

Abstract
Cadherin 23 (CDH23) is an important constituent of the hair cell tip link in the organ of Corti. Mutations in cdh23 are associated with age-related hearing loss (AHL). In this study, we proposed that the Cdh23nmf308/nmf308 mice with progressive hair cell loss had specific morphological changes and suffered a base to apex gradient and age-related hearing loss, and that mutations in cdh23 were linked to AHL. The Cdh23nmf308/nmf308 mice produced by the N-nitrosourea (ENU) mutagenesis program were used as an animal model to study AHL and progressive hair cell loss. RT-PCR was performed to confirm the cdh23 mutation in Cdh23nmf308/nmf308 mice and genetic analysis was used to map the specific mutation site. Distortion product otoacoustic emission (DPOAE) assay and acoustic brainstem evoked response (ABR) threshold analysis were carried out to evaluate the AHL. Cochlear histology was examined with scanning electron microscope (SEM) and transmission electron microscope (TEM), as well as the nuclear labeling by propidium iodide staining; terminal deoxynucleotidyl transferase mediated dUTP nick end labeling (TUNEL) assay and caspase-3 activities were examined to evaluate cell apoptosis. Genetic mapping identified the candidate gene linking AHL in Cdh23nmf308/nmf308 mice as cdh23. A mutation in exon3 (63 T>C) was screened as compared with the sequence of the same position of the gene from B6 (+/+) mice. The cochleae outer hair cells were reduced from 5–10% at one month to 100% at three months in the basal region. DPOAE and ABR exhibited an increasing threshold at high frequencies (≥16 kHz) from one month of age. Morphological and cellular analysis showed that Cdh23nmf308/nmf308 mice exhibited a time course of histological alterations and cell apoptosis of outer hair cells. Our results suggest that the cdh23 mutation may be harmful to the stereociliary tip link and cause the hair cell apoptosis. Due to the same cdh23 mutations in human subjects with presbycusis (Petit et al., 2001; Zheng et al., 2005), the Cdh23nmf308/nmf308 mouse is an excellent animal model for investigating the mechanisms involved in human AHL.
Keywords: Aged-related hearing loss, Cdh23, Cdh23nmf308/nmf308 mice, Tip link, Apoptosis
1. Introduction
Age-related hearing loss (AHL) occurs in humans as a consequence of advancing age. Mice are widely used as a model in order to screen the genes and alleles that promote the major forms of AHL and their combinations (Ohlemiller et al., 2006). The mouse ENU (N-ethyl-N-nitrosourea) mutagenesis program identified a series of independent mutations on mouse chromosome 10 that harbors several loci associated with hearing loss including modifier of deaf waddler (mdfw), age-related hearing loss (ahl) and waltzer (v) (Erway et al., 1993; Johnson et al., 1997; Johnson et al., 1997, 2000). The mdfw loci, which modify the hearing loss phenotype in mice resulting from mutations of the Atp2b2 (a member of the plasma membrane Ca2+ pump family of genes), may be allelic with ahl (Zheng and Johnson, 2001). Evidence indicates that inbred strain-specific alleles of the cadherin23 gene (cdh23) are responsible for the hearing loss attributed to the ahl/mdfw locus (Noben-Trauth et al., 2003).
Cadherin 23 (CDH23) is an important constituent of the hair cell tip link in the organ of Corti. Previous studies revealed that, by forming a complex with myosin-1c in the mechanotransduction apparatus, CDH23 regulates mechanoelectrical transduction and the mutations in cdh23 cause deafness and AHL (Noben-Trauth et al., 2003; Bolz et al., 2001; Bork et al., 2001; Holt et al., 2002). Together, CDH23 and myosin-1c regulate the activity of mechanically gated ion channels in hair cells (Slemens et al., 2004). CDH23 also interacts with harmonin b to anchor CDH23 intracellularly to the actin filaments (Boëda et al., 2001; Adato et al., 2005). More recently, investigations showed that the interaction between CDH23 and protocadherin 15 (Pcdh15) modulate tip link filaments in sensory hair cells (Alagramam et al., 2011). AHL, or presbycusis, is the slow loss of hearing that occurs as human subjects get older. Hearing loss is progressively worsened with age. Genetic hearing loss can be congenital or delayed onset, resulting from hereditary factors. AHL is frequently related to one or more gene mutations, especially CDH23 mutations (Noben-Trauth et al., 2003). AHL can be exacerbated by a variety of environmental insults including exposures to loud noises and ototoxic drugs, but at its core is a significant genetic component (DeStefano et al., 2003). Hence, the Cdh23nmf308/nmf308 mouse is an excellent animal model for investigating the mechanisms involved in human AHL.
Although much has been accomplished in uncovering the link between cdh23 and AHL, the exact mutation in cdh23 that causes AHL and the elaborate morphological profiling in mutated cdh23-mediated AHL mice remains unknown. Using Cdh23nmf308/nmf308 mice as an animal model, we systematically characterized AHL and progressive hair cell loss on a molecular level. We utilized genetic mapping to locate the candidate gene responsible for AHL and single mutation in cdh23 was identified. Furthermore, we demonstrated that the alteration of an amino acid in extracellular cadherin 1 (EC1) domain affects CDH23 structure and this structural change was linked to AHL. Using morphological and cellular analysis, we also showed that Cdh23nmf308/nmf308 mice exhibited a time course of histological alterations and cellular apoptosis of the outer hair cells. Our results systematically uncover the roles of cdh23 in AHL on the molecular, cellular and morphological levels.
2. Materials and methods
2.1. Experimental animals
All mice were obtained from research or production colonies at Case Western Reserve University (Cleveland, OH, USA). The Cdh23nmf308/nmf308 mutation was developed for the ENU mutagenesis program at The Jackson Laboratory (Bar Harbor, Maine, USA), and the nmf mutant colony was maintained by sibling mating. Mutant mice (nmf/nmf) were confirmed by the auditory-evoked brainstem response (ABR). All procedures were approved by Animal Care and Use Committee at Case Western Reserve University.
2.2. DNA isolation
Genomic DNA was prepared from mouse-tail tips using the alkaline lysis method. In brief, 2 mm of mouse-tail tip was transferred to a 0.5 ml Eppendorf tube containing 0.3 ml of 50 mM NaOH. The samples were heated at 95 °C for 10 min and 26 μl of 1 M Tris–HCl was then added to each tube. Samples were centrifuged at 12,000×g for 5 min to removed debris, and the supernatant was transferred to a new tube. DNA concentration was determined using a BioPhotometer (Eppendorf, Hamburg, Germany).
2.3. Genetic linkage analysis
C57BL/6(B6) mice were crossed with C3H/C3H (+/+) mice with normal hearing. The F1 progeny of B6/C3H (nm/+) were intercrossed to produce F2 siblings, after which the B6/B6 (nm/nm) mice were examined by ABR. The nmf mutation was genetically mapped using a previously described DNA pooling strategy (Taylor et al., 1997; Ackert-Bicknell et al., 2007). DNA samples from 13 B6/B (nm/nm) mutant F2 mice from an interspecific cross between [B6/C3H (nmf/+)-B6/C3H (nmf/+)] F1 hybrids were pooled for a genome wide screen, and the linkage was found with markers on mouse chromosome 10. To refine the map position of the mutation, genotypes of individual F2 progeny from this cross were obtained for multiple chromosomes 10 markers. The uniSTS database of National Center for Biotechnology Information (NCBI) was used to search MGI map position.
2.4. Allele test
To confirm the mutations in cdh23 responsible for the hearing loss, the B6/B6 (nm/nm) mice were crossed with v2J/v2J, which also have recessive hearing loss. Mice exhibiting higher ABR thresholds were confirmed as nmf 308/v2J at postnatal day 30.
2.5. DNA sequencing
According to the cdh23 sequence in Ensembl Mouse Genome Server (Integrated DNA Technologies), we designed and subsequently synthesized 69 pair of primers using Primers 3 software (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi). PCR for comparative DNA analysis of nmf 308mutant and control mice was performed. The PCR products were purified with the QIAquick PCR Purification kit (Qiagen, Valencia, CA, USA). DNA sequencing was confirmed using an Applied Biosystems 3730 DNA Analyzer (HITACHI, Tokyo, Japan) using the same primers as used for DNA amplification.
2.6. PCR and RT-PCR
Based on the mutation identified, we established a method to genotype the homozygote and heterozygote mice of Cdh23nmf308/nmf308 by PCR and exon digestion of the PCR products. The PCR reaction mixture contained 100 ng of genomic DNA in 20 μl of reaction buffer containing 50mMKCl, 10mMTris–HCl, pH 9.0 (at 25 °C), 0.01% Triton X-100 (v/v), 2 mM MgCl2, 250 nM of each primer (forward and reverse), 200 μM of each of four deoxyribonucleoside triphosphates, and 1.0 U of Taq DNA polymerase (New England BioLabs, CA). PCR was performed in a BioRad PTC-200 Peltier Thermal Cycler (Watertown, MA). PCR Amplification consisted of an initial denaturation at 95 °C for 3 min followed by 32 cycles, each consisting of 94 °C for 30 s, 60 °C for 40 s, and 72 °C for 45 s. After the 32 cycles, the PCR product was incubated for an additional 5 min at 72 °C. A 10 μl of enzyme digestion mixture was made which contained 1 μl of 10×NE Buffer 2 (NewEngland BioLabs, Ipswich, MA, USA), 4 μl of PCR products, 0.1 μl (1U) of BstNI and, and 0.1 μl of the acetyl BSA (100 μg/ml). The mixture was incubated at 60 °C for 2 h and subjected to 3% agarose gel electrophoresis.
Two-week Cdh23nmf308/nmf308 and B6-+ahl mice were sacrificed under Avertin anesthesia. The inner ears were quickly removed. Total RNA (DNA free) was prepared using the pure-Link™ Micro-to-midi Total RNA purification system (Invitrogen, Carlsbad, CA, USA). RT-PCR was carried out using PTC-200 PCR Amplification (MJ Research. Inc, Waltham, MA, USA). The PCR primers (CdhmF: 5′-TGACACGTACCTGCTCATCA- 3′; CdhmR: 5′-CCTTGGTGGTCACTGACAGA-3′)were designed at exon 2 and exon 4 of cdh23 and the PCR product was a 228 bp fragment that contained exon3 and its flanking regions (exon 2 and exon 4).
2.7. Measurement of auditory-evoked brainstem response
Both ears from all animals were assessed using ABR analysis. For this analysis, each animal was anaesthetized with Avertin and then placed in a soundproof room. A recording electrode was inserted into the calvaria. A reference electrode was inserted into the mastoid and a ground electrode was inserted into the opposite mastoid. The animals were stimulated with a click at 8, 16, or 32 kHz. ABR thresholds were measured with a Smart EP system (Intelligent Hearing Systems, Miami, FL, USA) and Wave III was used as a marker, as previously described (Zheng et al., 1999). Stimuli were presented at a rate of 25 ms and averaged across 512 sweeps. Responses were band-pass filtered at 100 and 3,000 Hz and amplified 100,000-fold. A threshold was defined as the minimal stimulus level that gave a recognizable waveform on a normalized scale and, the waveform was well repetitive at the same stimulation sound (Li et al., 2008).
2.8. DPOAE assay
Distortion product otoacoustic emission (DPOAE) was measured using HIS SmartEP3.30 USBez Software (Intelligent Hearing Systems, Miami, FL, USA). The measurements were conducted using pure tones from 2 to 36 kHz. An Etymotic 10B+ probe (Etymotic Research, Elk Grove Village, IL, USA) was inserted into the external ear canal and monitored using two different types of transducers depending on the range of stimulation frequency. For frequencies ranging from 2 to 16 kHz, Etymotic ER2 stimulus response signals were sampled at a rate of 128 kHz using a 16-bit D/A converter. L1 and L2 amplitudes were set to the same level. Frequencies were acquired with an F2–F1 ratio of 1:22. Stimuli were presented starting from the lowest frequency tested and increasing to the highest frequency. Five stimulation levels ranging from 25 to 65 dB sound pressure level (SPL), in 10 dB steps, were used. Each stimulation frequency was acquired by a running of “block module” consisting of 32 sweeps (Li et al., 2008).
2.9. Histology preparation
Animals were euthanized using an overdose of ketamine (100 mg/ml; 0.1mg/g) and xylazine (20 mg/ml; 0.2 mg/g). The right ear cochlea and vestibular organ were removed from the temporal bone. The round window and oval window were opened and holes were made in the apex. A basal perfusion of 1.0% silver nitrate solution was administered through the cochlea three times along the haustorial tube. The specimens were fixed in 10% formalin for 2 h and the slides were exposed to sunlight for approximately 1 h to enhance the silver stain. A Leica S6D stereo zoom microscope was used to micro-dissect the sensory epithelium. Then the left temporal bones were removed and perfused with fixative (2.5% glutaraldehyde, 4.0% paraformaldehyde in 0.1 M PBS) and then immersed in the same fixative overnight. The following day, the temporal bones and bullas were dissected leaving the bilateral middle and inner ears connected, after which time they were processed together for histology. Tissues were decalcified using a microwave plus 10% EDTA, embedded in the horizontal plane at 5 μm, serially mounted on glass slides, and stained with Harris’ hematoxylin. All qualitative and quantitative microscopic assessments were performed using a Zeiss Standard microscope (Carl Zeiss AG, Germany).
2.10. Sample preparation for scanning electron microscopy (SEM)
The cochlea was used for this procedure. Each animal was decapitated and the cochleae were quickly removed from the skull and immersed in 2.5% glutaldehyde (Sigma, St Louis, MO, USA) in 0.1 M PBS. The round and oval windows were opened immediately and a small hole was drilled in the apex and basal turn of the cochlea. Fixative was perfused through the cochlea via the oval window or round window. Specimens were post-fixed in the same fixative for 3 h to several days at 4 °C. After rinsing in buffer, the cochlea were micro-dissected and removed from the bone, vestibular membrane, and tectorial membrane using a surgical dissecting microscope (Olympus x10, Japan). Then the cochlea were treated with 1% OsO4 (Sigma, ST. Louis, MO, USA) for 1 h. Afterwards, samples were rinsed again, and dehydrated in increasing concentrations (30–100%) of ethanol. The basilar membrane was exposed at a critical point by drying and then coated with gold. All specimens were viewed with a H-600 SEM scanning electron microscope (Hitachi, Japan) and JSM-6700F field emission scanning electron microscope (JEOL Ltd., Japan) (Li et al., 2008).
2.11. Sample preparation for transmission electron microscopy (TEM)
The bilateral otic bulla was quickly removed under Avertin anesthesia. The round window and oval window membrane were opened and then holes on the wall of the apical and basal turn bone were made. Cochlea were fixed by perilymphatic perfusion with 2.5% glutaraldehyde in 0.1 M PBS (pH 7.4) with two buffer changes. Two hours after fixation, decalcification was carried out in 120 mM EDTA for 1–1.5 h. The lateral walls of the cochlear duct were removed to expose the organ of Corti, which was then dissected out with the aid of a dissection microscope. Each cochlea was divided into two longitudinal sections. Dissected specimens were washed three times with 0.1 M PBS and post-fixed in 1% OsO4 for 1 h. They were then dehydrated in an ascending series of ethanol and embedded in paraffin. After exposing the organ of Corti, the apical turn was carefully peeled away. After decalcification in 120 mM EDTA for 1–1.5 h, each cochlea was then transferred to PBS and divided into two longitudinal sections. The lateral wall was trimmed away and the tectorial membrane was removed to expose the reticular lamina and apical surface of sensory epithelium. Cochleae were prepared as described above. Semithin and ultrathin radial sections of the plastic embedded organ of Corti were cut from the basal and middle turns and observed using a Hitachi 7100 electron microscope (Hitachi, Tokyo, Japan). All thin sections were examined in a JEOL 1010 transmission electron microscope (JEOL Ltd., Japan). Photographic negatives of the micrographs were digitized on a flatbed scanner and the images were imported into Adobe Photoshop (Li et al., 2008).
2.12. Propidium iodide (PI) labeling of nuclei
The animals were anesthetized with CO2 and then sacrificed. The cochlea were quickly removed from the skull and gently perfused with 30 μl of PI-staining solution (5 μg/ml in Hanks solution; Molecular Probes, Oregon, USA) through the round window. The cochlea were incubated in the same staining solution for 10min at room temperature and then fixed with 10% buffered formalin. Fixed tissues were labeled with PI (5 μg/ml in 10mM PBS) for 10 min. After rinsing with PBS, the organs of corti were mounted on slides containing an antifade medium (Prolong TM Antifade kit, Molecular Probes, Oregon, USA).
2.13. TUNEL assay
TUNEL (Terminal dexynucleotidyl transferase (TdT)mediated dUTP Nick End Labeling, Roch Diagnostics, Germany) was used to identify apoptotic cells. Cochleae were immediately fixed with 4% paraformaldehyde (ACROS, New Jersey, USA) overnight at 4 °C. The cochleae surface preparations of organ of Corti were incubated in 0.5% Triton X-100 sodium citrate for 15 min at room temperature, then washed twice with PBS. Subsequently, 100 μl of TUNEL reaction mixture containing dUTP and TdT was added to each surface preparation and incubated in the dark for 60 min at 37 °C. Sections were subsequently incubated in working strength TdT enzyme (mixture of 100 μl reaction buffer and 30 μl of TdT enzyme (Terminal Deoxynucleotidyl Transferase, Recombinant). The surface preparations were then washed, incubated again with PI-staining solution (2–5 μg/ml PI in PBS) for 30 min at room temperature. Finally, the surface preparation was dissected, mounted and photographed with the laser confocal microscopy (Carl Zeiss, Inc, Germany) (Hu et al., 2006).
2.14. Detection of caspase-3
The cochlea were exposed by removing a piece of bone covering the apex, rupturing the round window membrane, extracting the stapes from the oval window, and breaking the bone between round and oval window to give access to basal cochlear turn and the vestibule. Fixative was perfused directly into the cochlea through these openings under the PBS and fixed with 4% paraformaldehyde overnight at 4 °C followed by two washes in PBS. After incubating with Triton X-100 at room temperature for 30 min, followed by two washes in PBS. The cochleae were then incubated in blocking solution (5% goat serum with 2% BSA in PBS) at room temperature for 1 h. Afterwards, the cochleae were incubated with anti-caspase-3 polyclonal antibody (Cell Signaling Technology, Danvers, MA, USA) at 4 °C overnight then stained with PI (5 μl/ml PI in PBS) for 30 min at room temperature. After washing twice with PBS, rabbit anti-rat second antibody (Cell Signaling Technology, Danvers, MA, USA) was added for 1 h. After washing twice with PBS, the cochlear surface preparations were dissected and mounted on glass sides.
2.15. Statistics
Chi-square test was used to confirm linkage; t test was used to compare the differences among ABR threshold values; Mann–Whitney rank sum test was used to compare difference in hair cell counts between Cdh23nmf308/nmf308 mice and its wild-type littermates with SPSS 12.0.1 software (SPSS Inc., Chicago, IL. USA).
3. Results
3.1. Novel mutation in cdh23 in Cdh23nmf308/nmf308 mice
To identify specific mutations in cdh23 linked to AHL, PCR was performed using genomic DNA from experimental mice and the PCR products were sequenced. DNA sequencing analysis showed that a mutation in exon 3 (63 T>C) or in the transcript sequence (208 T>C) in cdh23 occurred in Cdh23nmf308/nmf308 mice compared to B6 (+/+) and BALB/c mice (Fig. 1A). This mutation also caused the single amino acid change from Serine to Proline (70 S-P) in CDH23 (Fig. 1A). Alteration of the amino acid sequence in CDH23 (70 S-P) caused changes in the secondary structure of the protein as analyzed by GOR IV secondary structure prediction (Fig. 1B), which suggests the DNA mutation-mediated protein structural alterations that may be involved in AHL.
Fig. 1.

Novel mutation in cdh23 in Cdh23nmf308/nmf308 mice. (A) Nucleotide mutation (208T>C, arrow indicated) in cdh23 in Cdh23nmf308/nmf308 causing a CDH23 sequence alteration (70 S-P, red indicated). (B) Sequence alterations in CDH23 (70 S-P) induced a change in secondary structure of the protein as analyzed by GOR IV secondary structure prediction. Only the first 140 amino acids of the protein are shown, e: extended strand; c: random coil.
3.2. Genotype analysis of nmf308 mice
To further confirm the mutation of 63 T-C in exon3 in cdh23 in Cdh23nmf308/nmf308 mice, we used restrictive enzyme digestion to examine single base pair changes, since the mutation introduced a new end on the BstN I restriction site (CCTGG). A 219 bp fragment of exon 3 containing the restriction site was amplified by PCR and subjected to BstN I digestion. After BstN I digestion, 152 bp and 67 bp fragments were observed on a 3% agarose gel following electrophoresis (Fig. 2A, lanes 3 and 4).
Fig. 2.

Genotype analysis of nmf308 mice. (A) BstN I digestion confirmed the 63 T-C in exon3 in cdh23 in Cdh23nmf308/nmf308 mice. Genomic DNA from Cdh23nmf308/nmf308 mice were prepared and exon 3 of cdh23 was amplified followed by BstN I digestion. M: 100 bp DNA ladders; Lanes 1 and 2: PCR products using genomic DNA from mice of B6 +/+; Lanes 3 and 4: PCR products using genomic DNA from nmf/nmf 308 mice (ID, 298 and 299 respectively); Lanes 5 and 6: PCR products using genomic DNA from nmf/+308 mice (ID, 298 and 299 respectively). (B) Genomic DNA from two-week old Cdh23nmf308/nmf308 mice and two-week old B6 mice was prepared and exon 3 of cdh23 was amplified followed by BstN I digestion. M: 100 bp DNA ladders; Lanes 1–2: PCR products from inner ears of two-week-old Cdh23nmf308/nmf308 mice; Lanes 3–4: PCR products from inner ears of two-week- old B6 mice. (C) The sequencing results of RT-PCR products from Cdh23nmf308/nmf308 mouse (a) and B6-+ahl mouse (b).
Using genomic DNA from nmf/+308 mice, nmf308 mice were genotyped (B6+/+, nmf/nmf and nmf/+) using RT-PCR followed by BstN I digestion. After 3 months of age, Cdh23nmf308/nmf308 mice manifested hearing loss at wide range of sound frequencies. However, Cdh23nmf308/nmf308 mice less than three-months old had little hearing loss. We thus used the genomic DNA from two-week old Cdh23nmf308/nmf308 mice to examine the cdh23 mutation. After RTPCR and BstN I digestion, DNA were visualized by 1.8% agarose gel electrophoresis (Fig. 2B). Lanes 1–2 show the PCR products from inner ears of two-week old Cdh23nmf308/nmf308 mice and lanes 3–4 show the PCR products from inner ears of two-week old B6 mice. Neither of the amplified DNA could not be digested by BstN I, suggesting that there was no 63 T-C mutation in cdh23 mice of that age. The sequencing results of RT-PCR products from three-month old Cdh23nmf308/nmf308 mouse and B6-+ ahl mouse are presented in Fig. 2C, a and b and the mutation (63 T-C) in exon 3 was confirmed.
3.3. Cdh23nmf308/nmf308 mice exhibited hearing loss at wide range of sound frequencies
Cdh23nmf308/nmf308 mice were bred with C3H (+/+) (normal hearing) mice and F1 progenies (nm/+) were generated and crossed. Among the 51 F2 offspring (five-month old), 13 (nm/nm) had higher ABR thresholds and 38 (nm/+) had normal ABR thresholds when stimulated at different sound frequencies (Fig. 3A).
Fig. 3.

Cdh23nmf308/nmf308 mice exhibited hearing loss at wide range of sound frequencies. (A) By mating Cdh23nmf308/nmf308 mice with those of C3H (+/+), which had normal hearing, the F1 progenies (nm/+) were generated and then crossed. Among the 51 F2 offspring (five-month old), 13 (nm/nm) had higher ABR thresholds and 38 (nm/+) had normal ABR thresholds when stimulated with different sound frequencies. (B) Auditory-evoked brainstem response (ABRs) exhibited threshold elevation from 30 to 100 dB SPL at frequencies over 8 kHz in three-month old Cdh23nmf308/nmf308 mice. (C) Three-month old Cdh23nmf308/nmf308 mice manifested hearing loss at wide range of sound frequencies.
To confirm the mutations in cdh23 responsible for the hearing loss, B6/B6 (nm/nm) mice were crossed with v2J/v2J (recessive for hearing loss). The nmf 308/v2J mice (23 days) were identified by higher ABR thresholds. There were significant differences in the thresholds between the nm/nm and nm/+ genotype groups. ABR exhibited increasing threshold elevation (from 30 to 100 dB sound pressure level) at high frequencies (≥16 kHz) beginning at 3 months of age (Fig. 3B). At 3 months of age, the mice manifested hearing loss at wide range of sound frequencies. Cdh23 nmf 308/v2J mice manifested hearing loss at wide range of sound frequencies from 1 month to 9 months old (Fig. 3C).
3.4. Cdh23nmf308/nmf308 mice exhibited a time course of DPOAE
DPOAE exhibited increasing threshold elevation at high frequencies (≥16 kHz). At 5 weeks of age, Cdh23nmf308/nmf308 mice manifested hearing loss at wide range of sound frequencies compared to control B6 mice, which the same characteristics 22 weeks old. All Cdh23nmf308/nmf308 mice exhibited characteristic hearing loss, beginning with high frequencies at five-weeks old. The same pattern was evident in the DP-gram plots (Fig. 4A). The onset of such a hearing loss and its rate of progression are clearly related to the genetic background of a specific mutation. DP-grams for mouse strains were classified as AHL-like. The Cdh23nmf308/nmf308 mice were characterized by DPOAE loss beginning at the highest F2 frequencies and extending in varying degrees to the lower F2 frequencies. In the first plot for each strain, it is clear that these mice exhibited restricted frequency regions where the average DP-grams obtained at 5 weeks worsened when tested at 22 weeks of age (Fig. 4B).
Fig. 4.

Cdh23nmf308/nmf308 mice exhibited time course of DPOAE. (A) DPOAE exhibited increasing threshold at high frequencies (≥16 kHz). At five-weeks of age, the Cdh23nmf308/nmf308 mice manifested hearing loss at wide range of sound frequencies at 22 weeks of age compared with B6mice with same age. (B) At 3 months of age the Cdh23nmf308/nmf308 mice manifested hearing loss at wide range of sound frequencies.
3.5. Time course of histological alterations in Cdh23nmf308/nmf308 mice
Progressive hair cell loss was characterized in Cdh23nmf308/nmf308 mice. Two-week old Cdh23nmf308/nmf308 mice (n=4) were normal in the cochleae turn region. In one-month old, Cdh23nmf308/nmf308 mice, the inner hair cell (IHC) was in the basal region and the middle and apex turn. At two-months of age, outer hair cell (OHC) loss was commonly observed in nmf308 nmf/nmf mice (n=8). By 3 months of age, nearly all the OHCs except for a small percentage were in the apical third of the cochlea, and approximately 75% of IHC lost were from the extreme apex in comparison to IHC in the base (Fig. 5). In these studies, Cdh23nmf308/nmf308 mice with progressive hair cell loss presented base to apex gradient and age-related hearing loss. The cochlear outer hair cells reduced from 5–10% at 1 month to 100% at 3 months in the basal region. Loss of inner hair cells appeared at 2 months and the rate of loss decreased from 100% in the base to roughly 25% at the extreme apex at 3 months (Fig. 5).
Fig. 5.
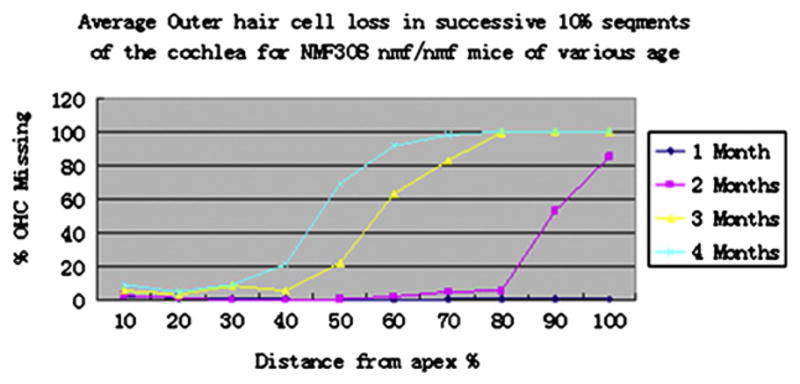
Time course of histology alterations in Cdh23nmf308/nmf308 mice. Cochleograms and the percentage of missing OHC from the apical to basal turn in various aged Cdh23nmf308/nmf308 mice.
3.6. Morphological analysis of outer and inner hair cells in Cdh23nmf308/nmf308 mice
To address the cellular alterations leading to AHL, we observed features of outer and inner hair cells in Cdh23nmf308/nmf308 mice with TEM and SEM. TEM micrographs of B6 mice revealed that outer hair cells had relatively light cytoplasmic staining and basally located nuclei. The cell membrane was smooth and lined by a thick layer of subsurface cisterna. A large number of mitochondria were preferentially located along the lateral membrane and in the sub-cuticular region. Some supporting cells had slender phalangeal processes, which project towards the luminal surface. Efferent nerve terminals were attached to the basal end of the cells (Fig. 6A, a). Efferent nerve terminals from one-month old Cdh23nmf308/nmf308 mice had a relatively normal shape, but abnormal mitochondria number. The nucleus staining was darker and the DNA showed early signs of clumping. The nuclei were positioned higher than normal within the cytoplasm. Efferent terminals were not clearly visualized on these outer hair cells. Supporting cells flanking the outer hair cells had phalangeal processes exhibited enlarged diameters, OHC have missing their stereocilia and shown an shrunken nuclei (Fig. 6A, b).
Fig. 6.

Morphological analysis of outer and inner hair cells in Cdh23nmf308/nmf308 mice. (A) Morphology of outer hair cell to B6/Colony (a) and one-month old Cdh23nmf308/nmf308 mice (b). (B) Morphological observation of cochlear OHC with SEM: scanning electron microscopy indicated in the apical and middle turns the three rows of organized outer hair cells had only a few residual outer hair cells remaining, which exhibited disorganized or V-shaped stereocilia (a–c). OHC loss observed in Cdh23nmf308/nmf308 mice (d). Cdh23nmf308/nmf308 mice had disorganized stereocilia on the top of each outer hair cell that were not arranged in a row and did not form the characteristic staircase-like pattern. Stereocilia were gradually missing leading to a missing tip link (e, f). In scanning electron microscopy, the hair cells at top turn and basal turn of cochlea outer hair cell were missing, and the stereocilia were disorganized and the bundles were broken in Cdh23nmf308/nmf308 at 25 days age (h, i). The hair cells at the top turn and basal turn of cochlea outer hair cell were missing and the stereocilia were gradually decreased and disorganized in Cdh23nmf308/nmf308 by 25 days of age.
Next, we used SEM to examine the morphological features of cochlear OHC in experimental mice. In 28 day B6 mice, the three rows of OHC and row of inner hair cells (IHCs) in the cochlea had normal highly organized stereocilia (Fig. 6B, a), with a normal pattern of stereocilia shapes and regular arrangement of adjacent stereocilia bundles (Fig. 6B, b), forming a staircase-like shape and tip link (Fig. 6B, c). Compared with stereocilia of Cdh23nmf308/nmf308 mutant mice (Fig. 6B, d–f), hair cell loss in the mutant cochlea underlied the disrupted appearance of the hair cell pattern. Scanning electron micrographs of the organ of Corti from 28-day old Cdh23nmf308/nmf308 mice, in the apical and middle turns, indicated only a few residual outer hair cells remaining, all of which exhibited disorganized or V-shaped stereocilia among the three rows of organized outer hair cells. OHC loss was also observed in Cdh23nmf308/nmf308 mice (Fig. 6B, d). Disorganized stereocilia on the top of each outer hair cell in Cdh23nmf308/nmf308 mice were not arranged in rows and did not form a staircase-like pattern. Stereocilia were dispersed, which resulted in the missing tip link (Fig. 6B, e and f). SEM revealed missing hair cells at the top turn and basal turn of cochlea outer hair cell, and the stereocilia were disorganized and the bundles were broken in Cdh23nmf308/nmf308 mice at 25 days of age (Fig. 6B, g–i). Together, all these observations demonstrated that there were unique cellular features in Cdh23nmf308/nmf308 mice.
3.7. Cdh23nmf308/nmf308 mice with AHL exhibited OHC cell apoptosis
We proposed that Cdh23nmf308/nmf308 mice had increased OHC apoptosis, which could be linked to AHL. We examined the caspase-3 staining and carried out TUNEL analysis on OHCs. Laser confocal inspection of 42 day Cdh23nmf308/nmf308 mice, indicated that most OHCs were caspase-3 positive, and some OHCs had abnormal morphologies without v-shaped stereocilia bundles. Furthermore, the cytoplasm was not clearly stained (Fig. 7A, a). OHCs in Cdh23ahl/ahl mice were not labeled by caspase-3 and no obvious assembly of caspase-3was observed (Fig. 7A, b). These results demonstrated that Cdh23nmf308/nmf308 mice with AHL exhibited extensive OHC cell apoptosis compared to Cdh23ahl/ahl mice. We next carried out double caspase-3 and PI staining of fixed sections from 57-day old Cdh23nmf308/nmf308 mice. As shown, caspase-3 positive cells were distributed on the organ of Corti, spiral ganglion (SG), and spiral ligament (SLg) (Fig. 7B, a). Caspase-3 expression was particularly dense in the cytoplasm of OHCs (Fig. 7B, d). PI stained positive cells were more widely distributed compared to caspase-3, especially in the nuclei of OHC, SG and stria vascularis (Fig. 7B, b and e). There were overlapping areas within the SG and OHCs that were both caspase-3 and PI positive. In contrast, the SLg only expressed caspase-3, while stria vascularis only expressed PI (Fig. 7B, c and f). Taken together, we concluded that at the molecular level, the mutation in cdh23 was associated with AHL. Our findings at the cellular level, indicating extensive cell apoptosis in OHCs from Cdh23nmf308/nmf308 mice strengthens this conclusion (Fig. 8). Therefore, the relationship between cdh23 mutation and cell apoptosis in OHCs should be investigated further to determine the elaborate mechanism leading to AHL.
Fig. 7.

Cdh23nmf308/nmf308 mice with AHL exhibited OHC cell apoptosis. (A) Laser confocal inspection of OHCs of Cdh23nmf308/nmf308 (a) and Cdh23ahl/ahl mice (b) at 42 days of age. (B) Detection of apoptosis by caspase-3 and PI staining in fixed sections from Cdh23nmf308/nmf308 mice at 57 days of age. Caspase-3 positive cells were distributed in the organ of Corti (OC), SG and SLg (a), especially in the cytoplasm of OHCs (d). SG and OHC were overlapping with both caspase-3 and PI positive cells. In contrast, the SLg only expressed cas-3, while StV was only stained by PI (c, f). Scale bar: 20 μm.
Fig. 8.

Cdh23nmf308/nmf308 mice with AHL exhibited OHC cell apoptosis with TUNEL staining. Cdh23nmf308/nmf308 mice (postnatal days 20) were sacrificed at 46 days of age and the inner ear was fixed. Fluorescence microscopy image of whole mount cochlea preparations using propidium iodine (PI, red) and TUNEL staining (green). Ears receiving no treatment exhibited significantly more TUNEL-positive apoptotic OHC in organ of Corti, most OHCs were already missing in the basal turn, but a few OHCs were also missing in apical and middle turn (arrows).
4. Discussion
CDH23, as a member of the cadherin superfamily of cell surface adhesion proteins, is encoded by 69 exons covering 350 kb with size from 3 bp (exon 33) to 927 bp (exon 69). Among these coding exons, 68 SNPs have been identified with various mutations that either alter CDH23 structure or generate premature proteins resulting in multiple diseases. For instance, mutations in cdh23 attribute to AHL, usher syndrome type 1 subtype D, and a form of autosomal recessive hearing loss designated DFNB12 (Noben-Trauth et al., 2003). Besides hearing loss, Cdh23nmf308/nmf308 mutation can lead to balance dysfunction and pigmented retinopathy in human and rats (Schultz, et al., 2011).
A multi-generation family with apparent autosomal recessive non-syndromic childhood hearing loss has been well documented. Previous studies revealed that patients from two branches of the multi-generation family (A and B) carry two new mutations in cdh23 (DFNB12) (Santos et al., 2005. Astuto et al., 2002). A homozygous CDH23 mutation has been found in branch A (D2148N) and in branch B (D1341N) (Astuto et al., 2002). Substituted aspartic acid residues are highly conserved and are part of the calcium-binding sites of the extracellular cadherin (EC) domains. Molecular modeling of the mutated EC domains of CDH23 demonstrated that calcium binding is impaired. The two mutations in DFNB12 are also likely to decrease CDH23 affinity to calcium. Since calcium provides rigidity to the elongated structure of cadherin molecules, enabling homophilic lateral interactions, these mutations may limit interactions of CDH23 either with CDH23 or with other proteins. Though the two mutations and some binding proteins of CDH23 have documented that cdh23 is a key factor to recessive hearing loss, it is still unclear whether there are other mutations in cdh23 that are involved in progressive aging-related hearing loss (AHL). To systematically investigate the role of cdh23 in AHL, we comprehensively characterized Cdh23nmf308/nmf308 mice for AHL and loss of hair cells. We demonstrated that cdh23 is a critical gene underlying AHL. A novel mutation (208 T>C) at exon3 of cdh23 was found, which leads to amino acid substitution of Ser 70 to Pro in the EC region and alteration in the second structure of CDH23 (Figs. 2, 3 and 6). We then reasoned that the mutation in cdh23 and the alteration in CDH23 structure might be responsible for AHL in nmf 308 mice.
In that nmf 308 mice have degeneration of the organ of Corti, with loss of hair cells along the base to apex (Fig. 6). More detailed analysis confirmed that the profound deafness was caused by disorganization and degeneration of the organ of Corti. Our study supports Schuknecht established criteria that sensory presbycusis is any uninterrupted loss of hair cells beginning at the basal end of the cochlea, which is at least 10 mm in length so as to involve the speech frequency area of cochlea (Schuknecht, 1964). Abnormalities of cochlear hair cell stereocilia were apparent in the mice at postnatal day 21 (P21). Disorganization of outer hair cell stereocilia was observed as early as P21 where the tip links were no longer observed, and using scanning electron microscopy and immunohistochemical staining, we observed that the hair cells at the top turn and basal turn were absent and the stereocilia became gradually dispersed disorganized in two-month old mice (Fig. 6B, e–f). This result suggested that the cdh23 mutation may be harmful to stereociliary tip link and causes hair cell apoptosis, leading to early onset age-related hearing loss in Cdh23nmf308/nmf308 mice.
Strong evidence indicates that CDH23 is a tip link component and that these tip link defects affect mechanosensory transduction in zebrafish, even in the absence of gross structural defects in hair cell bundles (Sollner et al., 2004). Since the tip link has been proposed as a part of the mechanotransduction system (Sollner et al., 2004), the loss of the tip link removes structural support necessary for normal stereociliary organization, producing stereociliary disarray (Fig. 6B). Mutations in cdh23 also result in stereociliary disarray as indicated by the lack of CDH23 staining in our study. CDH23 has been shown to coimmunoprecipitate with myosin 1c, a motor involved in the stereocilia mechotransduction system (Vollrath, et al., 2007). Functional studies, electron microscopy, and protein modeling support the hypothesis that CDH23 creates the tip link in mature animals. Cadherin 23 function in the mechanotransduction system may be evolutionarily conserved across species (Marcos, et al., 2010; Müller, 2008).
The importance of CDH23 is not limited to a component of tip link in the mechanotransduction system. It may also function by binding extracellular molecules and may also transmit signals outside of the cells through transmembrane and intracellular domains. The amino acid substitution in CHD23 occurring in nmf308 mice is apparent in the EC1 domain, which may influence the binding of CDH23 with other molecules and affect cell signal transduction. Caspase-3 is the most extensively studied apoptotic protein, and sensory hair cell degeneration is associated with activation of this pathway, even in response to different ototoxic stimuli (Cheng et al., 2005). Inhibition of caspase prevents or delays hair cell death and may preserve hearing/balance function. Inhibition of mitogen-activated protein kinase protects against noise-induced and aminoglycoside-induced, but not cisplatin-induced hair cell death, which suggests divergent upstream regulatory mechanisms. In this study we showed that in nmf308 mice at 2 months age, the cochlear outer hair cell had activated caspase in the basal turn (Fig. 7). These studies indicate that the activation of the apoptotic process is a central event in AHL. Therefore, investigation of the apoptotic process in aged cochlea and the regulation thereof may lead to promising therapeutic interventions that can prevent and rescue presbycusis.
Acknowledgments
This research was supported by the National Natural Science Foundation of China (39970785, 81170922 and 30440080), the International Collaborate Research Foundation of National Natural Science of China (322200462), and the National Institutes of Health (R01DC007392 and R01DC009246).
Abbreviations
- AHL
age-related hearing loss
- ENU
N-nitrosourea
- DPOAE
distortion product otoacoustic emission
- ABR
acoustic brainstem evoked response
- SEM
scanning electron microscope
- TEM
transmission electron microscope
- TUNEL
terminal deoxynucleotidyl transferase mediated dUTP nick end labeling
- EC1
cadherin 1
- IHCs
inner hair cells
References
- Ackert-Bicknell, et al. A chromosomal inversion within a quantitative trait locus has a major affect on adipogenesis and osteoblastogenesis. Ann NY Acad Sci. 2007;1116:291–305. doi: 10.1196/annals.1402.010. [DOI] [PubMed] [Google Scholar]
- Adato A, et al. Interactions in the network of Usher syndrome type 1 proteins. Hum Mol Genet. 2005;14:347–356. doi: 10.1093/hmg/ddi031. [DOI] [PubMed] [Google Scholar]
- Alagramam KN, et al. Mutations in protocadherin 15 and cadherin 23 affect tip links and mechanotransduction in mammalian sensory hair cells. PLoS One. 2011;6(4):e19183. doi: 10.1371/journal.pone.0019183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astuto LM, et al. CDH23 mutation and phenotype heterogeneity: a profile of 107 diverse families with Usher syndrome and nonsyndromic deafness. Am J Hum Genet. 2002;71:262–275. doi: 10.1086/341558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boëda B, Weil D, Petit C. A specific promoter of the sensory cells of the inner ear defined by transgenesis. Hum Mol Genet. 2001;15:1581–1589. doi: 10.1093/hmg/10.15.1581. [DOI] [PubMed] [Google Scholar]
- Bolz H, et al. Mutation of CDH23, encoding a new member of the cadherin gene family, causes Usher syndrome type 1D. Nat Genet. 2001;27:108–112. doi: 10.1038/83667. [DOI] [PubMed] [Google Scholar]
- Bork JM, et al. Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB12 are caused by allelic mutations of the novel cadherin-like gene CDH23. Am J Hum Genet. 2001;68:26–37. doi: 10.1086/316954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng AG, Cunningham LL, Rubel EW. Mechanisms of hair cell death and protection. Curr Opin Otolaryngol Head Neck Surg. 2005;6:343–348. doi: 10.1097/01.moo.0000186799.45377.63. [DOI] [PubMed] [Google Scholar]
- DeStefano AL, et al. Genomewide linkage analysis to presbycusis in the Framingham Heart Study. Arch Otolaryngol Head Neck Surg. 2003;129:285–289. doi: 10.1001/archotol.129.3.285. [DOI] [PubMed] [Google Scholar]
- Erway LC, Willott JF, Archer JR, Harrison DE. Genetics of age-related hearing loss in mice: I. Inbred and F1 hybrid strains. Hear Res. 1993;65:125–132. doi: 10.1016/0378-5955(93)90207-h. [DOI] [PubMed] [Google Scholar]
- Holt JR, et al. A chemical-genetic strategy implicates myosin-1c in adaptation by hair cells. Cell. 2002;108:371–381. doi: 10.1016/s0092-8674(02)00629-3. [DOI] [PubMed] [Google Scholar]
- Hu BH, Henderson D, Nicotera TM. Extremely rapid induction of outer hair cell apoptosis in the chinchilla cochlea following exposure to impulse noise. Hear Res. 2006;211:16–25. doi: 10.1016/j.heares.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Johnson KR, et al. A major gene affecting age-related hearing loss in C57BL/6J mice. Hear Res. 1997;114:83–92. doi: 10.1016/s0378-5955(97)00155-x. [DOI] [PubMed] [Google Scholar]
- Johnson KR, Zheng QY, Erway LC. A major gene affecting age-related hearing loss is common to at least ten inbred strains of mice. Genomics. 2000;70:171–180. doi: 10.1006/geno.2000.6377. [DOI] [PubMed] [Google Scholar]
- Li SL, et al. Assessment criteria for rotated stereociliary bundles in the guinea pig cochlea. Otol Neurotol. 2008;29:86–92. doi: 10.1097/mao.0b013e31815dbb5b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcos S, et al. Structural determinants of cadherin-23 function in hearing and deafness. Neuron. 2010;66:85–100. doi: 10.1016/j.neuron.2010.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller U. Cadherins and mechanotransduction by hair cells. Curr Opin Cell Biol. 2008;20:557–566. doi: 10.1016/j.ceb.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noben-Trauth K, Zheng QY, Johnson KR. Association of cadherin 23 with polygenic inheritance and genetic modification of sensorineural hearing loss. Nat Genet. 2003;35:21–23. doi: 10.1038/ng1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlemiller KK, Lett JM, Gagnon PM. Cellular correlates of age-related endocochlear potential reduction in a mouse model. Hear Res. 2006;220:10–26. doi: 10.1016/j.heares.2006.06.012. [DOI] [PubMed] [Google Scholar]
- Petit C, Levilliers J, Hardelin JP. Molecular genetics of hearing loss. Annu Rev Genet. 2001;35:589–646. doi: 10.1146/annurev.genet.35.102401.091224. [DOI] [PubMed] [Google Scholar]
- Santos RLP, et al. Low prevalence of Connexin 26 (GJB2) variants in Pakistani families with autosomal recessive non-syndromic hearing impairment. Clin Genet. 2005;67:61–68. doi: 10.1111/j.1399-0004.2005.00379.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuknecht HF. Further observation on the pathology of presbyacusis. Arch Otolaryngol. 1964;80:369–382. doi: 10.1001/archotol.1964.00750040381003. [DOI] [PubMed] [Google Scholar]
- Schultz JM, et al. Allelic hierarchy of CDH23 mutations causing non-syndromic deafness DFNB12 or Usher syndrome USH1D in compound heterozygotes. J Med Genet. 2011;48:767–775. doi: 10.1136/jmedgenet-2011-100262. [DOI] [PubMed] [Google Scholar]
- Slemens JS, Lillo C, Dumont RA. Cadherin 23 a component of the tip link in hair-cell stereocilia. Nature. 2004;428:950–954. doi: 10.1038/nature02483. [DOI] [PubMed] [Google Scholar]
- Sollner C, et al. Mutations in cadherin 23 affect tip links in zebrafish sensory hair cells. Nature. 2004;428:955–959. doi: 10.1038/nature02484. [DOI] [PubMed] [Google Scholar]
- Taylor BA, Bedigian HG, Meier H. Genetic studies of the Fv-1 locus in mice linkage with Gpd-1 in recombinant inbred lines. J Virol. 1997;34:106–109. doi: 10.1128/jvi.23.1.106-109.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollrath MA, Kwan KY, Corey DP. The micromachinery of mechanotransduction in hair cells. Annu Rev Neurosci. 2007;30:339–365. doi: 10.1146/annurev.neuro.29.051605.112917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng QY, Johnson KR. Hearing loss associated with the modifier of deaf waddler(mafw) locus corresponds with age-related hearing loss in 12 inbred strains of mice. Hear Res. 2001;154:45–53. doi: 10.1016/s0378-5955(01)00215-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng QY, Johnson KR, Erway LC. Assessment of hearing in 80 inbred strains of mice by ABR threshold analyses. Hear Res. 1999;130:94–107. doi: 10.1016/s0378-5955(99)00003-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng QY, et al. Digenic inheritance of deafness caused by mutations in genes encoding cadherin 23 and protocadherin 15 in mice and humans. Hum Mol Genet. 2005;14:103–111. doi: 10.1093/hmg/ddi010. [DOI] [PMC free article] [PubMed] [Google Scholar]