

Abstract
Objective
To investigate placental protein 13 (PP13) localization in relation to cytoskeleton and lipid rafts in preeclampsia and HELLP syndrome.
Study Design
Placental cryosections from patients with preeclampsia and HELLP, and controls were stained for PP13, actin, PLAP (lipid raft marker), and CD71 (nonraft marker). BeWo cells exposed to stress conditions were stained for PP13 and actin. Protein localization were investigated by confocal microscopy, PP13 concentrations by ELISA.
Results
PP13-actin colocalization was increased in syncytiotrophoblast juxtamembrane regions in term/preterm preeclampsia and HELLP. PP13-CD71 colocalization was decreased and PP13-PLAP proximity was increased in preterm but not term preeclampsia and HELLP. PP13-release from BeWo cells was inhibited by cytoskeleton disruption, and augmented by Ca2+-influx and ischemic stress.
Conclusion
The actin cytoskeleton, probably in connection with lipid rafts, controls trophoblastic “nonclassical” PP13 export. PP13 is released from the syncytiotrophoblast in preterm preeclampsia and HELLP, mimicked in BeWo cells by ischemic stress, suggesting PP13 is a placental alarmin.
Keywords: alarmin, brush border membrane, damage-associated molecular pattern (DAMP), danger signal, galectin, “nonclassical” secretory pathway
INTRODUCTION
Preeclampsia, one of the “great obstetric syndromes”,1,2 affects approximately 3–5% of pregnant women and is a major cause of maternal and perinatal morbidity and mortality. It is characterized by the new onset of hypertension and proteinuria after 20 weeks of gestation. In the most severe cases, seizures (eclampsia), multiorgan damage including cerebrovascular, hepatic or renal failure, and placental abruption and HELLP (Hemolysis, Elevated Liver enzymes and Low Platelets) syndrome may develop.3–5 HELLP syndrome, which affects 0.5–0.9% of all pregnancies and 2–12% of pregnancies complicated by preeclampsia, is thought to be more severe form of preeclampsia although approximately 15–20% of the patients do not develop hypertension and/or proteinuria.6–11
Both preeclampsia and HELLP syndrome are heterogeneous. The involvement of the placenta in the different pathogenic pathways is well-established because the only definitive treatment of these syndromes is delivery of the fetus and placenta. Preterm preeclampsia and HELLP syndrome are associated with abnormal placentation, impaired trophoblast invasion, and remodeling of the uterine spiral arteries.12–20 Failure of physiologic transformation of the spiral arteries in these cases is thought to result in intermittent, turbulent flow, damage of the placental architecture, fluctuations in placental oxygenation, and placental endoplasmic reticulum-, oxidative-, and nitrative stress.21–24 Placental ischemia-reperfusion injury triggers an increased release of apoptotic-necrotic syncytiotrophoblast microparticles (MPs), proinflammatory cytokines, and antiangiogenic factors, which in turn, lead to generalized maternal endothelial cell dysfunction and an exaggerated maternal systemic inflammatory response.3–5,22– 23,25–40
In normal pregnancy, MPs continuously shed from the syncytiotrophoblast, the outermost fused cell layer of the human placenta, to the maternal circulation as part of the natural renewal process of placental microvilli. The amount of circulating MPs increases with advancing gestational age in normal pregnancies and is significantly elevated in the blood of women with preterm preeclampsia. 34,41 Of note, we have previously shown that syncytiotrophoblast cytoplasm protrusions, membrane blebs, and MPs shed from the syncytiotrophoblast apical membrane have intense placental protein 13 (PP13) immunostaining in preterm preeclampsia and HELLP syndrome.42 This is consistent with the findings that maternal serum PP13 concentration increases during normal pregnancy with a peak in the third trimester,43–45 and it is higher in patients presenting with preterm preeclampsia and HELLP syndrome than in preterm controls. Surprisingly, we did not find such difference in placental PP13 staining and maternal serum PP13 concentrations in patients with term preeclampsia when compared with normal term control patients.42
In the context of these observations, it is important to note that PP13 is specifically expressed by the placenta in anthropoid primates, primarily in the syncytiotrophoblast.42,46–50 PP13 is a member of the evolutionarily conserved family of galectins (galectin-13), which are key regulatory proteins of immune-homeostasis and inflammation.49–53 Galectins are synthesized in the cytosol and are alternatively transported to the plasma membrane, avoiding the endoplasmic reticulum (ER) and Golgi vesicles,54 through a so-called “nonclassical” secretory pathway.55,56 On the cell surface and in the extracellular matrix, galectins can initiate leukocyte signaling and modify cell fate on binding to their cell-surface ligands, leading to either apoptosis or cell survival. Galectins can also have various effects on cell growth, cytokine secretion, cell-cell and cell-extracellular matrix interactions.50,57
A previous study showed that, similar to other galectins, PP13 accumulates in the cytoplasmic side of the plasma membrane, leading us to hypothesize that it is secreted to the syncytiotrophoblast surface by the actin filamental network.49 It has also been demonstrated that PP13 specifically binds to and colocalizes with anexin-II at the apical membrane of the syncytiotrophoblast.49 Of note, similar to placental alkaline phosphatase (PLAP), a protein highly expressed in the syncytiotrophoblast apical membrane, anexin-II is also associated with lipid rafts,58 which are highly dynamic, cholesterol- and sphingolipid-rich 10–200 nm microdomains in the mammalian cell membrane. Lipid rafts can grow to 1 μm and provide a stable platform for lipid-lipid and protein-lipid interactions on various stimuli, such as receptor cross-linking during signaling or endocytosis.59
Based on these findings, we hypothesized that: (1) the actin filamental network and lipid rafts in the syncytiotrophoblast apical membrane may play a role in the normal secretory and shedding mechanisms of PP13, and (2) the pathophysiologic processes affecting the syncytiotrophoblast in various phenotypes of preeclampsia and HELLP syndrome may lead to the increased secretion and/or shedding of PP13 from the villous tree. Therefore, this study was designed to investigate whether changes in the placental localization of PP13 and its spatial relationship to lipid rafts and the actin cytoskeleton could be detected in the placentas obtained from women with preeclampsia or HELLP syndrome and gestational age-matched controls using confocal imaging. To get insight into the possible cellular mechanisms of PP13-release from the trophoblast, the role of the actin cytoskeleton and different stress factors on PP13-release was studied in vitro in BeWo cells.
MATERIALS AND METHODS
Sample collection and patient groups
The study was approved by the Health Science Board of Hungary (ad.22—164/2007-1018EKU) and the Human Investigation Committee of Wayne State University (036410M1X). Written informed consistent was obtained from women before sample collection; specimens were coded and data were stored anonymously. Placental villous tissues were collected at the First Department of Obstetrics and Gynecology, Semmelweis University (Budapest, Hungary, Federalwide Assuarnce: FWA00002527) in the following gestational age-matched groups (n = 5 in each): (1) preterm preeclampsia (≤ 35 weeks), (2) preterm HELLP syndrome (≤ 35 weeks), (3) term preeclampsia (> 37 weeks), (4) preterm (≤ 35 weeks), and (5) term (> 37 weeks) controls. Patients with multiple pregnancies or with fetuses having congenital or chromosomal abnormalities were excluded. Demographic and clinical characteristics of patient groups are shown in the Table.
Table 1.
Demographic and clinical characteristics of the study groups
| Groups | Term controls | Term preeclampsia | Preterm controls | Preterm preeclampsia | Preterm HELLP syndrome |
|---|---|---|---|---|---|
| Number of casesa | 5 | 5 | 5 | 5 | 5 |
| Maternal age, yb | 30.8 (30.6–31.5) | 32.2 (30.3–33.6) | 31.6 (31.5–34.3) | 34.0 (30.2–34.5) | 28.1 (24.1–29.2) |
| Gestational age at delivery, wkb | 38.9 (38.7–39.0) | 38.4 (37.7–39.9) | 31.0 (30.9–34.0) | 32.6 (30.3–34.9) | 32.0 (29.3–33.1) |
| Primiparityc | 40 | 60 | 40 | 80 | 60 |
| Smokingc | 0 | 0 | 20 | 0 | 0 |
| Systolic blood pressure, mm Hgb | 130 (120–135) | 160 (153–170)d | 120 (120–120) | 160 (155–163)e | 170 (170–170)f |
| Diastolic blood pressure, mm Hgb | 80 (78–89) | 90 (90–100) | 80 (70–80) | 100 (98–101)e | 100 (90–110)f |
| Proteinuriac | 0 | 100 | 0 | 100 | 100 |
| Maternal BMI, kg/m2b | 26.7 (25.2–28.0) | 22.0 (20.0–23.3) | 23.4 (21.6–24.6) | 24.2 (22.6–24.9) | 21.4 (21.2–26.0) |
| Neonatal birthweight, gb | 3440 (3400–4030) | 2810 (2620–3200)d | 1990 (1640–2210) | 1100 (990–1200) | 1480 (990–1610) |
| Cesarean sectionc | 100 | 100 | 100 | 100 | 100 |
| Placental weight, gb | 518 (458–665) | 458 (431–476) | 301 (294–322) | 217 (211–224) | 302 (255–305) |
| Thrombocytes, M/mm3b | 178 (173–238) | 206 (191–250) | 227 (170–298) | 233 (187–360) | 80 (73–93)e |
| Hgb, g/100mL | 10 (10–11) | 12 (12–12) | 11 (11–11) | 13 (13–13)f | 13 (12–14) |
| SGOT, U/Lb,g | 25 (22–32) | 31 (25–80) | 333 (318–439) | ||
| SGPT, U/Lb,g | 17 (13–19) | 33 (23–80) | 337 (315–448) | ||
| LDH, U/Lb,g | 246 (196–332) | 293 (217–350) | 572 (510–646) | ||
| Bilirubin, μmol/Lb,g | 15 (12–15) | 5 (4–12) | 19 (17–21) |
All women were white.
BMI, body mass index; HELLP, hemolysis elevated liver enzymes and low platelets; LDH, lactate dehydrogenase; SGOT, serum glutamate oxaloacetate transaminase; SGPT, serum glutamate pyruvate transaminase.
Values are presented as number;
Values are presented as median (interquartile [IQR] range);
Values are presented as percentage;
P<.05 compared with gestational age matched, term controls;
P<.05 compared with gestational age matched, preterm controls;
P<.01;
Routinely not examined in normal pregnant women.
Balogh. Subcellular relocalizaiton of PP13 in preeclampsia and HELLP syndrome. Am J Obstet Gynecol 2011.
Term and preterm controls had no medical complications or clinical or histologic signs of chorioamnionitis, and delivered neonates of appropriate weight for gestational age.60 Preeclampsia was defined as hypertension that developed after 20 weeks of gestation (systolic or diastolic blood pressure ≥140 or ≥90mm Hg, respectively, measured at 2 different time points, 4 points, 4 points to 1 week apart) coupled with proteinuria (≥300mg in a 24-hour urine collection, or 2 random urine specimens obtained 4 hours to 1 week apart containing ≥1 + by dipstick or 1 dipstick of ≥2+ protein).5,61 HELLP syndrome was defined as hemolysis (serum LDH >600 IU/L; bilirubin >1.2mg/dL; presence of schistocytes in peripheral blood), elevated liver enzymes (serum ALT and/or AST >70 IU/L), and thrombocytopenia (platelet count <100,000/mm3).10
None of the patients had regular uterine contractions; cesarean delivery was performed in all cases because of previous cesarean section (term controls), or preterm premature rupture of the membranes and breech presentation (preterm controls), or severe clinical symptoms (patients with preeclampsia or HELLP syndrome).
Fluroescence immunohistochemistry
Fresh placental specimens were frozen in in isopentane (Sigma-Aldrich, St. Louis, MO); subsequently, 10 μm thick cryosections (2–4 cryosections/sample) were cut and placed on Superfrost Plus slides (Thermo Scientific, Walthem, MA), fixed with 4% paraformaldehyde and then kept at 4°C in phosphate-buffered saline (PBS) until staining. Nonspecific antibody was blocked with 5% bovine serum albumin (BSA) in PBS (30 minutes at room temperature). Samples were incubated with monoclonal anti-PP13 antibody (clone: 27-2-3; 5 μm/mL; Diagnostic Technologies Ltd, Yokneam, Israel) alone or in combination with anti-PLAP (1 μm/mL; Sigma-Aldrich), anti-CD71 (5 μm/mL; Biolegend, San Diego, CA), or antiactin (8 μm/mL; Sigma-Aldrich) antibodies in PBS containing 1% BSA. Samples were stained with anti-PP13 overnight, whereas in the case of other primary antibodies, the incubation time was 45 minutes. Washing was followed by incubation (45 minutes at room temperature) with Alexa Fluor488-conjugated antimouse IgG1 (2 μm/mL; Invitrogen-Molecular Probes, Eugene, OR) for anti-PP13, Alexa Fluor633-conjugated antimouse IgG2a (2 μm/mL; Invitrogen-Molecular Probes) for anti-PLAP and anit-CD71, or Alexa Fluor647-conjugated antirabbit IgG (2 μm/mL; Invitrogen-Molecular Probes) for antiactin. Isotype control antibodies were used as negative controls; mouse IgG1 (clone MOPC-21; BD Pharmingen, San Jose, CA) and mouse IgG2a (clone HOPC-1; Southern Biotech, Birmingham, AL). Nuclei were counterstained with propidium iodide (0.2 μm/mL; Sigma-Aldrich). Washed samples were assayed with an Olympus Fluoview 500 confocal microscope (Hamburg, Germany) equipped with 3 lasers and 4 optical channels, using a 60x (N.A.: 1.1) oil-immersion objective.
BeWo cell transfection
BeWo cells (American Type Culture Collection, Manassas, VA) were incubated in 2 T-75 flasks with F12 medium (Invitrogen-Gibco, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S) until reaching 80% confluency. PP13 vector DNA (9 μm; OriGene, Rockville, MD) was diluted in 450 μL of serum-free Opti-MEM I medium (Invitrogen-Gibco). Then, 36 μL FuGENE HD transfection reagent (Promega, Madision, WI) was added to the mix, which was incubated for 15 minutes at room temperature. Ten milliliters of fresh Opti-MEM I medium were added to 1 of the T-75 flasks after the cells were washed once with PBS. The transfection complex was then added to this flask and incubated for 6 hours before replacing the medium to F12 (supplemented with 10% FBS and 1% P/S). BeWo cells without PP13 vector transfection were used as control.
BeWo cell treatments
After 24 hours in culture, transfected and nontransfected BeWo cells were trypsinized and plated onto 6-well plates (2 × 105 cells/well) and 4-well Lab-Tek chamber slides (5 × 104 cells/well; Thermo Scientific), and were cultured for 24 hours in F12 medium supplemented with 10% FBS and 1% P/S. After medium change, 60-hour treatments started in the following regimes: (1) to inhibit actin polymerization, cells were treated with Latrunculin B (0.125 and 0.25 μM; EMD Chemicals, Gibbstown, NJ) between hours 48–60; (2) to stimulate nonclassical protein secretion by elevated cytoplasmic Ca2+ -level, cells were treated with ionophore A23187 (1 μM; Sigma-Aldrich) between hours 48–60; and (3) to induce cellular stress, cells were treated with either human recombinant TNFα (4ng/mL; R&D Systems, Minneapolis, MN) between hours 0–60, or kept under hypoxic (1%O2) or ischemic conditions (1% and 20% O2 in alternating, 12 hour cycles) between hours 0–60. Control cells were treated with vehicle (DMSO; Sigma-Aldrich).
RNA isolation and quantitative real-time reverse transcribe-polymerase chain reaction
After collecting supernatants, BeWo cells were harvested from 6-well plates and total RNA was isolated using Quick-Gene-Mini80 (Fujifilm, Allendale, NJ) according to the manufacturer’s protocol. Five hundred nanograms of total RNA was reverse transcribed with TaqMan Revers Transcription Reagent kit using random hexamers (Applied Biosystems, Foster City, CA). Quantitative reverse transcribe-polymerase chain reaction (qRT-PCR) was run in triplicates with LGALS13 TawMan assay (Hs00848811_m1) and RPLPO TaqMan Endogenous Control (4326314E) on a 7500 Fast Real-Time PCR System (Applied Biosystems) Ct values for LGALS13 and RPLPO were averaged over 3 technical replicates. −DCt value, a surrogate of log2 mRNA concentration, was obtained for each sample as: −DCt(LGALS13) = Ct(RPLPO) − Ct(LGALS13).
Protein isolation
After collecting supernatants, BeWo cells were harvested from 6-well plates, homogenized with 120 μL lysis buffer (50 mM Tris-HCl, pH 7.5; 150 mM NaCl; 1% NP40; 5 mM EDTA) and incubated for 1 hour on ice. Homogenates were centrifuged (15 minutes at 10,000 rpm) to pellet nonsoluble or solubilized proteins were collected, and their total protein contents were measured by BCA assay (Thermo Scientific).
PP13 immunoassay
PP13 content of BeWo cell lysates and supernatants was measured using a solid phase sandwich enzyme-linked immunosorbent assay (ELISA) (Diagnostic Technologies Ltd) with a pair of PP13-specific monoclonal antibodies, marked with amplified biotin-extravidin-horseradish-peroxidase complex, and developed with tetramethylbenzidine substrate, as described previously.43 The optical density was measured at 450 nm against a 650 nm background. PP13 concentrations were determined by extrapolation from a standard curve constructed using recombinant PP13 standards (0–500 pg/mL), and were normalized to total protein in cell lysates. The sensitivity of the assay was 9.6 pg/mL. The laboratory staff performing the assays was blinded to sample information.
Fluorescence immunocytochemistry
PP13-tranfected, treated or untreated BeWo cells on chamber slides were simultaneously fixed and permeabilized in PBS supplemented with 4% paraformaldehyde and 0.1% Trimeton X-100 (Sigma-Aldrich), followed by washing with PBS supplemented with 1% FBS and 0.1% sodium-azide. Nonspecific antibody binding was blocked with 5% BSA in PBS (30 minutes at room temperature). Samples were incubated overnight with monoclonal anti-PP13 antibody (clone:27-2-3; 20 μg/mL) alone or in combination with antiactin (7 μg/mL) antibody in PBS containing 5% BSA. Washing was followed by incubation (2 hours at room temperature) with Alexa Fluor488-conjugated antimouse IgG1 (2 μg/mL) for anti-PP13 and Alexa Fluor555-conjugated antirabbit IgG (2 μg/mL) for antiactin. Isotype control antibodies were used as negative controls. Alternatively, cells were stained with Alexa Fluor555-conjugated phalloidin (2 U/mL; Invitrogen-Molecular probes) to visualize filamentous actin. Nuclei were counterstained with DRAQ5 (1 μM; Biostatus, Leicestershire, UK). Washed samples were assayed with an Olympus Fluoview 500 confocal microscope equipped with 4 lasers and 4 optical channels, using 20X (N.A.: 0.7) of 40X (N.A.: 0.85) objectives.
Data analysis
Colocalization indices were determined from approximately 100 regions of interest (ROIs)/each group by the ImageJ software (http://rsbweb.nih.gov/ij. Accessed May 2, 2011.) using the Red Green Correlator plug-in (http://www.uhnresearch.ca/facilities/wcif/imagej/colour_analysis.htm. Accessed May 2, 2011.). The Pearson’s colocalization index (CI) provides a reliable estimate on the extent of protein colocalization. A CI value of −1 means the exclusive appearance of the 2 labeled specimens, its value close to zero indicates a random appearance, whereas CI values between 0 and +1 reflect a continuously increasing degree of colocalization, while CI = 1 corresponds to a full overlap between the 2 colors in each pixels of the image.62,63
Line scan analysis was carried out using the Fluoview software. Lones (approximately 200/group) were laid across placental microvilli stained for PP13 and actin or PLAP. Intensity values were sorted into membrane/juxtamembrane (3 μm at opposite sides of the villi) and cytoplasmic (internal) regions. Based on the staining pattern, 3 groups were formed for quantifying these intensity distributions: (1) PP13 and actin (or PLAP) were localized in the same region; (2) only PP13 was localized in the selected region; and (3) only actin (or PLAP) was localized in the selected region (based on the actual intensity exceeding the threshold level). The relative frequency of each staining pattern was then calculated from approximately 300 ROIs. Intensity distributions were also demonstrated on line scan histograms. The interactive 3-dimensional (3D) surface plot plug-in of ImageJ was also used to create another representation platform of the data on the different samples.
Statistical analysis
Demographic and clinical data were analyzed by Statistica8 (StatSoft Inc, Tulsa, OK). Comparisons among the groups were performed by X2 test and Fisher’s exact test for proportions and Kruskal-Wallis test, followed by Mann-Whitney test for continuous variables. GraphPad Prism 4 (GraphPad Software, La Jolla, CA) was used to analyze the differences in the colocalization, gene expression, and protein concentration values between the groups using Mann-Whitney test or t test. A P value of < .05 was considered statistically significant.
RESULTS
Localization of PP13 in the syncytiotrophoblast in preeclampsia and HELLP syndrome
The localization of PP13 in placental villous tissues from patients with preeclampsia of HELLP syndrome and gestational age-matched controls was determined with confocal microscopy using immunohistochemical staining. Similar to earlier data,42,49,50 PP13 was present in the endothelium of fetal vessels, and it was found in the membrane and the cytoplasm of the syncytiotrophoblast (Figure 1). There was a shift in the localization of PP13; stronger staining was observed in the plasma membrane rather than the cytoplasm in patients with term and preterm preeclampsia and HELLP syndrome in comparison to gestational age-matched controls (Figure 1). This finding is consistent with our previous report.42
Figure 1. The syncytiotrophoblastic localization of PP13 is altered in patients with preeclampsia and HELLP syndrome.

Representative confocal fluorescence-DIC composite images of cryosections of placentas from women with preterm preeclampsia, preterm HELLP syndrome, term preeclampsia, and gestational age-matched controls. Cryosections were stained with monoclonal anti-PP13 antibody (green) and PI for nuclei (red). There was a weaker cytoplasmic staining of the syncytiotrophoblast in term and preterm preeclampsia and HELLP syndrome when compared with gestational age-matched controls. Stars indicate PP13 staining of the endothelium of fetal vessels, arrows indicate the strong apical membrane staining of terminal villi in disease cases.
DIC, differential interface contrast; HELLP, hemolysis elevated enzymes and low platelets; PI, propidium iodide; PP13, placental protein 13.
Balogh. Subcellular relocalizaiton of PP13 in preeclampsia and HELLP syndrome. Am J Obstet Gynecol 2011.
Visualizaiton of the interaction patterns of PP13 with cortical actin cytoskeleton and the raft marker PLAP or the nonraft protein CD71 in placental cryosections
Next, we analyzed the interaction patterns of PP13 with the cortical actin cytoskeleton and with 2 membrane proteins, PLAP CD71. A relatively high colocalization was found between PP13 and actin in term and preterm controls (CIs; mean ± standard error of the mean [SEM]: 0.357 ± 0.009 and 0.357 ± 0.008, respectively). The colocalization of these 2 proteins (PP13 and actin) was significantly increased in all patient groups (CIs, 0.459 ± 0.008, P < .05 for term preeclampsia; 0.509 ± 0.009, P < .05; and 0.520 ± 0.010, P < .05 for preterm preeclampsia and HELLP syndrome, respectively) when compared with gestational age-matched controls (Figures 2, A and 3, A). The membrane localization of PP13 was also altered in preeclampsia and HELLP syndrome. Although PP13 highly colocalized with the lipid raft-associated membrane protein PLAP in both control groups (CIs, 0.468 ± 0.007, 0.481 ± 0.012), no significant changes in the colocalization of PP13 and PLAP could be seen in term and preterm preeclampsia or HELLP syndrome (CIs, 0.468 ± 0.009, 0.440 ± 0.009, and 0.485 ± 0.010, respectively) (Figures 2, B and 3, B). In contrast, PP13 highly colocalized with CD71 in term and preterm controls (CIs, 0.505 ± 0.009 and 0.466 ± 0.009, respectively) as well as in term preeclampsia (CI, 0.490 ± 0.010), but this colocalization was significantly reduced in preterm preeclampsia (CI, 0.327 ± 0.009, P < .05) and HELLP syndrome (CI, 0.350 ± 0.013, P < .05) (Figures 2, C and 3, C).
Figure 2. Colocalization of PP13 with actin, CD71 or PLAP in preeclampsia and HELLP syndrome compared to controls.

Cryosections of placentas taken from women with HELLP syndrome and controls were stained with A–C, anti-PP13 (green) in combination with A, anti-actin, B, anti-PLAP, or C, anti-CD71 (all in red), and analyzed by confocal microscopy. Cell nuclei were counterstained with PI (blue) on all representative confocal images.
HELLP, hemolysis elevated liver enzymes and low platelets; PI, propidium iodide; PLAP, placental alkaline phosphatase; PP13, placental protein 13.
Balogh, subcellular relocalizaiton of PP13 in preeclampsia and HELLP syndrome. Am J Obstet Gynecol 2011.
Figure 3. PP13 is redistributed in the placenta of patients with preeclampsia and HELLP syndrome.

Cryosections of placentas taken from women with preterm preeclampsia of HELLP syndrome, term preeclampsia, and gestational age-matched controls were stained with anti-PP13 in combination with antiactin, anti-PLAP or anti-CD71, and analyzed by confocal microscopy. Colocalization indices for PP13 (derived from approximately 100 ROIs/group) are shown as mean ± SEM with A, actin, B, PLAP, or C, CD71.
HELLP, hemolysis elevated liver enzymes and low platelets; PLAP, placental alkaline phosphatase; PP13, placental protein 13; ROI, regions of interest.
Balogh. Subcellular relocalizaiton of PP13 in preeclampsia and HELLP syndrome. Am J Obstet Gynecol 2011.
Spatial coexistence of cortical filamental actin and PP13: a statistical analysis of line scanned intensity distributions
Because one of the major questions of the current study was whether any redistribution of PP13 could be observed toward the cortical actin network, the relative frequency of the spatial coexistence of PP13 and actin (or the plasma membrane PLAP) in the membrane/juxtamembrane region and in the cytoplasm was also determined. This analysis was performed by laying a line across the apical microvillous membranes of the syncytiotrophoblast on the opposing sides of the villi. The intensity distribution profile of PP13 and actin was then analyzed along this line (Figure 4, A). As shown in Figure 4, B, in all pathologic samples the coexistence of PP13 and actin showed a dramatically increased membrane/juxtamembrane: cytoplasmic distribution ratio. Such a relationship between distribution profiles of actin or PP13 alone in either of these disease groups could not be observed (Figure 4, C and D).
Figure 4. Coexistence of PP13 and actin in the juxtamembrane area of the syncytiotrophoblast is increased in preeclampsia and HELLP syndrome.
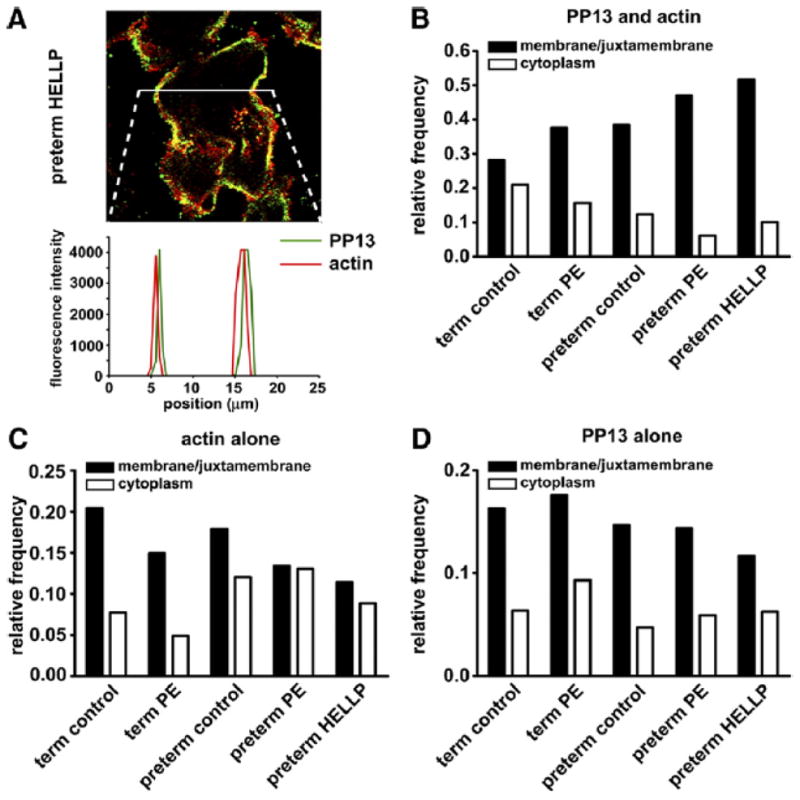
Cryosections of placenta taken from women with preeclampsia or HELLP syndrome and gestational age-matched controls were stained for PP13 and actin. A, Representative image and line scan intensity distribution are shown. Relative frequency of B, PP13-actin coexistence and of C, actin, or D, PP13 existence alone in membrane/juxtamembrane and in cytoplasmic regions, respectively, was calculated from approximately 200 ROIs in each group.
HELLP, hemolysis elevated liver enzymes and low platelets; PP13, placental protein 13; ROI, regions of interest.
Balogh. Subcellular relocalizaiton of PP13 in preeclampsia and HELLP syndrome. Am J Obstet Gynecol 2011.
Although no significant difference was detectable in PP13 and PLAP colocalization, analyzed in the plasma membrane region, the altered colocalization indices between PP13 and CD71 in preterm preeclampsia and HELLP syndrome reflect a shift in the membrane localization of PP13. Therefore, we also analyzed the spatial proximity between PP13 and PLAP by line scan and 3D surface plot analysis of the images. As the representative histograms and images show (Figure 5), PP13 is relocalized near lipid rafts in the juxtamembrane regions. Collectively, the data indicate redistribution of the PP13 in the syncytiotrophoblast of patients with preterm but not in term preeclampsia and HELLP syndrome, in comparison to the respective gestational age-matched control samples.
Figure 5. PP13 accumulates in the vicinity of PLAP-containing lipid rafts in preeclampsia and HELLP syndrome.

Samples stained for PP13 (green) and PLAP (red) were analyzed with confocal microscopy. Representative A, confocal images, B, 3D surface plots and, C, line scan intensity distributions of samples derived from preterm or term patients and controls.
HELLP, hemolysis elevated liver enzymes and low platelets; PLAP, placental alkaline phosphatase; PP13, placental protein 13.
Balogh, subcellular relocalizaiton of PP13 in preeclampsia and HELLP syndrome. Am J Obstet Gynecol 2011.
Disruption of cytoskeletal actin inhibits PP13-release from BeWo cells
Our confocal data suggested that cortical actin network may have a role in PP13 export from the trophoblast. To investigate this phenomenon in vitro, we treated BeWo trophoblast-like caells with Latrunculin B, a toxin that disrupts actin network by inhibiting actin polymerization. The treatment with Latrunculin B resulted in the disruption of actin filaments in BeWo cells visualized by phalloidin staining of F-actin (Figure 6, A). In parallel, intracellular accumulation and increased colocalization with actin were detected for PP13 in the juxtamembrane region of Latrunculin B-treated cells, in a concentration-dependent manner (Figure 6,B). Consistently, PP13 content of the cell culture supernatants gradually decreased with increasing Latrunculin B concentrations (mean ± SEM in pg/mL; untreated: 137.3 ± 13.5; 0.125 μM: 106.6 ± 9.5, P < .05 compared with untreated). There was no detectable PP13-release from nontransfected cells (Figure 6,C).
Figure 6. Disruption of the actin cytoskeleton results in intracellular accumulation of Pp13 in BeWo cells.
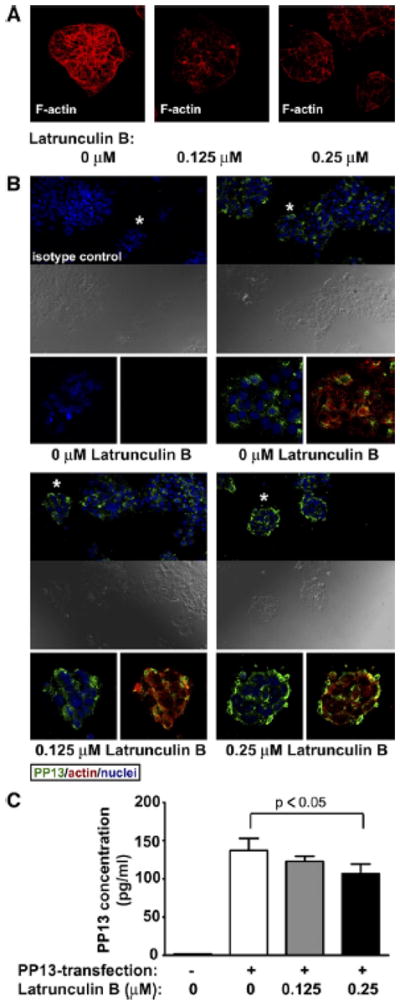
PP13-transfected, untreated or Latrunculin B-treated cells were stained with A, phalloidin or with B, anti-PP13 (green), and antiactin (red), followed by confocal microscopic analysis (40x or 20x magnifications, respectively). Cell nuclei were counterstained with DRAQ5 (Biostatus, Leicenstershire, UK) (blue). Asterisks denote enlarged areas. Yellow color reflects colocalization of PP13 and actin. C, PP13 content of cell culture supernatants of nontransfected controls, as well as PP13-transfected, untreated or Latrunculin B-treated cells were measured by ELISA (data are displayed as mean ± SEM of 3 independent experiments).
ELISA, enzyme-linked immunosorbent assay; PP13, placental protein 13.
Balogh. Subcellular relocalizaiton of PP13 in preeclampsia and HELLP syndrome. Am J Obstet Gynecol 2011.
PP13-release from BeWo cells is facilitated by Ca2+ influx or ischemic stress
Next, we attempted to explore the possible mechanisms promoting PP13-release from the trophoblast under normal and stress conditions. Calcium ionophore treatment led to decreased PP13 immunostaining compared with untreated cells (Figure 7, A and B), indicating that BeWo cells released their PP13 contents when cytoplasmic Ca2+ levels were elevated. From the applied stress conditions, TNFα and hypoxic stress did not reduce PP13 immunostaining; however, ischemic stress did markedly decrease it (Figure 7, C–E).
Figure 7. Calcium-influx or ischemic stress decreases PP13 immunostaining in BeWo cells.
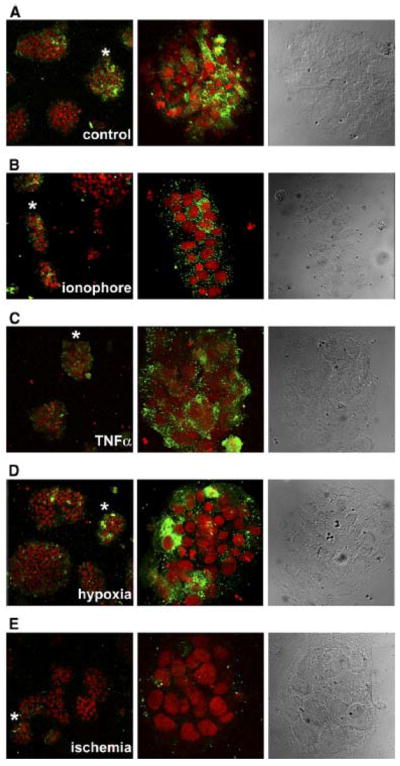
PP13-transfected BeWo cells were treated with either A, calcium ionophore to increase B, intracellular Ca2+ - level, or kept under stress, such as C, TNFα treatment, D, hypoxia, or E, ischemia to mimic placental milieu in preterm preeclampsia. B–E, Treated or A, untreated control cells were stained with anti-PP13 (green), nuclei were counterstained with DRAQ5 (Biostatus, Leicestershire, UK) (red). Representative confocal images of 3 independent experiments are displayed (20x magnifications). Asterisks denote enlarged areas.
PP13, placental protein 13.
Balogh. Subcellular relocalizaiton of PP13 in preeclampsia and HELLP syndrome. Am J Obstet Gynecol 2011.
To quantify PP13-release from BeWo cells under calcium ionophore treatment and ischemic stress, we determined PP13 contents of BeWo cells and their culture supernatants by ELISA. PP13 mRNA expression was negligible in nontransfected cells, but in transfected cells it reached the level previously detected in normal placentas at term50 (Figure 8, A). PP13 content (in pg, normalized to mg total protein; mean ± SEM) was 38.8 ± 2.6 pm/mg protein in nontransfected cells (Figure 8, C), whereas it was 851. ± 66.8 pg/mg protein in PP13-transfected, untreated cells. In transfected cells, ischemic stress or calcium ionophore treatment decreased PP13 content (215.5 ± 23.1 pg/mg protein, P < .001 or 134.1 ± 7.0 pg/mg protein, P < .001m respectively). In accord, PP13 concentration (mean ± SEM) of the supernatants of nontransfected cells was under the detection limit, whereas it was 130.9 ± 10.7 pg/mL in PP13-transfected, untreated cells. In transfected cells, ischemic stress or calcium ionophore treatment increased PP13 concentrations in supernatants (160.0 ± 13.4, P, = .06 or 185.9 ± 6.1, P < .001, respectively) (Figure 8, B).
Figure 8. Calcium-influx or ischemic stress promotes PP13-release from BeWo cells.
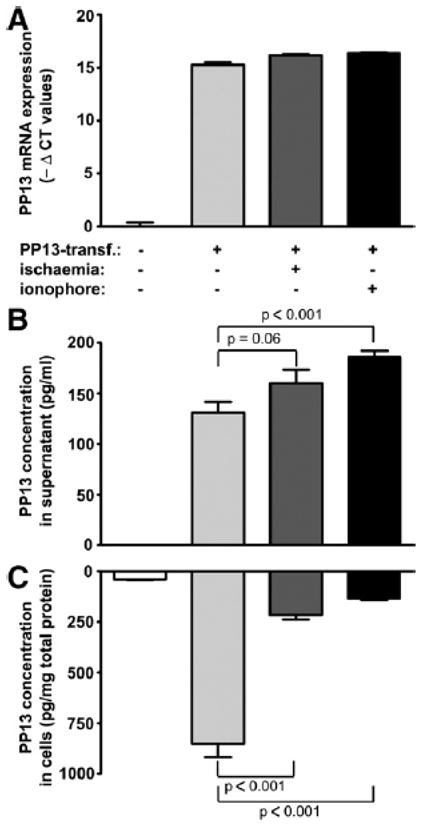
PP13-transfected cells were treated with calcium ionophore (black bars), kept under ischemic stress (dark gray bars), or left untreated (light gray bars). Nontransfected cells were used as negative controls (white bars). A, PP13 mRNA expression or B, PP13 protein concentration in culture supernatants and C, in cells were measured by qRT-PCR or ELISA, respectively. Data are displayed as mean ± SEM of 3 independent experiments.
ELISA, enzyme-linked immunosorbent assay; PP13, placental protein 13; qRT-PCR, quantitative reverse transcribe-polymerase chain reaction.
Balogh. Subcellular relocalizaiton of PP13 in preeclampsia and HELLP syndrome. Am J Obstet Gynecol 2011.
COMMENT
Principal findings of the study: (1) PP13 was localized in the cytoplasm and in the brush border membrane of the syncytiotrophoblast in normal placentas (term and preterm); (2) similarly, PP13 was colocalized with actin in the cortical cytoskeleton of the syncytiotrophoblast, as well as with placental alkaline phosphatase in the lipid raft regions of the syncytiotrophoblast and with CD71 in the nonlipid raft regions of the brush border membranes of this structure; (3) in term and preterm preeclampsia and HELLP syndrome, there was a stronger PP13 staining in the apical plasma membrane rather than in the cytoplasm, indicating a shift in the localization of PP13 in the syncytiotrophoblast; (4) similarly, a higher degree of colocalization of PP13 and actin was found in the apical juxtamembrane area of the syncytiotrophoblast in term and preterm preeclampsia and HELLP syndrome; (5) in contrast, a lower colocalization of PP13 and CD71 in nonlipid raft membrane regions was found in the syncytiotrophoblast; in addition, accumulation of PP13 in the vicinity of PLAP containing lipid rafts was noted in preterm but not in term preeclampsia and HELLP syndrome; (6) the inhibition of actin polymerization in trophoblast-like BeWo cells led to the intracellular accumulation of PP13 along with its increased colocalization with actin in the juxtamembrane region, and to a simultaneous decrease in PP13-release to culture supernatants; (7) Ca2+ influx mediated by calcium ionophore treatment increased “nonclassical” PP13-release from BeWo cells; and (8) ischemic stress resulted in increased PP13-release from BeWo cells. These data support the concept that the different pattern of localization of PP13 in the syncytiotrophoblast is related to distinct placental pathology in preterm and term preeclampsia, and that the cortical actin cytoskeleton, through lipid raft domains, has an active role in the increased secretion and/or shedding of PP13 on ischemic stress in preterm preeclampsia and HELLP syndrome.
Meaning of the study
Preeclampsia and HELLP syndrome are life-threatening pregnancy complications,1–11 previously attributed to defective hemochorial placentation12–18,20,22 placental stress and antiangiogenic state,21–24,29,30,32,33,35 generalized maternal endothelial dysfunction,3–5,38,39 and an excessive maternal systemic inflammatory response.4,25,26,64 Emerging evidence shows that the excessive placental secretion of antiangiogenic 29,30,32,33,35 and proinflammatory molecules,27,28,37 as well as the augmented shedding of microparticles and cellular debris from the syncytiotrophoblast surface could play a key role in triggering the exaggerated maternal systemic inflammatory response in preeclampsia and HELLP syndrome, especially in early-onset disease.31,34,37,39–41 It is noteworthy that patients with preterm (but not term) preeclampsia and HELLP syndrome have a higher concentration of PP13, a placenta-specific innate immune lectin, in the maternal circulation than women with normal pregnancy at the onset of disease.42 We have provided evidence that this change can partly be attributed to an excessive shedding of PP13 from the syncytiotrophoblast into the maternal circulation; however the mechanism responsible for this phenomenon remains to be dertmined.42
Therefore, this study was designed to investigate the changes in the localization of PP13 in the syncytiotrophoblast, focusing on its spatial relationship to the actin cytoskeleton and lipid rafts in the apical brush border membrane. We also aimed to reveal the potential mechanisms mediating PP13-release from the trophoblast in normal conditions and in preeclampsia. Because of (1) placental villous explants may disintegrate and their PP13 production is decreasing after 24–48 hours65,66; (2) primary trophoblasts can only be obtained from a single placenta in less quantity67 than needed for our parallel experiments; and (3) PP13 mRNA/protein expression is very low in trophoblastic cell lines,68 we used PP13 DNA-transfected BeWo cells for our in vitro studies. In these cells, PP13 mRNA expression was up-regulated by approximately 45,000-fold, much higher than in BeWo cells induced to fuse and differentiate (approximately 7-fold).68 reached the physiologic level of PP13 mRNA expression detected in term placentas.50
Actin network and the syncytiotrophoblast secretion/shedding of PP13
The findings reported herein about the cytoplasmic and brush border membrane localization of PP13 in the syncytiotrophoblast in normal term placentas are consistent with those reported with confocal microscopy and immunohistochemistry.42,45,49 The colocalization of PP13 and actin in the cortical cytoskeleton of the syncytiotrophoblast is consistent with data we have previously reported, demonstrating the specific binding of PP13 to β/γ-actin in placental tissue lysates.49 Importantly, PP13 also binds to annexin-II,49 which is a lipid raft-associated protein that interacts with the submembraneous actin network functions as an interface between lipid rafts and the actin cytoskeleton,69,70 and is involved in membrane trafficking events.71 As the colocalization of PP13 with annexin II on the apical membrane of the syncytiotrophoblast has also been reported,49,72 our past and present results suggest that actin, in conjunction with annexin-II, has a potentially important role in the membrane trafficking of PP13 on the apical surface of the syncytiotrophoblast. This concept is strongly supported by out in vitro data showing that the disruption of the actin filamental network leads to the intracellular accumulation and decreased release of PP13 from transfected BeWo cells.
Consistent with our previous observations with immunohistochemistry,42 we found a stronger apical membrane staining and a weaker cytoplasmic staining for PP13 in the syncytiotrophoblast in term and preterm preeclampsia and HELLP syndrome. This shift in the localization of PP13 in the syncytiotrophoblast in these cases was also observed as a higher colocalization of PP13 with the cortical actin cytoskeleton. As PP13 is released from the syncytiotrophoblast into the maternal circulation in significantly larger amounts in preterm but not in term preeclampsia and HELLP syndrome when compared with gestational age-matched controls,42 the shift in the cellular localization of PP13 itself is not sufficient to explain the increased apical membrane trafficking of this galectin in preterm cases. Therefore, we asked whether the altered membrane trafficking of PP13 in preterm preeclampsia and HELLP syndrome is related to lipid raft domains, which exist in epithelial brush borders70 and control and apical trafficking of some galectins and their glycoconjugates.73
The relationship of PP13 and lipid rafts in the syncytiotrophoblast apical membrane
Lipid rafts are highly dynamic, cholesterol-, and sphingolipid-rich micro-domains, which function in apical trafficking through interacting with the actin cytoskeleton.74,75 Of interest, galectin-4, an intestine-specific member of the galectin family, is a stabilizer of lipid rafts in the intestinal microvillar membranes through its interactions with lipid-raft associated proteins, such as alkaline phosphatase.76,77 The apical membrane of the syncytiotrophoblast is also rich in lipid rafts and raft-associated molecules, such as annexin-II, gangliosides, and the GPI-anchored PLAP.58 In addition to our previous data on the colocalization of PP13 with annexin II,49 in this study, we report a high colocalization of PP13 with PLAP and CD71 in the syncytiotrophoblast in control placentas, suggesting an equal distribution of PP13 in lipid raft and nonlipid raft regions of the syncytiotrophoblast brush border membrane in these cases.
Consistent with previous reports on the differences in placental pathology and global gene expression15,17,78 as well as in placental LGALS13 expression and PP13 immunostaining in preterm versus term preeclampsia,42,79 we observed a lower colocalization of PP13 with CD71 and the accumulation of PP13 in the vicinity of PLAP containing lipid rafts in preterm but not in term preeclampsia and HELLP syndrome. These findings are also consistent with reports showing similar changes in placental pathology and global transcriptome in preterm preeclampsia and HELLP syndrome.16,42,80 These data suggest that the apical, nonlipid raft localization of PP13 in the syncytiotrophoblast brush border is reduced, and the association of PP13 with the juxtamembrane cortical actin network and lipid raft domains is enhanced in preterm preeclampsia and HELLP syndrome by yet unknown mechanism, possibly facilitating the secretion and/or shedding of PP13 through lipid rafts and leading to the elevated PP13 concentrations in the maternal circulation.
Preeclampsia, galectins and “danger signals”
Because PP13 lacks a signal peptide required for the classical protein secretion pathway, it was hypothesized42,49 that PP13, similar to other galectins,55,56,81,82 is transported to the syncytiotrophoblast apical membrane and into the extracellular milieu through “nonclassical” secretory mechanism (e.g. via exovesicles or by the extrusion of membrane blebs), which circumvents the endoplasmic reticulum and the Golgi apparatus. Calcium ionophores induce the formation and membrane shedding of microvesicles, which contain actin, annexin II, and galectins, enhancing their “nonclassical” membrane transport.54 In this study, we tested the ionophore-effect on PP13-release from BeWo cells. Indeed, exposing BeWo cells to calcium ionophore treatment in vitro resulted in a marked release of PP13, suggesting microvesicle-mediated secretion of PP13 from the juxtamembrane region, where these vesicles are formed.54 These results are consistent with observations on the first trimester villous explant cultures in which calcium treatment increased PP13-release.66 Furthermore, our observations on clinical specimens suggest that PP13 uses carbohydrate structure sin lipid raft domains similar to other galectins,73,83–85 rather than CD71-marked compartments, for this apical membrane trafficking.
Our earlier42 and current clinical data indicate that there is an increased membrane trafficking of PP13 in preterm preeclampsia and HELLP syndrome, leading to increased PP13 concentrations in maternal circulation at the onset of clinical symptoms. This phenomenon could be mimicked in BeWo cells by ischemic stress, which is in accord with increasing evidence showing that not hypoxia but oxidative stress results in an increased trophoblastic release of antiangiogenic factors, proinflammatory debris and microparticles, triggering the exaggerated activation of the maternal immune system in preterm preeclampsia.3–5,21–39
Of importance, galectins can be increasingly secreted from inflamed tissues or after cellular stress and/or damage, and they have been implicated to act as “cell-stress sensors,”86,87 “danger signals,” or “alarmins.”88–91 “Alarmins” are endogenous danger signals secreted via “nonclassical” pathways by activated cells or passively released by dying cells undergoing necrosis. “Alarmins” signal tissue/cell damage to the immune system, eliciting effector responses from innate and adaptive immune cells, and contributing to the activation and resolution of immune responses.88–93 Indeed, because of their cytokine-like properties, galectins are powerful regulators of both arms of the immune system.57,82
“Danger signals” at the maternal-fetal interface have recently been proposed to create an abnormal placental cytokine milieu and link the activation of the innate immune system in preeclampsia, where there is considerable stress and damage to the interstitial trophoblast and syncytiotrophoblast.23,37,94–98 Because PP13 has proinflammatory properties on macrophages99 and can induce T cell death in vitro,50 we propose that PP13 may function as an endogenous danger signal or alarmin of the syncytiotrophoblast, and its increased release from the aponecrotic trophoblast upon ischemic stress may contribute to the exaggerated activation of the maternal immune system in preterm preeclampsia and HELLP syndrome.
Acknowledgments
We are grateful to Dr. Sándor Paku, Dr. Tibor Füle, and Nicolett Klucsik for their help with placental cryosections preparations (Semmelweis Univesrity), to Rona Wang, Lorri McLuckie, Hong Meng, and Sivasakthy Sivalogan for technical assistance with ELISA and cell transfection, to Adi Tarca for statistical advices, and to Sara Tipton for critical reading of the manuscript (Wayne State University).
This research was funded in part by grants from the European Union FP6 (Pregenesys/037244 to HM and NGT), Hungarian National Science Fund (OTKA CK 80935 to JM and AB) and by TÁMOP 4.2.1./B-09/1/KMR-2010-0003 grant supported partly by EU Social Fund (to JM and AB).
Footnotes
Author contributions: AB, JM, NGT designed the study; TV, MS, JR, HM, RR, ZP, NGT contributed with materials or clinical specimens; AB, JP, NGT performed research; AB, JP, JM, ZD, NGT analyzed and interpreted data; AB, JM, CJK, HM, RR, ZP, NGT wrote the paper.
References
- 1.Di Renzo GC. The great obstetrical syndromes. J Matern Fetal Neonatal Med. 2009;22:633–5. doi: 10.1080/14767050902866804. [DOI] [PubMed] [Google Scholar]
- 2.Romero R. Prenatal medicine: the child is the father of the man. J Matern Fetal Neonatal Med. 2009;22:636–9. doi: 10.1080/14767050902784171. [DOI] [PubMed] [Google Scholar]
- 3.Roberts JM, Lain KY. Recent Insights into the pathogenesis of pre-eclampsia. Placenta. 2002;23:359–72. doi: 10.1053/plac.2002.0819. [DOI] [PubMed] [Google Scholar]
- 4.Redman CW, Sargent IL. Latest advances in understanding preeclampsia. Science. 2005;308:1592–4. doi: 10.1126/science.1111726. [DOI] [PubMed] [Google Scholar]
- 5.Sibai B, Dekker G, Kupferminc M. Pre-eclampsia. Lancet. 2005;365:785–99. doi: 10.1016/S0140-6736(05)17987-2. [DOI] [PubMed] [Google Scholar]
- 6.Weinstein L. Syndrome of hemolysis, elevated liver enzymes, and low platelet count: a severe consequence of hypertension in pregnancy. AmJ Obstet Gynecol. 1982;142:159–67. doi: 10.1016/s0002-9378(16)32330-4. [DOI] [PubMed] [Google Scholar]
- 7.Romero R, Vizoso J, Emamian M, et al. Clinical significance of liver dysfunction in pregnancy- induced hypertension. Am J Perinatol. 1988;5:146–51. doi: 10.1055/s-2007-999675. [DOI] [PubMed] [Google Scholar]
- 8.Romero R, Mazor M, Lockwood CJ, et al. Clinical significance, prevalence, and natural history of thrombocytopenia in pregnancy-induced hypertension. Am J Perinatol. 1989;6:32–8. doi: 10.1055/s-2007-999540. [DOI] [PubMed] [Google Scholar]
- 9.Rath W, Faridi A, Dudenhausen JW. HELLP syndrome. J Perinat Med. 2000;28:249–60. doi: 10.1515/JPM.2000.033. [DOI] [PubMed] [Google Scholar]
- 10.Barton JR, Sibai BM. Diagnosis and management of hemolysis, elevated liver enzymes, and low platelets syndrome. Clin Perinatol. 2004;31:807–33. vii. doi: 10.1016/j.clp.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 11.Haram K, Svendsen E, Abildgaard U. The HELLP syndrome: clinical issues and management: a review. BMC Pregnancy Childbirth. 2009;9:8. doi: 10.1186/1471-2393-9-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khong TY, De Wolf F, Robertson WB, Brosens I. Inadequate maternal vascular response to placentation in pregnancies complicated by pre-eclampsia and by small-for-gestational age infants. BJOG. 1986;93:1049–59. doi: 10.1111/j.1471-0528.1986.tb07830.x. [DOI] [PubMed] [Google Scholar]
- 13.Kliman HJ. Uteroplacental blood flow: the story of decidualization, menstruation, and trophoblast invasion. Am J Pathol. 2000;157:1759–68. doi: 10.1016/S0002-9440(10)64813-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lyall F. The human placental bed revisited. Placenta. 2002;23:555–62. doi: 10.1053/plac.2002.0850. [DOI] [PubMed] [Google Scholar]
- 15.Moldenhauer JS, Stanek J, Warshak C, Khoury J, Sibai B. The frequency and severity of placental findings in women with preeclampsia are gestational age dependent. Am J Obstet Gynecol. 2003;189:1173–7. doi: 10.1067/s0002-9378(03)00576-3. [DOI] [PubMed] [Google Scholar]
- 16.Smulian J, Shen-Schwarz S, Scorza W, Kinzler W, Vintzileos A. A clinicohistopathologic comparison between HELLP syndrome and severe preeclampsia. J Matern Fetal Neonatal Med. 2004;16:287–93. doi: 10.1080/14767050400018015. [DOI] [PubMed] [Google Scholar]
- 17.Sebire NJ, Goldin RD, Regan L. Term preeclampsia is associated with minimal histopathological placental features regardless of clinical severity. J Obstet Gynaecol. 2005;25:117–8. doi: 10.1080/014436105400041396. [DOI] [PubMed] [Google Scholar]
- 18.Espinoza J, Romero R, Kim YM, et al. Normal and abnormal transformation of the spiral arteries during pregnancy. J Perinat Med. 2006;34:447–58. doi: 10.1515/JPM.2006.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.von Dadelszen P, Menzies JM, Payne B, Magee LA. Predicting adverse outcomes in women with severe pre-eclampsia. Semin Perinatol. 2009;33:152–7. doi: 10.1053/j.semperi.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 20.Brosens I, Pijnenborg R, Vercruysse L, Romero R. The ”Great Obstetrical Syndromes” are associated with disorders of deep placentation. Am J Obstet Gynecol. 2010;204:193–201. doi: 10.1016/j.ajog.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jauniaux E, Poston L, Burton GJ. Placental related diseases of pregnancy: involvement of oxidative stress and implications in human evolution. Hum Reprod Update. 2006;12:747–55. doi: 10.1093/humupd/dml016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burton GJ, Woods AW, Jauniaux E, Kingdom JC. Rheological and physiological consequences of conversion of the maternal spiral arteries for uteroplacental blood flow during human pregnancy. Placenta. 2009;30:473–82. doi: 10.1016/j.placenta.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burton GJ, Yung HW, Cindrova-Davies T, Charnock-Jones DS. Placental endoplasmic reticulum stress and oxidative stress in the pathophysiology of unexplained intrauterine growth restriction and early onset preeclampsia. Placenta. 2009;30 (Suppl A):S43–8. doi: 10.1016/j.placenta.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burton GJ. Oxygen, the Janus gas: its effects on human placental development and function. J Anat. 2009;215:27–35. doi: 10.1111/j.1469-7580.2008.00978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sacks GP, Studena K, Sargent K, Redman CW. Normal pregnancy and preeclampsia both produce inflammatory changes in peripheral blood leukocytes akin to those of sepsis. Am J Obstet Gynecol. 1998;179:80–6. doi: 10.1016/s0002-9378(98)70254-6. [DOI] [PubMed] [Google Scholar]
- 26.Gervasi MT, Chaiworapongsa T, Pacora P, et al. Phenotypic and metabolic characteristics of monocytes and granulocytes in preeclampsia. Am J Obstet Gynecol. 2001;185:792–7. doi: 10.1067/mob.2001.117311. [DOI] [PubMed] [Google Scholar]
- 27.Matthiesen L, Berg G, Ernerudh J, Ekerfelt C, Jonsson Y, Sharma S. Immunology of preeclampsia. Chem Immunol Allergy. 2005;89:49–61. doi: 10.1159/000087912. [DOI] [PubMed] [Google Scholar]
- 28.Jonsson Y, Ruber M, Matthiesen L, et al. Cytokine mapping of sera from women with preeclampsia and normal pregnancies. J Reprod Immunol. 2006;70:83–91. doi: 10.1016/j.jri.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 29.Levine RJ, Lam C, Qian C, et al. Soluble endoglin and other circulating antiangiogenic factors in preeclampsia. N Engl J Med. 2006;355:992–1005. doi: 10.1056/NEJMoa055352. [DOI] [PubMed] [Google Scholar]
- 30.Baumwell S, Karumanchi SA. Pre-eclampsia: clinical manifestations and molecular mechanisms. Nephron Clin Pract. 2007;106:c72–81. doi: 10.1159/000101801. [DOI] [PubMed] [Google Scholar]
- 31.Germain SJ, Sacks GP, Sooranna SR, Sargent IL, Redman CW. Systemic inflammatory priming in normal pregnancy and preeclampsia: the role of circulating syncytiotrophoblast microparticles. J Immunol. 2007;178:5949–56. doi: 10.4049/jimmunol.178.9.5949. [DOI] [PubMed] [Google Scholar]
- 32.Erez O, Romero R, Espinoza J, et al. The change in concentrations of angiogenic and anti- angiogenic factors in maternal plasma between the first and second trimesters in risk assessment for the subsequent development of preeclampsia and small-for-gestational age. J Matern Fetal Neonatal Med. 2008;21:279–87. doi: 10.1080/14767050802034545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maynard S, Epstein FH, Karumanchi SA. Preeclampsia and angiogenic imbalance. Annu Rev Med. 2008;59:61–78. doi: 10.1146/annurev.med.59.110106.214058. [DOI] [PubMed] [Google Scholar]
- 34.Redman CW, Sargent IL. Circulating microparticles in normal pregnancy and pre-eclampsia. Placenta. 2008;29 (Suppl A):S73–7. doi: 10.1016/j.placenta.2007.11.016. [DOI] [PubMed] [Google Scholar]
- 35.Romero R, Nien JK, Espinoza J, et al. A longitudinal study of angiogenic (placental growth factor) and anti-angiogenic (soluble endoglin and soluble vascular endothelial growth factor receptor-1) factors in normal pregnancy and patients destined to develop preeclampsia and deliver a small for gestational age neonate. J Matern Fetal Neonatal Med. 2008;21:9–23. doi: 10.1080/14767050701830480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Webster RP, Roberts VH, Myatt L. Protein nitration in placenta—functional significance. Placenta. 2008;29:985–94. doi: 10.1016/j.placenta.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Redman CW, Sargent IL. Placental stress and pre-eclampsia: a revised view. Placenta. 2009;30 (Suppl A):S38–42. doi: 10.1016/j.placenta.2008.11.021. [DOI] [PubMed] [Google Scholar]
- 38.Roberts JM, Hubel CA. The two stage model of preeclampsia: variations on the theme. Placenta. 2009;30 (Suppl A):S32–7. doi: 10.1016/j.placenta.2008.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cindrova-Davies T, Sanders DA, Burton GJ, Charnock-Jones DS. Soluble FLT1 sensitizes endothelial cells to inflammatory cytokines by antagonizing VEGF receptor-mediated signalling. Cardiovasc Res. 2010;89:671–9. doi: 10.1093/cvr/cvq346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aly AS, Khandelwal M, Zhao J, Mehmet AH, Sammel MD, Parry S. Neutrophils are stimulated by syncytiotrophoblast microvillous membranes to generate superoxide radicals in women with preeclampsia. Am J Obstet Gynecol. 2004;190:252–8. doi: 10.1016/j.ajog.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 41.Goswami D, Tannetta DS, Magee LA, et al. Excess syncytiotrophoblast microparticle shedding is a feature of early-onset pre-eclampsia, but not normotensive intrauterine growth restriction. Placenta. 2006;27:56–61. doi: 10.1016/j.placenta.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 42.Than NG, Abdul Rahman O, Magenheim R, et al. Placental protein 13 (galectin-13) has decreased placental expression but increased shedding and maternal serum concentrations in patients presenting with preterm pre-eclampsia and HELLP syndrome. Virchows Arch. 2008;453:387–400. doi: 10.1007/s00428-008-0658-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Burger O, Pick E, Zwickel J, et al. Placental protein 13 (PP-13): effects on cultured trophoblasts, and its detection in human body fluids in normal and pathological pregnancies. Placenta. 2004;25:608–22. doi: 10.1016/j.placenta.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 44.Gonen R, Shahar R, Grimpel YI, et al. Placental protein 13 as an early marker for preeclampsia: a prospective longitudinal study. BJOG. 2008;115:1465–72. doi: 10.1111/j.1471-0528.2008.01902.x. [DOI] [PubMed] [Google Scholar]
- 45.Huppertz B, Sammar M, Chefetz I, Neumaier-Wagner P, Bartz C, Meiri H. Longitudinal determination of serum placental protein 13 during development of preeclampsia. Fetal Diagn Ther. 2008;24:230–6. doi: 10.1159/000151344. [DOI] [PubMed] [Google Scholar]
- 46.Bohn H, Kraus W, Winckler W. Purification and characterization of two new soluble placental tissue proteins (PP13 and PP17) Oncodev Biol Med. 1983;4:343–50. [PubMed] [Google Scholar]
- 47.Than GN, Bohn H, Szabo D. Advances in pregnancy-related protein research. Boca Raton, FL: CRC Press; 1993. [Google Scholar]
- 48.Than NG, Sumegi B, Than GN, Berente Z, Bohn H. Isolation and sequence analysis of a cDNA encoding human placental tissue protein 13 (PP13), a new lysophospholipase, homologue of human eosinophil Charcot-Leyden Crystal protein. Placenta. 1999;20:703–10. doi: 10.1053/plac.1999.0436. [DOI] [PubMed] [Google Scholar]
- 49.Than NG, Pick E, Bellyei S, et al. Functional analyses of placental protein 13/galectin-13. Eur J Biochem. 2004;271:1065–78. doi: 10.1111/j.1432-1033.2004.04004.x. [DOI] [PubMed] [Google Scholar]
- 50.Than NG, Romero R, Goodman M, et al. A primate subfamily of galectins expressed at the maternal-fetal interface that promote immune cell death. Proc Natl Acad Sci U S A. 2009;106:9731–6. doi: 10.1073/pnas.0903568106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barondes SH, Cooper DN, Gitt MA, Leffler H. Galectins: structure and function of a large family of animal lectins. J Biol Chem. 1994;269:20807–10. [PubMed] [Google Scholar]
- 52.Than NG, Itakura A, Rao ChV, et al. Clinical applications of pregnancy-related proteins–a workshop report. Placenta. 2003;24(Suppl A):S60–4. doi: 10.1053/plac.2002.0947. [DOI] [PubMed] [Google Scholar]
- 53.Visegrady B, Than NG, Kilar F, Sumegi B, Than GN, Bohn H. Homology modelling and molecular dynamics studies of human placental tissue protein 13 (galectin-13) Protein Eng. 2001;14:875–80. doi: 10.1093/protein/14.11.875. [DOI] [PubMed] [Google Scholar]
- 54.Hughes RC. Secretion of the galectin family of mammalian carbohydrate-binding proteins. Biochim Biophys Acta. 1999;1473:172–85. doi: 10.1016/s0304-4165(99)00177-4. [DOI] [PubMed] [Google Scholar]
- 55.Nickel W. The mystery of nonclassical protein secretion: a current view on cargo proteins and potential export routes. Eur J Biochem. 2003;270:2109–19. doi: 10.1046/j.1432-1033.2003.03577.x. [DOI] [PubMed] [Google Scholar]
- 56.Nickel W. Unconventional secretory routes: direct protein export across the plasma membrane of mammalian cells. Traffic. 2005;6:607–14. doi: 10.1111/j.1600-0854.2005.00302.x. [DOI] [PubMed] [Google Scholar]
- 57.Rabinovich GA, Baum LG, Tinari N, et al. Galectins and their ligands: amplifiers, silencers or tuners of the inflammatory response? Trends Immunol. 2002;23:313–20. doi: 10.1016/s1471-4906(02)02232-9. [DOI] [PubMed] [Google Scholar]
- 58.Godoy V, Riquelme G. Distinct lipid rafts in subdomains from human placental apical syncytiotrophoblast membranes. J Membr Biol. 2008;224:21–31. doi: 10.1007/s00232-008-9125-5. [DOI] [PubMed] [Google Scholar]
- 59.Pike LJ. Rafts defined: a report on the Keystone Symposium on Lipid Rafts and Cell Function. J Lipid Res. 2006;47:1597–8. doi: 10.1194/jlr.E600002-JLR200. [DOI] [PubMed] [Google Scholar]
- 60.Papp C, Szabo G, Toth-Pal E, Papp Z. Fetal growth rate and its variations 1988/89. Orv Hetil. 1991;132:1865–6. 1869–70. [PubMed] [Google Scholar]
- 61.ACOG practice bulletin. Diagnosis and management of preeclampsia and eclampsia. Number 33, January 2002. Obstet Gynecol. 2002;99:159–67. doi: 10.1016/s0029-7844(01)01747-1. [DOI] [PubMed] [Google Scholar]
- 62.Adler J, Parmryd I. Quantifying colocalization by correlation: the Pearson correlation coefficient is superior to the Mander’s overlap coefficient. Cytometry A. 2010;77:733–42. doi: 10.1002/cyto.a.20896. [DOI] [PubMed] [Google Scholar]
- 63.Bolte S, Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224:213–32. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- 64.Redman CW, Sargent IL. Pre-eclampsia, the placenta and the maternal systemic inflammatory response—a review. Placenta. 2003;24 (Suppl A):S21–7. doi: 10.1053/plac.2002.0930. [DOI] [PubMed] [Google Scholar]
- 65.Crocker IP, Tansinda DM, Baker PN. Altered cell kinetics in cultured placental villous explants in pregnancies complicated by pre-eclampsia and intrauterine growth restriction. J Pathol. 2004;204:11–8. doi: 10.1002/path.1610. [DOI] [PubMed] [Google Scholar]
- 66.Grimpel YI, Kivity V, Cohen A, et al. Effects of calcium, magnesium, low-dose aspirin and low-molecular-weight heparin on the release of PP13 from placental explants. Placenta. 2011;32(Suppl):S55–64. doi: 10.1016/j.placenta.2010.11.019. [DOI] [PubMed] [Google Scholar]
- 67.Kliman HJ, Nestler JE, Sermasi E, Sanger JM, Strauss JF., 3rd Purification, characterization, and in vitro differentiation of cytotrophoblasts from human term placentae. Endocrinology. 1986;118:1567–82. doi: 10.1210/endo-118-4-1567. [DOI] [PubMed] [Google Scholar]
- 68.Orendi K, Gauster M, Moser G, Meiri H, Huppertz B. Effects of vitamins C and E, acetylsalicylic acid and heparin on fusion, betahCG and PP13 expression in BeWo cells. Placenta. 2010;31:431–8. doi: 10.1016/j.placenta.2010.02.017. [DOI] [PubMed] [Google Scholar]
- 69.Thiel C, Osborn M, Gerke V. The tight association of the tyrosine kinase substrate annexin II with the submembranous cytoskeleton depends on intact p11- and Ca(2_)-binding sites. J Cell Sci. 1992;103(Pt 3):733–42. doi: 10.1242/jcs.103.3.733. [DOI] [PubMed] [Google Scholar]
- 70.Danielsen EM, Hansen GH. Lipid raft organization and function in brush borders of epithelial cells. Mol Membr Biol. 2006;23:71–9. doi: 10.1080/09687860500445604. [DOI] [PubMed] [Google Scholar]
- 71.Danielsen EM, Hansen GH. Lipid rafts in epithelial brush borders: atypical membrane microdomains with specialized functions. Biochim Biophys Acta. 2003;1617:1–9. doi: 10.1016/j.bbamem.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 72.Aarli A, Kristoffersen EK, Jensen TS, Ulvestad E, Matre R. Suppressive effect on lymphoproliferation in vitro by soluble annexin II released from isolated placental membranes. Am J Reprod Immunol. 1997;38:313–9. doi: 10.1111/j.1600-0897.1997.tb00306.x. [DOI] [PubMed] [Google Scholar]
- 73.Delacour D, Koch A, Jacob R. The role of galectins in protein trafficking. Traffic. 2009;10:1405–13. doi: 10.1111/j.1600-0854.2009.00960.x. [DOI] [PubMed] [Google Scholar]
- 74.Simons K, van Meer G. Lipid sorting in epithelial cells. Biochemistry. 1988;27:6197–202. doi: 10.1021/bi00417a001. [DOI] [PubMed] [Google Scholar]
- 75.Ikonen E. Roles of lipid rafts in membrane transport. Curr Opin Cell Biol. 2001;13:470–7. doi: 10.1016/s0955-0674(00)00238-6. [DOI] [PubMed] [Google Scholar]
- 76.Braccia A, Villani M, Immerdal L, et al. Microvillar membrane microdomains exist at physiological temperature. Role of galectin-4 as lipid raft stabilizer revealed by ”superrafts”. J Biol Chem. 2003;278:15679–84. doi: 10.1074/jbc.M211228200. [DOI] [PubMed] [Google Scholar]
- 77.Lajoie P, Goetz JG, Dennis JW, Nabi IR. Lattices, rafts, and scaffolds: domain regulation of receptor signaling at the plasma membrane. J Cell Biol. 2009;185:381–5. doi: 10.1083/jcb.200811059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nishizawa H, Pryor-Koishi K, Kato T, Kowa H, Kurahashi H, Udagawa Y. Microarray analysis of differentially expressed fetal genes in placental tissue derived from early and late onset severe pre-eclampsia. Placenta. 2007;28:487–97. doi: 10.1016/j.placenta.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 79.Sekizawa A, Purwosunu Y, Yoshimura S, et al. PP13 mRNA expression in trophoblasts from preeclamptic placentas. Reprod Sci. 2009;16:408–13. doi: 10.1177/1933719108328615. [DOI] [PubMed] [Google Scholar]
- 80.Varkonyi T, Nagy B, Fule T, et al. Microarray profiling reveals that placental transcriptomes of early-onset HELLP syndrome and preeclampsia are similar. Placenta. 2010;32(Suppl):S21–9. doi: 10.1016/j.placenta.2010.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liu FT, Rabinovich GA. Galectins as modulators of tumour progression. Nat Rev Cancer. 2005;5:29–41. doi: 10.1038/nrc1527. [DOI] [PubMed] [Google Scholar]
- 82.Rabinovich GA, Toscano MA, Jackson SS, Vasta GR. Functions of cell surface galectinglycoprotein lattices. Curr Opin Struct Biol. 2007;17:513–20. doi: 10.1016/j.sbi.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stillman BN, Hsu DK, Pang M, et al. Galectin- 3 and galectin-1 bind distinct cell surface glycoprotein receptors to induce T cell death. J Immunol. 2006;176:778–89. doi: 10.4049/jimmunol.176.2.778. [DOI] [PubMed] [Google Scholar]
- 84.Carlsson S, Carlsson MC, Leffler H. Intracellular sorting of galectin-8 based on carbohydrate fine specificity. Glycobiology. 2007;17:906–12. doi: 10.1093/glycob/cwm059. [DOI] [PubMed] [Google Scholar]
- 85.Fajka-Boja R, Blasko A, Kovacs-Solyom F, Szebeni GJ, Toth GK, Monostori E. Co-localization of galectin-1 with GM1 ganglioside in the course of its clathrin- and raft-dependent endocytosis. Cell Mol Life Sci. 2008;65:2586–93. doi: 10.1007/s00018-008-8143-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Woolley JF, Al-Rubeai M. The isolation and identification of a secreted biomarker associated with cell stress in serum-free CHO cell culture. Biotechnol Bioeng. 2009;104:590–600. doi: 10.1002/bit.22408. [DOI] [PubMed] [Google Scholar]
- 87.Iwamoto M, Taguchi C, Sasaguri K, et al. The Galectin-1 level in serum as a novel marker for stress. Glycoconj J. 2010;27:419–25. doi: 10.1007/s10719-010-9288-z. [DOI] [PubMed] [Google Scholar]
- 88.Sato S, St-Pierre C, Bhaumik P, Nieminen J. Galectins in innate immunity: dual functions of host soluble beta-galactoside-binding lectins as damage-associated molecular patterns (DAMPs) and as receptors for pathogen-associated molecular patterns (PAMPs) Immunol Rev. 2009;230:172–87. doi: 10.1111/j.1600-065X.2009.00790.x. [DOI] [PubMed] [Google Scholar]
- 89.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 90.Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol. 2008;8:279–89. doi: 10.1038/nri2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sato S, Nieminen J. Seeing strangers or announcing ”danger”: galectin-3 in two models of innate immunity. Glycoconj J. 2004;19:583–91. doi: 10.1023/B:GLYC.0000014089.17121.cc. [DOI] [PubMed] [Google Scholar]
- 92.Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- 93.Matzinger P. Friendly and dangerous signals: is the tissue in control? Nat Immunol. 2007;8:11–3. doi: 10.1038/ni0107-11. [DOI] [PubMed] [Google Scholar]
- 94.Jones CJ, Fox H. An ultrastructural and ultrahistochemical study of the human placenta in maternal pre-eclampsia. Placenta. 1980;1:61–76. doi: 10.1016/s0143-4004(80)80016-6. [DOI] [PubMed] [Google Scholar]
- 95.de Luca Brunori I, Battini L, Brunori E, et al. Placental barrier breakage in preeclampsia: ultrastructural evidence. Eur J Obstet Gynecol Reprod Biol. 2005;118:182–9. doi: 10.1016/j.ejogrb.2004.04.024. [DOI] [PubMed] [Google Scholar]
- 96.Kim YM, Romero R, Oh SY, et al. Toll-like receptor 4: a potential link between ”danger signals,” the innate immune system, and preeclampsia? Am J Obstet Gynecol. 2005;193:921–7. doi: 10.1016/j.ajog.2005.07.076. [DOI] [PubMed] [Google Scholar]
- 97.Bonney EA. Preeclampsia: a view through the danger model. J Reprod Immunol. 2007;76:68–74. doi: 10.1016/j.jri.2007.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Than NG, Erez O, Wildman DE, et al. Severe preeclampsia is characterized by increased placental expression of galectin-1. J Matern Fetal Neonatal Med. 2008;21:429–42. doi: 10.1080/14767050802041961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kliman HJ, Sammar S, Lynch S, et al. PP13 (galectin-13) mediated zones of necrosis diverts the maternal immune response away from the decidual spiral arterioles (abstract) Hypertension in Pregnancy. 2008;27:578. [Google Scholar]