

Abstract
Pulmonary vasodilation is mediated through the activation of protein kinase G (PKG) via a signaling pathway involving nitric oxide (NO), natriuretic peptides (NP), and cyclic guanosine monophosphate (cGMP). In pulmonary hypertension secondary to congenital heart disease, this pathway is endogenously activated by an early vascular upregulation of NO and increased myocardial B-type NP expression and release. In the treatment of pulmonary hypertension, this pathway is exogenously activated using inhaled NO or other pharmacological agents. Despite this activation of cGMP, vascular dysfunction is present, suggesting that NO-cGMP independent mechanisms are involved and were the focus of this study. Exposure of pulmonary artery endothelial or smooth muscle cells to the NO donor, Spermine NONOate (SpNONOate), increased peroxynitrite (ONOO−) generation and PKG-1α nitration, while PKG-1α activity was decreased. These changes were prevented by superoxide dismutase (SOD) or manganese(III)tetrakis(1-methyl-4-pyridyl)porphyrin (MnTMPyP) and mimicked by the ONOO− donor, 3-morpholinosydnonimine N-ethylcarbamide (SIN-1). Peripheral lung extracts from 4-week old lambs with increased pulmonary blood flow and pulmonary hypertension (Shunt lambs with endogenous activation of cGMP) or juvenile lambs treated with inhaled NO for 24h (with exogenous activation of cGMP) revealed increased ONOO− levels, elevated PKG-1α nitration, and decreased kinase activity without changes in PKG-1α protein levels. However, in Shunt lambs treated with L-arginine or lambs administered polyethylene glycol conjugated-SOD (PEG-SOD) during inhaled NO exposure, ONOO− and PKG-1α nitration were diminished and kinase activity was preserved. Together our data reveal that vascular dysfunction can occur, despite elevated levels of cGMP, due to PKG-1α nitration and subsequent attenuation of activity.
Keywords: Peroxynitrite, cell signaling, pulmonary hypertension, nitration
INTRODUCTION
The mechanisms that contribute to pulmonary hypertension are muti-factorial and complex. Mounting evidence indicates that pulmonary vascular endothelial cell injury plays a critical role. Endothelial cell injury disrupts a complex homeostatic balance, resulting in an abnormal increase in vascular tone. Clinical and experimental studies have demonstrated alterations in the nitric oxide (NO)-cyclic guanosine monophosphate (cGMP) pathway, but the precise mechanisms, particularly the role of downstream mediators, remain unclear.
Previously, we have described alterations in pulmonary vascular endothelial function in two distinct models. In the first model, a large vascular graft (shunt) is placed between the aorta and pulmonary artery in late gestation fetal lambs (Reddy et al., 1995). After spontaneous delivery these lambs develop a significant left-to-right shunt, which exposes the pulmonary vasculature to increased blood flow and shear stress, leading to an upregulation of endothelial nitric oxide synthase (eNOS) and B-type natriuretic peptide (BNP). In the second model, 1-month old intact lambs are exposed to mechanical ventilation with 21% oxygen and inhaled NO for 24h (Black et al., 1999; McMullan et al., 2001). Due to endogenous and exogenous activation of pulmonary artery endothelial cells respectively, both models result in an increase in plasma and lung tissue cGMP levels. However, despite an increase in cGMP levels, both models display pulmonary vascular dysfunction that manifests as a selective impairment in endothelium-dependent pulmonary vascular relaxation in Shunt lambs and an abnormal increase in pulmonary arterial pressure and vascular resistance upon the acute withdrawal of inhaled NO in the second model. Furthermore, in both models endothelial dysfunction is also demonstrated by decreased eNOS activity and increased oxidative stress.
In response to NO and BNP, cGMP activates the downstream mediator protein kinase G (PKG) (Lohmann et al., 1997). PKG is a serine/threonine kinase that plays an important role in vascular relaxation (Hofmann et al., 2000; Walter, 1989). PKG exists in two forms: the soluble homodimer, PKG-I, and the membrane associated monomer, PKG-II (Walter, 1989). PKG-I has two isoforms: Iα (75KD) and Iβ (78KD), which are the products of alternate splicing of mRNA (Lincoln et al., 1988). PKG-Iα, predominantly found in the lungs, is more sensitive to activation by cGMP than PKG-Iβ and is the primary isoform involved in vasodilation (Geiselhoringer et al., 2004; Tamura et al., 1996). However, there is little information as to whether PKG-1α is dysregulated under conditions of endogenous or exogenous activation of cGMP. Recent reports do suggest that under hypoxic conditions there is a decrease in PKG-I activity due to peroxynitrite (ONOO−) mediated tyrosine nitration (Negash et al., 2007). Interestingly, our past investigations have shown that both Shunt lambs (Lakshminrusimha et al., 2007) and lambs exposed to inhaled NO (Oishi et al., 2006) have increased levels of protein nitration. Therefore, the purpose of the present study was to determine whether the nitration-induced decrease in PKG-1α kinase activity contributes to pulmonary vascular endothelial dysfunction secondary to endogenous (Shunt) and/or exogenous (inhaled NO) endothelial activation.
MATERIAL AND METHODS
Materials
Polyclonal anti-PKG-1α (goat) antibody was from Santa Cruz biotechnology (Santa Cruz, CA); Monoclonal anti-nitrotyrosine (mouse) antibody (Clone: CC22.8C7.3), monoclonal anti-pSer239VASP (mouse) antibody (Clone: 16C2), and ONOO− were from EMD Biosciences, Inc. (San Diego, CA); Monoclonal anti-VASP (mouse) antibody (Clone: IE273) was from Enzo life sciences (Plymouth Meeting, PA); Human BNP was from American Peptide Company (Sunnyvale, CA); Monoclonal anti-β-actin (mouse) antibody (Clone: AC-15), Polyethylene glycol-conjugated Superoxide Dismutase (PEG-S O D ) , P E G-Catalase, and Manganese(III)tetrakis(1-methyl-4-pyridyl)porphyrin (MnTMPyP) were from Sigma life sciences (St. Louis, MO); Cyclic GMP EIA Kit, Spermine NONOate (SpNONOate), 3-morpholinosydnonimine N-ethylcarbamide (SIN-1), and Dihydrorhodamine 123 (DHR) were from Cayman Chemicals (Ann Arbor, MI); Bovine PKG full length recombinant protein (alpha1 isozyme) and a non-radioisotopic kit for measuring PKG activity was from Cyclex Co., Ltd. (Nagano, Japan).
Animal Studies
Shunt lambs
Eighteen mixed-breed Western pregnant ewes (137–141 days of gestation, term = 145 days), anesthetized with the use of local anesthesia (2% lidocaine hydrochloride) and inhalational anesthesia (1–3% isoflorane), were operated on under sterile conditions to place an 8.0mm Gore-Tex vascular graft (~2mm long; W.L. Gore and Associates, Milpitas, CA) between the ascending aorta and the main pulmonary artery in the fetal lamb. This procedure has been previously described in detail (Reddy et al., 1995). Six lambs received, lifelong from the first day of life, 500mg/kg of L-arginine (L-arginine-Hydrochloride 21% Braun, B. Braun Melsungen AG, Melsungen, Germany), while a further six lambs received the equivalent amount of vehicle. Lambs were treated twice daily (8 AM and 8 PM) intravenously. L-arginine was mixed 1:1 with sterile isotonic NaCl solution (Sodio Cloruro 0,9%, Laboratori Diaco Biomedicali S.p.A., Trieste, Italy). Sterile isotonic NaCl solution (Sodio Cloruro 0,9%, Laboratori Diaco Biomedicali S.p.A., Trieste, Italy) was used as a vehicle. Three weeks after spontaneous delivery, Shunt and age-matched control lambs were fasted for 24h with free access to water. The lambs were then anesthetized with ketamine hydrochloride (15mg/kg IM). Under additional local anesthesia with 1% lidocaine hydrochloride, polyurethane catheters were placed in an artery and a vein of a hind leg. These catheters were advanced to the descending aorta and to the inferior vena cava, respectively. The lambs were then anesthetized with ketamine hydrochloride (0.3mg/kg/min), diazepam (0.002mg/kg/min), and fentanyl citrate (1.0μg/kg/h), intubated with a 7.0mm outer diameter cuffed endotracheal tube, and mechanically ventilated with 21% oxygen using a Healthdyne pediatric time-cycled pressure-limited ventilator. With the use of strict aseptic technique, a midsternotomy incision was then performed. Patency of the vascular graft was confirmed by inspection and changes in oxygen saturation. A side-biting vascular clamp was used to isolate peripheral lung tissue from randomly selected lobes, and the incisions were cauterized. Approximately 300mg of peripheral lung were obtained for each biopsy; four biopsies were obtained and stored at −80°C until analyzed. Blood was obtained from the femoral artery.
Exposure of lambs to inhaled NO
The surgical preparation used was previously described (Black et al., 1999; McMullan et al., 2001). After a 30min recovery, either PEG or PEG-SOD diluted in 5ml of saline was delivered through the left pulmonary artery catheter. PEG or PEG-SOD (1,000-2,000U/kg) was given every 6h to complete a total of four doses. The dose of PEG-SOD was based upon previous studies that demonstrate a sustained increase in plasma SOD activity (8, 32). Inhaled NO (40ppm) was delivered into the inspiratory limb of the ventilator (Ohmeda INOvent, Datex Ohmeda Inc., Madison, WI) and continued for 24h. The inspired concentration of NO and nitrogen dioxide were continuously quantified by electrochemical methodology (Ohmeda INOvent, Datex Ohmeda Inc., Madison, WI). The hemodynamic variables were monitored continuously. The systemic arterial blood gases were determined intermittently, and the ventilation was adjusted to achieve a PaCO2 between 35torr to 45torr and a PaO2>50torr. Sodium bicarbonate was administered intermittently to maintain a pH>7.30. Normal saline was administered intermittently to maintain stable atrial pressures throughout the study period. Peripheral lung biopsies were performed before and after 24h of inhaled NO.
At the end of each protocol, all lambs were euthanized with a lethal injection of pentobarbital sodium in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. All animal protocols and procedures were approved by the committee on Animal Research at the University of California, San Francisco, the Medical College of Georgia, Augusta, and the German Heart Center, Munich.
Cell culture
Primary cultures of ovine pulmonary artery endothelial cells (PAEC) were isolated as described previously (Wedgwood et al., 2001a). Cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) with 1gm glucose, L-glutamine, and sodium pyruvate, supplemented with 10% fetal bovine serum (FBS) (Hyclone, Logan, UT) and 1% antibiotic-antimycotic solution (Mediatech, Herndon, VA) at 37°C in a humidifier with 5% CO2 and 95% air. Cells used were seeded at 50% confluence and utilized when 100% confluent. Primary cultures of pulmonary artery smooth muscle cells (PASMC) from 4-week old lambs were isolated by the explant technique, as we have previously described (Wedgwood et al., 2001b). Briefly, a segment of the main pulmonary artery from a 4-week old lamb was excised and placed in a sterile 10cm dish containing DMEM, supplemented with 1gm/l glucose. The segment was stripped of adventitia with a sterile forceps. The main pulmonary artery segment was then cut longitudinally to open the vessel, and the endothelial layer was removed by gentle rubbing with a cell scraper. The vessel was then cut into 2mm segments, inverted, and placed on a collagen coated 35mm tissue culture dish. DMEM (~50μl) containing 10% FBS (Hyclone), antibiotics, and antimycotics (Media Tech) was then added to each segment, and the cells were grown overnight at 37°C in a humidified atmosphere with 5% CO2 and 95% air. The next day an additional 2ml of medium was added. The growth medium was subsequently changed every 2 days. When smooth muscle cell (SMC) islands were observed under the microscope, the tissue segment was removed, and the individual cell islands were subcloned using cloning rings. Identity was confirmed as PASMC by immunostaining (>99% positive) with SMC actin, caldesmon, and calponin. This was taken as evidence that the cultures were not contaminated with fibroblasts or endothelial cells. All culture for subsequent experiments was maintained in DMEM supplemented with 10% FBS, 1% antibiotics, and antimycotics at 37°C in a humidified atmosphere with 5% CO2 and 95% air. All experiments were conducted in cells between passages 5 and 15.
Measurement of Peroxynitrite Levels
The formation of peroxynitrite (ONOO−) was determined by the ONOO− dependent oxidation of dihydrorhodamine (DHR) 123 to rhodamine 123, as described previously (Song et al., 2007). Cultured PAEC or PASMC were pretreated with PEG-Catalase (100U, 30min) to reduce H2O2 dependent DHR 123 oxidation. The cells were then treated with either PEG-SOD (100U, 30min) or MnTMPyP (25μM, 30min) followed by incubation with SIN-1 (500μM, 30min) or SpNONOate (100μM, 4h) in phenol red-free media containing 5μmol/L DHR 123. Similarly, ONOO− levels were determined in peripheral lung tissue obtained from 4-week old control and Shunt lambs treated with or without L-arginine and the lambs exposed to inhaled NO with or without PEG-SOD. The tissue was pulverized; 10mg of tissue was placed in a microfuge tube, 100μl of 1xPBS was added, and the tissue was vortexed 3x for 10sec. The lysate was incubated with PEG-Catalase (100U) for 30min and then added to a 96 well black plate in the presence of 5μmol/L DHR 123 in 1xPBS for 1h. In both cases, the fluorescence of rhodamine 123 was measured at excitation 485nm and emission 545nm using a Fluoroskan Ascent Microplate Fluorometer.
Western Blot analysis
Cells or peripheral lung tissue were lysed in Triton X-100 lysis buffer (containing protease- and phosphatase- inhibitors), centrifuged at 6000g, and the supernatant was collected as previously described (Sharma et al., 2008; Sud et al., 2007). Tissue and cell extracts (25 μg) were resolved using 4-20% Tris-SDS-Hepes PAGE, electrophoretically transferred to Immuno-BlotTM PVDF membrane (Bio-Rad Laboratories, Hercules, CA), and then blocked with 5% nonfat dry milk in Tris-buffered saline. The membranes were probed with antibodies against PKG-1α (1:500 dilution) or pSer239VASP (1:500 dilution). Reactive bands were visualized using chemiluminescence (Pierce Laboratories, Rockford, IL) on a Kodak 440CF image station. Band intensity was quantified using Kodak 1D image processing software. Protein expression was normalized by re-probing with anti β-actin or total VASP.
Immunoprecipitation analysis
Cells or peripheral lung tissue were homogenized in 3x weight/volume of IP buffer (25mM Hepes, pH 7.5, 150mM NaCl, 1% NP-40, 10mM MgCl2, 1mM EDTA, 2% glycerol, supplemented with protease inhibitor). Homogenates were then centrifuged at 20,000g at 4°C for 10min, the tissue supernatant was collected, and protein concentration was quantified by the Bio-Rad DC Protein Assay (Bio-Rad Laboratories, Hercules, CA). To 1000μg of total protein, 4μg of antibody against PKG-1α was added; the volume was brought to 1ml with IP buffer, and the mixture was nutated at 4°C overnight. To precipitate the bound protein, 30μl of protein G plus agarose suspension (EMD biosciences, Inc., San Diego, CA) was added, and the samples were nutated for 2h at 4°C. To collect the bead-bound antibody, the samples were then centrifuged at 2000g for 5min, the supernatant was removed, and the beads were washed 3x with 500μl of IP buffer. To the samples, 30μl of 2x Laemmli buffer was added, and the samples were boiled for 5min and then resolved using 4-20% Tris-SDS-Hepes PAGE. The membrane was then probed for 3-nitrotyrosine (1:100 dilution), as described above. IP efficiency was normalized by re-probing with PKG-1α.
Determination of PKG activity
Total PKG (PKG-1 and -II) activity was determined using a non-radioactive immunoassay to measure PKG mediated phosphorylation of a synthetic substrate in cell lysates, peripheral lung extracts, or recombinant bovine PKG-1α protein, according to the manufacturer’s directions. 8-Br cGMP was used to activate PKG to ensure that endogenous cGMP was not a limiting factor, as per manufacturer’s instructions. The results were reported as pmols of phosphate incorporated into the GST-G substrate fusion protein in the presence of cGMP (10μM) per minute at 30°C per μg of PKG protein (pmol/min/μg).
Measurement of cGMP
PAEC and PASMC were grown to confluence and then serum starved overnight. Cells were treated with BNP (10μM) in two doses of 5μM, 30min apart for a total duration of 1h in the presence or absence of SIN-1 (500μM, 30min) pretreatment. The cells were then lysed in 0.1M HCl, and the supernatant was collected by centrifugation at 1000g for 10min. Subsequently, cGMP levels were measured using an immunoassay based EIA kit, according to the manufacturer’s protocol.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism version 4.01 (GraphPad Software, San Diego, CA). The mean ± SEM was calculated in all experiments, and statistical significance determined either by the unpaired t-test (for 2 groups) or ANOVA (for ≥ 3 groups). For the ANOVA analyses, Newman-Kuels post-hoc testing was employed. A value of p<0.05 was considered significant.
RESULTS
Previously, we have shown that Shunt lambs and lambs exposed to inhaled NO have increased levels of cGMP (Oishi et al., 2007; Ross et al., 2005). However, we have also observed a decrease in vascular relaxation in both these models, suggestive of vascular dysfunction (Oishi et al., 2007; Ross et al., 2005). To determine if there are NO-cGMP independent disruptions in signaling downstream of cGMP that are involved in the development of this vascular dysfunction, we initially performed in vitro studies using primary cultures of PAEC and PASMC. Exposing PAEC to the NO and superoxide donor, SIN-1 (500μM, 30min), which mimics the endothelial nitric oxide synthase (eNOS) uncoupling seen in our Shunt lambs (Steinhorn et al., 2001), increased ONOO− levels, as determined by the oxidation of DHR 123 to rhodamine 123 (Fig. 1 A). These elevated ONOO− levels correlated with an increase in PKG-1α nitration (Fig. 1 B) and a subsequent decrease in kinase activity (Fig. 1 C). However, the addition of PEG-SOD (100U) or the cell permeable SOD mimetic, MnTMPyP (25μM), attenuated the effects of SIN-1. Next, we determined if the addition of the NO donor, SpNONOate, which mimics the elevated exogenous NO levels seen in the 24h NO treated lambs, would impair cGMP/PKG-1α signaling. We exposed PAEC to SpNONOate (100μM, 4h) and found that ONOO− levels were elevated (Fig. 2 A), PKG-1α nitration was increased (Fig. 2 B), and kinase activity was decreased (Fig. 2 C). Similarly, we exposed PASMC to SIN-1 (500μM, 30min) and found increased ONOO− levels (Fig. 3 A), increased PKG-1α nitration (Fig. 3 B), and attenuated kinase activity (Fig. 3 C). Further, we were able to show that in PASMC, the SpNONOate-dependent increase in ONOO− levels (Fig. 4 A) and subsequent PKG-1α nitration (Fig. 4 B) resulted in decreased kinase activity (Fig. 4 C). The deleterious effects of both SIN-1 and SpNONOate on the cGMP/PKG-1α signaling pathway were mitigated by the pretreatment of PAEC & PASMC with PEG-SOD (100U) or MnTMPyP (25μM), suggesting that the effects on PKG-1α are the result of a nitration-mediated decrease in kinase activity. When recombinant bovine PKG-1α was exposed to authentic ONOO−, kinase activity was reduced, further confirming the key role of ONOO− in PKG-1α nitration and enzyme inhibition (Fig. 5). Finally, to investigate if the nitration induced decrease in kinase activity is independent of cGMP levels, we treated PAEC and PASMC with the guanylyl cyclase activator, BNP (10μM, 1h), in the presence or absence of SIN-1 (500μM, 30min). In PAEC, our results demonstrate that BNP treatment increases cGMP levels (Fig. 6 A) and PKG catalytic activity, as shown by the increased phosphorylation of VASP at Serine239 (Fig. 6 B). However, in the presence of SIN-1, PKG kinase activity is decreased, while cGMP levels are maintained. Similarly, using PASMC, we show that SIN-1 does not alter the cGMP levels induced by BNP (Fig. 6 C) but attenuates the levels of pSer239VASP (Fig. 6 D).
Fig. 1. Peroxynitrite (ONOO−) increases protein kinase G-1α (PKG-1α) nitration, and decreases PKG activity in pulmonary arterial endothelial cells (PAEC).

ONOO− formation induced by the nitric oxide (NO) and the superoxide donor, 3-morpholinosydnonimine N-ethylcarbamide (SIN-1), was assayed by monitoring the ONOO− dependent oxidation of dihydrorhodamine 123 (DHR 123) to rhodamine 123. The results are expressed as fold DHR oxidation compared to untreated control cultures. SIN-1 increases ONOO− levels in PAEC, and this was attenuated by the addition of polyethylene glycol conjugated superoxide dismutase (PEG-SOD) or manganese(III)tetrakis(1-methyl-4-pyridyl)porphyrin (MnTMPyP) (A). In addition, protein extracts (1000μg) were subjected to immunoprecipitation analysis using an antibody raised against PKG-1α. The immunoprecipitates were resolved using 4-20% Tris-SDS-Hepes PAGE and electrophoretically transferred to a PVDF membrane. The level of nitrated PKG-1α was then determined by probing the membranes with an antiserum raised against 3-nitrotyrosine. Blots were then stripped and re-probed for PKG-1α to normalize for the efficiency of the immunoprecipitation. A representative blot is shown. The exposure of PAEC to SIN-1 results in a significant increase in PKG-1α nitration (B). PEG-SOD or MnTMPyP both inhibit PKG-1α nitration (B). Using an ELISA based assay, we also found that SIN-1 attenuated total PKG activity, and this was prevented by pretreatment with PEG-SOD or MnTMPyP (C). Data are mean ± SEM, n=4, *p<0.05 vs. untreated, † p<0.05 vs SIN-1.
Fig. 2. Nitric oxide elevates ONOO− levels, increases PKG-1α nitration, and decreases PKG activity in pulmonary arterial endothelial cells.

ONOO− formation induced by the NO donor, Spermine NONOate (SpNONOate), was assayed by monitoring the ONOO− dependent oxidation of DHR 123 to rhodamine 123. The results are expressed as fold DHR oxidation compared to untreated control cultures. SpNONOate increases ONOO− levels in PAEC, and this was attenuated by the addition of PEG-SOD or MnTMPyP (A). In addition, protein extracts (1000μg) were subjected to immunoprecipitation analysis using an antibody raised against PKG-1α. The immunoprecipitates were resolved using 4-20% Tris-SDS-Hepes PAGE and electrophoretically transferred to a PVDF membrane. The level of nitrated PKG-1α was then determined by probing the membranes with an antiserum raised against 3-nitrotyrosine. Blots were then stripped and re-probed for PKG-1α to normalize for the efficiency of the immunoprecipitation. A representative blot is shown. The exposure of PAEC to SpNONOate results in a significant increase in PKG-1α nitration (B). PEG-SOD or MnTMPyP both inhibit PKG-1α nitration (B). Using an ELISA based assay, we also found that SpNONOate attenuated total PKG activity, and this was prevented by pretreatment with PEG-SOD or MnTMPyP (C). Data are mean ± SEM, n=4, *p<0.05 vs. untreated, † p<0.05 vs SpNONOate.
Fig. 3. Peroxynitrite increases PKG-1α nitration, and decreases PKG activity in pulmonary arterial smooth muscle cells (PASMC).

ONOO− formation induced by the NO and the superoxide donor, SIN-1, was assayed by monitoring the ONOO− dependent oxidation of DHR 123 to rhodamine 123. The results are expressed as fold DHR oxidation compared to untreated control cultures. SIN-1 increases ONOO− levels in PASMC, and this was attenuated by the addition of PEG-SOD or MnTMPyP (A). In addition, protein extracts (1000μg) were subjected to immunoprecipitation analysis using an antibody raised against PKG-1α. The immunoprecipitates were resolved using 4-20% Tris-SDS-Hepes PAGE and electrophoretically transferred to a PVDF membrane. The level of nitrated PKG-1α was then determined by probing the membranes with an antiserum raised against 3-nitrotyrosine. Blots were then stripped and re-probed for PKG-1α to normalize for the efficiency of the immunoprecipitation. A representative blot is shown. The exposure of PASMC to SIN-1 results in a significant increase in PKG-1α nitration (B). PEG-SOD or MnTMPyP both inhibit PKG-1α nitration (B). Using an ELISA based assay, we also found that SIN-1 attenuated total PKG activity, and this was prevented by pretreatment with PEG-SOD or MnTMPyP (C). Data are mean ± SEM, n=4, *p<0.05 vs. untreated, † p<0.05 vs SIN-1.
Fig. 4. Nitric oxide elevates ONOO− levels, increases PKG-1α nitration, and decreases PKG activity in pulmonary arterial smooth muscle cells.

ONOO− formation induced by the NO donor, SpNONOate, was assayed by monitoring the ONOO− dependent oxidation of DHR 123 to rhodamine 123. The results are expressed as fold DHR oxidation compared to untreated control cultures. SpNONOate increases ONOO− levels in PASMC, and this was attenuated by the addition of PEG-SOD or MnTMPyP (A). In addition, protein extracts (1000μg) were subjected to immunoprecipitation analysis using an antibody raised against PKG-1α. The immunoprecipitates were resolved using 4-20% Tris-SDS-Hepes PAGE and electrophoretically transferred to a PVDF membrane. The level of nitrated PKG-1α was then determined by probing the membranes with an antiserum raised against 3-nitrotyrosine. Blots were then stripped and re-probed for PKG-1α to normalize for the efficiency of the immunoprecipitation. A representative blot is shown. The exposure of PASMC to SpNONOate results in a significant increase in PKG-1α nitration (B). PEG-SOD or MnTMPyP both inhibit PKG-1α nitration (B). Using an ELISA based assay, we also found that SpNONOate attenuated total PKG activity, and this was prevented by pretreatment with PEG-SOD or MnTMPyP (C). Data are mean ± SEM, n=4, *p<0.05 vs. untreated, † p<0.05 vs SpNONOate.
Fig. 5. ONOO− decreases the kinase activity of recombinant bovine PKG1-α protein.
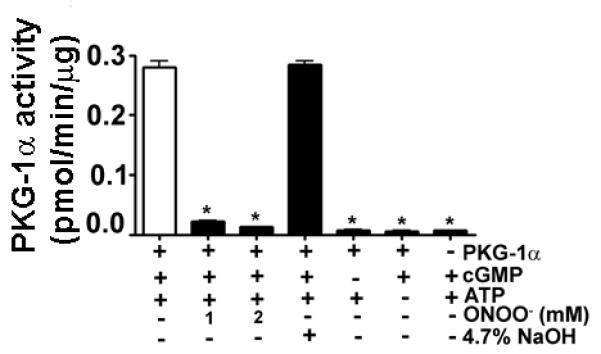
The kinase activity of purified recombinant bovine PKG-1α protein was measured by an ELISA in the presence of ONOO−. PKG1-α kinase activity is decreased in the presence of ONOO−. We also confirmed that the presence of 4.7% NaOH (the solvent for ONOO−) did not alter PKG-1α kinase activity. Data are mean ± SEM, n=3; *p<0.05 vs. untreated.
Fig. 6. The SIN-1 mediated attenuation of PKG kinase activity is independent of cellular cGMP levels.
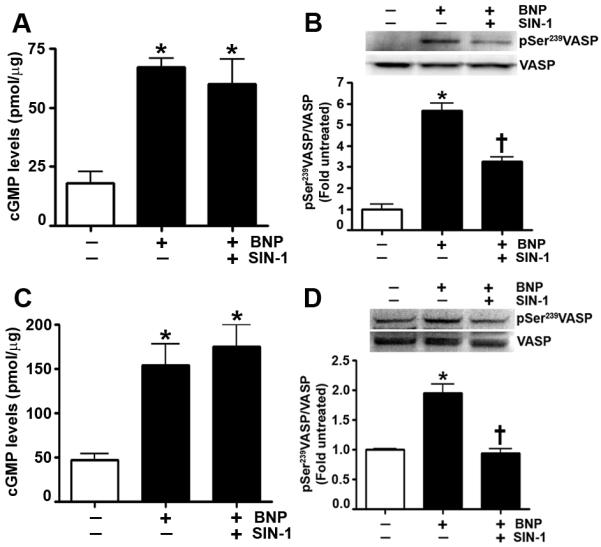
PAEC and PASMC were treated with B-type natriuretic peptide (BNP) (10μM) in two doses of 5μM, 30min apart for a total duration of 1h in the presence or absence of SIN-1 (500μM, 30min) pretreatment. Using an ELISA based assay, we demonstrate in PAEC that BNP increases cGMP levels in the presence or absence of SIN-1 (A). Immunoblot analysis shows that SIN-1 decreases the PKG dependent phosphorylation of vasodilator-stimulated phosphoprotein (VASP) at Serine239 induced by BNP (B). Similarly, our results show in PASMC that SIN-1 does not alter cGMP levels (C) but attenuates the BNP induced increase in pSer239VASP levels (D). Data are mean ± SEM, n=3, *p<0.05 vs. untreated, † p<0.05 vs BNP.
Next, we determined if a similar nitration-mediated inhibition of PKG-1α activity was occurring in our lamb models of endogenous and exogenous activation of cGMP. As mentioned previously, Shunt lambs have increased eNOS uncoupling, leading to an increase in reactive oxygen species (ROS) and reactive nitrogen species (RNS) production (Steinhorn et al., 2001). Therefore, we first demonstrated that Shunt lambs have higher ONOO− levels than age-matched control lambs (Fig. 7 A). However, when Shunt lambs were exposed to L-arginine to prevent eNOS uncoupling, there was a decrease in ONOO− levels. Further, our data indicated that PKG-1α protein levels were unchanged in peripheral lung tissue prepared from Shunt and control lambs (Fig. 7 B). However, in Shunts, there was an increase in PKG-1α nitration (Fig. 7 C), correlating with a decrease in kinase activity (Fig. 7 D). Treatment of Shunt lambs with L-arginine resulted in the attenuation of PKG-1α nitration, and the preservation of kinase activity. Finally, our data show that the lambs exposed to inhaled NO for 24h have increased DHR 123 oxidation, indicative of elevated ONOO− levels (Fig. 8 A). Although PKG-1α protein levels were unchanged in these lambs (Fig. 8 B), PKG-1α nitration was increased (Fig. 8 C), and kinase activity was decreased (Fig. 8 D). However, in lambs treated with PEG-SOD during inhaled NO exposure, ONOO− levels were diminished, PKG-1α nitration was reduced, and kinase activity was preserved.
Fig. 7. Shunt lambs have elevated ONOO− levels, increased PKG-1α nitration, and decreased PKG activity.

ONOO− levels were determined by monitoring the ONOO− dependent oxidation of DHR 123 to rhodamine 123 in the peripheral lung tissue obtained from 4-week old control and Shunt lambs treated with or without L-arginine. The results, expressed as fold DHR oxidation, show that vehicle treated Shunt lambs have elevated ONOO− levels compared to vehicle control lambs. However, in Shunts treated with L-arginine, ONOO− levels are comparable to vehicle control lambs (A). Protein extracts (25μg) prepared from the peripheral lung tissue of Shunt and age-matched control lambs were resolved using 4-20% Tris-SDS-Hepes PAGE, electrophoretically transferred to a PVDF membrane, and then subjected to Western blot analysis using a specific antiserum raised against PKG-1α. A representative blot is shown. Densitometric analysis indicated that the PKG-1α protein levels are unchanged between Shunt and control lambs (B). Protein levels were normalized by re-probing the membrane with an antibody to β-actin. In addition, protein extracts (1000μg) were subjected to immunoprecipitation analysis using an antibody raised against PKG-1α. The immunoprecipitates were resolved using 4-20% Tris-SDS-Hepes PAGE and electrophoretically transferred to a PVDF membrane. The level of nitrated PKG-1α was then determined by probing the membranes with an antiserum raised against 3-nitrotyrosine. Blots were then stripped and re-probed for PKG-1α to normalize for the efficiency of the immunoprecipitation. The immunoblot analysis shows that PKG-1α nitration is increased in vehicle treated Shunt lambs. However, there is a significant reduction in PKG-1α nitration in Shunts treated with L-arginine (C). Using an ELISA based assay, we also found that total PKG activity is attenuated in vehicle treated Shunt lambs. However, in Shunt lambs treated with L-arginine, there is no significant decrease in total PKG kinase activity (D). Data are mean ± SEM, n=4, *p<0.05 vs. vehicle control, † p<0.05 vs. vehicle treated Shunt lambs.
Fig. 8. Inhaled NO elevates ONOO− levels, increases PKG-1α nitration, and decreased PKG activity.

ONOO− levels were determined by monitoring the ONOO− dependent oxidation of DHR 123 to rhodamine 123 in the peripheral lung tissue obtained from 1-month old lambs prior to and post exposure to 24h of inhaled NO in the presence of either PEG or PEG-SOD. The results, expressed as fold DHR oxidation, show that in PEG treated lambs inhaled NO increases ONOO− levels. However, in lambs treated with PEG-SOD, ONOO− levels do not increase (A). Protein extracts (25μg) prepared from the peripheral lung tissue of these lambs were resolved using 4-20% Tris-SDS-Hepes PAGE, electrophoretically transferred to a PVDF membrane, and then subjected to Western blot analysis using a specific antiserum raised against PKG-1α. A representative blot is shown. Densitometric analysis indicates that the PKG-1α protein levels are unchanged by inhaled NO exposure (B). Protein levels were normalized by re-probing the membrane with an antibody to β-actin. In addition, protein extracts (1000μg) were subjected to immunoprecipitation analysis using an antibody raised against PKG-1α. The immunoprecipitates were resolved using 4-20% Tris-SDS-Hepes PAGE and electrophoretically transferred to a PVDF membrane. The level of nitrated PKG-1α was then determined by probing the membranes with an antiserum raised against 3-nitrotyrosine. Blots were then stripped and re-probed for PKG-1α to normalize for the efficiency of the immunoprecipitation. The immunoblot analysis shows that PKG-1α nitration is increased by inhaled NO. However, there is a significant reduction in PKG-1α nitration in the lambs treated with PEG-SOD (C). Using an ELISA based assay, we also found that total PKG activity is attenuated in PEG treated lambs exposed to inhaled NO. However, in PEG-SOD treated lambs, there is no significant decrease in total PKG kinase activity (D). Data are mean ± SEM, n=4, *p<0.05 vs. pre+PEG, † p<0.05 vs. post+PEG.
DISCUSSION
Numerous clinical and experimental studies have shown that a downregulation of NO-cGMP signaling is central to the development of pulmonary vascular dysfunction that occurs secondary to diverse disease processes (Celermajer et al., 1993; Reddy et al., 1996) (Abman, 2007; Bachiller et al., ; Berkenbosch et al., 2000; Black et al., 1998; Bland et al., 2003; Christou et al., 1997; Jiang et al., 2001; Negash et al., 2009; Resnik et al., 2006; Steinhorn et al., 1995; Turanlahti et al., 2001; Vermeersch et al., 2007; Wong et al., 2007; Ziegler et al., 1998). However, our understanding of the precise mechanisms remains incomplete, since in some of these conditions endothelial function is impaired, despite increased cGMP levels. For example, increased pulmonary blood flow, an endogenous stimulus for cGMP production, results in increased cGMP levels and impaired endothelial function. Likewise, pharmacologic therapies (such as inhaled NO), which exogenously stimulate cGMP production, may also be associated with the development of endothelial dysfunction that manifests as inconsistent or non-sustained responses to therapy, the development of tolerance, and/or disproportionate vasoconstriction upon the withdrawal of therapy (Atz et al., 1996; Black et al., 1999; Budts et al., 2001; Klodell et al., 2007; Ross et al., 2005).
In fact, we have previously demonstrated impaired pulmonary vascular endothelial function, despite increased cGMP levels, in two distinct lamb models. In the first model, chronic increased pulmonary blood flow was created by the placement of a large aortopulmonary shunt in fetal lambs. After birth, these Shunt lambs developed a progressive selective impairment in endothelial function, associated with a decrease in bioavailable NO (Oishi et al., 2008). However, cGMP levels are elevated due to an upregulation of BNP (Oishi et al., 2007). In the second model, intact lambs were exposed to inhaled NO for 24h, which increased cGMP levels. However, upon the withdrawal of inhaled NO, these lambs developed an abrupt increase in pulmonary arterial pressure and vascular resistance above their pretreatment baseline (Thelitz et al., 2004). These data suggest that, despite both endogenous and exogenous cGMP activation, there is a dysregulation of downstream effectors in these models.
Production of cGMP is catalyzed by the conversion of GTP to its cyclic isomer by the actions of the membrane bound and soluble isoforms of guanylyl cyclases in response to vasodilators, such as NP and NO, respectively (Harteneck et al., 1990; Kuno et al., 1986; Lincoln et al., 1988; Lohmann et al., 1997; Lucas et al., 2000). Of the two isoforms of PKG-1, PKG-1α is the principal mediator of cGMP dependent vasodilation (Arnold et al., 1977; Dhanakoti et al., 2000; Knowles et al., 1989) and is 10 times more sensitive to cGMP than PKG-1β (Ruth et al., 1991). In the present study, we found that total PKG activity is attenuated in both Shunt lambs and lambs exposed to NO for 24h. The changes in PKG activity seen in these lambs were independent of total PKG-1α protein levels. This is interesting as prior studies have demonstrated that the chronic exposure of PASMC to NO donors can decrease PKG expression (Gao et al., 2004). It is also worth noting that the decrease in PKG expression was due to the increased accumulation of cGMP, which led to the downregulation of PKG protein expression and activity in a negative feedback manner (Gao et al., 2004). However, our data demonstrates in vivo that increased cGMP has no effect on the decrease in PKG activity, suggesting that both transcriptional and post translational events can modulate PKG activity and that there are likely differences between the effect of increased cGMP on PKG expression and activity in vitro and in vivo. Indeed, our findings also show that there is an increase in the nitration of tyrosine residues in PKG-1α, which correlate with decreased kinase activity. Tyrosine nitration of proteins is recognized as an important post translational event, representing the shift from the physiological to the pathological actions of NO. The presence of 3-nitrotyrosine in specific tyrosine residues can alter protein function and/or structure (Sud et al., 2007) and is formed by RNS such as ONOO− (Beckman, 1996; Beckman and Koppenol, 1996). ONOO− is the product of the interaction between NO and superoxide (Beckman and Koppenol, 1996). In Shunt lambs, there is preferential production of the superoxide anion over NO, correlating to an increase in both total protein and eNOS nitration (Oishi et al., 2008). Shunt lambs also have higher superoxide due to the increased expression and activity of xanthine oxidase (Sharma et al., 2010), NADPH oxidase (Grobe et al., 2006; Sharma et al., 2010), and uncoupled eNOS (Grobe et al., 2006). Our present study shows that when the uncoupling of eNOS is attenuated in the Shunt lambs by treatment with L-arginine, ONOO− levels are decreased, PKG-1α nitration is reduced, and kinase activity is improved. We have also shown, using isolated pulmonary arteries from the Shunt lambs, that ROS scavenging proteins (SOD and catalase) restore endothelium dependent vascular relaxation (Steinhorn et al., 2001).
Lambs that are exposed to inhaled NO also have elevated levels of superoxide and ONOO− (Oishi et al., 2006). Although it is unclear what the source of the superoxide is in these lambs, our cell culture studies, in which we have exposed vascular cells to NO donors, indicate that there are again roles for xanthine oxidase (Sheehy et al., 1998), NADPH oxidase (Brennan et al., 2002), and uncoupled eNOS (Sheehy et al., 1998). We have also shown that in lambs treated with PEG-SOD during inhaled NO therapy that there was no increase in pulmonary vascular resistance upon the withdrawal of inhaled NO (Oishi et al., 2006). In agreement with this study, our present results demonstrate that PEG-SOD treated lambs have diminished ONOO− levels, decreased PKG-1α nitration, and preserved kinase activity. Together these results confirm that the underlying vascular dysfunction in these lambs is associated with increased oxidative and nitrosative stress. Our results are also consistent with a previous report that found that PKG-1 activity was decreased under hypoxic conditions due to ONOO− mediated nitration (Negash et al., 2007). In another recent study, patients with idiopathic pulmonary arterial hypertension had a loss of caveolin-1, which led to hyper-activation of eNOS and a subsequent nitration dependent impairment of PKG-1 activity (Zhao et al., 2009). In the latter study, a mutational analysis was used to define the tyrosine (Y) residues susceptible to nitration. The authors concluded that ONOO− attenuates PKG-1 activity via the nitration of Y345 and Y549 (Zhao et al., 2009). However, until mass spectroscopy confirms these data, caution should be exercised when interpreting the mutation data because it is unclear how the replacement of the Y residues with phenylalanine (F) may be altering the overall structure of the PKG-1 protein. It is also possible that the F mutation affects the ability of ONOO− to nitrate other Y residues within the protein.
In conclusion, this study confirms PKG-1α nitration as a mechanism leading to decreased kinase activity in two lamb models of pulmonary vascular endothelial dysfunction: a model of increased pulmonary blood flow and a model of exposure to inhaled NO. Our findings have potentially significant implications for the management of pulmonary vascular dysfunction in children with congenital heart diseases, and infants, children, and adults treated with inhaled NO for a variety of pulmonary vascular disorders. Furthermore, our data, in combination with recent studies (Negash et al., 2007; Zhao et al., 2009), predict that the impairment of the cGMP/PKG-1α signaling cascade may be a common mechanism underlying the decreased vascular relaxation in pulmonary hypertension. Further studies are warranted to identify the Y residues in PKG-1α that are susceptible to nitration, and we speculate that therapies based on preventing PKG-1α nitration may have clinical utility in the management of patients with pulmonary vascular disorders.
ACKNOWLEDGMENTS
This research was supported in part by the grants: HL60190 (to SMB), HL67841 (to SMB), HL084739 (to SMB), R21HD057406 (to SMB), HL61284 (to JRF) all from the National Institutes of Health, by a grant from the Foundation Leducq to (SMB, SF, and JRF), a pre-doctoral fellowship from the Southeast Affiliates of the American Heart Association (to SA), and Cardiovascular Discovery Institute Seed Awards (to SK).
Contract grant sponsor: National Institute of Health; Contract grant numbers: HL60190 (to SMB), HL67841 (to SMB), HL084739 (to SMB), R21HD057406 (to SMB), HL61284 (to JRF).
Contract grant sponsor: Foundation Leducq; Contract grant number: 05CVD04 (to SMB, SF, and JRF).
Contract grant sponsor: Southeast Affiliates of the American Heart Association; Contract grant number: 09PRE2400015 (to SA).
Contract grant sponsor: Cardiovascular Discovery Institute, Medical College of Georgia; Contract grant number: Seed Award (to SK).
REFERENCES
- Abman SH. Recent advances in the pathogenesis and treatment of persistent pulmonary hypertension of the newborn. Neonatology. 2007;91(4):283–290. doi: 10.1159/000101343. [DOI] [PubMed] [Google Scholar]
- Arnold WP, Mittal CK, Katsuki S, Murad F. Nitric oxide activates guanylate cyclase and increases guanosine 3′:5′-cyclic monophosphate levels in various tissue preparations. Proc Natl Acad Sci U S A. 1977;74(8):3203–3207. doi: 10.1073/pnas.74.8.3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atz AM, Adatia I, Wessel DL. Rebound pulmonary hypertension after inhalation of nitric oxide. The Annals of thoracic surgery. 1996;62(6):1759–1764. doi: 10.1016/s0003-4975(96)00542-5. [DOI] [PubMed] [Google Scholar]
- Bachiller PR, Nakanishi H, Roberts JD., Jr Transforming growth factor-beta modulates the expression of nitric oxide signaling enzymes in the injured developing lung and in vascular smooth muscle cells. American journal of physiology. 298(3):L324–334. doi: 10.1152/ajplung.00181.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckman JS. Oxidative damage and tyrosine nitration from peroxynitrite. Chemical research in toxicology. 1996;9(5):836–844. doi: 10.1021/tx9501445. [DOI] [PubMed] [Google Scholar]
- Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. The American journal of physiology. 1996;271(5 Pt 1):C1424–1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- Berkenbosch JW, Baribeau J, Perreault T. Decreased synthesis and vasodilation to nitric oxide in piglets with hypoxia-induced pulmonary hypertension. American journal of physiology. 2000;278(2):L276–283. doi: 10.1152/ajplung.2000.278.2.L276. [DOI] [PubMed] [Google Scholar]
- Black SM, Heidersbach RS, McMullan DM, Bekker JM, Johengen MJ, Fineman JR. Inhaled nitric oxide inhibits NOS activity in lambs: potential mechanism for rebound pulmonary hypertension. The American journal of physiology. 1999;277(5 Pt 2):H1849–1856. doi: 10.1152/ajpheart.1999.277.5.H1849. [DOI] [PubMed] [Google Scholar]
- Black SM, Johengen MJ, Soifer SJ. Coordinated regulation of genes of the nitric oxide and endothelin pathways during the development of pulmonary hypertension in fetal lambs. Pediatric research. 1998;44(6):821–830. doi: 10.1203/00006450-199812000-00001. [DOI] [PubMed] [Google Scholar]
- Bland RD, Ling CY, Albertine KH, Carlton DP, MacRitchie AJ, Day RW, Dahl MJ. Pulmonary vascular dysfunction in preterm lambs with chronic lung disease. American journal of physiology. 2003;285(1):L76–85. doi: 10.1152/ajplung.00395.2002. [DOI] [PubMed] [Google Scholar]
- Brennan LA, Wedgwood S, Bekker JM, Black SM. The overexpression of copper-zinc superoxide dismutase protects NOS III from nitric oxide-mediated inhibition. DNA and cell biology. 2002;21(11):827–838. doi: 10.1089/104454902320908478. [DOI] [PubMed] [Google Scholar]
- Budts W, Van Pelt N, Gillyns H, Gewillig M, Van De Werf F, Janssens S. Residual pulmonary vasoreactivity to inhaled nitric oxide in patients with severe obstructive pulmonary hypertension and Eisenmenger syndrome. Heart (British Cardiac Society) 2001;86(5):553–558. doi: 10.1136/heart.86.5.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celermajer DS, Cullen S, Deanfield JE. Impairment of endothelium-dependent pulmonary artery relaxation in children with congenital heart disease and abnormal pulmonary hemodynamics. Circulation. 1993;87(2):440–446. doi: 10.1161/01.cir.87.2.440. [DOI] [PubMed] [Google Scholar]
- Christou H, Adatia I, Van Marter LJ, Kane JW, Thompson JE, Stark AR, Wessel DL, Kourembanas S. Effect of inhaled nitric oxide on endothelin-1 and cyclic guanosine 5′-monophosphate plasma concentrations in newborn infants with persistent pulmonary hypertension. The Journal of pediatrics. 1997;130(4):603–611. doi: 10.1016/s0022-3476(97)70245-2. [DOI] [PubMed] [Google Scholar]
- Dhanakoti SN, Gao Y, Nguyen MQ, Raj JU. Involvement of cGMP-dependent protein kinase in the relaxation of ovine pulmonary arteries to cGMP and cAMP. J Appl Physiol. 2000;88(5):1637–1642. doi: 10.1152/jappl.2000.88.5.1637. [DOI] [PubMed] [Google Scholar]
- Gao Y, Dhanakoti S, Trevino EM, Wang X, Sander FC, Portugal AD, Raj JU. Role of cGMP-dependent protein kinase in development of tolerance to nitric oxide in pulmonary veins of newborn lambs. Am J Physiol Lung Cell Mol Physiol. 2004;286(4):L786–792. doi: 10.1152/ajplung.00314.2003. [DOI] [PubMed] [Google Scholar]
- Geiselhoringer A, Gaisa M, Hofmann F, Schlossmann J. Distribution of IRAG and cGKI-isoforms in murine tissues. FEBS letters. 2004;575(1-3):19–22. doi: 10.1016/j.febslet.2004.08.030. [DOI] [PubMed] [Google Scholar]
- Grobe AC, Wells SM, Benavidez E, Oishi P, Azakie A, Fineman JR, Black SM. Increased oxidative stress in lambs with increased pulmonary blood flow and pulmonary hypertension: role of NADPH oxidase and endothelial NO synthase. American journal of physiology. 2006;290(6):L1069–1077. doi: 10.1152/ajplung.00408.2005. [DOI] [PubMed] [Google Scholar]
- Harteneck C, Koesling D, Soling A, Schultz G, Bohme E. Expression of soluble guanylyl cyclase. Catalytic activity requires two enzyme subunits. FEBS letters. 1990;272(1-2):221–223. doi: 10.1016/0014-5793(90)80489-6. [DOI] [PubMed] [Google Scholar]
- Hofmann F, Ammendola A, Schlossmann J. Rising behind NO: cGMP-dependent protein kinases. Journal of cell science. 2000;113(Pt 10):1671–1676. doi: 10.1242/jcs.113.10.1671. [DOI] [PubMed] [Google Scholar]
- Jiang Z, Lei Y, Gu K, Xianghua J, Liming X, Kejian H. The influences of NO and Ach on cGMP levels in two patient populations. The Journal of extra-corporeal technology. 2001;33(1):23–26. [PubMed] [Google Scholar]
- Klodell CT, Jr., Morey TE, Lobato EB, Aranda JM, Jr., Staples ED, Schofield RS, Hess PJ, Martin TD, Beaver TM. Effect of sildenafil on pulmonary artery pressure, systemic pressure, and nitric oxide utilization in patients with left ventricular assist devices. The Annals of thoracic surgery. 2007;83(1):68–71. doi: 10.1016/j.athoracsur.2006.08.051. [DOI] [PubMed] [Google Scholar]
- Knowles RG, Palacios M, Palmer RM, Moncada S. Formation of nitric oxide from L-arginine in the central nervous system: a transduction mechanism for stimulation of the soluble guanylate cyclase. Proc Natl Acad Sci U S A. 1989;86(13):5159–5162. doi: 10.1073/pnas.86.13.5159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuno T, Andresen JW, Kamisaki Y, Waldman SA, Chang LY, Saheki S, Leitman DC, Nakane M, Murad F. Co-purification of an atrial natriuretic factor receptor and particulate guanylate cyclase from rat lung. The Journal of biological chemistry. 1986;261(13):5817–5823. [PubMed] [Google Scholar]
- Lakshminrusimha S, Wiseman D, Black SM, Russell JA, Gugino SF, Oishi P, Steinhorn RH, Fineman JR. The role of nitric oxide synthase-derived reactive oxygen species in the altered relaxation of pulmonary arteries from lambs with increased pulmonary blood flow. Am J Physiol Heart Circ Physiol. 2007;293(3):H1491–1497. doi: 10.1152/ajpheart.00185.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lincoln TM, Thompson M, Cornwell TL. Purification and characterization of two forms of cyclic GMP-dependent protein kinase from bovine aorta. The Journal of biological chemistry. 1988;263(33):17632–17637. [PubMed] [Google Scholar]
- Lohmann SM, Vaandrager AB, Smolenski A, Walter U, De Jonge HR. Distinct and specific functions of cGMP-dependent protein kinases. Trends in biochemical sciences. 1997;22(8):307–312. doi: 10.1016/s0968-0004(97)01086-4. [DOI] [PubMed] [Google Scholar]
- Lucas KA, Pitari GM, Kazerounian S, Ruiz-Stewart I, Park J, Schulz S, Chepenik KP, Waldman SA. Guanylyl cyclases and signaling by cyclic GMP. Pharmacological reviews. 2000;52(3):375–414. [PubMed] [Google Scholar]
- McMullan DM, Bekker JM, Johengen MJ, Hendricks-Munoz K, Gerrets R, Black SM, Fineman JR. Inhaled nitric oxide-induced rebound pulmonary hypertension: role for endothelin-1. Am J Physiol Heart Circ Physiol. 2001;280(2):H777–785. doi: 10.1152/ajpheart.2001.280.2.H777. [DOI] [PubMed] [Google Scholar]
- Negash S, Gao Y, Zhou W, Liu J, Chinta S, Raj JU. Regulation of cGMP-dependent protein kinase-mediated vasodilation by hypoxia-induced reactive species in ovine fetal pulmonary veins. American journal of physiology. 2007;293(4):L1012–1020. doi: 10.1152/ajplung.00061.2007. [DOI] [PubMed] [Google Scholar]
- Negash S, Narasimhan SR, Zhou W, Liu J, Wei FL, Tian J, Raj JU. Role of cGMP-dependent protein kinase in regulation of pulmonary vascular smooth muscle cell adhesion and migration: effect of hypoxia. Am J Physiol Heart Circ Physiol. 2009;297(1):H304–312. doi: 10.1152/ajpheart.00077.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oishi P, Grobe A, Benavidez E, Ovadia B, Harmon C, Ross GA, Hendricks-Munoz K, Xu J, Black SM, Fineman JR. Inhaled nitric oxide induced NOS inhibition and rebound pulmonary hypertension: a role for superoxide and peroxynitrite in the intact lamb. American journal of physiology. 2006;290(2):L359–366. doi: 10.1152/ajplung.00019.2005. [DOI] [PubMed] [Google Scholar]
- Oishi P, Sharma S, Grobe A, Azakie A, Harmon C, Johengen MJ, Hsu JH, Fratz S, Black SM, Fineman JR. Alterations in cGMP, soluble guanylate cyclase, phosphodiesterase 5, and B-type natriuretic peptide induced by chronic increased pulmonary blood flow in lambs. Pediatr Pulmonol. 2007;42(11):1057–1071. doi: 10.1002/ppul.20696. [DOI] [PubMed] [Google Scholar]
- Oishi PE, Wiseman DA, Sharma S, Kumar S, Hou Y, Datar SA, Azakie A, Johengen MJ, Harmon C, Fratz S, Fineman JR, Black SM. Progressive dysfunction of nitric oxide synthase in a lamb model of chronically increased pulmonary blood flow: a role for oxidative stress. American journal of physiology. 2008;295(5):L756–766. doi: 10.1152/ajplung.00146.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy VM, Meyrick B, Wong J, Khoor A, Liddicoat JR, Hanley FL, Fineman JR. In utero placement of aortopulmonary shunts. A model of postnatal pulmonary hypertension with increased pulmonary blood flow in lambs. Circulation. 1995;92(3):606–613. doi: 10.1161/01.cir.92.3.606. [DOI] [PubMed] [Google Scholar]
- Reddy VM, Wong J, Liddicoat JR, Johengen M, Chang R, Fineman JR. Altered endothelium-dependent responses in lambs with pulmonary hypertension and increased pulmonary blood flow. The American journal of physiology. 1996;271(2 Pt 2):H562–570. doi: 10.1152/ajpheart.1996.271.2.H562. [DOI] [PubMed] [Google Scholar]
- Resnik E, Herron J, Keck M, Sukovich D, Linden B, Cornfield DN. Chronic intrauterine pulmonary hypertension selectively modifies pulmonary artery smooth muscle cell gene expression. American journal of physiology. 2006;290(3):L426–433. doi: 10.1152/ajplung.00281.2005. [DOI] [PubMed] [Google Scholar]
- Ross GA, Oishi P, Azakie A, Fratz S, Fitzgerald RK, Johengen MJ, Harmon C, Hendricks-Munoz K, Xu J, Black SM, Fineman JR. Endothelial alterations during inhaled NO in lambs with pulmonary hypertension: implications for rebound hypertension. American journal of physiology. 2005;288(1):L27–35. doi: 10.1152/ajplung.00144.2004. [DOI] [PubMed] [Google Scholar]
- Ruth P, Landgraf W, Keilbach A, May B, Egleme C, Hofmann F. The activation of expressed cGMP-dependent protein kinase isozymes I alpha and I beta is determined by the different amino-termini. European journal of biochemistry / FEBS. 1991;202(3):1339–1344. doi: 10.1111/j.1432-1033.1991.tb16509.x. [DOI] [PubMed] [Google Scholar]
- Sharma S, Kumar S, Wiseman DA, Kallarackal S, Ponnala S, Elgaish M, Tian J, Fineman JR, Black SM. Perinatal changes in superoxide generation in the ovine lung: Alterations associated with increased pulmonary blood flow. Vascular pharmacology. 2010;53(1-2):38–52. doi: 10.1016/j.vph.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Sud N, Wiseman DA, Carter AL, Kumar S, Hou Y, Rau T, Wilham J, Harmon C, Oishi P, Fineman JR, Black SM. Altered carnitine homeostasis is associated with decreased mitochondrial function and altered nitric oxide signaling in lambs with pulmonary hypertension. American journal of physiology. 2008;294(1):L46–56. doi: 10.1152/ajplung.00247.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehy AM, Burson MA, Black SM. Nitric oxide exposure inhibits endothelial NOS activity but not gene expression: a role for superoxide. The American journal of physiology. 1998;274(5 Pt 1):L833–841. doi: 10.1152/ajplung.1998.274.5.L833. [DOI] [PubMed] [Google Scholar]
- Song P, Wu Y, Xu J, Xie Z, Dong Y, Zhang M, Zou MH. Reactive nitrogen species induced by hyperglycemia suppresses Akt signaling and triggers apoptosis by upregulating phosphatase PTEN (phosphatase and tensin homologue deleted on chromosome 10) in an LKB1-dependent manner. Circulation. 2007;116(14):1585–1595. doi: 10.1161/CIRCULATIONAHA.107.716498. [DOI] [PubMed] [Google Scholar]
- Steinhorn RH, Russell JA, Lakshminrusimha S, Gugino SF, Black SM, Fineman JR. Altered endothelium-dependent relaxations in lambs with high pulmonary blood flow and pulmonary hypertension. Am J Physiol Heart Circ Physiol. 2001;280(1):H311–317. doi: 10.1152/ajpheart.2001.280.1.H311. [DOI] [PubMed] [Google Scholar]
- Steinhorn RH, Russell JA, Morin FC., 3rd Disruption of cGMP production in pulmonary arteries isolated from fetal lambs with pulmonary hypertension. The American journal of physiology. 1995;268(4 Pt 2):H1483–1489. doi: 10.1152/ajpheart.1995.268.4.H1483. [DOI] [PubMed] [Google Scholar]
- Sud N, Sharma S, Wiseman DA, Harmon C, Kumar S, Venema RC, Fineman JR, Black SM. Nitric oxide and superoxide generation from endothelial NOS: modulation by HSP90. American journal of physiology. 2007;293(6):L1444–1453. doi: 10.1152/ajplung.00175.2007. [DOI] [PubMed] [Google Scholar]
- Tamura N, Itoh H, Ogawa Y, Nakagawa O, Harada M, Chun TH, Suga S, Yoshimasa T, Nakao K. cDNA cloning and gene expression of human type Ialpha cGMP-dependent protein kinase. Hypertension. 1996;27(3 Pt 2):552–557. doi: 10.1161/01.hyp.27.3.552. [DOI] [PubMed] [Google Scholar]
- Thelitz S, Bekker JM, Ovadia B, Stuart RB, Johengen MJ, Black SM, Fineman JR. Inhaled nitric oxide decreases pulmonary soluble guanylate cyclase protein levels in 1-month-old lambs. The Journal of thoracic and cardiovascular surgery. 2004;127(5):1285–1292. doi: 10.1016/j.jtcvs.2003.07.024. [DOI] [PubMed] [Google Scholar]
- Turanlahti M, Pesonen E, Pohjavuori M, Lassus P, Fyhrquist F, Andersson S. Plasma cyclic guanosine monophosphate reflecting the severity of persistent pulmonary hypertension of the newborn. Biology of the neonate. 2001;80(2):107–112. doi: 10.1159/000047128. [DOI] [PubMed] [Google Scholar]
- Vermeersch P, Buys E, Pokreisz P, Marsboom G, Ichinose F, Sips P, Pellens M, Gillijns H, Swinnen M, Graveline A, Collen D, Dewerchin M, Brouckaert P, Bloch KD, Janssens S. Soluble guanylate cyclase-alpha1 deficiency selectively inhibits the pulmonary vasodilator response to nitric oxide and increases the pulmonary vascular remodeling response to chronic hypoxia. Circulation. 2007;116(8):936–943. doi: 10.1161/CIRCULATIONAHA.106.677245. [DOI] [PubMed] [Google Scholar]
- Walter U. Physiological role of cGMP and cGMP-dependent protein kinase in the cardiovascular system. Reviews of physiology, biochemistry and pharmacology. 1989;113:41–88. doi: 10.1007/BFb0032675. [DOI] [PubMed] [Google Scholar]
- Wedgwood S, Bekker JM, Black SM. Shear stress regulation of endothelial NOS in fetal pulmonary arterial endothelial cells involves PKC. American journal of physiology. 2001a;281(2):L490–498. doi: 10.1152/ajplung.2001.281.2.L490. [DOI] [PubMed] [Google Scholar]
- Wedgwood S, McMullan DM, Bekker JM, Fineman JR, Black SM. Role for endothelin-1-induced superoxide and peroxynitrite production in rebound pulmonary hypertension associated with inhaled nitric oxide therapy. Circulation research. 2001b;89(4):357–364. doi: 10.1161/hh1601.094983. [DOI] [PubMed] [Google Scholar]
- Wong RC, Koh GM, Choong PH, Yip WL. Oral sildenafil therapy improves health-related quality of life and functional status in pulmonary arterial hypertension. International journal of cardiology. 2007;119(3):400–402. doi: 10.1016/j.ijcard.2006.07.170. [DOI] [PubMed] [Google Scholar]
- Zhao YY, Zhao YD, Mirza MK, Huang JH, Potula HH, Vogel SM, Brovkovych V, Yuan JX, Wharton J, Malik AB. Persistent eNOS activation secondary to caveolin-1 deficiency induces pulmonary hypertension in mice and humans through PKG nitration. The Journal of clinical investigation. 2009;119(7):2009–2018. doi: 10.1172/JCI33338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler JW, Ivy DD, Wiggins JW, Kinsella JP, Clarke WR, Abman SH. Effects of dipyridamole and inhaled nitric oxide in pediatric patients with pulmonary hypertension. American journal of respiratory and critical care medicine. 1998;158(5 Pt 1):1388–1395. doi: 10.1164/ajrccm.158.5.9710117. [DOI] [PubMed] [Google Scholar]