

Abstract
The anthrax protective antigen (PA) binds to the host cellular receptor capillary morphogenesis protein 2 (CMG2) with high affinity. To gain a better understanding of how pH may affect binding to the receptor, we have investigated the kinetics of binding as a function of pH to the full-length monomeric PA and to two variants: a 2-fluorohistidine-labeled PA (2-FHisPA), which is ∼1 pH unit more stable to variations in pH than WT, and an ∼1 pH unit less stable variant in which Trp346 in the domain 2β3-2β4 loop is substituted with a Phe (W346F). We show using stopped-flow fluorescence that the binding rate increases as the pH is lowered for all proteins, with little influence on the rate of dissociation. In addition, we have crystallized PA and the two variants and examine the influence of pH on structure. In contrast to previous X-ray studies, the domain 2β3-2β4 loop undergoes little change in structure from pH ∼8 to 5.5 for the WT protein, but for the 2-FHis labeled and W346F mutant there are changes in structure consistent with previous X-ray studies. In accord with pH stability studies, we find that the average B-factor values increase by ∼20–30% for all three proteins at low pH. Our results suggest that for the full-length PA, low pH increases the binding affinity, likely through a change in structure that favors a more “bound-like” conformation.
Keywords: protective antigen, anthrax, CMG2, pH stability, binding, stopped-flow
Introduction
The association of the full-length, monomeric 83 kDa anthrax protective antigen (PA) to the host receptor is a key step leading to the formation of the anthrax toxin. Upon binding to the host receptor,1–3 PA is cleaved by a cell-surface furin-like protease, releasing a 20 kDa fragment.4 This allows the remaining 63 kDa fragment, bound to the receptor, to oligomerize into heptameric5 or octameric6 complexes, called collectively the prepore. The prepore creates binding sites for the two enzymatic moieties of the toxin, edema factor (EF) and lethal factor (LF).7,8 The toxin is then endocytosed into the cell by receptor-mediated endocytosis,9,10 and subsequently trafficked to late endosomes which become acidic. In this low pH environment (pH = 5–6), the prepore undergoes a major conformational change, forming a 14-stranded β-barrel pore.11–13 It is through the β-barrel pore that EF and LF must translocate through, from the acidified endosome, into the cell cytosol.14 Once in the cytosol, EF functions as an adenylate cyclase15 which raises cellular cyclic-AMP levels, altering chemotaxis,16 and LF is a zinc-metalloproteinase that cleaves mitogen activated protein kinase kinases (MAPKKs),17 disrupting cell integrity.18
Based on the crystal structures of PA19 or the heptameric prepore (PA63)720 bound to the soluble von-Willebrand factor A (vWA) domain of the receptor, capillary morphogenesis protein 2 (CMG2), the contact interfaces between the two proteins are well defined. Domain 4 of PA has been shown to be required and sufficient for binding to CMG2.1,21 Domain 4 accounts for the largest binding interface (∼1300 Å2), and contributes an aspartic acid (D683) to the coordination of a magnesium ion that sits in a metal-ion dependent adhesion site (MIDAS) on the surface of CMG2. Domain 2 also contributes to the binding interface by inserting a small loop, the domain 2β3-2β4 loop, into a groove on the surface of CMG2. Residues in CMG2 that contact this loop, or that contact residues in domain 4, have been shown to modulate the pH dependence of pore formation.22,23
The first crystal structures of PA,24 solved at pH 7.5 and 6 by Petosa et al., indicate that the domain 2β3-2β4 loop becomes disordered at pH 6, suggesting that this loop is sensitive to pH. Because this loop has been shown to be an important determinant in binding to CMG2,22,23 the pH sensitivity of this loop may impact the rate of binding or dissociation of PA to the receptor. In addition, recent evidence by NMR suggests that receptor contacts to this loop may be the first contacts to break in the transition to forming a pore.25 To understand how pH affects PA binding to the vWA domain of CMG2, we have determined the rates of association and dissociation as a function of pH to WT PA and to two other variants of PA—one in which PA is biosynthetically labeled with 2-fluorohistidine (2-FHis), an analog of histidine with a reduced side-chain pKa,26 and another in which tryptophan 346 (W346) in the domain 2β3-2β4 loop is mutated to Phe (W346F). We show that lowering the pH increases the rate of association of all proteins to CMG2, with little effect on rates of dissociation. Further, we show that the Arrhenius activation barriers decrease as the pH is lowered, but the activation barriers at low pH (pH ∼6) are lowest for the least pH stable, slowest associating protein (W346F) and highest for the most pH stable, fastest associating protein (2-FHis). We have also determined the three-dimensional structures of the WT, 2-FHis and W346F mutant proteins using X-ray crystallography, at acidic and basic pH values. The proteins assume similar three-dimensional structures, at high and low pH values. In contrast to previous studies, we do not observe a significant change in the domain 2β3-2β4 loop for the WT protein at acidic pH. However, the overall main-chain B-factors for all proteins increase by ∼20–30% as the pH is lowered. Our studies suggest that low pH shifts an equilibrium between “unbound” and “bound” states, favoring the bound state, leading to a faster rate of association and an overall lowering of the KD for binding.
Results
Equilibrium binding and kinetics of association as a function of pH
Because of the observed high affinity of binding between PA and CMG2 (KD ∼170 pM), Wigelsworth et al. utilized surface plasmon resonance as well as fluorescence resonance energy transfer (FRET) to monitor association and dissociation kinetics of PA to CMG2, establishing on and off rates and using these values to determine a KD.27 In the experiments presented here, we also utilized FRET to monitor association of PA to CMG2 as a function of pH. In our case, we have labeled a mutant of PA, E712C, with the maleimide form of AlexaFluor350 (AF350), and have labeled the R40C mutant of CMG2 (CMG2C40)27 with AF488. These two fluorophores have a Förster distance (Ro) of ∼50 Å, and are sufficiently close in the bound state to allow FRET (∼44 Å). We chose E712C since E712, located in domain 4, is in close proximity to domain 2 and specifically W346 (∼17 Å), which is part of the domain 2β3-2β4 loop. The Förster distance between tryptophan and AlexaFluor 350 is predicted to be ∼20 Å.28 E712C is a nonstructurally perturbing mutation,29 and labeling this residue with a fluorophore would allow us, in future experiments, to report on changes in structure at the domain 2-domain 4 interface (Fig. 1).
Figure 1.
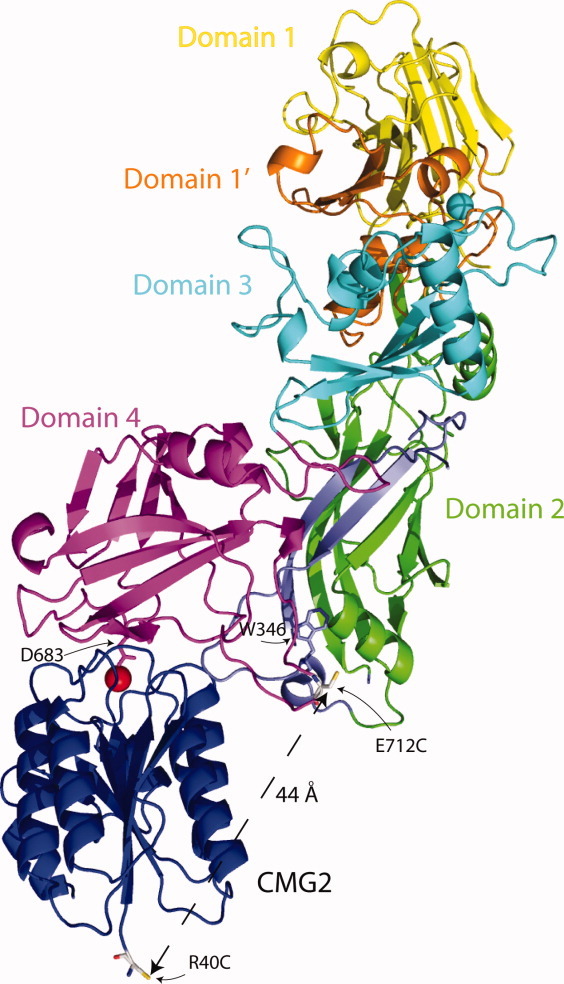
Structure of WT PA bound to CMG2 (PDB: 1T6B) with side chains of E712C and the R40C (CMG2—blue) shown as gray sticks. Domain 1 (1–167) is colored in yellow; domain 1′ (168–258) is colored orange; domain 2 (259–487) is colored green with the residues that comprise the pore (275–352) shown in blue; W346 within the domain 2β3-2β4 loop is shown as a blue stick figure; domain 3 (488–595) is colored cyan and domain 4 (596–735) is colored magenta. The manganese ion on the surface of CMG2 is colored red. The distance between E712C in domain 4 (magenta) and R40C in CMG2 is shown (44 Å). Figure was rendered using Pymol v1.3 (http://pymol.org/ep).
In a similar manner, we measured binding of CMG2 to two other variants of PA—one in which the E712C PA was uniformly, biosynthetically labeled with 2-fluorohistidine (2-FHis), an analog of histidine with a reduced side-chain pKa.26,30,31 Biosynthetic incorporation of 2-FHis results in a protein which is more stable to variations in pH than WT PA (Fig. 2).30 This protein was subsequently labeled at the E712C with AlexaFluor350 maleimide. We also generated a double mutant, E712C/W346F. W346F was made in the course of our studies to investigate the contribution of W346 to the overall quantum yield of fluorescence of PA (which contains seven tryptophans), and may serve as a fluorescent donor to the nearby E712C-AF350. This mutation decreases the pH stability by ∼1 pH unit (Fig. 2) relative to the WT PA protein. Despite differences in pH stability, both E712C and W346F mutants are able to mediate toxicity of CHO-K1 cells by LFN-DTA, a fusion containing the N-terminal PA binding domain of LF (LFN) and the catalytic A domain of diphtheria toxin (DTA) (Fig. 1, Supplementary Information), indicating that interactions with the host receptor, pore formation or translocation are not affected by these mutations.
Figure 2.
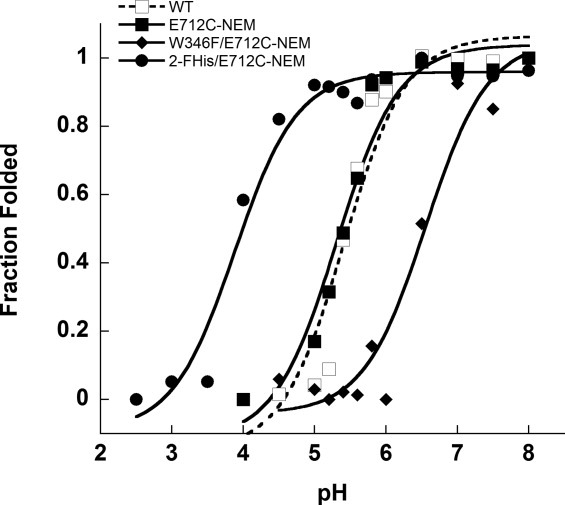
Peak fluorescence emission intensity (Ex. 280 nm) of WT (□), E712C-NEM (▪), 2-FHis/ E712C-NEM (•), and W346F/E712C-NEM (♦) as a function of pH. All measurements were carried out at 20°C in 20 mM Bis-Tris/HEPES/cacodylate/citrate on a Cary Eclipse fluorescence instrument. Fluorescence intensity was plotted against pH and the data were fitted to Henderson–Hasselbalch equation assuming two state protonation equilibrium: Fl(obs) = (Fl(N) + (10pH – pKapp) × Fl(U))/(1 + 10pH-pKapp) where pKapp represents an apparent pKa encompassing all titratable sites. The fits gave a pKapp of 5.4 ± 0.1 for WT, 5.3 ± 0.1 for E712C, 3.9 ± 0.1for 2-FHis-E712C, and 6.6 ± 0.2 for W346F PA. The data were normalized to give fraction folded molecules as a function of pH.
Equilibrium binding was determined to be 1:1.3 (PA E712C), 1:1 (E712C/W346F), and 1:1.2 (2-FHis E712C) (Fig. 2, Supplementary Information), indicating that binding followed a ∼1:1 stoichiometry, as observed previously.27 Because of the high affinity of binding of PA to CMG2 (∼170 pM27), determining the binding affinity using equilibrium methods by fluorescence was not possible. To determine the binding affinity as a function of pH, we monitored the rates of association between WT PA, 2-FHisPA, and W346F PA E712C-AF350 labeled proteins using stopped-flow, and manual mixing experiments to monitor dissociation, as was done previously.27 We refer hereafter to the WT PA, 2-FHisPA, and W346F E712C-AF350 variants as simply WT PA, 2-FHisPA, and W346F, unless otherwise specified. Similarly, we refer to CMG2C40-AF488 as CMG2, unless otherwise specified. Figure 3 shows the plots of the rate constants for association as a function of CMG2 concentration, measured at different pH values. The data were recorded under pseudo first-order conditions, and plots were fit to the linear equation kobs = ka[CMG2] + kd, where ka is the second-order rate of association (M−1 s−1), and kd is the first-order rate of dissociation.27
Figure 3.
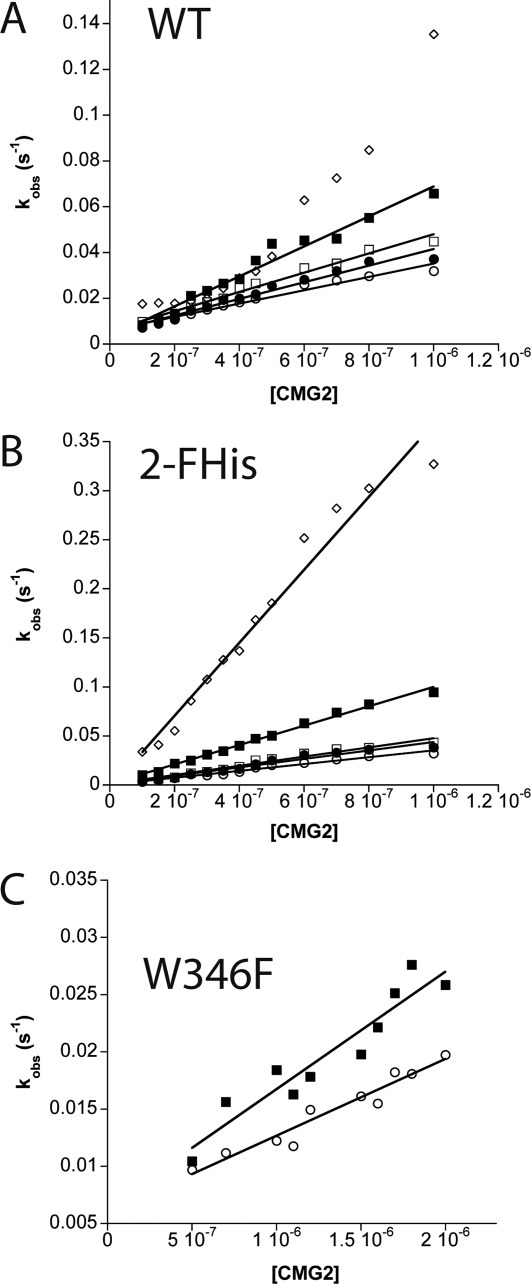
Kinetics of association as a function of CMG2C40-AF488 concentration for (A) WT PA, (B) 2FHis, and (C) W346F. The pH values measured include pH 8 (open circle), pH 7.5 (closed circle), pH 7 (open square), pH 6 (closed square), and pH 5 (open diamond) are shown. Association rate constants (ka) were calculated using the slope of the linear fits. All kinetic experiments were carried out at 20°C in a buffer system containing 20 mM Tris/MES/HEPES/acetic acid with 0.1 mg mL−1 BSA and 2 mM MgCl2.
The rate of association is pH-dependent, with rate constants for binding increasing as the pH is lowered. The rate constants for association of the WT PA vary approximately twofold from pH 8 to 6 (∼3 × 104 M−1 s−1 to ∼ 6 × 104 M−1 s−1), and ∼10 fold for the 2-FHisPA proteins (3.5 × 104 M−1 s−1 at pH 8 to 3.7 × 105 M−1 s−1 at pH 5). Although the rate constants versus pH data could be fit to a linear equation, the nonlinearity at pH 5 for WT PA made an accurate determination of the rate constant not possible. This may be due in part to the pH instability of WT PA at pH 5.0, which exhibits an overall pKapp of 5.3 (Fig. 2). For the W346F mutant, the increased rate of binding from pH 8 to 6 is small (∼1.6 fold), and the lowest pH that we were able to measure was 6, again due to the pH instability of this protein.
Receptor dissociation as a function of pH
To monitor the pH-dependence of receptor dissociation, we carried out experiments in a manner similar to that described previously,27 in which binding of a twofold excess of CMG2 to WT PA, 2-FHisPA, and W346F was carried out by monitoring the ratio of the acceptor fluorescence emission at 516 nm to the donor emission at 440 nm. Once the intensity remained constant for an extended period (∼10,000 s), an ∼20-fold excess of N-ethyl-maleimide (NEM) labeled CMG2C40 (CMG2C40-NEM) was added to initiate dissociation. Under these conditions, it is assumed that the displaced fluorescently labeled CMG2 cannot reassociate with the labeled PA, due to the large excess of nonfluorescent (NEM-labeled) CMG2, and thus the determined rate constant is the dissociation constant. Also, since R40C is distant from the binding interface, we assume that the association and dissociation kinetics for both the fluorescently labeled and NEM labeled proteins are similar. The dissociation data obtained at various pH values are shown in Figure 4. The data indicate that the rate of dissociation for WT PA is pH independent, from pH 8 to 5. For the 2-FHisPA, we observed little change in dissociation from pH 8 down to pH 6, but an approximately twofold increase in the rate of dissociation at pH 5. For W346F, we also observe little change in the rate of dissociation between pH 8 and 6, but a approximately eightfold increase in the rate of dissociation compared to either the WT PA or 2-FHisPA proteins. Again, we suspect that the decreased stability of the W346F protein leads to the increased rate of dissociation. A summary of all data, including association and dissociation rate constants and corresponding equilibrium dissociation constants as a function of pH, are shown in Table I.
Figure 4.
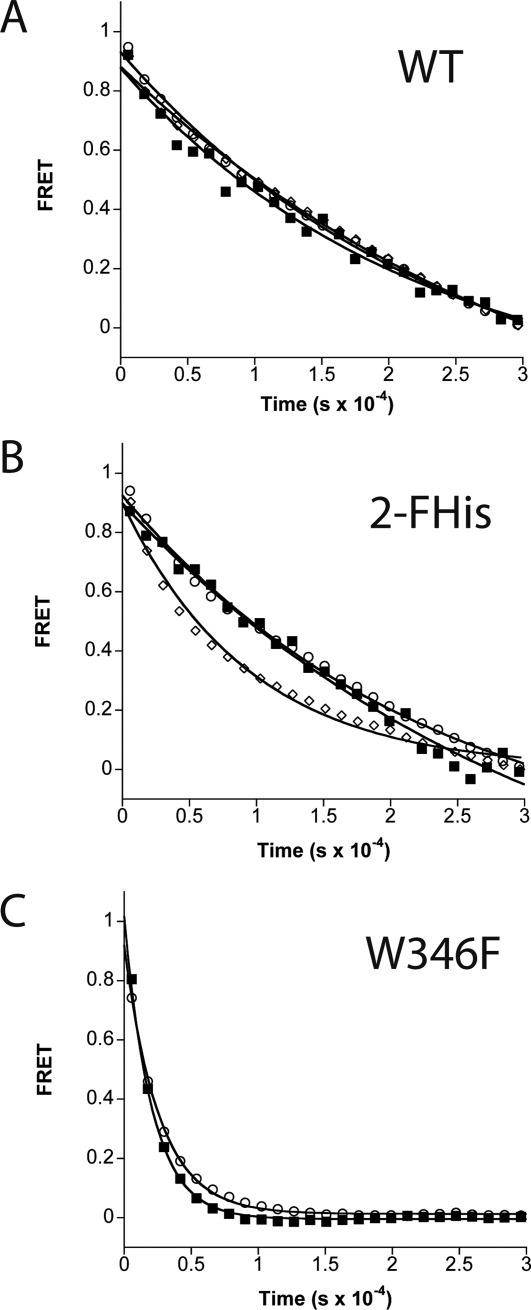
FRET-based dissociation kinetic experiments of WT PA (A), 2FHis (B) and W346F (C) at pH 8 (open circles), 6 (closed squares), and 5 (open diamonds). Dissociation was initiated by the addition of a 20-fold excess of CMG2C40-NEM (2 μM), and monitoring the loss of FRET signal. Lines through the data are best fits using nonlinear least squares analysis using a single-exponential function.
Table I.
Effect of pH on Kinetics and Thermodynamics of Binding PA to CMG2
| PA variant | pH | ka (×104) (M−1 s−1) | kd (×10−5) (s−1) | KDa (M) | EA (kJ mol−1) |
|---|---|---|---|---|---|
| WT | 8 | 2.90 (0.02)b | 4.00 (0.08) | 1.4 × 10−9 | 44.70 (0.07) |
| 8c | 3.00 (0.05) | ||||
| 7.5 | 3.60 (0.02) | 41.00 (0.13) | |||
| 7 | 4.20 (0.01) | 40.20 (0.05) | |||
| 6 | 6.50 (0.02) | 4.60 (0.16) | 7.0 × 10−10 | 30.20 (0.04) | |
| 6c | 5.90 (0.04) | ||||
| 5 | ND | 4.40 (0.08) | ND | 30.00 (0.12) | |
| W346F | 8 | 0.63 (0.03) | 38.40 (0.16) | 6.1 × 10−8 | 51.00 (0.10) |
| 6 | 1.00 (0.05) | 48.30 (0.21) | 4.8 × 10−8 | 23.80 (0.23) | |
| 2FHis | 8 | 3.50 (0.01) | 4.40 (0.08) | 1.3 × 10−9 | 49.20 (0.10) |
| 7.5 | 4.30 (0.02) | 48.20 (0.12) | |||
| 7 | 4.70 (0.02) | 41.50 (0.07) | |||
| 6 | 10.00 (0.004) | 3.10 (0.16) | 3.1 × 10−10 | 41.90 (0.02) | |
| 5 | 37.00 (0.02) | 10.50 (0.16) | 2.8 × 10−10 | 34.70 (0.15) |
The equilibrium dissociation constant is calculated from the association and dissociation rate constants according to KD = kd/ka.
Error is in parentheses
Measured using constant ionic strength conditions: 100 mM Tris/50 mM MES/50 mM Acetic acid.32
Activation barriers to association
To determine the energetic barriers to association, we monitored the temperature dependence of the rate of association as a function of pH. Figure 5 shows the Arrhenius plots for association, which are linear throughout a wide temperature range (5–23°C). For WT PA, the barrier decreases from a ΔG‡ ∼44 kJ mol−1 at pH 8 to ∼30 kJ mol−1 at pH 5.0. Similarly, for 2-FHisPA, the ΔG‡ decreases from ∼49 kJ mol−1 to ∼34 kJ mol−1 at pH 5.0. However, the W346F mutant, which is less stable to pH, exhibits a ΔG‡ from ∼49 kJ mol−1 at pH 8.0 to ∼23 kJ mol−1 at pH 6. The values for the activation barrier are lower than values reported for amide bond isomerization (70–90 kJ mol−133,34), suggesting that binding does not involve an amide bond isomerization process, but are consistent with activation barriers for loop formation found in small peptides (∼35 kJ mol−1).35
Figure 5.
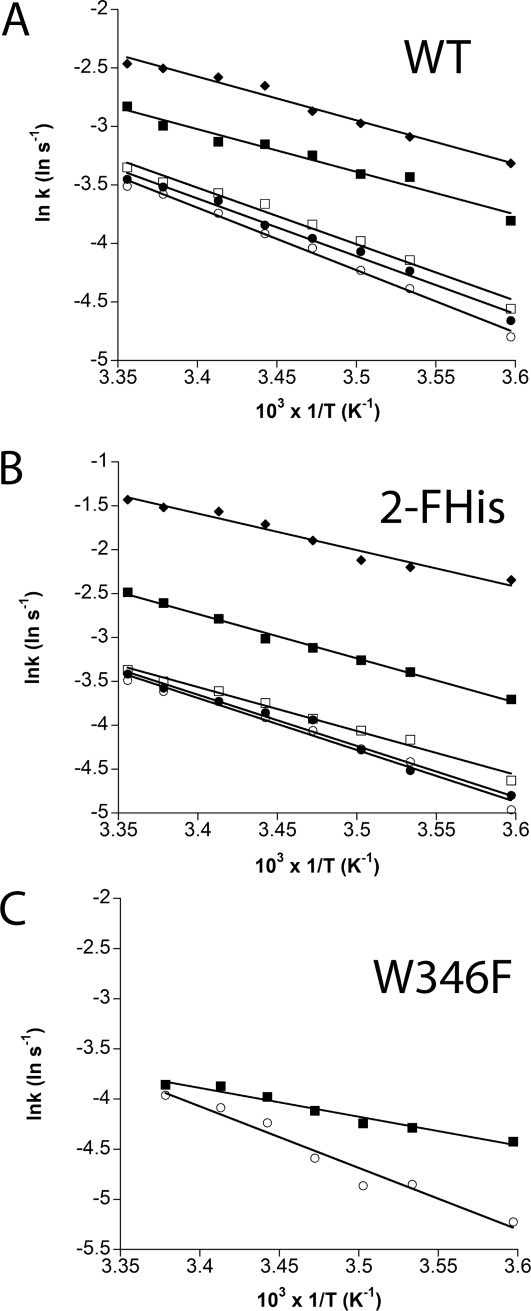
Arrhenius plots showing the effect of temperature on the rate of association at different pH values. Association rate constants of WT PA, 2FHis and W346F were determined at pH 8 (open circle), pH 7.5 (closed circle), pH 7 (open square), pH 6 (closed square), and pH 5 (open diamond). The lines through the data are best fits using linear regression.
Crystal structures of WT PA, 2-FHisPA and W346F as a function of pH
To address whether structural changes occur in the proteins at low pH that might account for the increased rate of association, we crystallized the WT PA, W346F, and 2-FHis PA (where E712 has not been mutated cysteine) at basic and acidic pH values, and determined their three-dimensional structures (Fig. 6). The crystallization parameters are shown in Table II. We have reported recently the structure of 2-FHisPA (PDB: 3MHZ), which was determined to 1.7 Å at pH 8.2,31 and report here the structure of 2-FHisPA at pH 5.8 solved to 2.10 Å. The structures of W346F were solved at pH 8.5 and 5.5, with resolutions of 2.10 and 2.85 Å, respectively. The structures of WT PA were solved at pH 9.0 and 5.5, with resolutions of 2.0 and 3.13 Å, respectively. We note that the previously determined structures of PA, crystal form 1 at pH 7.5 (2.5 Å) and crystal form 2 at pH 6.0 (2.1 Å),24 while both crystallized in the space group P212121, the unit cell parameters were different: crystal form 1, a = 119.8 Å, b = 73.8 Å, and c = 95.0 Å; crystal form 2, a = 99.1 Å, b = 93.7 Å, and c = 82.0 Å. The unit cell parameters for the proteins reported here, which also crystallized in the P212121 space group, are (mean ± std. dev.): a = 71.97 ± 0.45; b = 93.79 ± 0.08 and c = 116.13 ± 0.47. Because these crystals exhibit similar unit cell parameters, and the conditions for crystallization at a given pH are similar, the structural changes observed should be comparable from one protein to the other.
Figure 6.
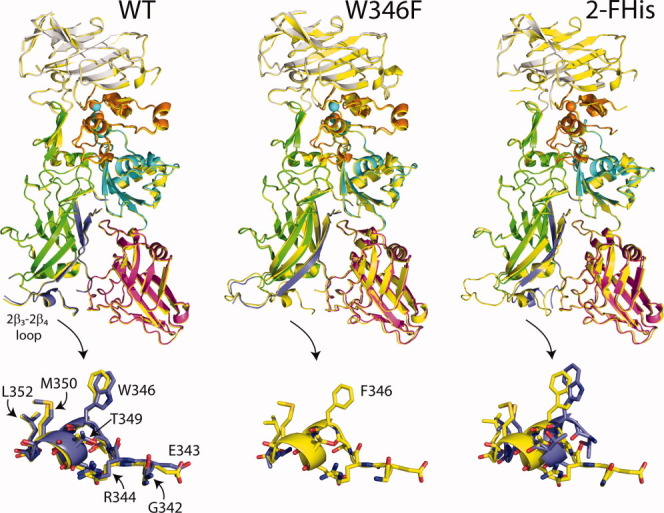
Overlay of WT PA, 2-FHis, and W346F crystal structures at basic (yellow) and acidic pH values. For the acidic pH structures, domain 1 is shown in grey, domain 1′ is orange, domain 2 is green, domain 3 is cyan and domain 4 is magenta. Residues 275–352, which comprise the β-barrel portion of the pore are shown in blue. Below each structure we have zoomed in on residues within the 2β3-2β4 loop (G342-L352). Note that no loop structure is observed for the W346F mutant at acidic pH. Overlay was carried out in Coot,36 and visualized using Pymol v1.3 (http://pymol.org/ep). An interactive view is available in the electronic version of the article.
Table II.
Crystallographic Data for PA Structures
| WT-PA (pH 9.0) | WT-PA (pH 5.5) | W346F-PA (pH 8.5) | W346F-PA (pH 5.5) | 2FHis-PA (pH 5.8) | |
|---|---|---|---|---|---|
| Data collection | |||||
| Unit-cell parameters (Å, °) | a = 71.38, b = 93.80, c = 115.73 | a = 70.84, b = 93.85, c = 114.88 | a = 71.37, b = 93.60, c = 117.21 | a = 72.67, b = 94.05, c = 117.24 | a = 73.29, b = 93.66, c = 115.57 |
| Space group | P212121 | P212121 | P212121 | P212121 | P212121 |
| Resolution (Å)a | 40.0–2.0 (2.11–2.00) | 114.88–3.13 (3.30–3.13) | 117.2–2.1 (2.21–2.10) | 35.00–2.85 (3.00–2.85) | 45.38–2.1 (2.21–2.10) |
| Wavelength (Å) | 1.0000 | 1.0000 | 1.0000 | 1.0000 | 1.0000 |
| Temperature (K) | 100 | 100 | 100 | 100 | 100 |
| Observed reflections | 362,189 | 87,134 | 296,114 | 128,366 | 344,018 |
| Unique reflections | 52,880 | 14,065 | 46,703 | 19,416 | 46,290 |
| Mean <I/σI>)a | 13.0 (2.8) | 11.3 (3.2) | 17.7 (3.1) | 16.5 (2.7) | 20.3 (3.0) |
| Completeness (%)a | 99.4 (98.5) | 99.7 (99.8) | 100 (100) | 99.9 (100) | 98.4 (98.1) |
| Multiplicitya | 6.3 (6.3) | 6.2 (6.8) | 6.3 (6.3) | 6.6 (6.7) | 7.4 (7.5) |
| Rmerge (%)a,b | 8.7 (68.7) | 10.9 (47.7) | 6.0 (49.0) | 9.0 (72.5) | 5.6 (74.6) |
| Rmeasc | 9.4 (74.4) | 13.2 (56.3) | 7.2 (58.9) | 9.8 (81.6) | 6.1 (80.1) |
| Rpimc | 3.6 (28.4) | 5.3 (21.4) | 2.8 (23.2) | 3.8 (31.4) | 2.2 (28.9) |
| Refinement | |||||
| Resolution (Å) | 39.20–2.00 | 56.54–3.13 | 45.29–2.10 | 34.71–2.85 | 43.40–2.10 |
| Reflections (working/test) | 50,123/2,690 | 13,305/696 | 44,130/2,352 | 18,367/991 | 43,869/2341 |
| Rfactor/Rfree (%)d | 20.7/25.8 | 23.6/27.9 | 19.4/24.8 | 20.9/27.1 | 22.2/26.7 |
| No. of atoms (protein/Ca2+/water) | 5290/2/219 | 5273/2/— | 5370/2/243 | 5242/2/— | 5052/2/26/124 |
| Model Quality | |||||
| R.M.S deviations | |||||
| Bond lengths (Å) | 0.012 | 0.007 | 0.012 | 0.007 | 0.011 |
| Bond angles (°) | 1.317 | 1.092 | 1.282 | 1.036 | 1.277 |
| Average B factor (Å2) | |||||
| All atoms | 42.6 | 75.8 | 44.9 | 68.1 | 53.0 |
| Protein | 42.7 | 75.8 | 45.0 | 68.1 | 53.2 |
| Ca2+ | 27.7 | 56.4 | 28.5 | 59.0 | 35.9 |
| Water | 39.2 | — | 42.6 | — | 44.5 |
| Coordinate error based on Maximum Likelihood (Å) | 0.27 | 0.36 | 0.31 | 0.45 | 0.34 |
| Ramachandran Plot | |||||
| Favored (%) | 97.1 | 89.1 | 97.2 | 92.9 | 95.5 |
| Allowed (%) | 2.7 | 9.7 | 2.7 | 6.0 | 3.7 |
Values in parenthesis are for the highest resolution shell.
Rmerge = ∑hkl∑i |Ii(hkl) − <I(hkl)>|/∑hkl∑i Ii(hkl), where Ii(hkl) is the intensity measured for the ith reflection and <I(hkl)> is the average intensity of all reflections with indices hkl.
Rmeas = redundancy-independent (multiplicity-weighted) Rmerge.37 Rpim = precision-indicating (multiplicity-weighted) Rmerge.38
Rfactor = ∑hkl ||Fobs (hkl) | − |Fcalc (hkl) ||/∑hkl |Fobs (hkl)|; Rfree is calculated in an identical manner using 5% of randomly selected reflections that were not included in the refinement.
For the structures reported here, lowering the pH decreases the resolution of all structures, and is likely due to an increase in vibrational motion, as determined by the Debye–Waller factors (B-factors). If we compare the average main chain B-factors between the structures solved at basic and acidic pH, we find that these values increase at acidic pH by ∼20–30% (Fig. 7). Interestingly, for the W346F and 2-FHis labeled PA proteins, we find that the B-factors of the α1 helix of domain 3 increase by ∼40% [Fig. 7(C)], and for the WT PA protein we observe a modest increase in this region [from ∼30 to 40%—Fig. 7(D)]. We note that in crystal form 1 solved at pH 7.524 the α1 helix is observed, but in crystal form 2 (pH 6.0) this helix is missing, consistent with our crystallographic studies indicating that this region exhibits more motion at acidic pH.
Figure 7.
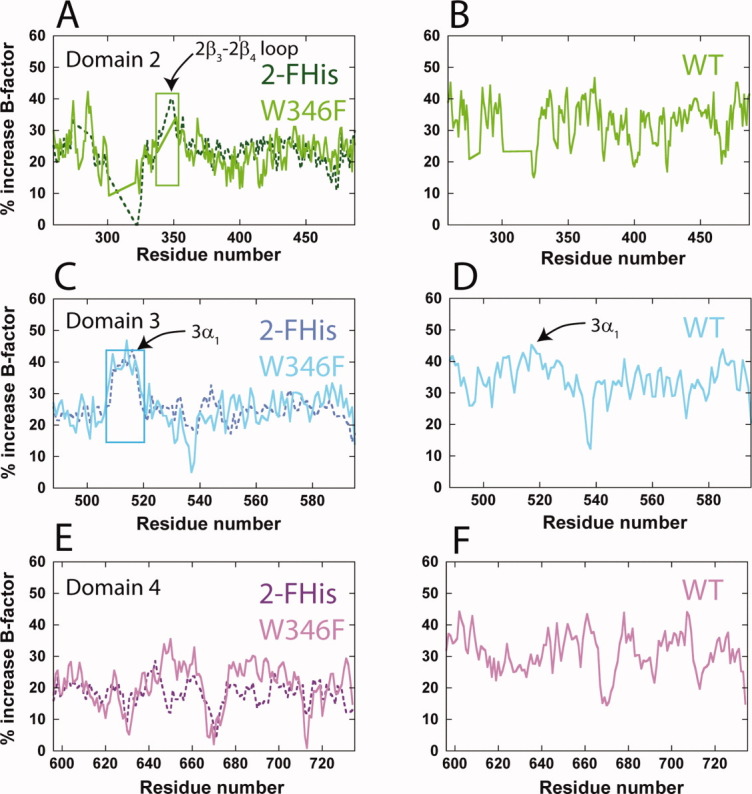
B-factor analysis of domains 2, 3, and 4. Shown are the percent increase in B-factors from basic to acidic pH values for domain 2 (A and B, green), 3 (C and D, cyan), and 4 (E and F, magenta). Panels B, D, and F are the percent increase in B-factors for the WT protein only, for comparative purposes. The color coding is similar to that in Figure 1. The regions encompassing the domain 2β3-2β4 (residues ∼339–352) and the domain 3α1 helix (residues 512–518) are indicated by boxes.
Structural changes in the domain 2β3-2β4 loop
The domain 2β3-2β4 loop has been postulated to be an important determinant in the binding of WT PA to CMG2. Although domain 4 constitutes the largest binding interface (∼1300 Å2), the domain 2β3-2β4 loop binds within a groove on the surface of CMG2, burying an additional 600 Å2.19 This loop has also been postulated by to be sensitive to pH—in the study by Petosa et al. comparing crystal form 1 to crystal form 2,24 the domain 2β3-2β4 loop becomes disordered at pH 6.0, suggesting that this region undergoes a structural change at acidic pH.
For the WT PA protein studied here, we find very little change in structure of the 2β3-2β4 loop upon lowering the pH from 9.0 to 5.5, with only residue L340 missing electron density at either pH (Fig. 6, lower panels). The B-factors for this region increase at acidic pH, but the increase is no different than the average percent increase throughout the protein [Fig. 7(B)]. This is unlikely to be due to altered crystal packing forces, or to changes in contacts between symmetry related molecules.
In contrast, for the 2-FHisPA, the electron density for all residues within the domain 2β3-2β4 loop is observed at pH 8.2, but at pH 5.8, electron density for residues G342 to R344 is missing (Fig. 6). For W346F, electron density for residues L340 to A341 is missing at pH 8.5, and from S339 to G351 at pH 5.5 (Fig. 6). In comparison to the other structures, the larger extent of missing electron density for the domain 2β3-2β4 loop of W346F, at either basic or acidic pH, suggests that the domain 2β3-2β4 loop may be less stable.
Discussion
The influence of pH on the kinetics of association and dissociation of the 83 kDa monomeric PA to the vWA domain of CMG2 was examined. We also examined the association and dissociation kinetics of two variants of PA, 2-FHisPA and W346F PA, whose pH stabilities differ from the WT protein by ∼1 pH unit (1 pH unit above for 2-FHis, and 1 pH unit below for W346F). Our data, using stopped-flow fluorescence to monitor the kinetics, indicate that the rate of association increases for all proteins as the pH is lowered, with little effect on the rate of dissociation. The Arrhenius activation barriers for association also decrease as the pH is lowered, consistent with an increased rate of association. However, the activation barriers at low pH are lowest for the least stable protein (W346F), and highest for the most pH stable protein (2-FHis).
Effects on the kinetics of association: protonation of residues at or near the binding interface
Is the effect we observe on the kinetics of association because of changes in the protonation state of residues at the binding interface, or to structural changes at the binding interface, or to protonation events/structural changes in the protein far from the interface? As mentioned, the binding interface between PA and CMG2 is well defined, and includes contributions from domain 4 and 2.19,20,23 Electrostatic interactions are expected to dominate the interactions between PA and CMG2. In domain 4, coordination of the metal ion at the MIDAS site is an important determinant of binding, where removal of the magnesium ion in the MIDAS site using mixtures of EDTA/EGTA, or changing the metal ion to calcium, slows the rate of association and increases the rate of dissociation.27 Mutation of D683 in PA, which coordinates the MIDAS magnesium, to Asn decreases the binding affinity.39 Coordination of the magnesium ion in the MIDAS site involves the titratable residues Asp 50 and Asp 148 from CMG2, and Asp 683 from PA, whose protonation states may change as the pH is lowered. However, one would expect binding to the metal ion to be weaker in the protonated state, and thus weaker association at lower pH values.
In the domain 2β3-2β4 loop, the salt bridge between R344 of PA and E122 of CMG2 has been shown experimentally to be an important determinant of the affinity between PA and CMG2.23 Molecular dynamics simulations have also shown that protonation of E122 of CMG2 would disrupt the salt bridge, which again would lead to weaker association at lower pH values.40 In addition, protonation of H121 of CMG2, which is adjacent to E122 and near R344, would aid in further disrupting this salt bridge.40 These studies also indicate that the calculated pKa of E122 and H121 is significantly reduced upon receptor binding, with the pKa of E122 decreasing from 7.4 to 2.3 when bound to CMG2, and H121 decreasing from −4.4 to −6.3. If the pKa values are reduced, this may explain the lack of a pH-dependence on receptor dissociation that we observe—that once bound, one must go to very low pH values (beyond pH 5) to break the salt bridge.
Our studies were carried out within the pH range of 8 to 5, certainly within the pKa range of histidine (∼6–6.5). There are a total of 10 histidine residues in PA, with two (H597 and H616) in domain 4 and five (H263, H299, H304, H310, and H336) located in domain 2, with H336 being the closest histidine in PA to the binding interface (∼12–15 Å). Labeling with 2-FHis, which has a reduced pKa (∼1), should prevent protonation of these histidine residues. The fact that we observe pH-dependent kinetics of association even with 2-FHis labeling suggests that histidine protonation is not a mechanism of increased association at low pH.
Surprisingly, the rate of dissociation of 2-FHisPA is approximately twofold faster at pH 5 than at pH 6, despite the fact that this protein is more stable to pH than the WT protein.30 Recent adiabatic compressibility studies using ultrasonic velocimetry41 indicate that the 2-FHisPA exhibits higher adiabatic compressibility than the WT protein at pH 5, suggesting that the protein is globally more dynamic at this pH than WT (Russ Middaugh and Lei Hu, personal communication). The exact mechanism for increased dynamics of 2-FHisPA at pH 5 is not understood. It may be that one or more of the 2-FHis residues in PA becomes protonated at pH 5, and that, because of the fluorine on the side chain, alters side-chain packing and increases protein dynamics. In any case, increased dynamics would likely contribute to an increased rate of dissociation.
Although we have not explicitly measured the effect of salt on the kinetics of association and dissociation (which would help determine the role of electrostatics on binding), we have measured kinetics in two separate buffer systems: 20 mM Tris/MES/HEPES/acetic acid, a buffer system used previously to monitor the kinetics of association,27 and a constant ionic strength (CIS) buffer (100 mM Tris/50 mM MES/50 mM acetic acid).32 In our case, changing the buffer did not influence the kinetics of association at pH 8 and 6, suggesting that the kinetic changes were not due to changes in ionic strength as the pH was lowered.
Effects on the kinetics of association: conformational changes
We also tried to correlate the kinetics of binding to structural changes in the proteins using X-ray crystallography, focusing initially on potential structural changes in the domain 2β3-2β4 loop. We find that, for the 2-FHis and W346F mutant proteins, the domain 2β3-2β4 loop becomes disordered at acidic pH, consistent with previous studies.24 W346 is located in the domain 2β3-2β4 loop, and while this residue does not make a direct contact to CMG2, mutation to Phe may increase the degree of disorder of the domain 2β3-2β4 loop and prevent the R344-E122 salt-bridge from forming. However, for the WT PA protein, we do not observe any changes in the structure of the domain 2β3-2β4 loop, and very little change in structure for any of the domains of PA between pH 8.5 and pH 5.5 (Fig. 6). We have previously shown that R344 forms a symmetry-related crystal contact between E224 located in domain 1′,31 a contact that is maintained in the acidic pH structure of the WT PA protein. Maintenance of this contact at acidic pH may help to explain the lack of disorder in this region. In any case, whether this loop becomes disordered at acidic pH remains somewhat inconclusive, and will require other methods (NMR for instance) for verification.
Although we could not adequately correlate the binding kinetics to changes in structure of the domain 2β3-2β4 loop, we do find that the B-factors increase by ∼20–30% for all proteins at acidic pH, suggesting a greater degree of vibrational motion throughout the protein structure. Because we observe an increase in disorder at low pH for the 2-FHisPA protein as well as the WT and W346F proteins, this further indicates that protonation of histidine residues is likely not the cause of the increase in B-factors. The increase in B-factors is consistent with a decrease in the stability of the proteins as the pH is lowered, but yet the binding affinity increases as the pH is lowered. Taking into account all of our data, the selected-fit model put forth by Weikl and von Deuster42 may help explain our observations.
Proposed model for the effect of pH on the binding of PA to CMG2
The selected-fit model, which contrasts the induced-fit binding model,43 suggests that there is a pre-equilibrium, prior to association, between a population that is in an “unbound” conformation, and a population that is in a “bound” conformation. It may be that, as the pH is lowered, the protein increases vibrational motion, relaxing regions required for binding. This effectively would shift the equilibrium from an “unbound” state that is perhaps more conformationally rigid, to a “bound-like” state that is more conformationally loose. In our case, conformational changes toward a “bound” state may involve the relaxation of one or more side-chains at the interface, within the domain 2β3-2β4 loop, or regions more distant from the binding interface (as mentioned above).
In addition, the selected fit model would also predict that, if pH does not affect the binding energy (bonds formed after association), that the rate of dissociation will be pH independent. Indeed, we observe that the rates of dissociation do not change significantly from pH 8 to 6. The fact that the dissociation rates do not change indicates that pH mainly influences the conformational equilibrium between the “unbound” and “bound” states, without influencing the energetics of the binding interface. However, for the W346F mutant, the rate constants for association and dissociation at pH 8 or 6 differ significantly from the comparative values of the WT and 2-FHis proteins—slower rates of association, and faster rates of dissociation. This may be a case in which the mutation W346F affects both the equilibrium between the “unbound” and “bound” conformational states, as well as the energetics of binding. As discussed above, mutation of W346 to Phe may increase the flexibility of the domain 2β3-2β4 loop (altering the equilibrium between “bound” and “unbound” states), and may explain in part the lower activation barrier to binding that we observe. The increased flexibility of this mutant may in turn affect the energetics of the binding interface (the stability of the R344-E122 salt bridge, for instance).
Can we extrapolate our data on the monomeric 83 kDa PA to the heptameric or octameric prepore states? After all, crystallographic studies have shown that the binding interface does not change significantly when CMG2 is bound to the monomeric 83 kD PA or when bound to the heptameric prepore.19,20 Our data suggests that the increased affinity as the pH is lowered may aid in keeping the receptor associated with the prepore until a critical pH is reached and pore formation ensues. This would be consistent with studies by Lacy and coworkers, who showed that binding of CMG2 to the heptameric prepore shifts the pH requisite for pore formation to lower pH values, suggesting that receptor binding delays the onset of pore formation until the prepore reaches the correct pH environment.20 However, recent NMR studies on the WT heptameric prepore bound to CMG2 indicate that low pH causes dissociation of the domain 2β3-2β4 loop from CMG2, which would favor a decreased rate of binding/increased rate of dissociation at low pH.25 The latter study suggests that there may not be a direct correlation between the monomeric and heptameric forms of PA in terms of binding, but the FRET methodology presented here and elsewhere27 should, in future experiments, clarify any potential differences.
In conclusion, binding of PA and two variants which are more (2-FHis) or less (W346F) stable to pH, to the host receptor CMG2 has been examined as a function of pH. We have found that the rate constants for binding for all three proteins increase as the pH is lowered, with little change in the rate of dissociation, effectively lowering the overall KD for binding. The work presented here provides a basis for future studies aimed at understanding how pH affects binding of CMG2 to the prepore state, and continues our biophysical characterization of 2-FHisPA which is currently under development as a potential therapeutic and/or safer vaccine against anthrax.
Experimental Procedures
Reagents, plasmids, and strains
All reagents for purification and analysis were either from Sigma (St. Louis, MO) or Fisher Scientific unless otherwise specified and were ≥ 99% pure. The fluorescent dyes Alexa350-C5 maleimide (AF350) and Alexa488-C5 maleimide (AF488) were purchased from Invitrogen. The histidine auxotroph UTH780 was obtained from the E. coli genetic stock center at Yale University (New Haven, CT). The protective antigen gene in the plasmid pQE80 was used for the production of PA as described previously.30 We used the forward primer (5′ CACTATTATTAATCCTAGTTGCAATGGGGATACTAGTACCAACGGGATG 3′) and forward primer (5′CTATCTCTAGCAGGGGAAAGAACTTTTGCTGAAACAATGGG3′), and the corresponding reverse strands to create mutants E712C and W346F, using the Quickchange mutagenesis kit from Stratagene. Sequences were verified at the Protein Nucleic Acid Laboratory (PNACL) at Washington University in St. Louis. Generation of the CMG2C40 mutant was carried out as described previously.27
Protein expression and purification
Recombinant PA E712C, E712C/W346F, 2-FHis-E712C, and CMG2C40 were produced in 650-mL cultures grown in the Fernbach shaker flasks using ECPM1 growth medium supplemented with 100 mg mL−1 ampicillin at 32°C for PA and 37°C for CMG2 until an OD600 of 2.5–3 was reached. The cultures were then induced with 1 mM IPTG for 3 h at 26°C for PA variants and 37°C for CMG2C40. The cells were harvested in a centrifuge equipped with a swinging bucket rotor (3000g) for 10 min. Cells were then placed on ice for immediate periplasm purification. For purification of CMG2C40, the cells were frozen at −20°C prior to purification. Labeling with 2-FHis was carried out in a manner similar to that previously described.30 Purification of PA variants was done according to previous reports,27,30 with slight modifications. The periplasmic extracts were buffered with 20 mM Tris-HCl pH 8 and supplemented with 1 mM dithiothreitol to prevent oxidation of the cysteine residue in PA. PA E712C, 2-FHis-E712C or E712C/W346F were purified by anion exchange chromatography (Q Sepharose) with a 0–30% NaCl gradient starting with 20 mM Tris-HCl/1 mM DTT, pH 8, 4°C. The proteins were further purified by Sephadex S-200 gel filtration column (GE-Healthcare) equilibrated in 20 mM Tris-HCl, 1 mM DTT, 150 mM NaCl pH 8. Fractions were pooled, concentrated and stored at −20°C. CMG2C40 was purified essentially as described previously.27 After elution of CMG2C40 from a Hi-Trap GST column in phosphate-buffered saline, the protein was concentrated and a 10-fold excess of TCEP was added to protect the free thiol group, and stored at 4°C.
Fluorescent probe labeling
An S-200 gel filtration column was pre-equilibrated with a 20 mM Tris-HCl, 0.5 mM TCEP, and 150 mM NaCl, pH 8 that had been degassed by sparging with N2 for 15 min. Approximately 5–10 mg of either WT E712C, 2-FHis-E712C or E712C/W346F were purified to remove excess DTT which would otherwise react with the maleimide activated fluorophores. A 10 mM stock solution of either AF350 or AF488 was prepared in N2 degassed water immediately prior to labeling. Sufficient protein modification reagents were added to each protein to give ∼15-fold molar excess of reagent for each mole of protein, where a 10-fold molar excess of TCEP to protein is present. The reaction was allowed to proceed for 2 h at room temperature with rocking. Upon completion of the reaction, a 10-fold molar excess to dye of β-mercaptoethanol was added to eliminate any excess reactive substances during the purification step. The labeled proteins were subsequently purified from the free dye by gel filtration (S-200 column) in 20 mM Tris–HCl, 150 mM NaCl, pH 8. A similar protocol was carried out with proteins labeled with NEM, using similar starting concentrations. Protein solutions were assessed for labeling efficiency using extinction coefficients 19,000 and 72,000 M−1 cm−1 for AF350 and AF488 fluorophores, respectively, at their respective absorbance maxima. Labeling efficiency was 95% or greater. Fractions containing pure protein with a labeling efficiency of 1:1 (since there is only one cysteine in the protein) were identified by SDS-PAGE, pooled and concentrated and kept on ice. Protein concentrations prior to labeling were determined using a calculated extinction coefficient at 280 nm of 80,220 M−1 cm−1 for E712C and 2-FHis-E712C, 74,720 M−1 cm−1 for E712C/W346F and 12,914 M−1 cm−1 for CMG2.
Equilibrium binding
Labeled PA proteins were diluted to 100 nM in 2 mL of a buffer containing 10 mM each Tris/MES/HEPES/acetic acid pH 8.0, supplemented with 0.1 mg mL−1 BSA and 2 mM MgCl2. An initial 100-μL aliquot was removed, and 5 μL of ∼2 μM CMG2C40-AF488, also in 10 mM each Tris/MES/HEPES/acetic acid pH 8.0 supplemented with 0.1 mg mL−1 BSA and 2 mM MgCl2, was added. From this, another 100 μL was removed, followed by the addition of 5 μL of ∼2 μM CMG2C40-AF488. This process was repeated until the CMG2C40-AF488 concentration was in an approximately sixfold excess to PA. Each 100-μL aliquot was incubated overnight at 4°C to ensure complete binding. Each aliquot was then diluted with 400 μL of 10 mM each Tris/MES/HEPES/acetic acid pH 8.0 supplemented with 0.1 mg mL−1 BSA and 2 mM MgCl2. Fluorescence emission spectra were recorded using a Cary-Eclipse fluorimeter at 20°C. The donor fluorophore was excited at 350 nm and the emission at 440 and 516 nm were recorded. The apparent FRET signal was defined by the ratio of the acceptor to donor emission (Em 516 nm/Em 440 nm).
Association kinetics using stopped-flow
All kinetic experiments were performed at 20°C using an Applied Photophysics SX.18MV instrument. Mixing ratios were 1:1 for all experiments, and proteins (PA and variants, CMG2) were incubated at the respective pH values for at least 1 h. Excitation was set at 350 nm and the emission was monitored above 475 nm using a cutoff filter. In each experiment, 1000 data points were recorded over the course of the reaction. Increase of the FRET signal was monitored for 200 s under pseudo-first order reaction conditions using minimum five-fold excess of CMG2.27 For measuring the temperature dependence on association, we used an ∼25-fold excess of CMG2C40-AF488 for the E712C and 2-FHis-E712C proteins (600 nM: 25 nM (final concentration), CMG2C40-AF488:E712C-AF350) and an ∼25-fold excess for the E712C/W346F (1000 nM: 40 nM (final concentration) CMG2C40-AF488:E712C/W346F-AF350). Total shot volumes were 250 μL and the instrument dead time was ∼2 ms, and each data point plotted represents an average of three to four kinetic traces.
Dissociation kinetics
Dissociation was monitored at 20°C using a Cary Eclipse fluorimeter. The methodology here is similar to that used by Wigelsworth et al. to determine the dissociation of CMG2 from PA.27 Briefly, a 1.5-mL sample in 10 mM each Tris/MES/HEPES/Acetic acid, at the respective pH values, containing 50 nM E712C-AF350, 2-FHis-E712C-AF350 or E712C-AF350/W346F was equilibrated at 20°C. To this, CMG2C40-AF488 was added to a final concentration of 100 nM, and the association, as monitored by the increase in the ratio of fluorescence Em440/Em516, was carried out until the fluorescence showed no change (∼10,000 s). At this time, CMG2C40-NEM was added manually to a final concentration of 2 μM, and the ratio of the fluorescence emission at 516 nm/440 nm was monitored as a function of time.
Kinetic and equilibrium data analysis
Kinetic traces were averaged and fit to a single exponential function to obtain the observed rate constants for each CMG2 concentration. Observed rates were plotted against different CMG2 concentrations. The slopes of the linear fits were used to calculate the rate constant of association, ka. The observed dissociation rate constants (kd) were obtained by fitting to a single exponential function. Equilibrium dissociation constants (KD) are calculated from kinetic measurements of the association and dissociation rate constants according to KD = kd/ka. The activation energy (Ea) for the ligand-receptor binding was determined from slope of the best fit line described by Arrhenius equation, ln ka = ln(A) − [(Ea/R)] × (1/T)], where A is pre-exponential factor, Ea is the Arrhenius activation energy, R is the gas constant, and T is temperature.
Crystallization, structure solution, and refinement
All samples were screened for crystallization in Compact Jr. (Emerald biosystems) sitting drop vapor diffusion plates using 0.5 μL of protein and using 0.5 μL of crystallization solution equilibrated against 100 μL of the latter at 20°C. Proteins samples of WT (16.5 mg mL−1), W346F (9.0 mg mL−1), and 2-FHis PA (16.5 mg mL−1) were concentrated in 150 mM NaCl, 20 mM Tris pH 8.0 for crystallization. Prismatic crystals of each PA construct were obtained within 24 h from the following conditions: WT: Wizard 4 screen condition #20 (Emerald biosystems, 25%(w/v) PEG 1500, 100 mM MMT Buffer pH 9.0) and Wizard 4 screen condition #17 (Emerald biosystems, 25%(w/v) PEG 1500, 100 mM SPG Buffer pH 5.5). W346F: Wizard 4 screen condition #18 (Emerald biosystems, 25%(w/v) PEG 1500, 100 mM SPG Buffer pH 8.5) and Wizard 4 screen condition #17 (Emerald biosystems, 25%(w/v) PEG 1500, 100 mM SPG Buffer pH 5.5). Single crystals of WT and W346F PA were transferred to a drop containing 80% crystallization solution and 20% PEG 400 before freezing in liquid nitrogen for data collection. Data were collected at the Advanced Photon Source IMCA-CAT beamline 17ID using a Dectris Pilatus 6M pixel array detector. 2FHis-PA: Crystals were grown from a solution containing 30% PEG 400, 100 mM MES pH 7.0. Single crystals were soaked for 26 h in 35% PEG 400, 100 mM MES pH 5.8 then were frozen in 40% PEG 400, 100 mM MES pH 5.8 for data collection. Data were collected at the Advanced Photon Source IMCA-CAT beamline 17BM using a Mar 165 CCD detector. Intensities for all data sets were integrated and scaled using the XDS44 and Scala37 packages, respectively. The coordinates from a previously determined structure of 2-FHis-labeled PA (PDB: 3MHZ) were used for initial refinement against the processed diffraction data. Structure refinement and manual model building were performed with Phenix45 and Coot,46 respectively. Structure validation was carried out using Molprobity.47 The structures have been deposited in the PDB under accession codes 3Q8A and 3Q8B, WT pH 5.5 and 9.0, respectively; 3Q8C and 3Q8E, W346F pH 5.5 and 8.5, respectively; 3Q8F, 2-FHisPA pH 5.8.
Acknowledgments
The authors thank Hans Peter Bächinger and Carl Frieden for their comments on the manuscript. Use of the IMCA-CAT beamline 17-BM at the Advanced Photon Source was supported by the companies of the Industrial Macromolecular Crystallography Association through a contract with the Hauptman-Woodward Medical Research Institute. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357.
Glossary
Abbreviations
- 2-FHis
2-fluorohistidine
- 2-FHisPA
the 83 kDa form of 2-FHis labeled PA
- AF350
AlexaFluor 350
- AF488
AlexaFluor488
- AF546
AlexaFluor546
- CMG2
capillary morphogenesis protein 2
- PA
protective antigen
- vWA
von Willebrand factor A domain
- WT
wild-type
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.Bradley KA, Mogridge J, Mourez M, Collier RJ, Young JA. Identification of the cellular receptor for anthrax toxin. Nature. 2001;414:225–229. doi: 10.1038/n35101999. [DOI] [PubMed] [Google Scholar]
- 2.Scobie HM, Rainey GJ, Bradley KA, Young JA. Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc Natl Acad Sci USA. 2003;100:5170–5174. doi: 10.1073/pnas.0431098100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martchenko M, Jeong SY, Cohen SN. Heterodimeric integrin complexes containing beta1-integrin promote internalization and lethality of anthrax toxin. Proc Natl Acad Sci USA. 2010;107:15583–15588. doi: 10.1073/pnas.1010145107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Molloy SS, Bresnahan PA, Leppla SH, Klimpel KR, Thomas G. Human furin is a calcium-dependent serine endoprotease that recognizes the sequence Arg-X-X-Arg and efficiently cleaves anthrax toxin protective antigen. J Biol Chem. 1992;267:16396–16402. [PubMed] [Google Scholar]
- 5.Mogridge J, Cunningham K, Collier RJ. Stoichiometry of anthrax toxin complexes. Biochemistry. 2002;41:1079–1082. doi: 10.1021/bi015860m. [DOI] [PubMed] [Google Scholar]
- 6.Kintzer AF, Thoren KL, Sterling HJ, Dong KC, Feld GK, Tang II, Zhang TT, Williams ER, Berger JM, Krantz BA. The protective antigen component of anthrax toxin forms functional octameric complexes. J Mol Biol. 2009;392:614–629. doi: 10.1016/j.jmb.2009.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mogridge J, Cunningham K, Lacy DB, Mourez M, Collier RJ. The lethal and edema factors of anthrax toxin bind only to oligomeric forms of the protective antigen. Proc Natl Acad Sci USA. 2002;99:7045–7048. doi: 10.1073/pnas.052160199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Melnyk RA, Hewitt KM, Lacy DB, Lin HC, Gessner CR, Li S, Woods VL, Jr, Collier RJ. Structural determinants for the binding of anthrax lethal factor to oligomeric protective antigen. J Biol Chem. 2006;281:1630–1635. doi: 10.1074/jbc.M511164200. [DOI] [PubMed] [Google Scholar]
- 9.Abrami L, Bischofberger M, Kunz B, Groux R, van der Goot FG. Endocytosis of the anthrax toxin is mediated by clathrin, actin and unconventional adaptors. PLoS Pathog. 2010;6:e1000792. doi: 10.1371/journal.ppat.1000792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zornetta I, Brandi L, Janowiak B, Dal Molin F, Tonello F, Collier RJ, Montecucco C. Imaging the cell entry of the anthrax oedema and lethal toxins with fluorescent protein chimeras. Cell Microbiol. 2010;12:1435–1445. doi: 10.1111/j.1462-5822.2010.01480.x. [DOI] [PubMed] [Google Scholar]
- 11.Benson EL, Huynh PD, Finkelstein A, Collier RJ. Identification of residues lining the anthrax protective antigen channel. Biochemistry. 1998;37:3941–3948. doi: 10.1021/bi972657b. [DOI] [PubMed] [Google Scholar]
- 12.Nassi S, Collier RJ, Finkelstein A. PA63 channel of anthrax toxin: an extended beta-barrel. Biochemistry. 2002;41:1445–1450. doi: 10.1021/bi0119518. [DOI] [PubMed] [Google Scholar]
- 13.Katayama H, Wang J, Tama F, Chollet L, Gogol EP, Collier RJ, Fisher MT. Three-dimensional structure of the anthrax toxin pore inserted into lipid nanodiscs and lipid vesicles. Proc Natl Acad Sci USA. 2010;107:3453–3457. doi: 10.1073/pnas.1000100107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krantz BA, Finkelstein A, Collier RJ. Protein translocation through the anthrax toxin transmembrane pore is driven by a proton gradient. J Mol Biol. 2006;355:968–979. doi: 10.1016/j.jmb.2005.11.030. [DOI] [PubMed] [Google Scholar]
- 15.Leppla SH. Anthrax toxin edema factor: a bacterial adenylate cyclase that increases cyclic AMP concentrations of eukaryotic cells. Proc Natl Acad Sci USA. 1982;79:3162–3166. doi: 10.1073/pnas.79.10.3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hong J, Doebele RC, Lingen MW, Quilliam LA, Tang WJ, Rosner MR. Anthrax edema toxin inhibits endothelial cell chemotaxis via Epac and Rap1. J Biol Chem. 2007;282:19781–19787. doi: 10.1074/jbc.M700128200. [DOI] [PubMed] [Google Scholar]
- 17.Duesbery NS, Webb CP, Leppla SH, Gordon VM, Klimpel KR, Copeland TD, Ahn NG, Oskarsson MK, Fukasawa K, Paull KD, Vande Woude GF. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science. 1998;280:734–737. doi: 10.1126/science.280.5364.734. [DOI] [PubMed] [Google Scholar]
- 18.Lehmann M, Noack D, Wood M, Perego M, Knaus UG. Lung epithelial injury by B. anthracis lethal toxin is caused by MKK-dependent loss of cytoskeletal integrity. PLoS ONE. 2009;4:e4755. doi: 10.1371/journal.pone.0004755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Santelli E, Bankston LA, Leppla SH, Liddington RC. Crystal structure of a complex between anthrax toxin and its host cell receptor. Nature. 2004;430:905–908. doi: 10.1038/nature02763. [DOI] [PubMed] [Google Scholar]
- 20.Lacy DB, Wigelsworth DJ, Melnyk RA, Harrison SC, Collier RJ. Structure of heptameric protective antigen bound to an anthrax toxin receptor: a role for receptor in pH-dependent pore formation. Proc Natl Acad Sci USA. 2004;101:13147–13151. doi: 10.1073/pnas.0405405101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Williams AS, Lovell S, Anbanandam A, El-Chami R, Bann JG. Domain 4 of the anthrax protective antigen maintains structure and binding to the host receptor CMG2 at low pH. Protein Sci. 2009;18:2277–2286. doi: 10.1002/pro.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scobie HM, Marlett JM, Rainey GJ, Lacy DB, Collier RJ, Young JA. Anthrax toxin receptor 2 determinants that dictate the pH threshold of toxin pore formation. PLoS ONE. 2007;2:e329. doi: 10.1371/journal.pone.0000329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu S, Leung HJ, Leppla SH. Characterization of the interaction between anthrax toxin and its cellular receptors. Cell Microbiol. 2007;9:977–987. doi: 10.1111/j.1462-5822.2006.00845.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Petosa C, Collier RJ, Klimpel KR, Leppla SH, Liddington RC. Crystal structure of the anthrax toxin protective antigen. Nature. 1997;385:833–838. doi: 10.1038/385833a0. [DOI] [PubMed] [Google Scholar]
- 25.Pilpa RM, Bayrhuber M, Marlett JM, Riek R, Young JA. A receptor-based switch that regulates anthrax toxin pore formation. PLoS Pathog. 2011;7:e1002354. doi: 10.1371/journal.ppat.1002354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yeh HJC, Kirk KL, Cohen LA, Cohen JS. 19F and 1H nuclear magnetic resonance studies of ring-fluorinated imidazoles and histidines. J Chem Soc Perkins Trans. 1975;2:928–934. [Google Scholar]
- 27.Wigelsworth DJ, Krantz BA, Christensen KA, Lacy DB, Juris SJ, Collier RJ. Binding stoichiometry and kinetics of the interaction of a human anthrax toxin receptor, CMG2, with protective antigen. J Biol Chem. 2004;279:23349–23356. doi: 10.1074/jbc.M401292200. [DOI] [PubMed] [Google Scholar]
- 28.Villamil Giraldo AM, Lopez Medus M, Gonzalez Lebrero M, Pagano RS, Labriola CA, Landolfo L, Delfino JM, Parodi AJ, Caramelo JJ. The structure of calreticulin C-terminal domain is modulated by physiological variations of calcium concentration. J Biol Chem. 2010;285:4544–4553. doi: 10.1074/jbc.M109.034512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mourez M, Yan M, Lacy DB, Dillon L, Bentsen L, Marpoe A, Maurin C, Hotze E, Wigelsworth D, Pimental RA, Ballard JD, Collier RJ, Tweten RK. Mapping dominant-negative mutations of anthrax protective antigen by scanning mutagenesis. Proc Natl Acad Sci USA. 2003;100:13803–13808. doi: 10.1073/pnas.2436299100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wimalasena DS, Cramer JC, Janowiak BE, Juris SJ, Melnyk RA, Anderson DE, Kirk KL, Collier RJ, Bann JG. Effect of 2-fluorohistidine labeling of the anthrax protective antigen on stability, pore formation, and translocation. Biochemistry. 2007;46:14928–14936. doi: 10.1021/bi701763z. [DOI] [PubMed] [Google Scholar]
- 31.Wimalasena DS, Janowiak BE, Lovell S, Miyagi M, Sun J, Zhou H, Hajduch J, Pooput C, Kirk KL, Battaile KP, Bann JG. Evidence that histidine protonation of receptor-bound anthrax protective antigen is a trigger for pore formation. Biochemistry. 2010;49:6973–6983. doi: 10.1021/bi100647z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ellis KJ, Morrison JF. Buffers of constant ionic strength for studying pH-dependent processes. Methods Enzymol. 1982;87:405–426. doi: 10.1016/s0076-6879(82)87025-0. [DOI] [PubMed] [Google Scholar]
- 33.Schiene-Fischer C, Fischer G. Direct measurement indicates a slow cis/trans isomerization at the secondary amide peptide bond of glycylglycine. J Am Chem Soc. 2001;123:6227–6231. doi: 10.1021/ja0042480. [DOI] [PubMed] [Google Scholar]
- 34.Bachinger HP, Bruckner P, Timpl R, Engel J. The role of cis-trans isomerization of peptide bonds in the coil leads to and comes from triple helix conversion of collagen. Eur J Biochem. 1978;90:605–613. doi: 10.1111/j.1432-1033.1978.tb12641.x. [DOI] [PubMed] [Google Scholar]
- 35.Krieger F, Moglich A, Kiefhaber T. Effect of proline and glycine residues on dynamics and barriers of loop formation in polypeptide chains. J Am Chem Soc. 2005;127:3346–3352. doi: 10.1021/ja042798i. [DOI] [PubMed] [Google Scholar]
- 36.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Evans P. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 38.Diederichs K, Karplus PA. Improved R-factors for diffraction data analysis in macromolecular crystallography. Nat Struct Biol. 1997;4:269–275. doi: 10.1038/nsb0497-269. [DOI] [PubMed] [Google Scholar]
- 39.Scobie HM, Wigelsworth DJ, Marlett JM, Thomas D, Rainey GJ, Lacy DB, Manchester M, Collier RJ, Young JA. Anthrax toxin receptor 2-dependent lethal toxin killing in vivo. PLoS Pathog. 2006;2:e111. doi: 10.1371/journal.ppat.0020111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gao M, Schulten K. Onset of anthrax toxin pore formation. Biophys J. 2006;90:3267–3279. doi: 10.1529/biophysj.105.079376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ramsey JD, Gill ML, Kamerzell TJ, Price ES, Joshi SB, Bishop SM, Oliver CN, Middaugh CR. Using empirical phase diagrams to understand the role of intramolecular dynamics in immunoglobulin G stability. J Pharm Sci. 2009;98:2432–2447. doi: 10.1002/jps.21619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weikl TR, von Deuster C. Selected-fit versus induced-fit protein binding: kinetic differences and mutational analysis. Proteins. 2009;75:104–110. doi: 10.1002/prot.22223. [DOI] [PubMed] [Google Scholar]
- 43.Bosshard HR. Molecular recognition by induced fit: how fit is the concept? News Physiol Sci. 2001;16:171–173. doi: 10.1152/physiologyonline.2001.16.4.171. [DOI] [PubMed] [Google Scholar]
- 44.Kabsch W. Automatic indexing of rotation diffraction patterns. J Appl Crystallogr. 1988;21:67–72. [Google Scholar]
- 45.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 47.Lovell SC, Davis IW, Arendall WB, III, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.