

Abstract
Acute myocardial infarction and its sequelae are leading causes of morbidity and mortality worldwide. Nitroglycerin remains a first-line treatment for angina pectoris and acute myocardial infarction. Nitroglycerin achieves its benefit by giving rise to nitric oxide, which causes vasodilation and increases blood flow to the myocardium. However, continuous delivery of nitroglycerin results in tolerance, limiting the use of this drug. Nitroglycerin tolerance is due, at least in part, to inactivation of aldehyde dehydrogenase 2 (ALDH2), an enzyme that converts nitroglycerin to the vasodilator, nitric oxide. We have recently found that, in addition to nitroglycerin’s effect on the vasculature, sustained treatment with nitroglycerin negatively affects cardiomyocyte viability following ischemia, thus resulting in increased infarct size in a myocardial infarction model in animals. Co-administration of Alda-1, an activator of ALDH2, with nitroglycerin improves metabolism of reactive aldehyde adducts and prevents the nitroglycerin-induced increase in cardiac dysfunction following myocardial infarction. In this review, we describe the molecular mechanisms associated with the benefits and risks of nitroglycerin administration in myocardial infarction. (167 of 200).
Keywords: aldehyde dehydrogenase, nitric oxide, nitroglycerin tolerance, cardiomyocyte, cell death
Benefits of nitroglycerin (glyceryl trinitrate or GTN)
Despite advances in pharmacological therapies, ischemic heart disease and acute myocardial infarction (MI) continue to be a major cause of morbidity and death worldwide. According to the World Health Organization, over 7 million people die of ischemic heart disease every year. Consequently, novel pharmacological and non-pharmacological strategies need to be explored to benefit MI patients.1–3
Since its discovery over 150 years ago, GTN has become the most common treatment for patients with unstable angina pectoris, myocardial infarction and heart failure.4–8 The ability of GTN to promote vasodilation as well as tolerance was clearly noted during the GTN industry ascension in the 20th century. Factory workers, usually exposed to high levels of organic nitrites, often complained of headaches on Mondays that disappeared over the weekends. Indeed, factory workers suffering from angina pectoris or heart failure often experienced relief from chest pain during the work week, but which recurred on weekends. Both effects were attributed to the vasodilator action of GTN, which quickly became apparent to physicians. The phenomenon of nitrate tolerance became famously recognized by the onset of ‘Monday disease’ and of nitrate withdrawal/overcompensation by ‘Sunday Heart Attacks’.9
The positive effects of GTN arise from its ability to promote vasodilation, resulting in increased blood flow to the heart.10 GTN effects are also evident in systemic veins where the venodilator effect reduces cardiac preload and further decreases myocardial wall stress.11 GTN is extremely effective in restoring the equilibrium of oxygen and nutrients supply-demand in the ischemic heart. However, sustained GTN administration causes tolerance and is associated with pro-oxidant effects, endothelial dysfunction and increased sensitivity to vasoconstrictors.12–14 Other nitrates used in clinical practice include isosorbide-dinitrate, its active metabolite isosorbide-mononitrate, pentaerythrytol tetranitrate, erythrytyl-tetranitrate, and nicorandil.15
History of the therapeutic use of nitroglycerin
In 1847, working in Theophile-Jules Pelouze’s laboratory in Turin, Ascanio Sobrero discovered GTN. Sobrero first noted the aggressive headache for several hours produced by GTN.4 Two years later, knowing of Sobrero’s reports of headache, the German scientist Constantin Hering tested GTN in healthy volunteers and observed that headache was caused with much precision.4 Alfred Nobel joined Pelouze in 1851 and recognized the scientific and financial potential of GTN. Years later, he began manufacturing GTN in Sweden. Nobel suffered from poor health for most of his life. In later life, he suffered from intense pain and angina pectoris. It is therefore ironic that in 1890, his physicians recommended GTN for his heart compliant.4
During the second half of the 19th century, several British scientists became interested in the newly discovered amyl nitrite, recognized as a powerful vasodilator. Lauder Brunton used the compound to relieve angina in 1867, and first reported the pharmacological resistance to repeated doses.16, 17 Following Brunton’s work, scientists concentrated on recording the effects of nitrite-containing compounds on several pathological systems, which include angina pectoris, myocardial infarction, hypertension and heart failure.17–21 Finally, GTN was established as a treatment for the relief of angina at the end of the 19th century. However, the mechanism of action of GTN-induced benefit was discovered only 80 years later.
In the late 1970s, the vasodilator effect of GTN was discovered to be mediated by nitric oxide, which was apparently generated from GTN in vascular smooth muscle.22–24 Years later, it was discovered that mammalian cells synthesize nitric oxide.25 In 1998, approximately 130 years after the invention of dynamite by Alfred Nobel and the first observed clinical benefit of GTN, the Nobel Prize in Medicine or Physiology was awarded for “Nitric Oxide as a Signaling Molecule in the Cardiovascular System” to Robert Furchgott, Louis Ignarro and Ferid Murad. GTN remains the treatment of choice for relieving angina; other organic esters and inorganic nitrates are also used, but the rapid action of GTN and its established efficacy make it the mainstay of angina pectoris relief.4
ALDH2 in nitroglycerin bioactivation
The vasodilator action of GTN was discovered as a process mediated by nitric oxide.22, 24 Subsequent studies discovered that a chemical reaction between GTN (or other nitro compounds) and a thiol generate an intermediate S-nitrosothiol, which resulted in further production of nitric oxide.23 Nowadays, it is commonly assumed that GTN is converted in smooth muscle cells to nitric oxide which activates soluble guanylate cyclase to generate cyclic GMP which, in turn, results in vascular smooth muscle relaxation (Figure 1).26
Figure 1.
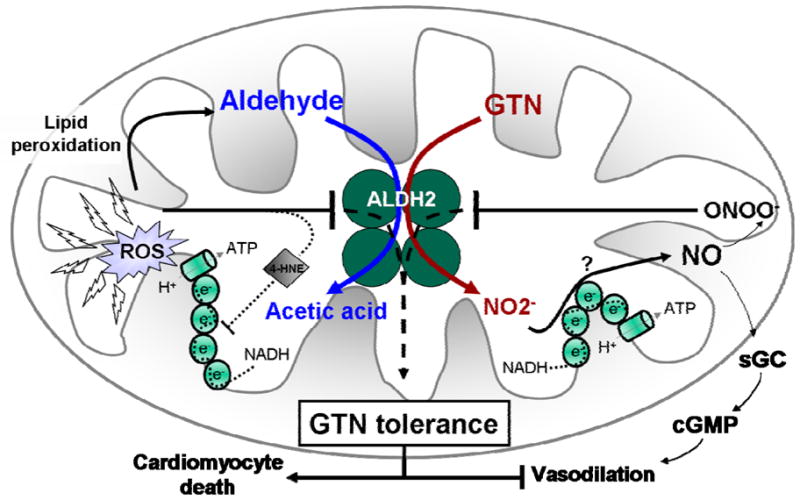
Tolerance to GTN (nitroglycerin) is mainly caused by oxidative inactivation of the reductase activity of ALDH2, which results in decreased GTN bioactivation, diminished levels of nitrite (NO2-) and nitric oxide (NO). This response abrogates the GTN-induced vasodilation phenomenon due to insufficient activation of soluble guanylate cyclase and production of cyclic guanosine monophosphate (cGMP) in the vascular smooth muscle. Besides its reductase activity, the dehydrogenase activity of ALDH2 is also down-regulated by GTN tolerance, leading to accumulation of toxic aldehydes (i.e. 4-hydroxy-2-nonenal; 4-HNE) inside the mitochondria, which further disrupts the mitochondrial respiratory chain and results in reactive oxygen species generation. GTN-mediated ALDH2 inactivation triggers apoptosis in cardiomyocytes as well as abolishes the vasodilator effect of GTN vascular smooth muscle.
Despite intense basic and clinical research, the molecular mechanism by which NO is generated from GTN remained elusive. In fact, it has been proposed that one or more enzymatic reactions might be involved in GTN bioactivation.27–29 This research was complicated because GTN bioactivation is mediated by either non-enzymatic or enzymatic reactions. Endogenous reductants (i.e. thiols and ascorbate) can cause non-enzymatic GTN bioactivation; however, their contribution to GTN-mediated vasodilation appears limited.30 Several enzymatic reactions are also involved in GTN bioactivation including glutathione-S-transferases 31, xanthine oxidoreductase 32 and the cytochrome P450 system.26, 33, 34 However, because none of these enzymes could catalyze the selective formation of 1,2- glyceryl dinitrite (1,2-GDN) from GTN, the search for the GTN metabolizing enzyme continued. In 2004, Stamler and collaborators identified mitochondrial ALDH2 as a key enzyme catalyzing GTN bioconversion to 1,2- glyceryl dinitrite (1,2-GDN).26, 35
ALDH2 is a mitochondrial enzyme and a member of the NAD(P)+-dependent aldehyde dehydrogenase family composed of 17 isozymes expressed with different tissue distributions.36 ALDH2 is a tetrameric enzyme known mainly for its role in ethanol metabolism, catalyzing aldehyde oxidation to form acetic acid.36–38 However, in addition to its dehydrogenase activity, ALDH2 also has esterase and reductase activities, at least in vitro.26 Based on the esterase activity of ALDH2, the reaction with GTN would be expected to generate an S-nitrosothiol intermediate that would be hydrolyzed to produce nitrate. However, nitrite formation indicates that S-nitrosothiol is reduced and not hydrolyzed.26 Therefore, it has been suggested that thiols potentiate GTN activity through formation of active S-nitrosothiol.39 In fact, purified ALDH2 catalyzes 1,2,-GDN formation only in the presence of reducing agents.35
The role of ALDH2 as the GTN-reductase enzyme as well as its contribution to GTN-induced vasodilation has been widely demonstrated over the last decade.13, 35, 40 Purified mitochondrial ALDH2 catalyzes predominantly 1,2-GDN formation from low levels of GTN (1 μM). In the presence of NAD+, however, the rate of GTN metabolism as well as the 1,2-GDN/1,3-GDN ratio drastically increases in either ALDH2 purified from bovine liver or overexpressed human ALDH2.35, 41 Using different animal models, it has been shown that inhibition of ALDH2 by chloral hydrate, disulfiram or cyanamide attenuates hypotension induced by intravenous injection of GTN.35, 42 Treatment of aortic rings with ALDH2 inhibitors or substrates inhibits GTN-induced vasodilation.35, 41, 43, 44 Of interest, these inhibitors do not attenuate the sodium nitroprusside-induced relaxation (which directly activates guanylate cyclase), suggesting that GTN-mediated effects are mainly through specific inhibition of vascular ALDH2 activity.35 In ALDH2 knockout mice, the hypotensive effect of GTN infusion was abrogated at low doses and substantially reduced at high doses.45 As expected, GTN bioactivation to 1,2-GDN was mainly eliminated in ALDH2 knockout mice. Together, all these studies support a critical role for ALDH2 in GTN bioactivation.
ALDH2 contains three cysteine residues within the catalytic site (Cys 301, 302 and 303). Stamler and collaborators proposed a specific mechanism for ALDH2 reductase activity based on the formation of a disulfide between Cys 302 (the Cys that participates in the catalysis) and one of the two adjacent Cys residues.35 Therefore, Mayer and collaborators proposed that GTN denitration and bioactivation reflect two separate pathways of ALDH2-catalyzed GTN biotransformation, both of which involve formation of a thionitrate intermediate at Cys302 as initial reaction step.46 However the molecular mechanisms underlying this fractional contribution to ALDH2-catalyzed GTN bioconversion are still unknown.
The hypothesis that nitrite produced by GTN-reductase activity of mitochondrial ALDH2 is further reduced to generate nitric oxide, which activates soluble guanylate cyclase and promotes vasodilation is well accepted (Figure 1). However, the molecular mechanisms of nitrite conversion to nitric oxide remain controversial. Chen and collaborators demonstrated that incubation of isolated mitochondria from fibroblasts with different concentrations of GTN resulted in the dose-dependent generation of nitric oxide, where ALDH2 inhibition blocked this response.45 It has been reported that mammalian mitochondria present a nitrite reductase activity, associated with complex III and IV of the mitochondrial respiratory chain.47, 48 However, the mechanism for the three-electron reduction of GTN to generate nitric oxide remains unknown. Recent studies demonstrated that mitochondria respiratory chain is not involved in bioactivation of GTN-derived nitrite.41, 49
Mechanisms involved in nitroglycerin tolerance
GTN bioactivation in vascular smooth muscle is required to promote effective vasorelaxation treatment of angina pectoris and congestive heart failure. However, the usefulness of GTN is widely limited by the development of tolerance to the drug. Sustained GTN therapy-induced tolerance is at least partially attributed to impaired bioactivation of GTN.44 Moreover, GTN therapy causes a cross-tolerance, which results in impaired relaxation by other nitrosovasodilators. Despite its multifactorial causes, including desensitization of soluble guanylate cyclase, cGMP-dependent kinase and myosin light chain phosphorylation, GTN tolerance is mainly mediated by the inability of ALDH2 to catalyze the conversion of GTN to 1,2-GDN and nitrite within mitochondria 26. For example, chloral hydrate, a substrate analog ALDH2 inhibitor, and the specific ALDH2 inhibitor, daidzin, suppress 1,2-GDN formation by purified ALDH2.35, 41, 44 Also, acetaldehyde competitively inhibits GTN turnover by ALDH2 both in vitro and in vivo.35, 44
The mechanism involved in ALDH2 inactivation may be a result of oxidation and formation of a disulfide bond that includes the active site Cys thiol (Figure 1).26 As mentioned above, the catalytic sites of ALDH2 contain three adjacent sulphydryl groups that can form intramolecular disulfide bonds 50, which leads to inactivation of ALDH2.51 Active site disulfide formation underlies the inhibition of ALDH2 by GTN, nitric oxide, its substrates such as 4-hydroxynonenal as well as by other ALDH inhibitors.35,50, 51 Analysis of purified ALDH2 in vitro indicates that reduction of the oxidized enzyme by exogenous thiols or other reductants restores ALDH2 activity.35, 52
Mitochondrial ALDH2 also become a convergence point of the superoxide hypothesis of GTN tolerance. Superoxide and peroxynitrite directly inhibit ALDH2 activity.44 Daiber et al. (2005) observed that GTN-stimulated superoxide production correlated well with decreases in ALDH2 activity.53 Moreover, reactive oxygen species production, mediated by mitochondrial respiration blockade, was associated with mitochondrial ALDH2 inactivation.54 Sydow and collaborators reported that progression of GTN tolerance in an animal model was directly associated with inhibition of vascular ALDH-2 activity, disruption of GTN metabolism, and increased reactive oxygen species production by mitochondria.44 Of interest, the increase in cGMP in response to GTN was blunted in cultured endothelial cells deficient in mitochondria. These findings support the hypothesis that the reactive oxygen species release and further accumulation of reactive aldehydes such as 4-hydroxy-2-nonenal may contribute directly to GTN tolerance, either by oxidative inhibition of ALDH2 55 or by oxidizing enzyme cofactors.56 In fact, superoxide-induced inhibition of key enzymes related to bioactivation of other organic nitrates, or of downstream elements in the vasorelaxation signaling pathways, may contribute to GTN-mediated cross-tolerance.26 Of interest, incubation of tolerant tissue with different reducing agents or antioxidant molecules improved vascular ALDH2 activity, thus supporting the hypothesis that enzyme oxidation leads to GTN tolerance.
It was recently shown that Alda-1 (a selective ALDH2 activator) may slightly inhibit GTN bioconversion to NO, in vitro 57, which is not attractive considering the medical benefit of NO in patients with angina pectoris. However, we found that Alda-1 given concomitantly with GTN in vivo did not inhibit vasodilatation 52, indicating that Alda-1 does not inhibit GTN bioactivation to NO in vivo. Moreover, we found that Alda-1 prevented ALDH2 inactivation due to prolonged treatment with GTN (Figure 2).
Figure 2.
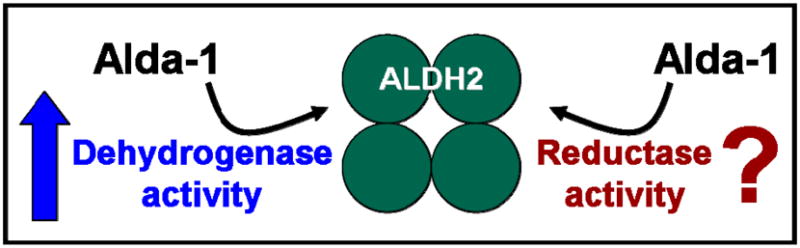
Alda-1 (a selective ALDH2 activator) increases dehydrogenase activity of ALDH2. The contribution of Alda-1 to the reductase activity of ALDH2 has not yet been determined.
Effects of GTN on cardiac cells
The effects of GTN in the vasculature have been widely investigated, but relatively little is known about GTN’s effect on cardiac cells. We have recently demonstrated that sustained treatment with GTN resulted in an increase in infarct size and cardiac dysfunction after myocardial infarction in rats.52, 55 GTN tolerance-mediated deleterious effects in the heart are associated with ALDH2 inactivation.55 As previously reported, GTN treatment drastically inhibits the dehydrogenase activity of recombinant ALDH2, in vitro and in vivo.52 We also found that co-incubation with GTN and Alda-1 (a selective ALDH2 activator that we have identified) completely prevented GTN-induced recombinant ALDH2 inactivation. Further, sustained treatment with GTN significantly reduced mitochondrial ALDH2 activity in the rat myocardium, resulting in increased cardiac damage and ventricular dysfunction after a myocardial infarction event.52 Of interest, co-treatment with GTN and Alda-1 restored ALDH2 activity, resulting in smaller infarct size and improved cardiac function in rats.52 We suggest that GTN tolerance is the main process involved in increased cardiac damage following MI, since the use of isosorbide dinitrate, an alternative NO donor often used in a sustained fashion to treat angina, did not cause ALDH2 inactivation and further cardiac damage in vivo.52 Similarly, Sydow et al. observed that in vivo treatment with GTN leads to reduced cardiac GTN biotransformation by mitochondrial ALDH2 and resulted in accumulation of reactive oxygen species, where incubation of mitochondria from tolerant animals with reducing agents restored ALDH2 function.44 These findings suggest that patients under continuous GTN treatment are at risk for increased cardiac damage.
Cellular side effects of GTN
The deleterious effects of organic nitrate therapy on mitochondrial were first described 55 years ago, where acute GTN exposure was described to induce mitochondrial swelling, to stimulated oxygen consumption and to caused loss of respiratory control of rat liver and heart mitochondria.58 More recently, it was demonstrated that GTN infusion resulted in mitochondrial dysfunction-induced oxidative stress in both animal and human blood vessels.59–61 An excessive reactive oxygen species production along with reduction of more than 50% in ALDH2 activity was observed in isolated mitochondria using the complex III inhibitor antimycin A.62, 63 Mitochondrial-target antioxidants prevent complex I inhibition mediated by GTN treatment.54 Therefore, accumulation of reactive aldehydes derived from oxidative stress may disrupt GTN bioactivation by negatively targeting ALDH2 function. Altogether, these findings suggest that GTN bioactivation requires functionally active mitochondria, since increased reactive oxygen species production due to mitochondrial dysfunction results in impaired ALDH2 activity and further GTN conversion.35, 44, 54
We have recently found that sustained GTN treatment significantly decreases aldehyde dehydrogenase activity in the failing heart.52 The fact that mammalian ALDH2 functions as a GTN reductase may explain the inhibitory effect of GTN on the aldehyde dehydrogenase activity. These findings point to a possible alcohol-GTN drug interaction through ALDH2 inactivation, which can result in a devastating phenomena induced by accumulation of reactive aldehydes inside the cell. However, this hypothesis needs to be better explored.
ALDH2*2 mutation and nitroglycerin metabolism
Over 40% of East Asians carries a common ALDH2*2 mutation. The mutation is caused by a single nucleotide substitution in exon 12 of the ALDH2 gene on chromosome 12, resulting in an amino acid change from glutamate to lysine at position 487 (E487K) of the mature enzyme.64 The E487K amino acid substitution at the interface of the tetrameric enzyme leads to a reduction of the catalytic ALDH2 activity due to a disruption of co-enzyme NAD binding.65 Heterozygous ALDH2*1/*2 and homozygous ALDH*2/*2 show approximately 40% and 5% of wild-type ALDH2 activity, respectively.66 Epidemiological studies have indicated that ALDH2 is crucial in cardiovascular diseases, where the ALDH*2/*2 allele has been linked to higher incidences of GTN tolerance 40, myocardial infarction 67, 68 and hypertension.69, 70
Considering the contribution of ALDH2 to the bioactivation of GTN, it is expected that the significant decrease of ALDH2 activity in Asians carrying the E487K mutation would lead to a decreased vasodilation response to GTN treatment. In fact, in vitro experiments demonstrated that GTN biotransformation to 1,2-GDN was drastically decreased in the E487K mutant enzyme.57 Subjects carrying the ALDH*2/*2 mutation have a significantly reduced vasodilatory response to GTN, where larger doses of GTN were required to achieve satisfactory vasodilation.71 Of interest, treatment of non-ALDH2 mutant individuals with the ALDH2 inhibitor, disulfiram, blocked the vasodilatory effect of GTN, but not that promoted by sodium nitroprusside.71 These findings confirm that ALDH2 is crucial to GTN bioactivation in humans and that individuals with the ALDH*2/*2 mutation display a reduced vasodilatory response to GTN and are probably more prone to develop GTN tolerance-associated cardiotoxicity.
Clinical Implications/summary
The findings that ALDH2 is crucial to GTN bioactivation may have important clinical implications. Considering that ALDH2 activation positively correlates with cardioprotection during ischemic events 55, therapies which reduce ALDH2 activity (i.e. sustained GTN administration) may worsen outcome during an ischemic episode.
GTN is among the most commonly used drugs in the treatment of angina, ischemia and heart failure.4–8 However, there is discussion regarding the deleterious effect of chronic GTN treatment.12, 36, 72 Given the importance of ALDH2 in cytoprotective signaling in different tissues (including the heart), GTN tolerance should not be considered as a simple loss of drug efficacy. The current clinical practice of GTN treatment in patients at elevated risk for an ischemic event should be reviewed. Sustained exposure to GTN should be avoided, not only because its benefit wears out, but perhaps more importantly, because it decreases the activity of ALDH2 and thus exacerbates damage associated with ischemia.
A drug that can enhance GTN bioconversion and prevent the inhibitory effect of GTN on ALDH2 will be very helpful, in particular for ALDH*2/*2 individuals. Activators of ALDH2, such as Alda-1, may have therapeutic potential due to their ability to prevent GTN-induced ALDH2 inactivation. The fact that Alda-1 restores activity of mutant ALDH*2/*2 suggests that these individuals may become more responsive to GTN treatment following Alda-1 administration (Figure 3).
Figure 3.
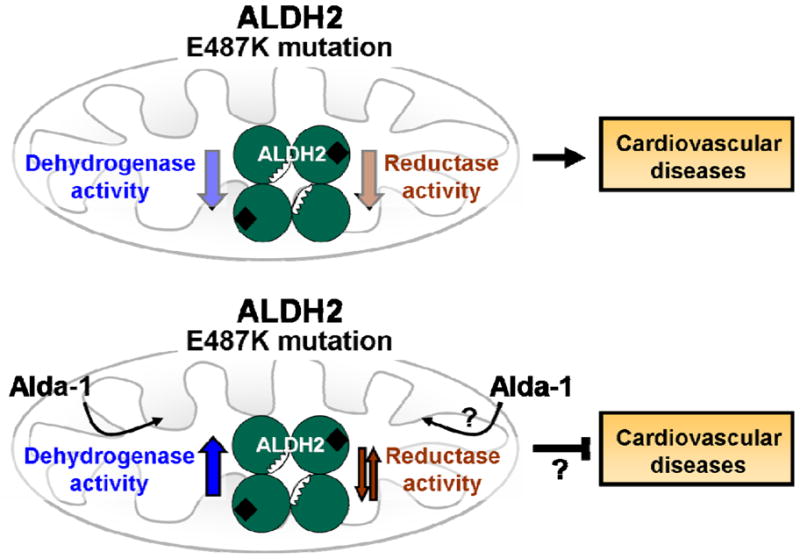
The ALDH2 mutation caused by a single amino acid change from glutamate to lysine at position 487 (E487K) leads to a reduction in both the dehydrogenase and reductase activities of ALDH2. Alda-1 significantly improves dehydrogenase activity of the mutant enzyme, causing an allosteric change that corrects the structural defect in the enzyme.73 The effect of Alda-1 on the reductase activity of ALDH2 has not yet been determined.
Finally, further studies to clarify the molecular mechanisms of GTN biotransformation and GTN tolerance, as well as clinical studies re-evaluating the use of sustained GTN treatment in patients with cardiovascular diseases, especially in East Asian patients, are needed.
Acknowledgments
Funding
This study was supported by National Institute of Health Grant AA11147 and HL52141 to DMR. JCF holds a post-doctoral fellowship from Fundação de Amparo a Pesquisa do Estado de São Paulo - Brasil (FAPESP 2009/03143-1).
Footnotes
Conflict of interest: D.M.-R. is the founder of KAI Pharmaceuticals, Inc. However, none of the research in her laboratory is supported by or is in collaboration with the company. J.C.B.F. has no disclosure.
References
- 1.Palaniyandi SS, Ferreira JC, Brum PC, Mochly-Rosen D. PKCbetaII inhibition attenuates myocardial infarction induced heart failure and is associated with a reduction of fibrosis and pro-inflammatory responses. J Cell Mol Med. 2010;15:1769–1777. doi: 10.1111/j.1582-4934.2010.01174.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Palaniyandi SS, Qi X, Yogalingam G, Ferreira JC, Mochly-Rosen D. Regulation of mitochondrial processes: a target for heart failure. Drug Discov Today Dis Mech. 2010;7:e95–e102. doi: 10.1016/j.ddmec.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pereira MG, Ferreira JC, Bueno CR, Jr, Mattos KC, Rosa KT, Irigoyen MC, et al. Exercise training reduces cardiac angiotensin II levels and prevents cardiac dysfunction in a genetic model of sympathetic hyperactivity-induced heart failure in mice. Eur J Appl Physiol. 2009;105:843–850. doi: 10.1007/s00421-008-0967-4. [DOI] [PubMed] [Google Scholar]
- 4.Marsh N, Marsh A. A short history of nitroglycerine and nitric oxide in pharmacology and physiology. Clin Exp Pharmacol Physiol. 2000;27:313–319. doi: 10.1046/j.1440-1681.2000.03240.x. [DOI] [PubMed] [Google Scholar]
- 5.Abrams J. Beneficial actions of nitrates in cardiovascular disease. Am J Cardiol. 1996;77:31C–37C. doi: 10.1016/s0002-9149(96)00186-5. [DOI] [PubMed] [Google Scholar]
- 6.Antman EM, Anbe DT, Armstrong PW, Bates ER, Green LA, Hand M, et al. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction; A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Revise the 1999 Guidelines for the Management of patients with acute myocardial infarction) J Am Coll Cardiol. 2004;44:E1–E211. doi: 10.1016/j.jacc.2004.07.014. [DOI] [PubMed] [Google Scholar]
- 7.Braunwald E, Antman EM, Beasley JW, Califf RM, Cheitlin MD, Hochman JS, et al. ACC/AHA 2002 guideline update for the management of patients with unstable angina and non-ST-segment elevation myocardial infarction--summary article: a report of the American College of Cardiology/American Heart Association task force on practice guidelines (Committee on the Management of Patients With Unstable Angina) J Am Coll Cardiol. 2002;40:1366–1374. doi: 10.1016/s0735-1097(02)02336-7. [DOI] [PubMed] [Google Scholar]
- 8.Watanabe H, Kakihana M, Ohtsuka S, Sugishita Y. Preventive effects of angiotensin-converting enzyme inhibitors on nitrate tolerance during continuous transdermal application of nitroglycerin in patients with chronic heart failure. Jpn Circ J. 1998;62:353–358. doi: 10.1253/jcj.62.353. [DOI] [PubMed] [Google Scholar]
- 9.Ignarro LJ. After 130 years, the molecular mechanism of action of nitroglycerin is revealed. Proc Natl Acad Sci U S A. 2002;99:7816–7817. doi: 10.1073/pnas.132271799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abrams J. Hemodynamic effects of nitroglycerin and long-acting nitrates. Am Heart J. 1985;110:216–224. [PubMed] [Google Scholar]
- 11.Kojima S, Matsui K, Sakamoto T, Ishihara M, Kimura K, Miyazaki S, et al. Long-term nitrate therapy after acute myocardial infarction does not improve or aggravate prognosis. Circ J. 2007;71:301–307. doi: 10.1253/circj.71.301. [DOI] [PubMed] [Google Scholar]
- 12.Kosugi M, Nakagomi A, Shibui T, Kato K, Kusama Y, Atarashi H, et al. Effect of long-term nitrate treatment on cardiac events in patients with vasospastic angina. Circ J. 2011;75:2196–2205. doi: 10.1253/circj.cj-11-0056. [DOI] [PubMed] [Google Scholar]
- 13.Munzel T, Daiber A, Mulsch A. Explaining the phenomenon of nitrate tolerance. Circ Res. 2005;97:618–628. doi: 10.1161/01.RES.0000184694.03262.6d. [DOI] [PubMed] [Google Scholar]
- 14.Klemenska E, Beresewicz A. Bioactivation of organic nitrates and the mechanism of nitrate tolerance. Cardiol J. 2009;16:11–19. [PubMed] [Google Scholar]
- 15.Csont T, Ferdinandy P. Cardioprotective effects of glyceryl trinitrate: beyond vascular nitrate tolerance. Pharmacol Ther. 2005;105:57–68. doi: 10.1016/j.pharmthera.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 16.Brunton L. On the use of nitrite amyl in angina pectoris. Lancet. 1867;ii:97–88. [Google Scholar]
- 17.Knot HJ. Nitrate tolerance in hypertension: new insight into a century-old problem. Circ Res. 2003;93:799–801. doi: 10.1161/01.RES.0000100846.76792.C2. [DOI] [PubMed] [Google Scholar]
- 18.Nesbitt SD. Nitrates as adjunct hypertensive treatment: a possible answer to resistant systolic hypertension. Hypertension. 2005;45:352–353. doi: 10.1161/01.HYP.0000156749.83009.5b. [DOI] [PubMed] [Google Scholar]
- 19.Gorlin R, Brachfeld N, Macleod C, Bopp P. Effect of nitroglycerin on the coronary circulation in patients with coronary artery disease or increased left ventricular work. Circulation. 1959;19:705–718. doi: 10.1161/01.cir.19.5.705. [DOI] [PubMed] [Google Scholar]
- 20.Venkataraman K, Durairaj SK, de Guzman M, Haywood LJ. Isosorbide dinitrate in acute and chronic heart failure. Clin Pharmacol Ther. 1979;25:43–50. doi: 10.1002/cpt197925143. [DOI] [PubMed] [Google Scholar]
- 21.Kattus AA, Alvaro AB, Zohman LR, Coulson AH. Comparison of placebo, nitroglycerin, and isosorbide dinitrate for effectiveness of relief of angina and duration of action. Chest. 1979;75:17–23. doi: 10.1378/chest.75.1.17. [DOI] [PubMed] [Google Scholar]
- 22.Arnold WP, Mittal CK, Katsuki S, Murad F. Nitric oxide activates guanylate cyclase and increases guanosine 3′:5′-cyclic monophosphate levels in various tissue preparations. Proc Natl Acad Sci U S A. 1977;74:3203–3207. doi: 10.1073/pnas.74.8.3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ignarro LJ, Lippton H, Edwards JC, Baricos WH, Hyman AL, Kadowitz PJ, et al. Mechanism of vascular smooth muscle relaxation by organic nitrates, nitrites, nitroprusside and nitric oxide: evidence for the involvement of S-nitrosothiols as active intermediates. J Pharmacol Exp Ther. 1981;218:739–749. [PubMed] [Google Scholar]
- 24.Murad F, Mittal CK, Arnold WP, Katsuki S, Kimura H. Guanylate cyclase: activation by azide, nitro compounds, nitric oxide, and hydroxyl radical and inhibition by hemoglobin and myoglobin. Adv Cyclic Nucleotide Res. 1978;9:145–158. [PubMed] [Google Scholar]
- 25.Ignarro LJ. Biological actions and properties of endothelium-derived nitric oxide formed and released from artery and vein. Circ Res. 1989;65:1–21. doi: 10.1161/01.res.65.1.1. [DOI] [PubMed] [Google Scholar]
- 26.Chen Z, Stamler JS. Bioactivation of nitroglycerin by the mitochondrial aldehyde dehydrogenase. Trends Cardiovasc Med. 2006;16:259–265. doi: 10.1016/j.tcm.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 27.McGuire JJ, Anderson DJ, McDonald BJ, Narayanasami R, Bennett BM. Inhibition of NADPH-cytochrome P450 reductase and glyceryl trinitrate biotransformation by diphenyleneiodonium sulfate. Biochem Pharmacol. 1998;56:881–893. doi: 10.1016/s0006-2952(98)00216-0. [DOI] [PubMed] [Google Scholar]
- 28.Tsuchida S, Maki T, Sato K. Purification and characterization of glutathione transferases with an activity toward nitroglycerin from human aorta and heart. Multiplicity of the human class Mu forms. J Biol Chem. 1990;265:7150–7157. [PubMed] [Google Scholar]
- 29.Yeates RA, Schmid M, Leitold M. Antagonism of glycerol trinitrate activity by an inhibitor of glutathione S-transferase. Biochem Pharmacol. 1989;38:1749–1753. doi: 10.1016/0006-2952(89)90408-5. [DOI] [PubMed] [Google Scholar]
- 30.Kollau A, Beretta M, Gorren AC, Russwurm M, Koesling D, Schmidt K, et al. Bioactivation of nitroglycerin by ascorbate. Mol Pharmacol. 2007;72:191–196. doi: 10.1124/mol.107.035642. [DOI] [PubMed] [Google Scholar]
- 31.Lau DT, Chan EK, Benet LZ. Glutathione S-transferase-mediated metabolism of glyceryl trinitrate in subcellular fractions of bovine coronary arteries. Pharm Res. 1992;9:1460–1464. doi: 10.1023/a:1015867031004. [DOI] [PubMed] [Google Scholar]
- 32.O’Byrne S, Shirodaria C, Millar T, Stevens C, Blake D, Benjamin N. Inhibition of platelet aggregation with glyceryl trinitrate and xanthine oxidoreductase. J Pharmacol Exp Ther. 2000;292:326–330. [PubMed] [Google Scholar]
- 33.Nakahira A, Minamiyama Y, Takemura S, Hirai H, Sasaki Y, Okada S, et al. Coadministration of carvedilol attenuates nitrate tolerance by preventing cytochrome p450 depletion. Circ J. 2010;74:1711–1717. doi: 10.1253/circj.cj-10-0149. [DOI] [PubMed] [Google Scholar]
- 34.Schroder H. Cytochrome P-450 mediates bioactivation of organic nitrates. J Pharmacol Exp Ther. 1992;262:298–302. [PubMed] [Google Scholar]
- 35.Chen Z, Zhang J, Stamler JS. Identification of the enzymatic mechanism of nitroglycerin bioactivation. Proc Natl Acad Sci U S A. 2002;99:8306–8311. doi: 10.1073/pnas.122225199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Budas GR, Disatnik MH, Mochly-Rosen D. Aldehyde dehydrogenase 2 in cardiac protection: a new therapeutic target? Trends Cardiovasc Med. 2009;19:158–164. doi: 10.1016/j.tcm.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Petersen DR, Doorn JA. Reactions of 4-hydroxynonenal with proteins and cellular targets. Free Radic Biol Med. 2004;37:937–945. doi: 10.1016/j.freeradbiomed.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 38.Klyosov AA, Rashkovetsky LG, Tahir MK, Keung WM. Possible role of liver cytosolic and mitochondrial aldehyde dehydrogenases in acetaldehyde metabolism. Biochemistry. 1996;35:4445–4456. doi: 10.1021/bi9521093. [DOI] [PubMed] [Google Scholar]
- 39.Needleman P, Jakschik B, Johnson EM., Jr Sulfhydryl requirement for relaxation of vascular smooth muscle. J Pharmacol Exp Ther. 1973;187:324–331. [PubMed] [Google Scholar]
- 40.Li Y, Zhang D, Jin W, Shao C, Yan P, Xu C, et al. Mitochondrial aldehyde dehydrogenase-2 (ALDH2) Glu504Lys polymorphism contributes to the variation in efficacy of sublingual nitroglycerin. J Clin Invest. 2006;116:506–511. doi: 10.1172/JCI26564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kollau A, Hofer A, Russwurm M, Koesling D, Keung WM, Schmidt K, et al. Contribution of aldehyde dehydrogenase to mitochondrial bioactivation of nitroglycerin: evidence for the activation of purified soluble guanylate cyclase through direct formation of nitric oxide. Biochem J. 2005;385:769–777. doi: 10.1042/BJ20041354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang J, Chen Z, Cobb FR, Stamler JS. Role of mitochondrial aldehyde dehydrogenase in nitroglycerin-induced vasodilation of coronary and systemic vessels: an intact canine model. Circulation. 2004;110:750–755. doi: 10.1161/01.CIR.0000138105.17864.6B. [DOI] [PubMed] [Google Scholar]
- 43.de la Lande IS, Stepien JM, Philpott AC, Hughes PA, Stafford I, Horowitz JD. Aldehyde dehydrogenase, nitric oxide synthase and superoxide in ex vivo nitrate tolerance in rat aorta. Eur J Pharmacol. 2004;496:141–149. doi: 10.1016/j.ejphar.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 44.Sydow K, Daiber A, Oelze M, Chen Z, August M, Wendt M, et al. Central role of mitochondrial aldehyde dehydrogenase and reactive oxygen species in nitroglycerin tolerance and cross-tolerance. J Clin Invest. 2004;113:482–489. doi: 10.1172/JCI19267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen Z, Foster MW, Zhang J, Mao L, Rockman HA, Kawamoto T, et al. An essential role for mitochondrial aldehyde dehydrogenase in nitroglycerin bioactivation. Proc Natl Acad Sci U S A. 2005;102:12159–12164. doi: 10.1073/pnas.0503723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wenzl MV, Beretta M, Gorren AC, Zeller A, Baral PK, Gruber K, et al. Role of the general base Glu-268 in nitroglycerin bioactivation and superoxide formation by aldehyde dehydrogenase-2. J Biol Chem. 2009;284:19878–19886. doi: 10.1074/jbc.M109.005652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kozlov AV, Staniek K, Nohl H. Nitrite reductase activity is a novel function of mammalian mitochondria. FEBS Lett. 1999;454:127–130. doi: 10.1016/s0014-5793(99)00788-7. [DOI] [PubMed] [Google Scholar]
- 48.Brudvig GW, Stevens TH, Chan SI. Reactions of nitric oxide with cytochrome c oxidase. Biochemistry. 1980;19:5275–5285. doi: 10.1021/bi00564a020. [DOI] [PubMed] [Google Scholar]
- 49.Kollau A, Beretta M, Russwurm M, Koesling D, Keung WM, Schmidt K, et al. Mitochondrial nitrite reduction coupled to soluble guanylate cyclase activation: lack of evidence for a role in the bioactivation of nitroglycerin. Nitric Oxide. 2009;20:53–60. doi: 10.1016/j.niox.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 50.DeMaster EG, Redfern B, Quast BJ, Dahlseid T, Nagasawa HT. Mechanism for the inhibition of aldehyde dehydrogenase by nitric oxide. Alcohol. 1997;14:181–189. doi: 10.1016/s0741-8329(96)00142-5. [DOI] [PubMed] [Google Scholar]
- 51.Shen ML, Lipsky JJ, Naylor S. Role of disulfiram in the in vitro inhibition of rat liver mitochondrial aldehyde dehydrogenase. Biochem Pharmacol. 2000;60:947–953. doi: 10.1016/s0006-2952(00)00435-4. [DOI] [PubMed] [Google Scholar]
- 52.Sun L, Ferreira JC, Mochly-Rosen D. ALDH2 activator inhibits increased myocardial infarction injury by nitroglycerin. Science Translational Medicine. 2011 doi: 10.1126/scitranslmed.3002067. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Daiber A, Oelze M, Sulyok S, Coldewey M, Schulz E, Treiber N, et al. Heterozygous deficiency of manganese superoxide dismutase in mice (Mn-SOD+/−): a novel approach to assess the role of oxidative stress for the development of nitrate tolerance. Mol Pharmacol. 2005;68:579–588. doi: 10.1124/mol.105.011585. [DOI] [PubMed] [Google Scholar]
- 54.Esplugues JV, Rocha M, Nunez C, Bosca I, Ibiza S, Herance JR, et al. Complex I dysfunction and tolerance to nitroglycerin: an approach based on mitochondrial-targeted antioxidants. Circ Res. 2006;99:1067–1075. doi: 10.1161/01.RES.0000250430.62775.99. [DOI] [PubMed] [Google Scholar]
- 55.Chen CH, Budas GR, Churchill EN, Disatnik MH, Hurley TD, Mochly-Rosen D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science. 2008;321:1493–1495. doi: 10.1126/science.1158554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Carbone DL, Doorn JA, Petersen DR. 4-Hydroxynonenal regulates 26S proteasomal degradation of alcohol dehydrogenase. Free Radic Biol Med. 2004;37:1430–1439. doi: 10.1016/j.freeradbiomed.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 57.Beretta M, Gorren AC, Wenzl MV, Weis R, Russwurm M, Koesling D, et al. Characterization of the East Asian variant of aldehyde dehydrogenase-2: bioactivation of nitroglycerin and effects of Alda-1. J Biol Chem. 2010;285:943–952. doi: 10.1074/jbc.M109.014548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Needleman P, Hunter FE., Jr Effects of organic nitrates on mitochondrial respiration and swelling: possible correlations with the mechanism of pharmacologic action. Mol Pharmacol. 1966;2:134–143. [PubMed] [Google Scholar]
- 59.Hink U, Daiber A, Kayhan N, Trischler J, Kraatz C, Oelze M, et al. Oxidative inhibition of the mitochondrial aldehyde dehydrogenase promotes nitroglycerin tolerance in human blood vessels. J Am Coll Cardiol. 2007;50:2226–2232. doi: 10.1016/j.jacc.2007.08.031. [DOI] [PubMed] [Google Scholar]
- 60.Daiber A, Oelze M, Coldewey M, Kaiser K, Huth C, Schildknecht S, et al. Hydralazine is a powerful inhibitor of peroxynitrite formation as a possible explanation for its beneficial effects on prognosis in patients with congestive heart failure. Biochem Biophys Res Commun. 2005;338:1865–1874. doi: 10.1016/j.bbrc.2005.10.106. [DOI] [PubMed] [Google Scholar]
- 61.Mulsch A, Oelze M, Kloss S, Mollnau H, Topfer A, Smolenski A, et al. Effects of in vivo nitroglycerin treatment on activity and expression of the guanylyl cyclase and cGMP-dependent protein kinase and their downstream target vasodilator-stimulated phosphoprotein in aorta. Circulation. 2001;103:2188–2194. doi: 10.1161/01.cir.103.17.2188. [DOI] [PubMed] [Google Scholar]
- 62.Daiber A, Oelze M, Wenzel P, Wickramanayake JM, Schuhmacher S, Jansen T, et al. Nitrate tolerance as a model of vascular dysfunction: roles for mitochondrial aldehyde dehydrogenase and mitochondrial oxidative stress. Pharmacol Rep. 2009;61:33–48. doi: 10.1016/s1734-1140(09)70005-2. [DOI] [PubMed] [Google Scholar]
- 63.Daiber A, Wenzel P, Oelze M, Schuhmacher S, Jansen T, Munzel T. Mitochondrial aldehyde dehydrogenase (ALDH-2)--maker of and marker for nitrate tolerance in response to nitroglycerin treatment. Chem Biol Interact. 2009;178:40–47. doi: 10.1016/j.cbi.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 64.Chen CH, Sun L, Mochly-Rosen D. Mitochondrial aldehyde dehydrogenase and cardiac diseases. Cardiovasc Res. 2010;88:51–57. doi: 10.1093/cvr/cvq192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Larson HN, Weiner H, Hurley TD. Disruption of the coenzyme binding site and dimer interface revealed in the crystal structure of mitochondrial aldehyde dehydrogenase “Asian” variant. J Biol Chem. 2005;280:30550–30556. doi: 10.1074/jbc.M502345200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Seitz HK, Matsuzaki S, Yokoyama A, Homann N, Vakevainen S, Wang XD. Alcohol and cancer. Alcohol Clin Exp Res. 2001;25:137S–143S. doi: 10.1097/00000374-200105051-00024. [DOI] [PubMed] [Google Scholar]
- 67.Takagi S, Iwai N, Yamauchi R, Kojima S, Yasuno S, Baba T, et al. Aldehyde dehydrogenase 2 gene is a risk factor for myocardial infarction in Japanese men. Hypertens Res. 2002;25:677–681. doi: 10.1291/hypres.25.677. [DOI] [PubMed] [Google Scholar]
- 68.Jo SA, Kim EK, Park MH, Han C, Park HY, Jang Y, et al. A Glu487Lys polymorphism in the gene for mitochondrial aldehyde dehydrogenase 2 is associated with myocardial infarction in elderly Korean men. Clin Chim Acta. 2007;382:43–47. doi: 10.1016/j.cca.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 69.Amamoto K, Okamura T, Tamaki S, Kita Y, Tsujita Y, Kadowaki T, et al. Epidemiologic study of the association of low-Km mitochondrial acetaldehyde dehydrogenase genotypes with blood pressure level and the prevalence of hypertension in a general population. Hypertens Res. 2002;25:857–864. doi: 10.1291/hypres.25.857. [DOI] [PubMed] [Google Scholar]
- 70.Ohsawa I, Kamino K, Nagasaka K, Ando F, Niino N, Shimokata H, et al. Genetic deficiency of a mitochondrial aldehyde dehydrogenase increases serum lipid peroxides in community-dwelling females. J Hum Genet. 2003;48:404–409. doi: 10.1007/s10038-003-0046-y. [DOI] [PubMed] [Google Scholar]
- 71.Mackenzie IS, Maki-Petaja KM, McEniery CM, Bao YP, Wallace SM, Cheriyan J, et al. Aldehyde dehydrogenase 2 plays a role in the bioactivation of nitroglycerin in humans. Arterioscler Thromb Vasc Biol. 2005;25:1891–1895. doi: 10.1161/01.ATV.0000179599.71086.89. [DOI] [PubMed] [Google Scholar]
- 72.Gori T, Parker JD. Nitrate-induced toxicity and preconditioning: a rationale for reconsidering the use of these drugs. J Am Coll Cardiol. 2008;52:251–254. doi: 10.1016/j.jacc.2008.04.019. [DOI] [PubMed] [Google Scholar]
- 73.Perez-Miller S, Younus H, Vanam R, Chen CH, Mochly-Rosen D, Hurley TD. Alda-1 is an agonist and chemical chaperone for the common human aldehyde dehydrogenase 2 variant. Nat Struct Mol Biol. 2010;17:159–164. doi: 10.1038/nsmb.1737. [DOI] [PMC free article] [PubMed] [Google Scholar]