

Abstract
The intrinsically disordered C-terminal domain (NTAIL) of the measles virus (MeV) nucleoprotein undergoes α-helical folding upon binding to the C-terminal X domain (XD) of the phosphoprotein. The NTAIL region involved in binding coupled to folding has been mapped to a conserved region (Box2) encompassing residues 489–506. In the previous studies published in this journal, we obtained experimental evidence supporting a KD for the NTAIL–XD binding reaction in the nM range and also showed that an additional NTAIL region (Box3, aa 517–525) plays a role in binding to XD. In striking contrast with these data, studies published in this journal by Kingston and coworkers pointed out a much less stable complex (KD in the μM range) and supported lack of involvement of Box3 in complex formation. The objective of this study was to critically re-evaluate the role of Box3 in NTAIL–XD binding. Since our previous studies relied on NTAIL-truncated forms possessing an irrelevant Flag sequence appended at their C-terminus, we, herein, generated an NTAIL devoid of Box3 and any additional C-terminal residues, as well as a form encompassing only residues 482–525. We then used isothermal titration calorimetry to characterize the binding reactions between XD and these NTAIL forms. Results effectively argue for the presence of a single XD-binding site located within Box2, in agreement with the results by Kingston et al., while providing clear experimental support for a high-affinity complex. Altogether, the present data provide mechanistic insights into the replicative machinery of MeV and clarify a hitherto highly debated point.
Keywords: isothermal titration calorimetry, intrinsically disordered proteins, folding coupled to binding, entropic penalty, entropic stabilization, paramyxoviruses, nucleoprotein, phosphoprotein
Introduction
Measles virus (MeV) is a member of the Morbillivirus genus. It has a nonsegmented, single-stranded, negative sense ribonucleic acid (RNA) genome that is encapsidated by the nucleoprotein (N). This N:RNA complex, rather than naked RNA, is the template for both transcription and replication. The viral polymerase (L) does not directly bind viral genomic RNA during the transcription and genome replication, but, instead, is tethered onto the nucleocapsid template via the viral phosphoprotein (P). The P protein simultaneously binds to L and to the exposed C-terminal disordered domain of N (NTAIL, amino acids 400–525) via its C-terminal X domain (XD, amino acids 459–507).1–9 Progressive movement of the polymerase along its template is thus thought to require cycles of NTAIL–XD binding and release.10,11
NTAIL is an intrinsically disordered domain,12,13 that is, it exists as a dynamic ensemble of interconverting conformers under physiological conditions of pH and salinity.14–19 Using computational approaches,13 an α-helical molecular recognition element (α-MoRE, aa 488–499 of N) has been identified within one (namely Box2, aa 489–506) of three NTAIL regions (referred to as Box1, Box2, and Box3) that are conserved within Morbillivirus members20 [see Fig. 1(A)]. MoREs are short-order prone regions within intrinsically disordered domains that have a propensity to undergo induced folding (i.e., a disorder-to-order transition) upon binding to a partner.22–25 This α-MoRE is involved in binding to XD, and the NTAIL region encompassing residues 486–502 adopts an α-helical conformation in the bound form.1–9,13,21 Binding of XD to Box2 also induces a reduction in the conformational flexibility of the downstream Box3 region (aa 517–525).4,6,8 The reduced flexibility does not arise from the establishment of stable contacts between Box3 and XD,4 instead being attributed to transient, nonspecific interactions.7 Support for a role of Box3 in NTAIL binding to XD comes from four independent lines of experimental evidence. First, heteronuclear NMR studies carried out on 15N-labeled NTAIL showed that the addition of XD triggered both α-helical folding of Box2 and a minor, though significant, magnetic perturbation within Box3.2,7 Second, the low-resolution structural model of the NTAIL–XD complex that was derived using small angle X-ray scattering showed lack of a C-terminal appendage exposed to the solvent, suggesting that beyond Box2, Box3 could also be involved in binding to XD.2 Third, intrinsic fluorescence spectroscopy studies revealed a dose-dependent impact of XD on the fluorescence of a trp residue inserted within Box3.2 Incidentally, those studies also allowed estimation of the equilibrium dissociation constant (KD) that was found to be in the submicromolar range (e.g., ∼100 nM) and in close agreement with the value determined by either surface plasmon resonance (SPR)2 or isothermal titration calorimetry (ITC).26 Finally, those previous SPR studies showed that removal of Box3 results in a strong increase in the KD, with this latter increasing from 80 nM to 12 μM.2 When SPR experiments were carried out using synthetic peptides mimicking Box2 and Box3, a Box2 peptide (aa 487–507), was found to display an affinity for XD that was similar (KD = 20 nM) to that between XD and NTAIL (KD = 80 nM) consistent with the role of Box2 as the primary binding site.11 Box3 peptide exhibits an insignificant affinity for XD (KD of ∼1 mM),11 consistent with a role for Box3 only in the context of NTAIL and not in isolation. Therefore, we proposed a model where Box3 and Box2 would be functionally coupled in the binding of NTAIL to XD, with the burying of the hydrophobic side of the α-MoRE at the XD interface providing the primary driving force in the NTAIL–XD interaction, while Box3 would act to further stabilize the bound conformation. In contrast to our model, recent data by the group of Kingston argue for an NTAIL–XD binding mechanism in which Box3 is dispensable. In those studies, the authors used both SPR and ITC to characterize binding reactions between XD and synthetic peptides corresponding to amino acids 477–505 (including Box2) or 477–525 (including both Box2 and Box3) of N, and derived a KD that was either 7.4 or 15 μM, respectively.27 Those results argue for a much less tight complex than suggested by our previous studies2,26 in addition to supporting the lack of involvement of Box3.
Figure 1.
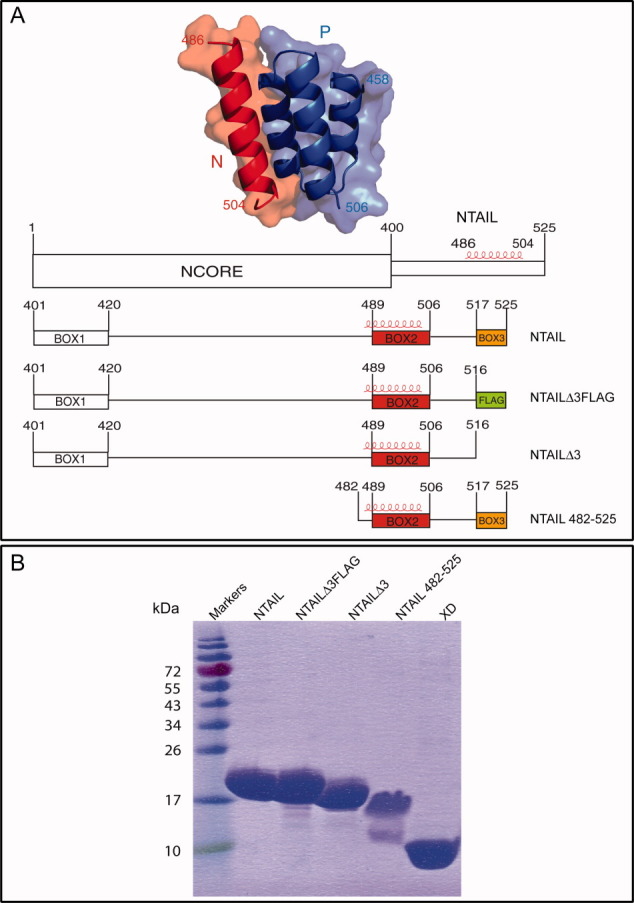
(A) Crystal structure of the chimera between XD (blue) and the α-MoRE of NTAIL (red) (pdb code 1T6O)21 and schematic representation of the NTAIL constructs used in this study. The three conserved NTAIL regions within Morbillivirus members are shown. Box2 and Box3 are shown in red and orange, respectively. The Flag sequence is shown as a green box. The α-helix observed in the crystal structure of the chimera is shown in red. All NTAIL constructs bear an N-terminal hexahistidine tag. (B) 15% SDS-PAGE of purified proteins followed by Coomassie blue staining. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
The objective of this study was to critically re-evaluate the role of Box3 in the NTAIL–XD interaction. As our previous studies2 made use of a truncated form of NTAIL (hereafter referred to as NTAILΔ3Flag) that was devoid of Box3 but did however possess an irrelevant Flag sequence (DYKDDDDK) appended at its C-terminus that might have confounded results, we herein generated a truncated form devoid of Box3 in which the Flag sequence was eliminated (referred to as NTAILΔ3). In addition, to directly assess the possibility that the basis for the discrepancy between our data and those of the group of Kingston in terms of binding affinity could reflect use of full-length NTAIL versus NTAIL peptides, respectively, we also designed and purified an NTAIL construct encompassing residues 482–525 (referred to as NTAIL482–525). We then characterized the binding reactions between XD and the various NTAIL forms using ITC. The experimental design allowed us to both assess the role of Box3 and the impact of an irrelevant sequence appended downstream the primary binding site, to NTAIL–XD interactions.
Results
The recombinant NTAILΔ3 protein and the NTAIL protein bearing a Tobacco Etch virus (TEV) protease cleavage site after residue 481, designed to allow purification of the 482–525 region of NTAIL, were all readily expressed in E. coli and their solubility was high, thus allowing recovery from the soluble fraction of the bacterial lysate. They were purified to homogeneity (>95%) in two steps, namely immobilized metal affinity chromatography (IMAC) and size exclusion chromatography (SEC), as was the case of the NTAIL and NTAILΔ3Flag proteins2 [Fig. 1(B)]. The NTAILΔ3 and NTAILΔ3Flag proteins were eluted from the gel filtration S200 column with a profile quite similar to that observed for the full-length NTAIL protein (data not shown), consistent with similar conformational properties.
After digestion with TEV protease of the purified NTAIL protein bearing a TEV protease cleavage site, the (untagged) NTAIL region encompassing residues 482–525 was recovered in the unretained fraction of an additional IMAC step. The final purified NTAIL482–525 product consists of two bands: a major band with an apparent molecular mass of ∼17 kDa, and a minor band with an apparent molecular mass of ∼12 kDa [Fig. 1(B)]. The identity of the NTAILΔ3 and NTAIL482–525 proteins was confirmed by mass spectrometry analysis of the tryptic fragments obtained after digestion of the purified proteins excised from sodium docecyl sulfate (SDS)-polyacrylamide gels, which yielded a high-sequence coverage and showed that the minor band observed in the NTAIL482–525 sample corresponds to a degradation product (data not shown). Note that the identity of the full-length NTAIL and of NTAILΔ3Flag proteins had been already confirmed in previous studies.2,12
All the NTAIL proteins migrate in SDS-polyacrylammide gel electrophoresis (PAGE) with an apparent molecular mass higher than expected (expected molecular masses are approximately as follows: 15 kDa for full-length NTAIL, 14.5 kDa for NTAILΔ3Flag, 13.5 kDa for NTAILΔ3, and 4.9 kDa for NTAIL482–525), with this abnormal migration being particularly pronounced in the case of NTAIL482–525 [Fig. 1(B)]. This peculiar migratory behavior is well documented for parental NTAIL12 and has also been reported for all NTAIL variants described so far.4,26 Such an anomalous electrophoretic mobility is frequently observed in intrinsically disordered proteins and is to be ascribed to a relatively high content in acidic residues,28,29 and/or to a high degree of protein extension in solution.30
We then investigated the binding abilities of the NTAIL proteins using ITC. The purified NTAIL proteins were loaded into the calorimeter sample cell, and titrated with XD (Fig. 2). The data, following integration and correction for the heats of dilution, were fit with a standard model allowing for a set of independent and equivalent binding sites (Fig. 2). The KD was derived from the KA, as the reciprocal of this latter. The estimates for the binding parameters of NTAIL, NTAILΔ3, and NTAILΔ3Flag revealed a stoichiometry of ∼0.7 (see Fig. 2 and Table I). Errors in the estimates of XD concentration are the most plausible explanation for this subunity stoichiometry. An even lower stoichiometry (0.52) was found in the case of the binding reaction with NTAIL482–525 (Table I), and this may reflect inability of part of the NTAIL482–525 sample to interact with XD, in agreement with the presence of a degradation product (possibly not competent for binding) in the purified NTAIL482–525 sample [Fig. 1(B)].
Figure 2.

ITC studies of complex formation between XD and the various NTAIL proteins. Data are representative of at least two independent experiments. The concentrations of NTAIL proteins in the microcalorimeter cell was 20 μM and that of XD in the microsyringe was 200 μM. Graphs shown in the bottom of each panel correspond to integrated and corrected ITC data fit to a single set of sites model (all sites identical and equivalent). The filled squares represent the experimental data, whereas the solid line corresponds to the model. The derived equilibrium dissociation constant (KD) as well as the stoichiometric number are shown.
Table I.
Equilibrium Dissociation Constants and Binding Parameters, as Derived From ITC Studies, for the Binding Reactions Between MeV XD and Four MeV NTAIL Proteins, namely the Full-Length Form, a Form Devoid of Box3 (NTAILΔ3), a Form in Which the Native Box3 Sequence is Replaced by the Flag Sequence (NTAILΔ3Flag) and a Form-Encompassing Residues 482–525 (NTAIL482–525)
| Stoichiometry, n | KD (nM) | Binding enthalpy, ΔH (cal mol−1) | Binding entropy, ΔS (cal mol−1 deg−1) | |
|---|---|---|---|---|
| NTAIL | 0.78 ± 0.005 | 170 ± 20 | −13,537 ± 164 | −14.4 |
| NTAILΔ3 | 0.72 ± 0.009 | 330 ± 50 | −10,635 ± 242 | −6.0 |
| NTAILΔ3Flag | 0.74 ± 0.006 | 186 ± 25 | −12,525 ± 154 | −11.2 |
| NTAIL482–525 | 0.52 ± 0.003 | 389 ± 24 | −9560 ± 125 | −3.3 |
Data are representative of two independent trials.
The obtained parameters also revealed that the binding reactions are all enthalpy-driven, being all characterized by a small unfavorable entropic contribution (Table I), which is consistent with the entropic penalty associated to the disorder-to-order transition of NTAIL upon binding.
The measured KD for the binding reaction between XD and full-length NTAIL is 170 ± 20 nM (Fig. 2 and Table I). Notably, removal of Box3 causes only a slight decrease in the affinity toward XD, with the KD increasing twofolds but still remaining in the nM range (Fig. 2 and Table I). This finding argues for a minimal impact of Box3 on binding to XD. Interestingly, replacing the native Box3 region by an irrelevant Flag sequence yields a KD close to the value observed for the full-length form (Fig. 2 and Table I). This observation supports a weak stabilizing role of the Flag sequence and suggests that amino acids downstream Box2 have a slight effect on binding, with this effect being independent of sequence. Finally, a KD of 389 ± 24 nM was found for the binding reaction involving NTAIL482–525, which corresponds to (only) a twofold increase with respect to the KD observed with full-length NTAIL (Fig. 2 and Table I). The persistence of a KD in the nanomolar range with a short NTAIL construct encompassing as few as 44 residues, rules out the possibility that the discrepancy between our previous studies2 and those obtained by the group of Kingston27 might arise from differences in the nature of the reactants (i.e., from the use of full-length NTAIL vs. NTAIL peptides).
Discussion and Conclusion
The present studies support at best only a minimal effect of Box3 on binding to XD, as illustrated by the fact that removal of Box3 leads to only a twofold increase in the KD. These findings therefore support the data reported by Kingston27 and are hence not compatible with the two-site binding model that we previously proposed based on SPR data.2 A possible explanation for the discrepancy between our previous SPR data and the present ITC data may reside in possible limitations of the former approach when it comes to the analysis of very fast binding reactions such as those involving truncated NTAIL forms, a point that was already raised by Kingston.27 In light of the present ITC data, previous NMR studies showing a magnetic perturbation within Box3 associated to NTAIL–XD binding,7 as well as previous electron paramagnetic resonance (EPR) data documenting a gain of rigidity in Box3 upon binding to XD,4 should be interpreted as reflecting only a modified Box3 environment resulting from the α-helical transition taking place in the neighboring Box2 region and not a contribution of Box3 to NTAIL–XD binding.
Sequences downstream of Box2 do however contribute to NTAIL–XD binding: indeed, appending an irrelevant sequence downstream of the primary binding site leads to a KD close to that observed with full-length NTAIL. This weakly stabilizing effect does not reflect entropic stabilization,31 as the entropic penalty of the binding reaction with NTAILΔ3Flag is more pronounced than that observed with NTAILΔ3. This is consistent with a scenario where Box2 is more flexible (i.e., less tightly bound to XD) in the absence of a downstream region and would explain the lower entropic penalty of NTAILΔ3 as compared to both full-length NTAIL and NTAILΔ3Flag (Table I).
Results of this study are consistent with previous work, in which the KD of the NTAIL–XD binding reaction is in the nM range.2,26 In further support of a tight NTAIL–XD complex is the relatively long half-life of active MeV P-L transcriptase complexes tethered on the nucleocapsid template, which has been determined to be well over 6 h.32 Moreover, such a high affinity is consistent with the ability to readily purify nucleocapsid-P complexes using rather stringent techniques such as CsCl isopycnic density centrifugation.33–36
A tight NTAIL–XD complex is not compatible with progressive movement of the viral polymerase complex along the nucleocapsid template. In fact, recent studies show that MeV polymerase activity (transcriptase elongation rate) is relatively constant over a wide range of NTAIL–XD binding affinities, with this having been shown using Box2 mutations that either maintained parent NTAIL–XD binding affinity or reduced this latter by as much as 30-fold.26 That the P-NTAIL binding affinity is not necessarily the primary determinant of a relatively slow polymerase elongation rate is further supported by observations in Sendai virus, where the polymerase elongation rate is even lower (i.e., 1.7 nt/s) than that of MeV (3.3 nt/s)32 and yet the NTAIL–XD complex has a KD of 60 μM.37 The finding that a relatively high MeV NTAIL–XD binding affinity does not impose constraint on (i.e., does not slow down) viral transcriptase function26 can be accounted for by two possible scenarios. In the first one, binding and release would not be required for transcription and replication: the P protein would be permanently bound to the nucleocapsid template and the polymerase would jump between adjacent P molecules. The so-called “jumping” mechanism has been proposed in the case of rabies virus (see Ref.38 and references therein cited), where (i) multimerization of P was shown to be dispensable for transcription39 and (ii) a similarly high affinity (KD = 160 nM) between N:RNA rings and the C-terminal domain of the phosphoprotein was observed.40 In this model, progress of the polymerase along the nucleocapsid template would rely on the dynamic breaking and reforming of contacts between L and P. The second possible scenario would rely on cellular cofactor(s) that might modulate the strength of the NTAIL–XD interaction and thereby facilitate cartwheeling. The major inducible heat shock protein (hsp70) was shown to stimulate MeV transcription and replication and to compete with XD for NTAIL binding.41–44 Based on these findings, it has been proposed that the basis for the hsp70-dependent stimulation of transcription and replication would reside in the ability of hsp70 to destabilize the NTAIL–XD complex, thereby promoting cycles of binding and release of the polymerase complex.10,11
In conclusion, the present study supports binding of one NTAIL molecule per XD molecule, as already reported for the full-length NTAIL protein,2 and shows that the MeV NTAIL–XD binding reaction is characterized by a submicromolar affinity. As we consistently obtained a KD in the nanomolar range irrespective of whether we used full-length or NTAIL peptides, we can rule out differences in the length of the reactants as a possible basis for the discrepancy with the data obtained by the group of Kingston.27 We can only speculate that the presence of the non-native TS dipeptide appended at the N-terminus of the NTAIL peptides used in the ITC studies by Kingston and coworkers is a possible basis for the experimentally observed differences, thereby leading to the calculation of a KD in the micromolar range.
Materials and Methods
Generation of NTAIL constructs and purification of recombinant NTAIL proteins
The coding region of the NTAILΔ3 construct was obtained by polymerase chain reaction (PCR) using Phusion (Finnzymes) polymerase, the plasmid pDEST14/NTAIL-HN, which encodes residues 401–525 of the Edmonston B MeV N protein with an N-terminal hexahistidine tag,2 as template and a couple of mutagenic primers (Operon) designed to introduce a TAA stop codon at position 517. After digestion with DpnI (New England Biolabs) to remove the methylated DNA template, CaCl2-competent E. coli TAM1 cells (Active Motif) were transformed with the amplified PCR product.
The NTAILTEV481 construct, encoding residues 401–525 of the Edmonston B MeV N protein with a TEV cleavage site (ENLYFQG) inserted after residue 481 and with a hexahistidine tag fused to its N-terminus, was obtained by PCR using as template the pDEST14/NTAILHN construct.2 Forward F-1 primer was designed to amplify an NTAIL-encoding fragment (fragment A) including the AttB1 sequence, while reverse R-1 primer was designed to introduce a TEV site after the codon encoding Ser481 of N. Concomitantly, fragment B was obtained by an independent PCR, using forward F-2 primer, designed to introduce a TEV site before codon encoding Ser482 of N, and reverse R-2 primer designed to amplify the downstream NTAIL fragment including the AttB2 sequence. Finally, a third PCR was carried out by using fragments A and B as template, and F-1 and R-2 as primers to yield the final amplification product. After purification (PCR Purification Kit, Qiagen), this latter was cloned into the pDEST14 vector (Invitrogen) using the Gateway recombination system (Invitrogen). The sequence of the coding region of all the expression plasmids was verified by sequencing (GATC Biotech) and found to conform to expectations.
The E. coli strains Rosetta [DE3] pLysS (Novagen) was used for expression of all recombinant proteins as already described,1,2 except that 2YT was used instead of LB medium. The induced cells were harvested, washed, and collected by centrifugation (5000 g, 10 min). The resulting bacterial pellets were frozen at −20°C.
Purification of histidine-tagged NTAIL and XD proteins was carried out as previously described,1,2 except that in the case of XD a washing step in the presence of increasing NaCl concentrations (from 0.5 up to 2M) was added before elution from the IMAC column. In the case of the NTAIL protein bearing a TEV protease site, the eluate from IMAC was desalted by using a HiPrep 26/10 desalting column (GE Healthcare), and the protein was eluted in buffer A (50 mM sodium phosphate pH 7, 300 mM NaCl, 10 mM imidazole). Cleavage with TEV protease was carried out overnight at 4°C using 1 mg of TEV per 10 mg of target protein. The TEV protease, bearing a hexahistidine tag, was purified by IMAC followed by SEC as already described45 (data not shown). After TEV digestion, the sample was incubated 1 h with gentle shaking with 4-mL chelating sepharose fast flow resin preloaded with Ni2+ ions (GE Healthcare), previously equilibrated in buffer A. The flow-through was then loaded onto a Superdex 200 16/60 column (GE Healthcare) and eluted in 10 mM sodium phosphate pH 7. The protein was concentrated using Centricon Plus-20 (molecular cutoff: 3000 Da) (Millipore). All purified proteins were stored in 10 mM sodium phosphate buffer pH 7 at −20°C. All purification steps, except for gel filtrations, were carried out at 4°C. Apparent molecular mass of proteins eluted from gel filtration columns was deduced from a calibration carried out with LMW and HMW calibration kits (GE Healthcare). Protein concentrations were calculated using the theoretical absorption coefficients ε (mg mL−1 cm−1) at 280 nm as obtained using the program ProtParam at the EXPASY server (http://www.expasy.ch/tools).
Mass spectrometry (MALDI-TOF)
The identity of the purified NTAILΔ3 and NTAIL482–525 proteins was confirmed by mass spectral analysis of tryptic fragments. The latter was obtained by digesting (0.25 μg trypsin) 1 μg of purified recombinant protein obtained after separation onto SDS-PAGE. The tryptic peptides were analyzed using an Autoflex II TOF/TOF. Spectra were acquired in the linear mode. Samples (0.7 μL containing 15 pmol) were mixed with an equal volume of sinapinic acid matrix solution, spotted on the target, then dried at room temperature for 10 min. The tryptic fragments were analyzed in the Autoflex matrix-assisted laser desorption ionization/time of flight (MALDI-TOF) (Bruker Daltonics, Bremen, Germany). Peptide fingerprints were obtained and compared with in silico protein digest (Biotools, Bruker Daltonics, Germany). The mass standards were either autolytic tryptic peptides or peptide standards (Bruker Daltonics).
Isothermal titration calorimetry
ITC experiments were carried out on a ITC200 isothermal titration calorimeter (Microcal, Northampton) at 20°C. In these studies, the concentration of NTAIL proteins was adjusted to 20 μM in the microcalorimeter cell (0.2 mL). The XD protein (stock solution at 200 μM) was added from a computer-controlled 40-μL microsyringe via a total of 19 injections of 2 μL each at intervals of 150 s. Note that the same XD sample was used in all these studies. Whatever the binding reaction being studied, the pair of proteins used in each binding assay were dialyzed against a 10 mM sodium phosphate buffer pH 7 to minimize undesirable buffer-related effects. The dialysis buffer was used in all preliminary equilibration and washing steps.
Heat dilution of the ligand was taken into account from peaks measured after full saturation of the protein sample contained in the microcalorimeter cell by the ligand. A theoretical titration curve was fitted to the experimental data using the ORIGIN software (Microcal). This software uses the relationship between the heat generated by each injection and ΔH° (enthalpy change in cal mol−1), KA (association binding constant in M−1), n (number of binding sites per monomer), total protein concentration, and free and total ligand concentrations. The variation in the entropy (ΔS in cal mol−1 deg−1) of each binding reaction was inferred from the variation in the free energy (ΔG), where this latter was calculated from the following relation: ΔG= −RT Ln 1/KA.
Acknowledgments
The authors thank Christophe Flaudrops and Nicholas Armstrong from the mass spectrometry platform of the IFR48 of Marseille for mass spectrometry analyses. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Johansson K, Bourhis JM, Campanacci V, Cambillau C, Canard B, Longhi S. Crystal structure of the measles virus phosphoprotein domain responsible for the induced folding of the C-terminal domain of the nucleoprotein. J Biol Chem. 2003;278:44567–44573. doi: 10.1074/jbc.M308745200. [DOI] [PubMed] [Google Scholar]
- 2.Bourhis JM, Receveur-Bréchot V, Oglesbee M, Zhang X, Buccellato M, Darbon H, Canard B, Finet S, Longhi S. The intrinsically disordered C-terminal domain of the measles virus nucleoprotein interacts with the C-terminal domain of the phosphoprotein via two distinct sites and remains predominantly unfolded. Protein Sci. 2005;14:1975–1992. doi: 10.1110/ps.051411805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morin B, Bourhis JM, Belle V, Woudstra M, Carrière F, Guigliarelli B, Fournel A, Longhi S. Assessing induced folding of an intrinsically disordered protein by site-directed spin-labeling EPR spectroscopy. J Phys Chem B. 2006;110:20596–20608. doi: 10.1021/jp063708u. [DOI] [PubMed] [Google Scholar]
- 4.Belle V, Rouger S, Costanzo S, Liquiere E, Strancar J, Guigliarelli B, Fournel A, Longhi S. Mapping alpha-helical induced folding within the intrinsically disordered C-terminal domain of the measles virus nucleoprotein by site-directed spin-labeling EPR spectroscopy. Proteins. 2008;73:973–988. doi: 10.1002/prot.22125. [DOI] [PubMed] [Google Scholar]
- 5.Bernard C, Gely S, Bourhis JM, Morelli X, Longhi S, Darbon H. Interaction between the C-terminal domains of N and P proteins of measles virus investigated by NMR. FEBS Lett. 2009;583:1084–1089. doi: 10.1016/j.febslet.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 6.Bischak CG, Longhi S, Snead DM, Costanzo S, Terrer E, Londergan CH. Probing structural transitions in the intrinsically disordered C-terminal domain of the measles virus nucleoprotein by vibrational spectroscopy of cyanylated cysteines. Biophys J. 2010;99:1676–1683. doi: 10.1016/j.bpj.2010.06.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gely S, Lowry DF, Bernard C, Ringkjobing-Jensen M, Blackledge M, Costanzo S, Darbon H, Daughdrill GW, Longhi S. Solution structure of the C-terminal X domain of the measles virus phosphoprotein and interaction with the intrinsically disordered C-terminal domain of the nucleoprotein. J Mol Recognit. 2010;23:435–447. doi: 10.1002/jmr.1010. [DOI] [PubMed] [Google Scholar]
- 8.Kavalenka A, Urbancic I, Belle V, Rouger S, Costanzo S, Kure S, Fournel A, Longhi S, Guigliarelli B, Strancar J. Conformational analysis of the partially disordered measles virus NTAIL-XD complex by SDSL EPR spectroscopy. Biophys J. 2010;98:1055–1064. doi: 10.1016/j.bpj.2009.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ringkjøbing Jensen M, Communie G, Ribeiro ED, Jr, Martinez N, Desfosses A, Salmon L, Mollica L, Gabel F, Jamin M, Longhi S, Ruigrok RW, Blackledge M. Intrinsic disorder in measles virus nucleocapsids. Proc Natl Acad Sci USA. 2011;108:9839–9844. doi: 10.1073/pnas.1103270108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Longhi S. Nucleocapsid structure and function. Curr Top Microbiol Immunol. 2009;329:103–128. doi: 10.1007/978-3-540-70523-9_6. [DOI] [PubMed] [Google Scholar]
- 11.Longhi S, Oglesbee M. Structural disorder within the measles virus nucleoprotein and phosphoprotein. Prot Peptide Lett. 2010;17:961–978. doi: 10.2174/092986610791498894. [DOI] [PubMed] [Google Scholar]
- 12.Longhi S, Receveur-Brechot V, Karlin D, Johansson K, Darbon H, Bhella D, Yeo R, Finet S, Canard B. The C-terminal domain of the measles virus nucleoprotein is intrinsically disordered and folds upon binding to the C-terminal moiety of the phosphoprotein. J Biol Chem. 2003;278:18638–18648. doi: 10.1074/jbc.M300518200. [DOI] [PubMed] [Google Scholar]
- 13.Bourhis J, Johansson K, Receveur-Bréchot V, Oldfield CJ, Dunker AK, Canard B, Longhi S. The C-terminal domain of measles virus nucleoprotein belongs to the class of intrinsically disordered proteins that fold upon binding to their physiological partner. Virus Res. 2004;99:157–167. doi: 10.1016/j.virusres.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 14.Wright PE, Dyson HJ. Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J Mol Biol. 1999;293:321–331. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- 15.Dunker AK, Oldfield CJ, Meng J, Romero P, Yang JY, Chen JW, Vacic V, Obradovic Z, Uversky VN. The unfoldomics decade: an update on intrinsically disordered proteins. BMC Genomics. 2008;9(Suppl 2):S1. doi: 10.1186/1471-2164-9-S2-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dunker AK, Silman I, Uversky VN, Sussman JL. Function and structure of inherently disordered proteins. Curr Opin Struct Biol. 2008;18:756–764. doi: 10.1016/j.sbi.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 17.Turoverov KK, Kuznetsova IM, Uversky VN. The protein kingdom extended: ordered and intrinsically disordered proteins, their folding, supramolecular complex formation, and aggregation. Prog Biophys Mol Biol. 2010;102:73–84. doi: 10.1016/j.pbiomolbio.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Uversky VN. The mysterious unfoldome: structureless, underappreciated, yet vital part of any given proteome. J Biomed Biotechnol. 2010;2010:568068. doi: 10.1155/2010/568068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Uversky VN, Dunker AK. Understanding protein non-folding. Biochim Biophys Acta. 2010;1804:1231–1264. doi: 10.1016/j.bbapap.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diallo A, Barrett T, Barbron M, Meyer G, Lefevre PC. Cloning of the nucleocapsid protein gene of peste-des-petits-ruminants virus: relationship to other morbilliviruses. J Gen Virol. 1994;75:233–237. doi: 10.1099/0022-1317-75-1-233. [DOI] [PubMed] [Google Scholar]
- 21.Kingston RL, Hamel DJ, Gay LS, Dahlquist FW, Matthews BW. Structural basis for the attachment of a paramyxoviral polymerase to its template. Proc Natl Acad Sci USA. 2004;101:8301–8306. doi: 10.1073/pnas.0402690101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oldfield CJ, Cheng Y, Cortese MS, Romero P, Uversky VN, Dunker AK. Coupled folding and binding with alpha-helix-forming molecular recognition elements. Biochemistry. 2005;44:12454–12470. doi: 10.1021/bi050736e. [DOI] [PubMed] [Google Scholar]
- 23.Mohan A, Oldfield CJ, Radivojac P, Vacic V, Cortese MS, Dunker AK, Uversky VN. Analysis of molecular recognition features (MoRFs) J Mol Biol. 2006;362:1043–1059. doi: 10.1016/j.jmb.2006.07.087. [DOI] [PubMed] [Google Scholar]
- 24.Fuxreiter M, Tompa P, Simon I. Local structural disorder imparts plasticity on linear motifs. Bioinformatics. 2007;23:950–956. doi: 10.1093/bioinformatics/btm035. [DOI] [PubMed] [Google Scholar]
- 25.Vacic V, Oldfield CJ, Mohan A, Radivojac P, Cortese MS, Uversky VN, Dunker AK. Characterization of molecular recognition features, MoRFs, and their binding partners. J Proteome Res. 2007;6:2351–2366. doi: 10.1021/pr0701411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shu Y, Habchi J, Costanzo S, Padilla A, Brunel J, Gerlier D, Oglesbee M, Longhi S. Plasticity in structural and functional interactions between the phosphoprotein and nucleoprotein of measles virus. J Biol Chem. 287:11951–11967. doi: 10.1074/jbc.M111.333088. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yegambaram K, Kingston RL. The feet of the measles virus polymerase bind the viral nucleocapsid protein at a single site. Protein Sci. 2010;19:893–899. doi: 10.1002/pro.354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iakoucheva LM, Kimzey AL, Masselon CD, Smith RD, Dunker AK, Ackerman EJ. Aberrant mobility phenomena of the DNA repair protein XPA. Protein Sci. 2001;10:1353–1362. doi: 10.1110/ps.ps.40101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tompa P. Intrinsically unstructured proteins. Trends Biochem Sci. 2002;27:527–533. doi: 10.1016/s0968-0004(02)02169-2. [DOI] [PubMed] [Google Scholar]
- 30.Blocquel D, Habchi J, Gruet A, Blangy S, Longhi S. Compaction and binding properties of the intrinsically disordered C-terminal domain of Henipavirus nucleoprotein as unveiled by deletion studies. Mol Biosyst. 2012;8:392–410. doi: 10.1039/c1mb05401e. [DOI] [PubMed] [Google Scholar]
- 31.Blenner MA, Shur O, Szilvay GR, Cropek DM, Banta S. Calcium-induced folding of a beta roll motif requires C-terminal entropic stabilization. J Mol Biol. 2010;400:244–256. doi: 10.1016/j.jmb.2010.04.056. [DOI] [PubMed] [Google Scholar]
- 32.Plumet S, Duprex WP, Gerlier D. Dynamics of viral RNA synthesis during measles virus infection. J Virol. 2005;79:6900–6908. doi: 10.1128/JVI.79.11.6900-6908.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robbins SJ, Bussell RH. Structural phosphoproteins associated with purified measles virions and cytoplasmic nucleocapsids. Intervirology. 1979;12:96–102. doi: 10.1159/000149074. [DOI] [PubMed] [Google Scholar]
- 34.Stallcup KC, Wechsler SL, Fields BN. Purification of measles virus and characterization of subviral components. J Virol. 1979;30:166–176. doi: 10.1128/jvi.30.1.166-176.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Robbins SJ, Bussell RH, Rapp F. Isolation and partial characterization of two forms of cytoplasmic nucleocapsids from measles virus-infected cells. J Gen Virol. 1980;47:301–310. doi: 10.1099/0022-1317-47-2-301. [DOI] [PubMed] [Google Scholar]
- 36.Oglesbee M, Tatalick L, Rice J, Krakowka S. Isolation and characterization of canine distemper virus nucleocapsid variants. J Gen Virol. 1989;70:2409–2419. doi: 10.1099/0022-1317-70-9-2409. [DOI] [PubMed] [Google Scholar]
- 37.Houben K, Marion D, Tarbouriech N, Ruigrok RW, Blanchard L. Interaction of the C-terminal domains of sendai virus N and P proteins: comparison of polymerase-nucleocapsid interactions within the paramyxovirus family. J Virol. 2007;81:6807–6816. doi: 10.1128/JVI.00338-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leyrat C, Gerard FC, de Almeida Ribeiro ED, Jr, Ivanov I, Ruigrok RW, Jamin M. Structural disorder in proteins of the rhabdoviridae replication complex. Protein Pept Lett. 2010;17:979–987. doi: 10.2174/092986610791498939. [DOI] [PubMed] [Google Scholar]
- 39.Jacob Y, Real E, Tordo N. Functional interaction map of lyssavirus phosphoprotein: identification of the minimal transcription domains. J Virol. 2001;75:9613–9622. doi: 10.1128/JVI.75.20.9613-9622.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ribeiro ED, Jr, Leyrat C, Gerard FC, Albertini AA, Falk C, Ruigrok RW, Jamin M. Binding of Rabies virus polymerase cofactor to recombinant circular nucleoprotein-RNA complexes. J Mol Biol. 2009;394:558–575. doi: 10.1016/j.jmb.2009.09.042. [DOI] [PubMed] [Google Scholar]
- 41.Zhang X, Glendening C, Linke H, Parks CL, Brooks C, Udem SA, Oglesbee M. Identification and characterization of a regulatory domain on the carboxyl terminus of the measles virus nucleocapsid protein. J Virol. 2002;76:8737–8746. doi: 10.1128/JVI.76.17.8737-8746.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang X, Bourhis JM, Longhi S, Carsillo T, Buccellato M, Morin B, Canard B, Oglesbee M. Hsp72 recognizes a P binding motif in the measles virus N protein C-terminus. Virology. 2005;337:162–174. doi: 10.1016/j.virol.2005.03.035. [DOI] [PubMed] [Google Scholar]
- 43.Carsillo T, Zhang X, Vasconcelos D, Niewiesk S, Oglesbee M. A single codon in the nucleocapsid protein C terminus contributes to in vitro and in vivo fitness of Edmonston measles virus. J Virol. 2006;80:2904–2912. doi: 10.1128/JVI.80.6.2904-2912.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oglesbee M. Nucleocapsid protein interactions with the major inducible 70 kDa heat shock protein. In: Longhi S, editor. Measles virus nucleoprotein. Hauppage, NY: Nova; 2007. pp. 53–98. [Google Scholar]
- 45.Vincentelli R, Bignon C, Gruez A, Canaan S, Sulzenbacher G, Tegoni M, Campanacci V, Cambillau C. Medium-scale structural genomics: strategies for protein expression and crystallization. Acc Chem Res. 2003;36:165–172. doi: 10.1021/ar010130s. [DOI] [PubMed] [Google Scholar]