpirole, when quinpirole was used at a concentration that was determined to produce maximal effect. A quinpirole dose response curve for the inhibition of forskolin dependent stimulation of adenylyl cyclase in human D2 receptors is shown in Figure 2A.
Figure 1.

Chemical structures of IABN, SV 293 and SV-III-130s.
Table 1. Pharmacological Profile of D2 Dopamine Receptor Selective Compoundsa.
|
Ki values (nM) |
|||
|---|---|---|---|
| compd | D2 (nM) | D3 (nM) | % D2 efficacy |
| SV 293 | 5.50 ± 0.1 (3) | 580 ± 65 (3) | –8.0 ± 4.4 (3) |
| SV-III-130s | 0.22 ± 0.01 (3) | 13.1 ± 2.3 (3) | 61.2 ± 4.4 (3) |
| quinpirole | 4400 ± 724 (3) | 182 ± 29 (3) | 100 |
| haloperidol | 1.1 ± 0.1 (3) | 12.7 ± 3.9 (3) | –0.3 ± 1.7 (43) |
The affinity of the compounds was determined from competitive radioligand binding studies using human D2 or D3 receptors expressed in stably transfected HEK cells using the radioligand 125I-IABN. Ki values are presented in nanomolar units as the mean ± SEM. The number of independent experiments is shown in parentheses. The efficacy is presented as a percent relative to the activity of the full agonist quinpirole (1 μM) using a forskolin-dependent (100 μM) adenylyl cyclase inhibition assay. Binding data for SV 293 and SV-III-130s is taken from refs (26) and (27), respectively. The adenylyl cyclase data for SV-III-130s is taken from ref (27).
Figure 2.

SV 293 inhibits the effect of the full agonist quinpirole at human D2 dopamine receptor using an adenylyl cyclase inhibition assay. (Left) The dose response curve for the inhibition of forskolin-dependent activation of adenylyl cyclase by quinpirole. (Right) (A) Activation of adenylyl cyclase in stably transfected HEK cells expressing human D2long dopamine receptors by forskolin (100 μM) (B) is inhibited approximately 60% using quinpirole (10 nM). (C) SV 293 at a final concentration of 100 nM results in a complete attenuation of the quinpirole-dependent inhibition of adenylyl cyclase activity. Data in the bar graph is presented the mean (n = 3) independent experiments ± SEM.
Intrinsic Efficacy of D2 Receptor Selective Compounds Using an Adenylyl Cyclase Assay
SV 293, as well as the other indole phenylpiperadines that we developed, showed no efficacy when tested alone in the adenylyl cyclase inhibition assay. SV 293 exhibits no intrinsic activity in this assay at a concentration approximately 20-fold higher than the Ki value. To further verify that SV 293 is an antagonist at D2 dopamine receptors, we evaluated the ability of SV 293 to attenuate quinpirole inhibition of cyclase activity. We found that when SV 293 was used at a concentration of approximately 20× its Ki value for D2 receptor, the inhibitory effect of 10 nM quinpirole was diminished (Figure 2B). In a previous publication,27 we reported that SV-III-130s was a partial agonist at D2 dopamine receptors.
Intrinsic Efficacy of D2 Receptor Selective Compounds Using a pERK1/2 Assay
We then evaluated our two D2 receptor selective compounds for efficacy in a phospho-ERK assay. As with the adenylyl cyclase inhibition assay, the efficacy of our compounds was compared to the response observed using the full agonist quinpirole. A quinpirole dose response curve was obtained for the phosphorylation of ERK1/2 using the same stably tranfected HEK cells expressing D2long receptor (Figure 3A) that were used for the adenylyl cyclase assays. First, we found that there was a 100-fold difference between the IC50 value obtained for the inhibition of adenylyl cyclase (Figure 2A) and the EC50 value obtained for ERK1/2 phosphorylation (Figure 3A). Second, when we evaluated both SV 293 and SV-III-130s for D2 receptor-dependent MAPKinase activity, we found that neither compound exhibited intrinsic activity over the concentration range tested, which included doses in excess of 10× the Ki values (Figure 3B). Therefore, we would classify each of the test compounds as antagonists in this functional assay. This finding is consistent with the results from the adenylyl cyclase assay for SV 293, but is inconsistent with the results obtained for SV-III-130s.
Figure 3.

ERK1/2 phosphorylation in human D2long dopamine receptors expressed in stably transfected HEK cells. (A) Quinpirole dose response curve for the phosphorylation of ERK1/2. Each point is the mean ± SEM for n > 3 determinations, where each determination was performed in at least quadriplicate. The data for this experiment was fit to a one site fit model where the curve was constrained to zero (vehicle control) and 100%, where it was assumed that the mean value for the response at a dose of 10–5 Molar quinpirole was the maximum response. For this analysis an EC50 value of 149 nM was obtained. (B) Similar dose responses were performed for SV 293 (●) and SV-III-130s (■). The dose range for the two test ligands included concentrations ≥ 10× the Ki values for binding at D2 dopamine receptors.
Intrinsic Efficacy of D2 Receptor Selective Antagonists Using a GIRK Channel Assay
We then evaluated SV 293 and SV-III-130s for the ability to activate GIRK channels using a cell line stably cotransfected with genes for the human D2long receptor and the GIRK2 channel subunit. As in the previously described cyclase and ERK phosphorylation assays, we began by evaluating the activity of the full agonist quinpirole. A representative profile of the ability of quinpirole to stimulate GIRK channel activity and the resultant dose response curve are shown in Figure 4A and B, respectively. An EC50 value of 38 nM was found for quinpirole activation of D2/GIRK2, which is approximately 25× higher than the EC50 value obtained for the cyclase inhibition assay and 4× lower than the EC50 value obtained for ERK1/2 phosphorylation.
Figure 4.

Activation of GIRK channels in HEK cells stably transfected with human D2long and GIRK2 channel subunit by quinpirole. (A) Representative channel activation profile for quinpirole using D2long/GIRK2 HEK cells. The concentration of quinpirole is presented as nanomolar. (B) Composite dose response curve for the activation of GIRK2 channels (current amplitude) as a function of quinpirole concentration. GIRK current amplitude is normalized to the maximal current (assigned as 100%). Each point is the mean value for n ≥ 3 ± SEM. Curve shown is the best fit of the data to the logistic equation (see Methods). An EC50 value of 38 ± 4 nM was determined with a Hill coefficient of 1.1, indicating a simple one site interaction for channel activation.
We proceeded by examining the effect of SV 293 and SV-III-130s on GIRK2 channels coupled to D2 receptors. We observed that SV 293 was not able to activate GIRK2 channels (Figure 5A) and that it was capable of blocking quinpirole activity (Figure 5B and C). Similar results were observed for SV-III-130s (Figure 6).
Figure 5.

Evaluation of the effect of SV 293 on GIRK channel activation in HEK cells stably transfected with human D2long and GIRK2 channel subunit. (A) Representative channel activation profile for the effect of quinpirole (40 nM) and SV 293 (60 nM) on GIRK2 channel activation. Essentially no activity is observed when SV 293 is applied alone. (B) Representative channel activation profile for the ability of SV 293 (60 nM) to attenuate the effect of the full agonist quinpirole (40 nM). (C) Finally, a bar is shown that summarizes the relative effects of quinipirole (40 nM), SV 293 (60 nM), and the combination of quinpirole and SV 293 on the ability to activate GIRK2 channels in HEK cells expressing dopamine D2long receptors and GIRK2 channels. Values for each bar represent the mean current amplitude relative to quinpirole control value ± SEM for n = 4 independent experiments.
Figure 6.

Evaluation of the effect of SV-III-130s on GIRK channel activation in HEK cells stably transfected with human D2long and GIRK2 channel subunit. (A) Representative channel activation profile for the effect of quinpirole (40 nM) and SV-III-130s (2 nM) on GIRK2 channel activation. Essentially no activity is observed for SV-III-130s. (B) Representative channel activation profile for the ability of SV-III-130s (2 nM) to attenuate the effect of the full agonist quinpirole (40 nM). (C) Finally, a bar graph is shown that summarizes the relative effects of quinipirole (40 nM), SV-III-130s (2 nM), and the combination of quinpirole and SV-III-130s on the ability to activate GIRK2 channels in HEK cells coexpressing the human dopamine D2long receptor. Values for each bar represent the mean of the current amplitude relative to quinpirole control value ± SEM for n = 4 independent experiments.
In summary, we found that SV 293 appears to be an antagonist at D2 dopamine receptors using three different assays: cyclase inhibition, phosphorylation of ERK1/2, and GIRK channel activation. However, SV-III-130s binding to D2 receptors exhibits functional selectivity in that it appears to be a partial agonist in the cyclase assay while acting as an antagonist in the pERK and GIRK assays.
Binding Studies Using Wild Type and Chimeric Receptors
We began to investigate the molecular basis for the dopamine D2 versus D3 receptor subtype binding selectivity of SV 293 and SV-III-130s by determining their binding affinity to a panel of D2/D3 receptor chimeric proteins. A series of D3/D2 dopamine receptor chimeric genes were constructed in which there is a sequential substitution of human D3 dopamine receptor sequence, 5′ to 3′, onto the human D2long receptor gene. The chimeric genes were each completely sequenced to verify that there were no (a) insertions or deletions of amino acids or (b) frame shifts. After construction, each chimeric receptor DNA was used to generate stably transfected HEK-293 cells.
A schematic representation of the initial series of D2/D3 chimeras, chimeras A–F, is shown in Table 2. Direct radioligand binding studies were performed using 125I-IABN to determine the Kd value for the binding of the radioligand to wild type human D2 and D3 receptors, as well as for the chimeric receptors. Competitive radioligand binding studies were subsequently performed to obtain Ki values using the stably transfected cells with (a) 125I-IABN as the radioligand and (b) SV 293 or SV-III-130s as the competitive inhibitor (Table 2).
Table 2. Affinity of D3 Selective Compounds at Human Dopamine D2, D3, and Chimeric Receptorsa.
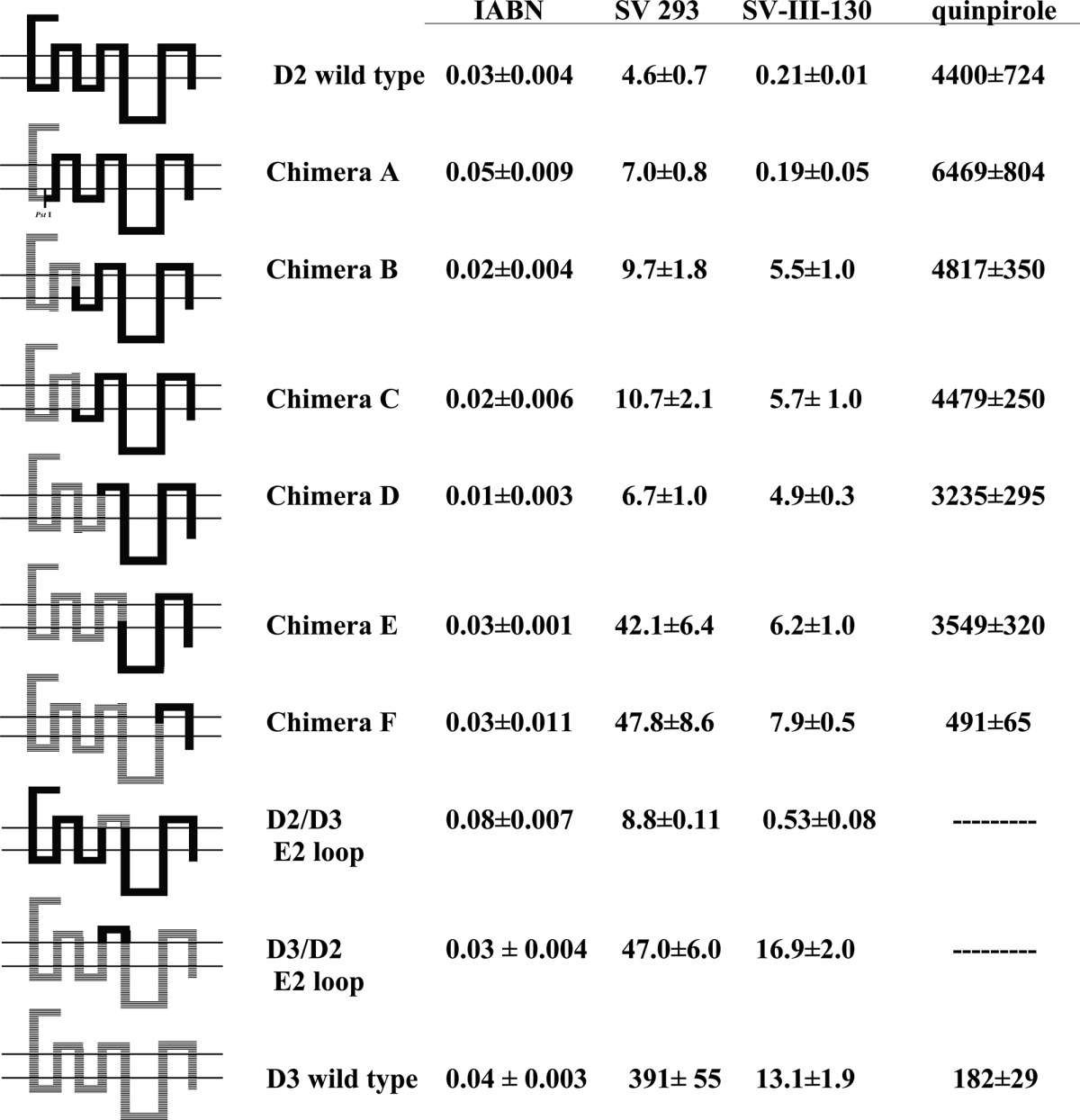
The schematic on the left represents the preparation of the D3/D2 chimeric receptor genes based upon DNA sequence. Because of the high degree of homology within the third helical transmembrane spanning region of the D2 and the D3 dopamine receptor subtypes, chimeras B and C have the same amino acid sequence. All of the dissociation constants are expressed as nanomolar and are the mean ± SEM. For IABN, Kd values were obtained by direct binding studies, and values for SV 293 and SV-III-130s are Ki values obtained from competitive radioligand binding analysis. The number of independent experiments performed to obtain the mean values is n ≥ 3 for the chimeric receptors. For the binding of 125I-IABN to D2 wild type, n = 10, and for the D3 wild type n = 13.
A bar graph representation of the affinity of each wild type and chimeric receptor for SV 293 and quinpirole is shown in Figure 7. We found very little variation (Kd values from 0.01 to 0.05 nM) in the binding affinity of the wild type and chimeric receptors for 125I-IABN (Table 2). However, there was a decrease in the SV 293 affinity with the addition of D3 receptor sequence in the chimeras. As shown in Figure 7, this change in affinity appeared to be quantal (stepwise manner) rather than incremental (gradual). For example, we found a <2-fold difference between the SV 293 affinity for the D2 wild type (4.6 nM) and chimera A (7.0 nM). The SV 293 affinity for chimeras A–D was essentially invariant. The SV 293 affinity for chimeras E and F was approximately 4- to 5-fold lower than that observed for chimeras A–D and ≥4-fold higher than the value found for the wild type D3 receptor. Therefore, there appeared to be two critical break points which influenced SV 293 binding selectivity: (1) the substitution of D3 residues corresponding to the second extracellular loop (E2) and the fifth TMS (TMS V) resulted in the difference in binding for chimeras D and E, and (2) substitution of residues within the extracellular half of the sixth TMS (TMS VI) through to the carboxy terminus were responsible for the difference in affinity observed between chimera F and the wild type D3 receptor subtype.
Figure 7.

Semilog Plot of the affinity of D2 dopamine receptor compounds for D3/D2 chimeric receptors. The log10 values for the affinity of the radioligand 125I-IABN (Kd values) (left) and the D2 selective compounds SV 293 (Ki values) (middle) and SV-III-130s (Ki values) (right) are plotted for the human D2 and D3 dopamine receptor, as well as for the D3/D2 chimeric receptors A–F (see Table 2). The data is presented as the mean value ± the SEM for n ≥ 3 independent experiments.
To further investigate how differences in the primary sequence between the D2 and D3 receptor subtypes might influence the selectivity of SV 293 and SV-III-130s, we also constructed two chimeric receptor genes containing a substitution of the second extracellular (E2) loop: a) D2 receptor with a D3 receptor E2 loop (D2/D3E2) and b) D3 receptor with a D2 receptor E2 loop (D3/D2E2) (Table 2). As with the first panel of chimeric receptors, there appeared to be little effect (<3-fold) of this E2 loop substitution on the binding of IABN. However, there was an 8-fold change in the binding affinity of SV 293 to the wild type D3 receptor subtypes when compared to the corresponding D3/D2 E2 loop substituted chimera. In addition, this change in affinity was in the appropriate direction. No change in affinity was observed between the D3 receptor and the D3/D2 E2 loop construct for SV-III-130s. An approximate 2-fold difference in affinity for both SV 293 and SV-III-130s was observed between the D2 receptor and the D2/D3 E2 loop construct. This result suggests that when SV 293 binds within the helical transmembrane spanning region of the D3 receptor it may also be interacting with the E2 loop.
Molecular Modeling Studies
Our binding studies with the chimeric dopamine receptors suggested that SV 293 and SV-III-130s were exploiting a different array of structural elements within the binding sites of the D2 and D3 receptors to achieve D2 vs D3 receptor binding selectivity. Therefore, computational studies were initiated to attempt to identify which molecular elements might be responsible for the D2/D3 receptor subtype binding selectivity of SV 293 and SV-III-130s.
The human D2 and D3 receptor models have been previously described.24 Docking calculations were carried out using the docking program GOLD, and all the protein residues were treated as rigid. A hydrogen bond constraint between the highly conserved TMS III Asp carbonyl group and the protonated ligand amine (a conserved salt bridge interaction) was specified.
IABN
Our initial studies focused on the binding of the nonselective D2/D3 receptor radioligand IABN. IABN contains a partially rigid bicyclo[3.3.1]nonane group that can adopt multiple conformations. The computer modeling program Macromodel was used for a conformational search of the bicyclo[3.3.1]nonane using automatic setups (MCMM, Monte Carlo) and energy comparisons. Macromodel identified three different energetically stable conformations for the bicyclo[3.3.1]nonane. However, the conformation with both 6-membered rings adopting chair conformations was found to be the most energetically stable. The conformational energies found by Macromodel for the three different conformations were (a) chair–chair, 46.48 kJ/mol; (b) chair–boat, 66.25 kJ/mol; and (c) boat–boat, 96.10 kJ/mol. Since there appeared to be a substantial energy advantage to adopting the chair–chair conformation, this conformation was used as the starting confirmation. The substituent groups were added at the ring terminals to create the final structure of IABN. After examining different combinations of the substituent positions, a total of eight different diastereomers were generated. The remaining fully rotatable bonds were assumed to be flexible. The prerequisite for the binding model included the formation of a salt bridge between the ligand protonated nitrogen and the highly conserved Asp in TMS III of both receptors. The orientation of IABN within the two binding sites was found to be essentially the same for both receptor subtypes (Figure 8), which is consistent with the experimental binding data indicating that IABN is a nonselective ligand for D2 and D3 receptors.
Figure 8.
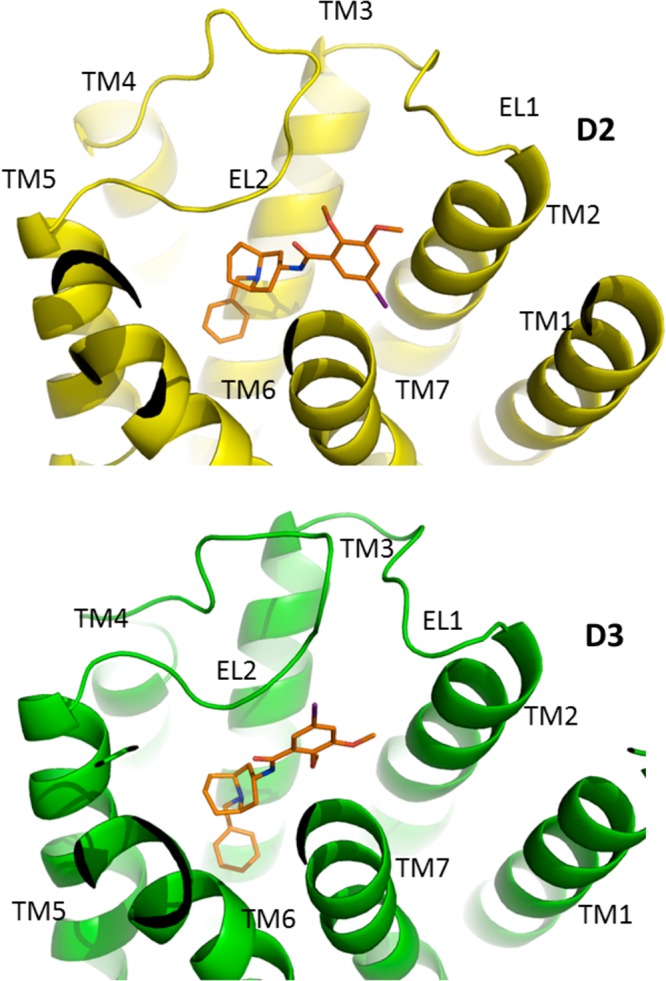
Binding of IABN at the human D2 and D3 dopamine receptor subtypes. IABN is shown in a stick model format with the carbon atoms in orange, nitrogen in blue, and iodine in purple. The receptors are shown in cartoon representation with the transmembrane helices and the first two extracellular loops (EL1 and EL2) labeled.
SV 293 versus SV-III-130s
Molecular modeling/docking techniques were then used to investigate the molecular parameters that might be responsible for the selective binding of SV 293 (27) (Figures 9–11). Since SV-III-130s has a longer saturated carbon chain than SV 293 (4 carbons vs 2 carbons), it extends toward the cleft between the first and second extracellular loops (E1 and E2, respectively). In contrast, this cleft is much smaller in the D3 receptor. Since the binding site is also deeper in the D3 receptor,24 SV-III-130s binds to D3 with the quinolinone moiety oriented toward TMS VII and adopts flexible conformations.
Figure 9.

Binding of SV 293 and SV-III-130s at the human D2 and D3 dopamine receptor subtypes. SV 293 and SV-III-130s are shown in a stick model format, with the carbon atoms in cyan for SV 293 and magenta for SV-III-130s. (Left) The receptors are shown in cartoon representation with the D2 (top) receptor subtype in yellow and D3 (bottom) subtype in green. (Right) The receptors are shown as surface models.
Figure 11.
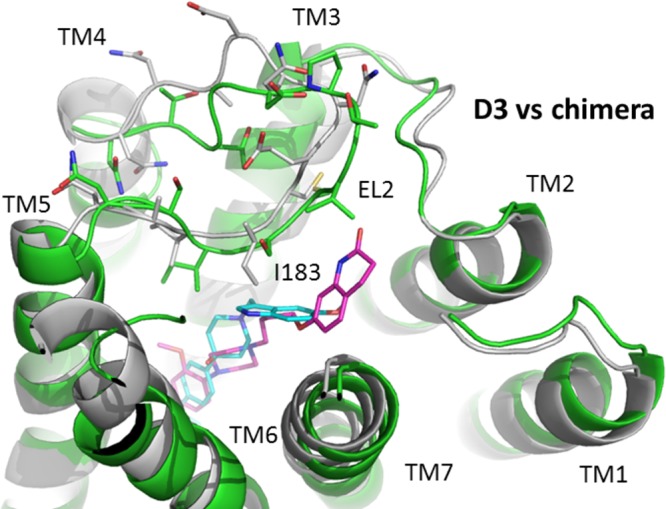
Comparison of ligand binding at the D3/D2 E2 loop chimeric receptor and the wild type D3 dopamine receptor. The binding of SV 293 (cyan) and SV-III-130s (magenta) at the D3 receptor, in alignment with the D3/D2E2 loop chimera, is shown. The receptors are shown in cartoon representation with D3 in green and chimera in gray. The side chains of the second extracellular (EL2) loops in both receptors are shown in stick model, with important structural features noted.
It is likely that at the D2 receptor E1/E2 cleft the quinolinone moiety makes aromatic/hydrophobic interactions with residues within the TMS II/EL1 region of the D2 receptor, including amino acid residue (a) L94 in TMS II, (b) W100 in E1, and (c) the disulfide bridge between C182 in EL2 and C107 in TMS III (Figure 10). Our computational studies indicate that SV 293 does not interact with this portion of the D2 receptor.
Figure 10.
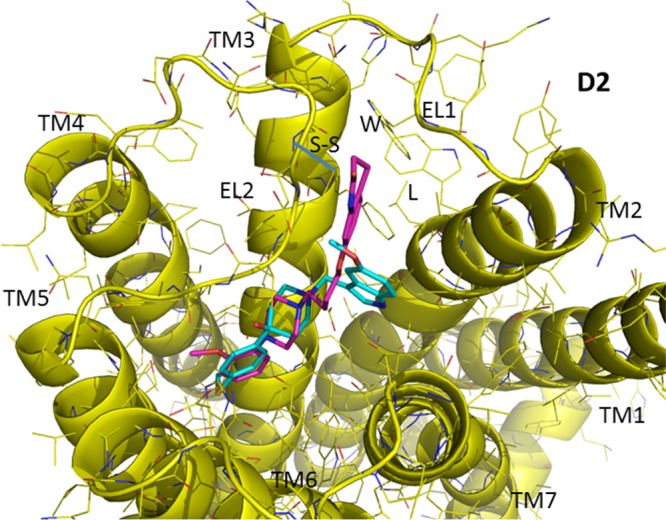
View of the ligand binding at the human D2 dopamine receptor. SV 293 and SV-III-130s are shown in a stick model format, with the carbon atoms in cyan for SV 293 and magenta for SV-III-130s. Several potential contact amino acid residues for the quinolinone moiety of SV-III-130s in the D2 E1/E2 cleft are labeled, including, L94 in TMS II, W100 in E1, and the disulfide bridge between C182 in EL2 and C107 in TMS III. Residue numbering is based upon the sequence of the human D2 dopamine receptor.
Experimental evidence is provided indicating that the E2 loop contributes to the D2 receptor subtype selectivity observed for compound SV 293 (Table 2). The magnitude of the difference in affinity between wild type receptors and chimeric receptors, in which the E2 loops are interchanged, was 4-fold. In addition, for both of the E2 loop chimeras, the affinity changed in the appropriate direction. To further investigate this issue, a model for the D3/D2E2 loop receptor chimera protein was constructed (Figure 11). Not only does the sequence of two E2 loops differ substantially between D2 (NNADQNECIIAN) and D3 (NTTGDPTVCSISN) receptors, the D2 E2 loop is one amino acid residue shorter than the D3 E2 loop. As a result, the D2 E2 loop likely adopts a different conformation in the two receptor models (D2 receptor vs the D3/D2 E2loop chimera). In the D3/D2 E2 loop chimeric receptor model, residues I183 and I184 (within the D2 E2 loop) are likely posed deeper inside the binding pocket because of the rigidity of the disulfide bond formed between TMS III C103 (D3 receptor numbering) and E2 loop C182 (D2 receptor numbering). This is especially true for residue I183 because it is oriented toward the indole moiety of SV 293 (assuming the binding mode of SV 293 is the same as when it is docked to the wild type D3 receptor) (Figure 11). The interaction between SV 293 and residue I183 within the D2 E2 loop of the D3/D2 E2 loop chimera likely involves interactions that are not possible when SV 293 binds to the D3 receptor wild type.
For SV-III-130s, these interactions appear to be weaker because this ligand and residue I183 would be further apart. Therefore, our modeling studies suggest that the substitution of the D2 E2 loop into the D3 receptor is responsible for the observed 8-fold increase in the binding affinity of SV 293 to the D3/D2 E2 loop chimera compared to the wild type D3 receptor. However, this change in amino acid sequence seems to have little to no effect upon the affinity of SV-III-130s for either the (a) D3 wild type receptor or (b) D3/D2 E2 loop chimeric receptor (Table 2).
Discussion
After the cloning and expression of the D3 dopamine receptor gene, it was recognized that there was a high degree of amino acid sequence homology between the D2 and D3 receptor subtypes and that the pharmacologic properties of these two receptor subtypes are quite similar.17,30 Identification of compounds that bind selectively to either the D2 or D3 dopamine receptor has been a difficult task because of this high degree of sequence homology, especially within the helical transmembrane spanning regions.20 However, we have reported studies on the development of both D226,27 and D3 dopamine receptor subtype selective compounds, with varying intrinsic activity.22,31
Development of D2 and D3 receptor subtype selective compounds would provide the pharmacological tools to enable the neuroscience community to better understand the role of these two receptor subtypes in complex behavioral and physiological processes. Furthermore, D2-like receptor subtype selective compounds of varying intrinsic activity have the potential for development of (a) pharmacotherapeutic agents for the treatment of neurological disorders,6 neuropsychiatric disease,32 and psychostimulant abuse,33 and (b) neuroimaging agents to study the differential expression and regulation of D2-like dopamine receptors.34,35
We initially assumed that ligand binding selectivity would be achieved by developing compounds capable of exploiting differences in contact residues between these two structurally related receptor subtypes. However, the results of early receptor modeling studies,36,37 as well as our site-directed mutagenesis studies (unpublished data), suggest that D2 versus D3 receptor subtype binding selectivity is likely due to differences in secondary structure, rather than direct effects on ligand subtype-specific residue interactions. In this hypothesis, the majority of contact residues are conserved residues in both the D2 and D3 receptor subtypes, while subtle changes in the topography/contour of the two binding sites leads to differences in binding affinity at D2 and D3 receptors.
The crystal structure of the human dopamine D3 receptor–T4 lysozyme in complex with the antagonist eticlopride was recently reported.25 As expected, these studies confirm that the D3 receptor consists of the seven transmembrane spanning bundle of helices, which resembles previously solved GPCR structures.38−42 The coordinates of the D3 receptor determined by X-ray diffraction were independently found by the Community-Wide GPCR Dock 2010 Assessment to be in excellent agreement with our model of the D3 receptor,43 which was a homology model constructed prior to the publication of the X-ray diffraction D3 receptor model. This validation provides confidence in the model building strategies for our model of the D2 receptor.
Our computational molecular modeling studies appear to be consistent with the receptor chimera binding data. First, for SV-III-130s there is a transition point between Chimera A and Chimera B. The structural difference between these two chimeric receptors involves the substitution of TMS regions II and III. The modeling studies identify these two TMS regions as containing important contact residues within the D2 E1/E2 cleft for the quinolinone moiety of SV-III-130s. No comparable binding transition point is found for SV 293 because this ligand is smaller and does not extend into this region. Second, the major transition point for SV 293 binding occurs between Chimeras D and E. The structural difference between these two chimeric receptors involves the substitution of the E2 loop and TMS region V. In our initial modeling studies,24 we proposed that the binding cavity of the D2 receptor was shallower than that of the D3 receptor. It is likely that the substitution of TMS V causes the Chimera E to transition to a deeper (more D3-like) binding cavity, thereby changing the interaction between the ligand and the initial contact residues. A second transition is observed between Chimera F and the wild type D3 receptor. The structural difference between these two receptors involves the substitution of the third extracellular loop (E3), part of TMS region VI and TMS VII. Since our modeling studies indicate very little direct interaction of these structural elements with SV 293, we propose that there is a second topographical transition that occurs, where the depth of the binding cavity is equivalent to that found for the wild type D3 receptor.
Experimental evidence is provided indicating that the E2 loop contributes to the D2 receptor subtype selectivity observed for compound SV 293 (Table 2). The magnitude of the difference in affinity between wild type receptors and chimeric receptors, in which the E2 loops are interchanged, was 4-fold. In addition, for both of the E2 loop chimeras, the affinity changed in the appropriate direction. A contribution of the E2 loop to ligand binding is consistent with studies suggesting the anchoring of the E2 loop to TMS III may impose a structural constraint at the entrance of the ligand binding site, thereby changing the kinetics and/or affinity of ligand binding.44,45 The E2 loop is highly divergent among GPCRs and between structurally homologous subtypes. While it is not likely playing a major role in selectivity of the endogenous bioamines, contributions to binding selectivity might be achieved by ligands capable of interacting with E2 loop residues. SV 293 may be an example of such a ligand.
Finally, the results of our signaling studies using the forskolin-dependent adenylyl cyclase inhibition assay indicate that both IABN28 and SV 29326 are neutral antagonists at D2 receptors, whereas SV-III-130s was found to be a partial agonist.27 However, both SV 293 and SV-III-130s were found to be antagonists when the phosphorylation of ERK1/2 and coupling to GIRK channels was evaluated. This observation suggests a functional selectivity for SV-III-130s.31,46−49
In conclusion, a combination of molecular pharmacologic, molecular genetic, and molecular modeling strategies have been employed to begin to explore the molecular basis for the D2 versus D3 dopamine receptor subtype binding selectivity of SV 293 and SV-III-130s. The current status of GPCR modeling and docking, as reflected by the Community-Wide GPCR Dock 2010 Assessment, indicates that computational techniques can be used to accurately predicted the structure of GPCRs when closely related templates are available.24,43 When experimental biophysical, pharmacological, and QSAR information is also incorporated into the model building process, a more accurate and precise picture of how GPCR receptors function can be achieved. These receptor models will need to be developed in the context of theories describing a multistate model of GPCR activation and functionally selective ligands.
It is hoped that the insights gained from these structural and functional studies, using ligands that exhibit binding and functional selectivity for structurally and pharmacologically related receptors subtypes, will be useful in future computer-assisted designed of novel (a) receptor subtype selective ligands that can be used to define the function of structurally related receptor subtypes, including the D2 and D3 dopamine receptors, (b) novel pharmacotherapeutic agents, and (c) in vitro and in vivo imaging agents.
Methods
Tissue Culture
Transfected HEK-293 cells were grown in DMEM media with 10% bovine calf serum in the presence of the appropriate antibiotic. Cells were grown in a humidified 5% CO2 incubator at 37 °C. HEK-293 cells were transfected with pIRES plasmid constructs that were introduced into the cells using lipofectin (Gibco/Life Sciences, Grand Island, NY) or Fugene (Roche, Nutley, NJ), and stably transfected cells were obtained by selection using G418. This vector contains an internal ribosomal entry site (IRES) of the encephalomyocarditis virus which permits the translation of two open reading frames (inserted gene of interest and neomycin resistance) from one mRNA. The translation of the receptor gene and the antibiotic gene are closely linked, thereby reducing the probability of recombination/deletion events with a high proportion of the surviving cells stably expressing the gene of interest following selection with G418 (400 μg/mL).
Radioligand Preparation
Peracetic acid was used to radio-iodinate 125I-IABN, and product was purified using a PRP-1 reverse phase HPLC column for use in the radioligand binding experiment with D2-like dopamine receptors, as previously described.28
Dopamine Receptor Binding Assays
A filtration binding assay was used to characterize the binding properties of the D2, D3, and chimeric D2/D3 dopamine receptors. Direct binding and competition curves were performed using 125I-IABN with dopamine receptors stably expressed in HEK 293 cells. Stably transfected cells were harvested by centrifugation, and the cell pellet was resuspended in cold (4 °C) homogenization buffer (50 mM Tris-HCl, pH 7.4, with 10 mM EDTA, 150 mM NaCl) by vortexing and then homogenizing with a Polytron instrument (Brinkmann Instruments, Westbury, NY). The homogenate was centrifuged at 12 000g at 4 °C, and the membrane pellet resuspended in buffer and kept at −80 °C. Tissue homogenates (50 μL) were suspended in 50 mM Tris-HCl/150 mM NaCl/10 mM EDTA buffer, pH 7.5 and incubated with 50 μL of 125I-IABN at 37 °C for 60 min. Nonspecific binding was defined using 2 μM (+)-butaclamol. For direct binding experiments, the concentration of radioligand ranged from approximately 2-fold below to 5-fold higher than the equilibrium dissociation constant (Kd) value with the receptor concentration adjusted to bind ≤10% of the ligand. For competition experiments, the radioligand concentration was generally equal to the Kd value and the concentration of the competitive inhibitor ranged over 5 orders of magnitude. Binding was terminated by addition of cold wash buffer (10 mM Tris-HCl/150 mM NaCl, pH 7.5) and filtration over a glass-fiber filter (Whatman No. 32, Piscataway, NJ). Filters were washed, and the radioactivity was measured using a Packard gamma counter with an efficiency of 75%. The protein concentration of the membranes was determined using a BCA reagent (Pierce, Rockford, IL) and BSA as the protein standard.
Estimates of the Kd and maximum binding sites (Bmax) were obtained using unweighted linear regression analysis of data.50 Data from competitive inhibition experiments were modeled using nonlinear regression analysis to determine the concentration of inhibitor that inhibits 50% of the specific binding of the radioligand (IC50 value). Since transfected cells expressing receptor were used for this study, competition curves will be modeled for a single site using
where B is the amount of ligand bound to tissue, B0 is the amount of ligand bound in the absence of competitive inhibitor, L is the concentration of the competitive inhibitor, Bns is the nonspecific binding of the radioligand (defined using a high concentration of a structurally dissimilar competitive inhibitor), and IC50 is the concentration of competitive inhibitor that inhibits 50% of the total specific binding. Data from competition dose response curves was analyzed using Tablecurve program (Jandel/Systat Software Inc., San Jose, CA). IC50 values were converted to equilibrium dissociation constants (Ki values).51
Adenylyl Cyclase Assays and Data Analysis
Whole cell cyclic AMP accumulation was measured by an adaptation of the method of Shimizu and co-workers.52 Transfected HEK-293 cells were treated with serum-free medium containing 2,8-3H-adenine (ICN), and cells were incubated at 37 °C for 75 min. The media was replaced with serum-free media containing 0.1 mM 3-isobutyl-1-methylxanthine (Sigma-Aldrich, St. Louis, MO) and drugs to a total volume of 500 uL and incubated at 37 °C for 20 min. The reaction was stopped by addition of 500 μL of 10% trichloroacetic acid and 1 mM cyclic AMP. After centrifugation, the supernatants were fractionated using Dowex AG1-X8 and neutral alumina to separate the 3H-ATP and the 3H-cyclic AMP. Individual samples were corrected for column recovery by monitoring the recovery of the cyclic AMP using spectrophotometric analysis at OD 259 nm.
Cell Based Phospho-MAPK (pERK1/2) Assay
The same stably transfected HEK cells expressing either the human D2 or D3 dopamine receptor gene that were used for the cyclase studies were also used for the ERK 1/2 phosphorylation studies. The formation of p-ERK 1/2 was monitored using an AlphaScreen SureFire assay (Perkin-Elmer, Waltham, MA). Cells are plated at a density of 80 000 cells/well in a 96-well plate. After 20 h in culture (DMEM with 0.5% fetal calf serum), cells were stimulated with the full agonist quinpirole or test compound for 5 min at 37 °C. After addition of lysis buffer (room temp 2 h and −20 °C overnight), supernates (4 μL) were added to the reader plate containing acceptor beads for 1 h at room temp. After addition of donor beads (room temp, overnight), plates were read using an EnSpire Alpha 2390 Multilabel Reader (Perkin-Elmer, Waltham, MA).
GIRK Activation Electrophysiology Assay
Agonist activation of G-protein-coupled inward-rectifying potassium (GIRK) channel currents was measured using a whole-cell patch clamp technique.12 Whole-cell patch recordings of GIRK currents were made at room temperature (22–25 °C) at a holding potential of −70 mV. Patch pipettes of borosilicate glass (M1B150F, World Precision Instruments, Inc., Sarasota, FL) were pulled (Flaming/Brown, P-87/PC, Sutter Instrument Co., Novato, CA) to a tip resistance of 7–8 MΩ. The pipet solution contained (in mM) the following: 130 KCl, 20 NaCl, 1 EGTA, 10 HEPES, 10 d-glucose, 5 Mg-ATP; 1 MgCl2, 0.1 Na3-GTP, pH 7.2. The HEK-293 cells were superfused (7–10 mL/min) with extracellular solution containing (in mM) the following: 120 NaCl, 30 KCl, 1 MgCl2, 2 CaCl2, 10 HEPES, 10 d-glucose, pH 7.3. Quinpirole with or without testing compound was prepared in extracellular solution and was applied to cells via gravity flow using a Y-shaped tube positioned near the target cell. GIRK currents from the whole-cell configuration were obtained using a patch clamp amplifier (Axopatch 200B, Molecular Devices, Sunnyvale, CA) equipped with a CV203BU headstage. The currents were low-pass filtered at 5 kHz, monitored on an oscilloscope and a chart recorder (Gould TA240), and stored on a computer (pClamp 9.0, Molecular Devices, Sunnyvale, CA) for subsequent analysis. Since there was a possibility that access resistance changed over time or during different experimental conditions, at the initiation of each recording the current response, a 5 mV voltage pulse, was measured and stored on a digital oscilloscope. This stored trace was continually referenced throughout the recording. If a change in access resistance was observed throughout the recording period, the patch was aborted and the data were not included in the analysis. Quinpirole (Sigma-Aldrich, St. Louis, MO) and testing compounds were prepared in dimethyl sulfoxide (DMSO). The final concentration of DMSO was <0.05% (v/v).
For the concentration–response analysis, peak GIRK currents induced by quinpirole were normalized to the initial response (100%). Quinpirole (QP) concentration–response profiles were fitted to the following equation using Origin 5.0 (Microcal Software Inc., Northampton, MA):
where I and Imax represent the normalized quinpirole-activated current at a given concentration and the maximum current induced by a saturating concentration of quinpirole, respectively, EC50 is the half-maximal effective quinpirole concentration, and n is the slope factor. All of the patch clamp data are presented as means ± SEM, and Student’s t test (paired or unpaired) was used to determine statistical significance (p < 0.05).
Generation of Chimeric Receptors
Four methods were utilized to construct the receptor chimeras discussed in this paper. The first method exploits a unique Pst1 restriction site in the first intracellular loop (I1) that is common to both the human D2 and human D3 receptor cDNAs. Digested fragments were gel purified, ligated into the pIRESneo2 expression vector, and transformed into competent DH5-alpha E. coli cells. Constructs were sequenced to verify the authenticity of the chimeras and then transfected into HEK-293 cells. The second method involved making chimeric D2/D3 receptors.53 A human D3 receptor cDNA and a human D2 receptor cDNA were cloned in tandem into the pIRESneo2 vector with a unique restriction site (Hpa1) located between the two receptor cDNAs. The construct was digested at this site, and the linear construct was transformed into competent cells. DNA was prepared from individual colonies and sequenced to verify the chimeric gene formation and then transfected into HEK-293 cells for expression. A third method was devised to create chimeras that previous methods had not produced, specifically with junctions at the helical TMS regions TMS IV and TMS V. Site-directed mutagenesis (Quick-Change Site-Directed Mutagenesis Kit, Stratagene/Agilent Technologies, Santa Clara, CA) was performed on D2 and D3 clones in pGEM7Z to create a unique restriction site to both cDNAs at the two different TMS regions (Xho1 at TMS IV and Xba1 at TMS V). Clones were then digested with the appropriate enzyme, and fragments were gel purified and ligated to create chimeras that transitioned within TMS IV and TMS V. Site-directed mutagenesis was performed on these new chimeras to delete the previously added restriction sites, and the DNA sequence was verified for each mutant. Chimeric receptors were then cloned into the pIRES vector and were transfected into HEK-293 cells. Expression of the receptor construct was verified using a radioligand binding assay. Finally, the D2/D3E2 and D3/D2E2 receptor loop chimeras were prepared using the Quick Change Kit strategy with synthetic oligonucleotides encoding the E2 loop with the appropriate 5′ and 3′ flanking regions. Both wild type receptor genes were in the pIRES expression vector (Clontech, Mountain View, CA). The size of the oligonucleotide for preparation of the D2/D3E2 chimera was 69 bases and 66 bases for the D3/D2E2 loop. The chimeric receptors were transfected into HEK-293 cells. The authenticity of the chimeric receptor was verified by DNA sequencing and the expression of the receptor construct in HEK-293 cells was verified by radioligand binding using 125I-IABN.
Molecular Modeling and Ligand Docking
The models for both D2 and D3 receptors were obtained as reported in previous work.24 Briefly, homology models were developed for both D2 and D3 in complex with the nonselective antagonist haloperidol by comparative modeling in the program MODELLER9.254 using the crystal structure of the human β2-adrenergic GPCR (PDB code: 2RH1) and bovine rhodopsin (PDB code: 1F88) as the templates. An extensive model refinement was carried out by multiple molecular dynamics (MD) simulations55 in explicit membrane/solvent environments, within the program NAMD with the all-atom CHARMM27 force field following a published protocol.56 The resulting D3 model has been compared and found to be in excellent agreement with the later published X-ray structure,25,43 with a heavy atom root-mean-square-deviation (RMSD) at 2.88 Å, lower than the resolution of the ray structure (2.89 Å). This result provides confidence that the D2 model should have a similar quality and both models could be used toward the docking calculations to study receptor–ligand interactions.
All of the docking calculations were performed with the docking program GOLD 5.1.57 For all the ligands, the piperidine rings were treated as rigid in a chair conformation. All the other rotatable bonds were set as flexible. The nitrogen atom connected with alkyl chain spacer was protonated. A hydrogen bond constraint was specified between the proton and the carboxyl oxygen of the highly conserved Asp in TMS III to embody the highly conserved salt bridge interactions. All the protein atoms were set as rigid. Each molecule was docked 20 times with early termination if the top three poses are within 1.5 Å RMSD. Each pose was ranked according to its ChemPLP fitness function, which uses a piecewise linear potential for hydrophobic and noncomplementary interactions and Chemscore terms for hydrogen bonding and internal energy.58 For each ligand, the highest scoring pose was chosen as the binding conformation and used for further analysis.
Glossary
Abbreviations
- E1
first extracellular loop
- E2
second extracellular loop
- I3
third intracellular loop
- IABN
2,3-dimethoxy-5-iodo-N-(9-benzyl-9-azabicyclo(3.3.1)nonan-3yl)benzamide
- IC50 value
the concentration of competitive inhibitor that inhibits 50% of the total specific binding
- IRES
internal ribosomal entry site
- Kd values
equilibrium dissociation constant determined using direct radioligand binding techniques
- Ki values
equilibrium dissociation constant determined using competitive radioligand binding techniques
- MD
molecular dynamics
- SV-III-130s
7-(4-(4-(2-methoxyphenyl)piperazin-1-yl)butoxy)-3,4-dihydroquinolin-2(1H)-one
- SV 293
1-((5-methoxy-1H-indol-3-yl)methyl)-4-(4-(methylthio)phenyl)piperidin-4-ol
- TMS
transmembrane spanning
Author Present Address
Glenn H. Dillon: Department of Physiology and Pharmacology, Center for Neuroscience, West Virginia University Health Science Center, Morgantown, WV 36506.
Author Contributions
R.R.L.: design of binding and functional experimental studies, analysis, and manuscript preparation. Y.M.: performed binding studies, data analysis, and manuscript preparation. Q.W.: performed computational modeling studies and manuscript preparation. S.A.G.: generated chimeric receptor gene constructs, transfection of cell lines and tissue culture, and manuscript preparation. C.B.-H.: performed electrophysiology studies and data analysis. M.T.: performed adenylyl cyclase and pERK functional studies, data analysis and tissue culture, and manuscript preparation. S.V.: ligand design, synthesis and structural characterization of the test ligands, and manuscript preparation. G.H.D.: designed and supervised electrophysiology studies. R.-Q.H.: designed and performed electrophysiology studies and manuscript preparation. D.E.R.: supervised, designed, and performed computational modeling studies and manuscript preparation. R.H.M.: ligand design, supervised computational modeling studies, supervised all data analysis, and manuscript preparation.
This work was supported by National Institute of Mental Health [Grant R21 NS050658] and National Institute on Drug Abuse [R01 DA13584].
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Kapur S.; Mamo D. (2003) Half a century of antipsychotics and still a central role for dopamine D2 receptors. Prog. Neuropsychopharmacol. Biol. Psychiatry 7, 1081–1090. [DOI] [PubMed] [Google Scholar]
- Nieoullon A. (2002) Dopamine and the regulation of cognition and attention. Prog. Neurobiol. 67, 53–83. [DOI] [PubMed] [Google Scholar]
- Jardemark K.; Wadenberg M. L.; Grillner P.; Svensson T. H. (2002) Dopamine D3 and D4 receptor antagonists in the treatment of schizophrenia. Curr. Opin. Invest. Drugs 3, 101–105. [PubMed] [Google Scholar]
- Cunnah D.; Besser M. (1991) Management of prolactinomas. Clin. Endocrinol. (Oxford, U.K.) 34, 231–235. [DOI] [PubMed] [Google Scholar]
- Korczyn A. D. (2003) Dopaminergic drugs in development for Parkinson’s disease. Adv. Neurol. 91, 267–271. [PubMed] [Google Scholar]
- Kumar R.; Riddle L. R.; Griffin S. A.; Chu W.; Vangvervong S.; Neissewander J.; Mach R. H.; Luedtke R. R. (2009) Evaluation of D2 and D3 dopamine receptor selective compounds on L-dopa dependent abnormal involuntary movements in rats. Neuropharmacology 56, 956–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow N. D.; Fowler J. S.; Wang G. J. (2002) Role of dopamine in drug reinforcement and addiction in humans: results from imaging studies. Behav. Pharmacol. 13, 355–366. [DOI] [PubMed] [Google Scholar]
- Nader M. A.; Morgan D.; Gage H. D.; Nader S. H.; Calhoun T. L.; Buchheimer N.; Ehrenkaufer R.; Mach R. H. (2006) PET imaging of dopamine D2 receptors during chronic cocaine self-administration in monkeys. Nat. Neurosci. 9, 1050–1056. [DOI] [PubMed] [Google Scholar]
- Herve D.; Le Moine C.; Corvol J. C.; Belluscio L.; Ledent C.; Fienberg A. A.; Jaber M.; Studler J. M.; Girault J. A. (2001) Galpha(olf) levels are regulated by receptor usage and control dopamine and adenosine action in the striatum. J. Neurosci. 21, 4390–4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sealfon S. C.; Olanow C. W. (2000) Dopamine receptors: from structure to behavior. Trends Neurosci. 23, S34–40. [DOI] [PubMed] [Google Scholar]
- Vallone D.; Picetti R.; Borrelli E. (2000) Structure and function of dopamine receptors. Neurosci. Biobehav. Rev. 24, 125–132. [DOI] [PubMed] [Google Scholar]
- Kuzhikandathil E. V.; Westrich L.; Bakhos S.; Pasuit J. (2004) Identification and characterization of novel properties of the human D3 dopamine receptor. Mol. Cell. Neurosci. 26, 144–155. [DOI] [PubMed] [Google Scholar]
- Senogles S. E. (2003) D2s dopamine receptor mediates phospholipase D and antiproliferation. Mol. Cell. Endocrinol. 209, 61–69. [DOI] [PubMed] [Google Scholar]
- Neve K.; Seamans J. K.; Trantham-Davidson H. (2004) Dopamine signaling. J. Recept. Signal Transduction Res. 24, 165–205. [DOI] [PubMed] [Google Scholar]
- Wong S.K.-F. (2004) A 384-well cell-based phospho-ERK assay for dopamine D2 and D3 receptors. Anal. Biochem. 333, 265–272. [DOI] [PubMed] [Google Scholar]
- Beom S.; Cheong D.; Torres G.; Caron M. G.; Kim K. M. (2004) Comparative studies of molecular mechanisms of dopamine D2 and D3 receptors for the activation of extracellular signal-regulated kinase. J. Biol. Chem. 279, 28304–28314. [DOI] [PubMed] [Google Scholar]
- Sokoloff P.; Giros B.; Martres M. P.; Bouthenet M. L.; Schwartz J. C. (1990) Molecular cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature 347, 146–151. [DOI] [PubMed] [Google Scholar]
- Joyce J. N. (2001) Dopamine D3 receptor as a therapeutic target for antipsychotic and antiparkinsonian drugs. Pharmacol. Ther. 90, 231–259. [DOI] [PubMed] [Google Scholar]
- Kim K.; Valenzano K. J.; Robinson S. R.; Yao W. D.; Barak L. S.; Caron M. G. (2001) Differential regulation of the dopamine D2 and D3 receptors by G protein-coupled receptor kinase and β-arrestins. J. Biol. Chem. 276, 37409–37414. [DOI] [PubMed] [Google Scholar]
- Luedtke R. R.; Mach R. H. (2003) Progress in developing D3 dopamine receptor ligands as potential therapeutic agents for neurological and neuropsychiatric disorders. Curr. Pharm. Des. 9, 643–671. [DOI] [PubMed] [Google Scholar]
- Mach R. H.; Huang Y.; Freeman R. A.; Wu L.; Blair S.; Luedtke R. R. (2003) Synthesis of 2-(5-bromo-2,3-dimethoxyphenyl)-5-(aminomethyl)-1H-pyrrole analogues and their binding affinities for dopamine D2, D3 and D4 receptors. Bioorg. Med. Chem. 11, 225–233. [DOI] [PubMed] [Google Scholar]
- Chu W.; Tu Z.; McElveen E.; Xu J.; Taylor M.; Luedtke R. R.; Mach R. H. (2005) Synthesis and in vitro binding of N-phenyl piperazine analogs as potential dopamine D3 receptor ligands. Bioorg. Med. Chem. 13, 77–87. [DOI] [PubMed] [Google Scholar]
- Grundt P.; Carlson E. E.; Cao J.; Bennett C. J.; McElveen E.; Taylor M.; Luedtke R. R.; Hauck-Newman A. (2005) Novel heterocyclic trans olefin analogues of N-{4-[4-(2,3-dichlorophenyl)piperazin-1-yl]butyl}arylcarboxamides as selective probes with high affinity for the dopamine D3 receptor. J. Med. Chem. 48, 839–848. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Mach R. H.; Luedtke R. R.; Reichert D. E. (2010) Subtype selectivity of dopamine receptor ligands: insights from structure and ligand-based methods. J. Chem. Inf. Model. 50, 1970–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien E..Y.; Liu W.; Zhao Q.; Katritch V.; Han G. W.; Hanson M. A.; Shi L.; Newman A. H.; Javitch J. A.; Cherezov V.; Stevens R. C. (2010) Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 330, 1091–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vangveravong S.; McElveen E.; Taylor M.; Griffin S. A.; Luedtke R. R.; Mach R. H. (2006) Identification and pharmacologic characterization of D2 dopamine receptor selective antagonists. Bioorg. Med. Chem. 14, 815–825. [DOI] [PubMed] [Google Scholar]
- Vangveravong S; Zhang Z.; Taylor M.; Bearden M.; Xu J.; Cui J.; Wang W.; Luedtke R. R.; Mach R. H. (2011) Synthesis and characterization of selective dopamine D2 receptor ligands using aripiprazole as the lead compound. Bioorg. Med. Chem. 19, 3502–351132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luedtke R. R.; Freeman R. A.; Boundy V. A.; Martin M. W.; Mach R. H. (2000) Characterization of 125I-IABN, a novel azabicyclononane benzamide selective for D2-like dopamine receptors. Synapse 38, 438–449. [DOI] [PubMed] [Google Scholar]
- Mach R. H.; Huang Y.; Freeman R. A.; Wu L.; Vangveravong S.; Luedtke R. R. (2004) Conformationally-flexible benzamide analogues as dopamine D(3) and sigma(2) receptor ligands. Bioorg. Med. Chem. Lett. 14, 195–202. [DOI] [PubMed] [Google Scholar]
- Boundy V. A.; Luedtke R. R.; Galitano A. L.; Smith J. E.; Filtz T. M.; Kallen R. G.; Molinoff P. (1993) Expression and characterization of the rat D3 dopamine receptor: Pharmacologic properties and development of antibodies. J. Pharmacol. Exp. Ther. 264, 1002–1011. [PubMed] [Google Scholar]
- Taylor M.; Griffin S. A.; Grundt P.; Newman A. H.; Luedtke R. R. (2010) Novel dopamine D3 receptor selective ligands with varying intrinsic activities at D3 dopamine receptor mediated signaling pathways. Synapse 64, 251–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber M.; Chang W.-L.; Durbin J. P.; Park P. E.; Luedtke R. R.; Mach R. H.; Swerdlow N. R. (2009) Using prepulse inhibition to detect functional D3 receptor antagonism: Effects of WC10 and WC44. Pharmacol., Biochem. Behav. 93, 141–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung T. H.; Nolan B. C.; Hammerslag L. R.; Weber S. M.; Durbin J. P.; Peartree N. A.; Mach R. H.; Luedtke R. R.; Neisewander J. L. (2012) Phenylpiperazine derivatives with selectivity for dopamine D3 receptors modulate cocaine self-administration in rats. Neuropharmacology 63, 1346–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J.; Chu W.; Vangveravong S.; Jones L. A.; Lewis J.; Luedtke R. R.; Perlmutter J. S.; Mintun M. A.; Mach R. H. (2009) [3H]4-(Dimethylamino)-N-[4-(4-(2-methoxyphenyl)piperazin-1-yl)butyl]benzamide, a selective radioligand for dopamine D3 receptors. I. In vitro characterization. Synapse 63, 717–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mach R. H.; Tu Z.; Xu J.; Li S.; Jones L. A.; Taylor M.; Luedtke R. R.; Derdeyn C. P.; Perlmutter J. S.; Mintun M. A. (2011) Endogenous dopamine competes with the binding of radiolabeled D3 partial agonists in vivo: A positron emission tomography study. Synapse 65, 724–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalani M. Y.; Vaidehi N.; Hall S. E.; Trabanino R. J.; Freddolino P. L.; Kalani M. A.; Floriano W. B.; Kam V. W.; Goddard W. A. 3rd. (2004) The predicted 3D structure of the human D2 dopamine receptor and the binding site and binding affinities for agonists and antagonists. Proc. Natl. Acad. Sci. U.S.A. 101, 3815–3820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobrath J. V.; Wang S (2006) Computational elucidation of the structural basis of ligand binding to the dopamine 3 receptor through docking and homology modeling. J. Med. Chem. 49, 4470–4476. [DOI] [PubMed] [Google Scholar]
- Palczewski K.; Kumasaka T.; Hori T.; Behnke C. A.; Motoshima H.; Fox B. A.; Le Trong I.; Teller D. C.; Okada T.; Stenkamp R. E.; Yamamoto M.; Miyano M. (2000) Crystal structure of rhodopsin: A G protein-coupled receptor. Science 289, 739–745. [DOI] [PubMed] [Google Scholar]
- Xu F.; Wu H.; Katritch V.; Han G. W.; Jacobson K. A.; Gao Z. G.; Cherezov V.; Stevens R. C. (2011) Structure of an agonist-bound human A2A adenosine receptor. Science 332, 322–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherezov V.; Rosenbaum D. M.; Hanson M. A.; Rasmussen S. G.; Thian F. S.; Kobilka T. S.; Choi H. J.; Kuhn P.; Weis W. I.; Kobilka B. K.; Stevens R. C. (2007) High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 318, 1258–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaakola V. P.; Griffith M. T.; Hanson M. A.; Cherezov V.; Chien E. Y.; Lane J. R.; Ijzerman A. P.; Stevens R. C. (2008) The 2.6 Å crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 322, 1211–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B.; Chien E. Y.; Mol C. D.; Fenalti G.; Liu W.; Katritch V.; Abagyan R.; Brooun A.; Wells P.; Bi F. C.; Hamel D. J.; Kuhn P.; Handel T. M.; Cherezov V.; Stevens R. C. (2010) Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 330, 1066–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kufareva I., Rueda M., Katritch V.. GPCR Dock 2010 participants, Stevens, R. C., and Abagyan, R. (2011) Status of GPCR modeling and docking as reflected by Community-wide GPCR Dock 2010 Assessment. Structure 19, 1108–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olah M. E.; Jacobson K. A.; Stiles G. L. (1994) Role of the second extracellular loop of adenosine receptors in agonist and antagonist binding. Analysis of chimeric A1/A3 adenosine receptors. J. Biol. Chem. 269, 24692–24698. [PMC free article] [PubMed] [Google Scholar]
- Shi L; Javitch J. A. (2004) The second extracellular loop of the dopamine D2 receptor lines the binding-site crevice. Proc. Natl. Acad. Sci. U.S.A. 101, 440–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gay E. A.; Urban J. D.; Nichols D. E.; Oxford G. S.; Mailman R. B. (2004) Functional selectivity of D2 receptor ligands in a Chinese hamster ovary hD2L cell line: Evidence for induction of ligand-specific receptor states. Mol. Pharmacol. 66, 97–105. [DOI] [PubMed] [Google Scholar]
- Clarke W. P. (2005) What’s for lunch at the conformational cafeteria?. Mol. Pharmacol. 67, 1819–1821. [DOI] [PubMed] [Google Scholar]
- Urban J. D.; Clarke W. P.; von Zastrow M.; Nichols D. E.; Kobilka B.; Weinstein H.; Javitch J. A.; Roth B. L.; Christopoulos A.; Sexton P. M.; Miller K. J.; Spedding M.; Mailman R. B. (2007) Functional selectivity and classical concepts of quantitative pharmacology. J. Pharmacol. Exp. Ther. 320, 1–13. [DOI] [PubMed] [Google Scholar]
- Kenakin T.; Miller L. J. (2010) Seven transmembrane receptors as shapeshifting proteins: The impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol. Rev. 62, 265–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGonigle P., and Molinoff P. B. (1989) Quantitative aspects of drug-receptor interactions. In Basic Neurochemistry: Molecular, Cellular, and Medical Aspects (Siegel, Agranoff, Albers, and Molinoff, Eds.), Raven Press: New York, Chapter 9; pp 183–201. [Google Scholar]
- Cheng Y. C.; Prusoff W. H. (1972) Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 122, 3099–3108. [DOI] [PubMed] [Google Scholar]
- Shimizu H; Daly J. W.; Creveling C. R. (1969) A radioisotopic method for measuring the formation of adenosine 3′,5′-cyclic monophosphate in incubated slices of brain. J. Neurochem. 16, 1609–1619. [DOI] [PubMed] [Google Scholar]
- Moore K. R.; Blakely R. D. (1994) Restriction site-independent formation of chimeras from homologous neurotransmitter-transporter cDNAs. Biotechniques 17, 130–137. [PubMed] [Google Scholar]
- Marti-Renom M. A.; Stuart A. C.; Fiser A.; Sanchez R.; Melo F.; Sali A. (2000) Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 29, 291–325. [DOI] [PubMed] [Google Scholar]
- Caves L. S.; Evanseck J. D.; Karplus M. (1998) Locally accessible conformations of proteins: multiple molecular dynamics simulations of crambin. Protein Sci. 7, 649–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H. L.; Cheng X.; Taylor P.; McCammon J. A.; Sine S. M. (2008) Control of cation permeation through the nicotinic receptor channel. PLoS Comput. Biol. 4, e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdonk M. L.; Cole J. C.; Hartshorn M. J.; Murray C. W.; Taylor R. D. (2003) Improved protein-ligand docking using gold. Proteins: Struct., Funct., Genet. 52, 609–623. [DOI] [PubMed] [Google Scholar]
- Korb O.; Stutzlek T.; Exner T. E. (2009) Empirical scoring functions for advanced protein-ligand docking with PLANTS. J. Chem. Inf. Model. 49, 84–96. [DOI] [PubMed] [Google Scholar]