

Abstract
Type VI Secretion Systems (T6SS) are bacterial virulence-associated nanomachines composed of proteins that are evolutionarily related to components of bacteriophage tails. Here we show that protein secretion by the T6SS of Vibrio cholerae requires the action of a dynamic intracellular tubular structure that is structurally and functionally homologous to contractile phage tail sheath. Time-lapse fluorescence light microscopy revealed that T6SS sheaths cycle between assembly, quick contraction, disassembly and re-assembly. Whole-cell electron cryotomography further showed that T6SS sheaths appear as long tubular structures in either extended or contracted conformations that are connected to the inner membrane by a distinct basal structure. These data support a model in which the contraction of the T6SS sheath provides the energy needed to translocate proteins out of effector cells and into adjacent target cells.
Secretion systems allow bacteria to transport macromolecules such as proteins out of effector cells or into either target host cells during pathogenesis or target bacterial cells during competition in various ecological settings. The type 6 secretion systems (T6SS) are encoded by a cluster of 15–20 genes that is present in at least one copy in about 25% of all sequenced Gram-negative bacteria. Although linked to virulence during host infection, recent studies showed that T6SS of Pseudomonas, Burkholderia and Vibrio species can kill bacterial as well as eukaryotic target host cells 1–7. T6SS+ bacterial effector cells are thought to kill target cells through delivery of toxic effector proteins in a cell-cell contact-dependent process 1–3,8. Little is known, however, about how T6SS transport toxic proteins through their own cell membranes or across target cell membranes.
Bioinformatic 9 and structural analyses 10,11 have indicated that some T6SS components are structural homologs of components present in contractile phage tails. For example, secreted VgrG proteins are structural homologs of the T4 phage needle or spike complex 9,10 and secreted Hcp is a structural homolog of a phage tail tube protein 10,11. Another highly conserved T6SS gene product is predicted to be a homolog of gp25, a major component of the T4 phage tail base plate 10,12,13. Two T6SS gene products of V. cholerae, VipA and VipB, form tubular structures in E. coli that can be depolymerized by another T6SS gene product, ClpV 14–16. Leiman et al. 10 noted that VipA/VipB tubules visually resemble T4 contracted tail sheath and were the first to propose that a sheath-like structure might power T6SS translocation by a phage tail-like contraction mechanism. Here we show that T6SS-dependent secretion of Hcp and killing of E. coli by V. cholerae correlates with the activity of a dynamic intracellular structure that indeed appears structurally and functionally related to contractile phage tail sheath.
Fluorescence microscopic imaging of the T6SS
To test the hypothesis that the T6SS apparatus is a dynamic structure we constructed a C-terminal fusion of VipA protein with superfolder GFP (sfGFP) 17. As shown in Supplementary Fig. S1a, VipA-sfGFP complements a chromosomal in-frame deletion of vipA for Hcp secretion when the fusion protein is expressed from pBAD24 plasmid to the same level as the wild type allele. Visualization of cells expressing the functional sfGFP fusion protein by fluorescence microscopy revealed that the VipA-sfGFP fusion is associated with long straight structures in the cytosol that spread throughout the width and length of the cell. The number of visible structures in a single cell varied from 0 to 5 in WT background cells (Supplementary Fig. 2a). Critically, these structures were not visible in vipB mutant cells (Supplementary Fig. 2b), suggesting that the fluorescent structures could be the T6SS sheath structures hypothesized to form in part by interaction of VipA with VipB 10. Because VipA is not secreted and resides within cellular fractions (Supplementary Fig. S3) and the expression level of VipA-sfGFP was comparable to VipA under the conditions used to visualize these sheath structures (Supplementary Fig. S4), we conclude that the fluorescent structures were within the cytosol of imaged cells.
Time-lapse imaging revealed these putative sheath structures to be highly dynamic. As shown in Fig. 1a and Supplementary Videos 1 and 2, the VipA-sfGFP labelled sheaths extended within 10’s of seconds in different subcellular locations and then contracted and disassembled also within 10’s of seconds. Most of the extended sheath structures visible in cells stretched from one lateral side of the cell to the other, perpendicular to the membrane, and thus had lengths approximately equal to the width of the cell (about 0.75 to 1 μm). As shown in Fig. 1a, these sheaths assembled at speeds of ~20–30s per μm. Contraction was very fast, occurring in ~5 milliseconds or less (unresolvable at frame rates of ~200 frames per second, see Fig. 1b–c, Supplementary Fig. S5 and Supplementary Video 3). Sheaths contracted to about 50% of their extended length (Supplementary Fig. S6), and then disassembled over the next 30–60s (Fig. 1a). The disassembly of the contracted sheath is most likely a ClpV-dependent event because ClpV is known to disassemble VipA/VipB tubules in vitro in the presence of ATP 14 and clpV mutants do not disassemble VipA-sfGFP-labeled contracted sheaths (Supplementary Fig. S2d, Supplementary Video 8). A similar number of VipA-sfGFP-labeled sheaths were seen in strains 2740–80 and V52 and at various levels of VipA-sfGFP expression (Supplementary Fig. S2c, S2g and Videos 4, 5 and 6), suggesting that some other component is the limiting factor in their formation. The sheaths in wild type cells displayed the same extension-contraction-disassembly cycles as sheaths observed in complemented vipA mutant cells (Supplementary Videos 4 and 5) and when mCherry2 18 was substituted for sfGFP (Supplementary Fig. S2f, Supplementary Video 7). Thus, the dynamic behavior observed is not just a property of sheaths that contain exclusively VipA-sfGFP fusion protein, but also of sheaths composed largely of wild type VipA or other VipA fusion proteins. VCA0109 encodes a member of a family of phage base plate proteins 10. In a VCA0109 deletion background most of the VipA-sfGFP was dispersed in the cytosol with only a rare, VipA-sfGFP-containing structure ever visible (Supplementary Fig. S2e, Supplementary Video 9). VCA0109 therefore plays a critical role in the formation of functional T6SS sheaths.
Figure 1. Fluorescence light microscopy of VipA-sfGFP.
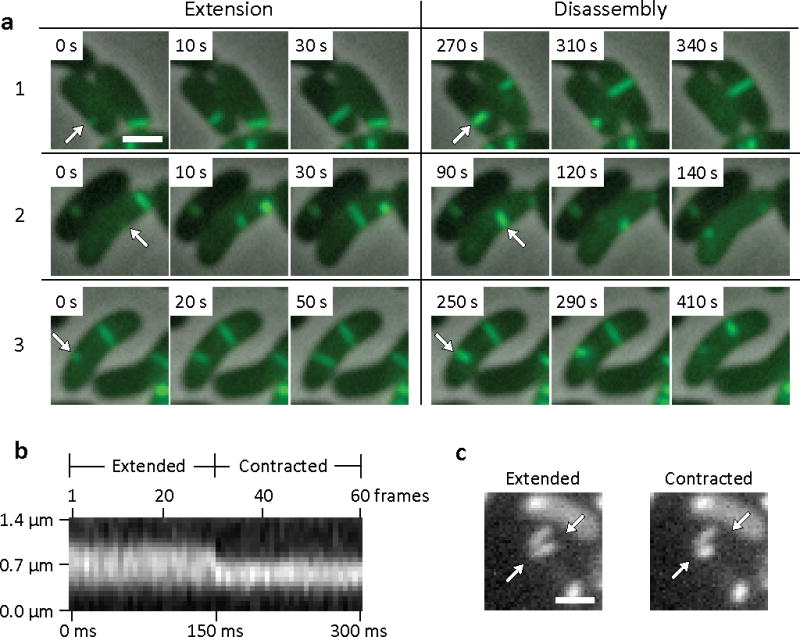
(a) Individual 3×3 μm frames from a time-lapse imaging with a frame rate of 10 sec per frame show 3 frames of extension of VipA-sfGFP structure in ΔVipA background from one side of the cell to another (arrows) followed by a contraction event and apparent disassembly (shown on 3 frames) of the contracted VipA-sfGFP structure (arrows). Bar shown on the first frame represents 1 μm. The whole 10 minute time-lapse sequence is shown in Supplementary Video 1 together with another 17 similar events, larger field of cells is shown in Supplementary Video 2. (b) Kymogram illustrating rapid change in the length of VipA-sfGFP structure. Projection of signal intensity in time at a rate of 200 frames per second along the axis of the maximal intensity on an extended structure (30 frame average shown on the panel (c) left) showing a contraction in length and increase in maximal intensity of the contracted structure (30 frame average shown on the panel (c) right). Arrows indicate contracting VipA-sfGFP structure and mark start and end of a line for generating the kymogram. Bar shown on the average frames is 1 μm long. Gaussian blur filter (sigma radius = 1) was applied to individual frames prior to generating the kymogram. All 60 frames of the time-lapse sequence are shown in Supplementary Video 3 (video segment number 3) together with 4 more contraction events imaged at the same or lower frame rate.
Electron cryotomographic imaging of the T6SS
In order to visualize the sheaths directly, we imaged wild type and mutant whole cells with electron cryotomography (ECT). ECT has been shown to preserve and reveal bacterial cytoskeletal structures directly in 3-D in a near-native, life-like state 19. ECT analyses of wild type 2740–80 cells showed straight, tubular structures which appeared to exist in two different conformations: a longer, thinner ‘extended’ conformation (Fig. 2a–d) and a shorter, wider, ‘contracted’ conformation (Fig. 2e–h). Both structures were oriented roughly perpendicular to the cytoplasmic membrane and were clearly located exclusively in the cytosol (Supplementary Video 10). Tubular structures were observed in 26 of 90 imaged wild type cells. Some cells exhibited more than one tubule and on occasion both extended and contracted conformations were seen in the same cell (see Supplementary Table 1 and Fig. S7). No tubular structures were observed in a vipB mutant (0 of 53 cells), a VCA109 mutant (0 of 10 cells), and a VCA0109/ClpV double mutant (0 of 8 cells), strongly suggesting that both types of tubules are T6SS-related structures.
Figure 2. Electron cryotomographic imaging of T6SS structures inside intact cells.

Shown are different tomographic slices (19 nm in a, e, c, g; 9.5 nm in b,f; 190 nm in d, h) of an extended (a–d) and a contracted (e–h) structure imaged in two different wild type cells (contracted/extended structures, T6SS; IM, inner membrane; OM, outer membrane; F, flagellum; R, putative ribosome; SG, polyphosphate storage granule). (b) and (f), each show three slices at the same orientation but at different Z-heights. Compared to extended structures, contracted structures are shorter (b, f), have a helical surface pattern (pitch angle of 87°) and a smaller diameter (indicated in the perpendicular views in d, h). (c) and (g) are segmentations of densities observed in the extended (c) and contracted (g) structures. Densities shown in (h) originate from a contracted structure from a different tomogram. Segmented are putative densities corresponding to sheath (green), baseplate (pink and yellow) and membranes (blue). Bar in (a) 100 nm (applies to a, e), bar in (b) 100 nm (applies to b, f), bar in (c) 20 nm (applies to c, d, g, h).
Consistent with the dynamic sheaths in the 2-D fluorescence projection images, the extended and contracted tubes seen in the 3-D cryotomograms had lengths of 667 nm +/−83 (n = 13) and 372 nm +/− 56 (n = 16), respectively. While extended tubes had diameters of 11.6 +/− 0.7 nm, dense interiors, and a homogeneous surface, we observed that contracted tubes were thicker (14.6 nm +/− 0.7 diameter), hollow, and had helical ridges (87 pitch angle) spaced 6 nm apart (Fig. 2). The tubular structures of both conformations were almost always found with one end in close proximity to the cytoplasmic membrane in a near perpendicular orientation (Fig. 2, Supplementary Fig. S8). The tubes did not contact the membrane directly, however, but instead appeared to be connected to it by a flared bell-shaped base (Figs. 2c and 2g, pink highlights). Distal to the flared base of extended, but not contracted tubes, there was an additional conical-shaped density (Fig. 2c, yellow highlights) that crossed the periplasm and protruded through the outer membrane. Given that various T6SS components have been localized to the inner membrane, periplasm and outer membrane including a lipoprotein unique to T6SS as well as proteins related to orthologs IcmF, DotU, and OmpA in other organisms 20–22, we propose that all these densities simply be called the “T6SS base plate” complex.
Purification of T6SS sheath from V. cholerae
In order to prove that the dynamic fluorescent structures observed in VipA-sfGFP expressing cells and the tubes observed by ECT were indeed T6SS sheath related structures, we sought to purify the corresponding structures from disrupted cells. Negative stain electron microscopic (EM) analysis of macromolecular fractions purified from cell lysates of wild type cells revealed straight, hollow tubular structures similar to, but more uniform than the VipA/VipB tubules produced previously in E. coli 14 and distinct from the V. cholerae flagellum (Fig. 3a left and Supplementary Fig. S1b left). No sheath-like structures were detected in identically prepared samples from mutants defective in VipA or VipB, although flagella were clearly present (data not shown). EM analysis of sheath preparations made from the VipA-sfGFP complemented vipA mutant strain revealed sheath structures similar to those produced by wild type cells except that a diffuse coat was laterally displayed on the filament’s surface, likely corresponding to the sfGFP moiety on the fusion protein (Figure 3a right and S1b right).
Figure 3. Images of purified VipA/VipB sheaths and comparison with phage tails.
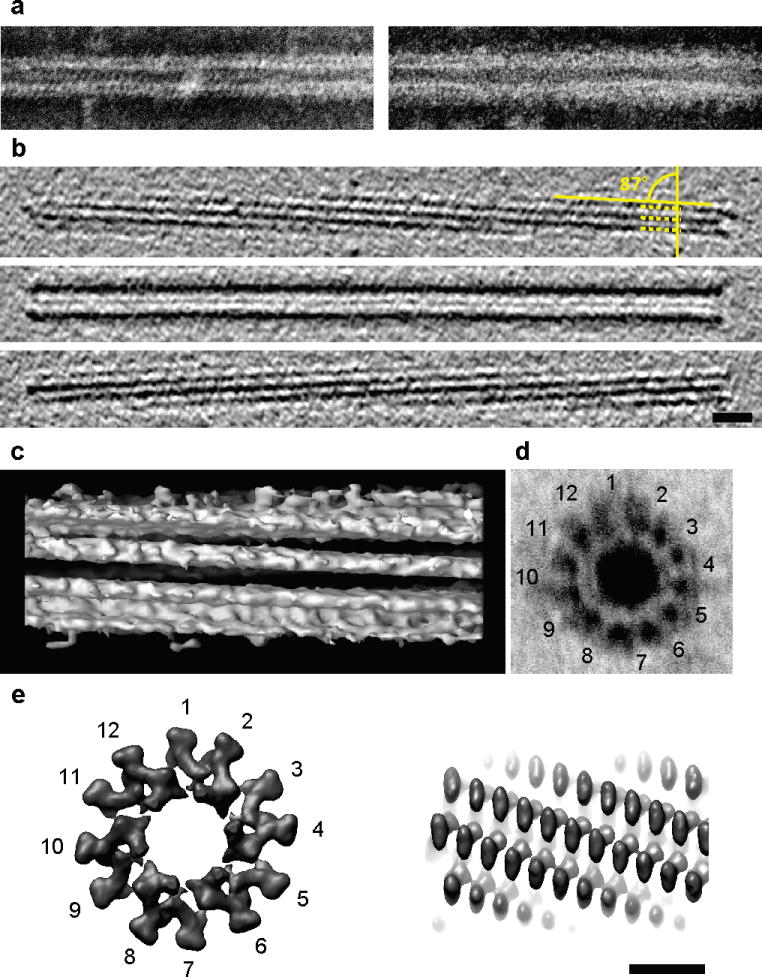
Negative staining (a) images of purified wild type sheath (left) and VipA-sfGFP-labeled sheath (right) are highly similar except for flared extra densities on the outside of the VipA-sfGFP-labeled structure. Cryotomograms of wild type sheath (b, shown three 12.6-nm slices at different Z-heights) were highly similar to contracted structures imaged in vivo (Figure 2f). Note the matching surface pitch angle of 87° seen in tomographic slices (b) and an isosurface of a subtomogram average (c). The negatively stained perpendicular view of a purified wild type sheath showed the characteristic “cog-wheel” like structure with 12 paddles (d) and is similar to the perpendicular view of a contracted T4 phage sheath (e, left; two rings of six gp18 subunits, created in Chimera from EMDB 1086 map). Similar to T6SS sheath (c), also the surface of the contracted T4 phage sheath appears helical (e, right) though with a different pitch angle. Bar in (b) 20 nm (applies to a,b), bar in (e) 10 nm (applies to c–e). Note that protein densities appear white in negative stain images and black in cryotomograms.
To identify proteins that were associated with these sheaths, the structures were purified from a non-flagellated mutant (flgG) of V. cholerae 2740–80 (Supplementary Fig. S9a). Two major proteins were enriched in these preparations with apparent molecular weights of 55 and 20 kDa, respectively (Supplementary Fig. S9b). Mass spectrometry analysis revealed that the 55 kDa band was VipB and the 20 kDa band was VipA (Supplementary Table 2). Interestingly, we also identified four additional T6SS proteins in the sheath samples: ClpV, VCA0109 (a gp25-like protein), and two other proteins within the T6SS gene cluster encoded by genes VCA0111 and VCA0114. ClpV was recently shown to interact directly with VipB, most strongly in its polymerized state with VipA 15. As noted earlier, VCA0109 is a homolog of T4 base plate protein gp25 10 and a T6SS gp25-like protein was recently shown to localize to the cytoplasm of Pseudomonas aeruginosa 13. The function of VCA0111 and VCA0114 are currently unknown, but they are essential components of the V. cholerae T6SS 7.
The VipA/VipB sheath preparations purified from wild type 2740–80 cells were also imaged by ECT (Fig. 3b and c), by which they were recognized as the contracted tubes seen previously inside cells (14.4-nm diameter, hollow interior, and helical surface ridges spaced 6 nm apart with pitch angle 87 ). Interestingly, in addition to helical surface ridges, purified sheaths exhibited cogwheel-like cross-sections with 12 surface “paddles” per rotation (Fig. 3d, seen most clearly after negative staining) and thus are structurally similar to contracted T4 phage sheaths 12 (Fig. 3e and Supplementary Fig. S10). We conclude that the two tubular structures seen in vivo by ECT correspond to extended and contracted states of the dynamic VipA-sfGFP labeled T6SS sheath that was visualized using fluorescence light microscopy.
Contractile phage tails consist of a contractile outer sheath and an inner tube that is projected into a target host cell during phage infection 23. The T6SS of V. cholerae is known to possess antibacterial activity against E. coli that correlates with its ability to secrete the T6SS substrate protein Hcp 4,7. As shown in Supplementary Figs. S1a and S11a, V. cholerae strain 2740–80 secretes abundant Hcp and this secretion is completely abolished by deletion of T6SS genes vipA, vipB and VCA0109 (which encodes a gp25-like protein), as has been previously shown for V. cholerae strain V52 7,14. While the material inside the extended tubule visualized with ECT (Figs. 2d, S10a) could not be resolved as a separate “inner tube” per se, its diameter was similar to the diameter of Hcp tubes described at either the crystallographic or microscopic level 16,24. Furthermore contracted tubes were clearly hollow (Fig. 2h and S10c-f). Thus, we propose that the thinner extended tubule found in whole cells is an un-contracted “extended T6SS sheath” whose VipA/VipB subunits are likely wrapped around a thinner inner tube composed of Hcp protein. Unfortunately, the un-contracted, extended T6SS sheaths could not be purified from V. cholerae cells for further analysis perhaps because of spontaneous sheath contraction during cellular disruption and purification. Because Hcp was not found in purified contracted T6SS sheaths, we conclude that the postulated inner Hcp tube of extended sheaths is largely expelled from the cell at the moment of contraction.
ClpV and T6SS sheath recycling
Like strain V52 4,7, V. cholerae 2740–80 also rapidly kills E. coli when co-cultivated on agar in a vipA-, vipB-, and VCA0109-dependent fashion (Supplementary Fig. S11b). Consistent with published results in strain V52 7, the ClpV mutant of 2740–80 showed 90% loss of T6SS-dependent killing in 2 hr assays but retained bacteriocidal activity well above background during a 4 hr incubation. Thus, ClpV is not essential for T6SS function in V. cholerae. Because ClpV has been shown to disassemble in vitro a tubular structure that is produced in E. coli expressing VipA and VipB 14, we asked whether ClpV affected the dynamics of T6SS sheath imaged with the VipA-sfGFP fusion. We did not observe any polymerization or disassembly events in the ClpV mutant background, but rather found that the majority of VipA-sfGFP existed in static punctate structures (Supplementary Fig. S2d); these punctate structures were likely contracted T6SS sheaths because ClpV mutants produce contracted sheath-like structures (Supplementary Fig. S1b).
Because our fluorescence microscopic analysis showed that contracted sheath forms from extended sheath, it follows that ClpV may not play a role in T6SS sheath assembly or secretion function but rather is responsible for recycling VipA and VipB from contracted T6SS sheath structures through disassemble. The ClpV-mediated disassembly process frequently begins with its dislocation of the contracted sheath from the original site of extension (i.e., the T6SS base plate complex) and then continues in a random fashion throughout the cell cytosol; VipA-sfGFP released by this process can be seen to quickly be reassembled into new extended sheaths in many cells (Supplementary Videos 1 and 2). Based on these observations we propose a detailed model of the dynamic steps in the functional cycle of the T6SS apparatus that will serve as a framework for further studies (Fig. 4). While analogous to translocation events mediated by contractile phage tails, the proposed T6SS process is different because it occurs in a topologically reversed orientation and compartmentalization (within the cytosol), and further undergoes efficient recycling through the action of other T6SS components such as ClpV. Collectively, the data presented here provide strong evidence that energy captured from conformational changes in polymeric structures can rapidly transport proteins through cell membranes.
Figure 4. Model of T6SS action.
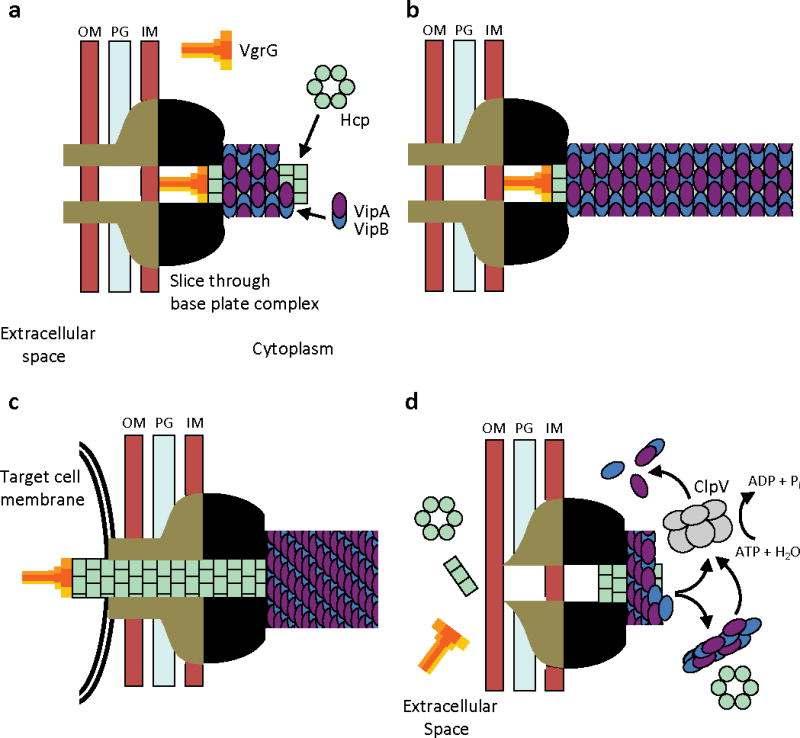
IM – inner membrane, PG – peptidoglycan, OM – outer membrane. (a) Assembly - First step is a base plate complex formation that initiates the Hcp tube polymerization. The base plate complex is likely composed of gp25, VgrG and other T6SS proteins that define a bell-shaped cytoplasmic component (black objects) and periplasmic component (brown objects) which together span the inner membrane, peptidoglycan, and outer membrane. Second step is polymerization of the sheath (from VipA/VipB heterodimers) around the Hcp tube in an extended conformation. (b) Extended T6SS apparatus in extended “ready to fire” conformation. The membrane distal end may be capped by an unknown protein or VipAB conformational state. (c) Contraction - Upon an unknown extracellular signal a conformational change in the base plate complex triggers sheath contraction that leads to the translocation (secretion) of the VgrG/Hcp tube complex through effector cell membranes and penetration of adjacent target cell membrane. Translocation of additional effector proteins might then follow using the Hcp tube as a conduit. (d) Disassembly - Contracted sheath is detached and disassembled by ClpV ATPase. VipA/B dimers released are recycled into a new extended T6SS apparatus at either the original or a newly formed base plate complex. In the absence of target cell penetration (see panel c), Hcp and VgrG proteins are released into the extracellular space as secreted proteins.
Methods
Bacterial strains
V. cholerae 2740–80 is a nontoxinogenic El Tor strain isolated in 1980 from a patient in Florida, United States 25. A streptomycin resistant, lacZ− derivative of 2740–80 was used as the wild type parental strain. E. coli DH10β and Sm10 λpir were used for cloning and conjugation, respectively. Gentamicin resistant E. coli MG1655 strain was used in bacterial killing assays. V. cholerae V52 and its deletion variants were described previously 7. Antibiotic concentrations used were streptomycin (100 μg/ml), gentamicin (15 μg/mL), and carbenicillin (100 μg/ml). Luria-Bertani (LB) broth was used for all growth conditions. Liquid cultures were grown with shaking (250 rpm) at 37 °C.
DNA manipulations
To generate an in-frame deletion in vipA, vipB, clpV, flgG or VCA0109, the corresponding surrounding DNA was amplified by overlap extension polymerase chain reaction and cloned into pWM91 35 for subsequent sacB-mediated allelic exchange as described 26. Primers were designed such that each deletion resulted in the replacement the entire open reading frame, with the exception of first and last seven codons. Gene deletion was confirmed by PCR with primers outside of the replaced region. For construction of variants of pBAD24, full-length vipA gene was amplified from chromosomal DNA and sfGFP or mCherry2 genes were amplified from plasmids carrying respective genes. Full length vipA gene, or vipA genes that had been fused with either sfGFP or mCherry2 genes (both separated by a DNA linker encoding 3xAla 3xGly), were cloned into plasmid pBAD24 27. All cloning products were sequence-verified.
Bacterial killing assay
V. cholerae 2740–80 strains and E. coli MG1655 strain were incubated for 14–18 hours at 37 °C in LB, then washed in fresh LB and diluted 10× in LB. Optical density at 600nm (OD600) of the culture was adjusted to 0.4 for both V. cholerae and E. coli. V. cholerae was mixed with E. coli in 10:1 ratio and 10 μL of the mixture was spotted on a dry LB agar plate. After 2 and 4 hours bacterial spots were cut out and the cells were re-suspended in 0.5 mL LB. The cellular suspension was serially diluted in LB and 5 μL of the suspensions was spotted on selective plates (gentamicin for E. coli, and streptomycin 100 μg/mL V. cholerae). Colonies were detected after ~16 h incubation at 30 °C.
Cell fractionation and immunoblot analysis
Cells from overnight cultures were washed with fresh LB and diluted 1:100 in 1.5 mL of fresh LB (supplemented with appropriate antibiotics and arabinose to indicated concentrations), cultivated for 2.5–3.0 hours to OD600 about 1.0. Cells were collected by centrifugation 21.000×g for 1 min and re-suspended in 250 μL SDS-PAGE loading buffer, 15 μl was loaded for western blot analysis. Cell free supernatants (1.0 mL) were precipitated by 10% trichloroacetic acid (TCA) for 1 hour on ice. Precipitated proteins were collected by centrifugation for 15 minutes at 21.000xg, washed with 100% acetone and re-suspended in 60 μL SDS-PAGE loading buffer. 20 μL was loaded on a SDS-PAGE for western blot analysis. Cell and supernatant protein samples were boiled for 5 min, separated by 10–20% pre-cast polyacrylamide gels (Biorad) and transferred to nitrocellulose membrane (Biorad). Membrane was blocked by 5% milk in Tris buffered saline (pH 7.4) containing Tween 0.05% (TBST), incubated with primary peptide antibody for 2 hours, washed with TBST, incubated for 1 h with horseradish peroxidase labeled anti-rabbit antibody (Jackson Lab), washed with TBST, and peroxidase was detected by SuperSignal West Pico Chemiluminescent Substrate (PIERCE).
Sheath preparations
Overnight culture was diluted 1:200 into 200 mL of fresh LB and then shaken at 37 °C for 2.5–3.0 hours to reach an OD600 of 1.0–1.5. Cells were cooled on ice, centrifuged for 10 min 7000×g and lysed in 12 mL lysis buffer (150 mM NaCl, 50 mM Tris, pH 7.4, lysozyme 200 μg/mL, DNAse I 50 μg/mL, 1 mM phenylmethylsulfonyl fluoride, 0.5× CelLytic B (Sigma), 1% Triton X-100). Cell lysis was complete after 5–10 minutes incubation at 37 °C. After cell lysis, samples were cooled on ice and intact cells and cell debris was removed by centrifugation for 15 min at 15.000×g. Cleared lysates were subjected to ultraspeed centrifugation at 150,000×g for 1 h at 4 °C. Pellets were re-suspended in 0.5 mL of 50mM Tris, 150mM NaCl, pH 7.4 supplemented with protease inhibitor cocktail Complete Mini (Roche) and stored at 4 °C or −20 °C for electron microscopy analysis.
Preparation of sheath for mass spectrometry (MS) analysis
Sheath for MS analysis was prepared from an flgG in-frame deletion mutant of the parental 2740–80 strain. Cells were prepared and lysed as described above. To separate the sheath from soluble proteins, the pellet obtained by ultracentrifugation was re-suspended in 12 mL of TN buffer (50 mM Tris, 150 mM NaCl, pH 7.4) and insoluble material removed by a 2 minute 15.000×g centrifugation step. The sheath was then collected by sequential ultracentrifugation at 150.000×g for 1 h. The sheath pellet was again re-suspended in 12 mL TN buffer and subjected to another ultracentrifugation step. After 3 successive ultracentrifugations, samples typically showed only 2 major bands on a 10–20% SDS-PAGE. The two detectable bands (20 and 50 kDa) and the areas above and below the bands were cut out from the gel and analyzed by tandem MS for peptide identity (Taplin Biological Mass Spectrometry Facility, Harvard).
Peptide specific antibodies
Antigen purified rabbit polyclonal antibodies raised against an Hcp peptide (QSGQPSGQRVHKPF) and VipA peptide (MSKEGSVAPKERIN) were obtained commercially (GenScript, USA). Specificity of the antibodies was tested on V. cholerae V52 strains expressing or lacking Hcp protein or V. cholerae 2740–80 strains expressing or lacking VipA.
Fluorescence microscopy
Overnight cultures of V. cholerae 2740–80 or V52 strains carrying plasmid pBAD24-VipA-sfGFP or pBAD24-VipA-mCherry2 were diluted 1:100 into fresh LB supplemented with carbenicillin and arabinose (concentration 0.01 %, 0.003 %, or as indicated) and cultivated for 2.5 – 3 hours to OD about 1.0. Cells from 100 μL of the culture were re-suspended in 5 μL phosphate buffered saline (PBS), spotted on a thin pad of 1% agarose in PBS, covered with a cover slip and immediately imaged at room temperature.
Fluorescence and phase contrast micrographs were captured using a Nikon TE2000 inverted microscope outfitted with a Nikon Intensilight illuminator, a Coolsnap HQ2 charge-coupled device camera from Photometrics, and CFI Plan Apo DM 100 objective lens (1.4 NA). The sfGFP images were taken by using ET-GFP filter set (Chroma #49002). The mCherry2 images were taken by using the ET-mCherry filter set (Chroma #49008). Images were captured using Nikon Elements software. Images were collected every 6 or 10 s, using an exposure time of 100 to 600 ms for fluorescence and about 10–20 ms for phase contrast. Phase contrast imaging was used to automatically refocus between individual time points. Contrast on images for phase and fluorescence channels were adjusted identically for compared image sets and merged using ImageJ 1.45 software (http://rsb.info.nih.gov/ij/). Small movement of whole field in time was corrected by registering individual frames using StackReg plugin for ImageJ (“Rigid Body” transformation). The pixel-size was 60 nm.
High frame rate fluorescent images were collected with Nikon Ti-E inverted motorized microscope equipped with 100× Plan Apo NA 1.4 objective lens and the Perfect Focus System for continuous maintenance of focus. VipA-sfGFP fluorescence was excited using Prior Lumen200Pro metal halide epi-fluorescence light source, selected with an ET490/20× filter (Chroma) and collected with ET535/30m filter (Chroma). Two different cameras and acquisition setting were used to collect images. A Hamamatsu ORCA-R2 cooled CCD camera was used to acquire images every 118 ms (exposure time of 50 ms, with a continuous illumination). A Hamamatsu ORCA-Flash2.8 cooled CMOS camera was used to acquire images every 20 ms (no analog gain) or 5 ms (8× on-chip analog gain) under continuous illumination light. Both cameras were controlled with Molecular Devices MetaMorph v7.7 software. Contrast was adjusted identically for compared image sets. All image processing and analysis was done using ImageJ 1.45 software. The pixel-size was 67 nm for ORCA-R2 and 78 nm for ORCA-Flash2.8 camera.
Plunge-freezing
For electron cryotomography, V. cholerae 2740–80 wild type and mutant strains were grown aerobically with shaking (200 rpm) at 37 °C in LB medium. A 5 ml overnight-culture was diluted 1000-fold and grown to an OD600 of 1.5–2.2. Copper/rhodium EM grids (R2/2, Quantifoil) were glow-discharged for 1 min. A 20×-concentrated bovine serum albumin-treated solution of 10 nm colloidal gold (Sigma) was added to the sample (1:4 v/v) immediately before plunge freezing. A 4-μl droplet of the mixture was applied to the EM grid, then automatically blotted and plunge-frozen into a liquid ethane-propane mixture 28 using a Vitrobot (FEI Company) 36. The grids were stored in liquid nitrogen.
Negative stain electron microscopy (EM)
Samples were incubated on carbon-coated grids for about 1 min. Grids were washed in water and stained by 1% uranyl formate. The grids were examined in a JEOL 1200EX Transmission electron microscope and images were recorded with an AMT 2k CCD camera.
Electron cryotomography
Tilt series were collected using a Polara 300 kV FEG transmission electron microscope (FEI Company) equipped with an energy filter (slit width 20 eV; Gatan) on a lens-coupled 4k by 4k UltraCam (Gatan). Pixels on the CCD represented 0.95 nm (22500×) or 0.63 nm (34000×) at the specimen level. Typically, tilt series were recorded from −60° to +60° with an increment of 1° at 10 μm under-focus. The cumulative dose of a tilt-series was 180–220 e-/Å2 (for whole cells) or 80–100 e-/Å2 (for sheath preparations). Leginon 29 or UCSF Tomo 30 was used for automatic tilt-series acquisition. Three-dimensional reconstructions were calculated using the IMOD software package 31 or Raptor 32.
Sub-tomogram averaging
IMOD 31 was used to model the center of the sheath. The program addModPts was run to fill in model points every 8 nm along the tube axis. The PEET software package 33 was used to align and average repeating sub-volumes. Isosurface rendering of the sub-volume averages was carried out using Chimera 34.
Supplementary Material
Acknowledgments
We thank Dr. Thomas G. Bernhardt and Nick T. Peters for assistance with fluorescence microscopy, useful discussions, and for a kind gift of plasmids carrying sfGFP and mCherry2 genes. We thank the Nikon Imaging Center at Harvard Medical School for help with fluorescence microscopy and companies Research Precision Instruments and Hamamatsu for lending ORCA-Flash2.8 camera. We thank the Harvard Medical School EM Facility for help and supervision of transmission electron microscopy. We thank Prof. Matthew K. Waldor for a V. cholerae 2740–80 strain and helpful discussions. We thank to Dr. D. Ewen Cameron for a knockout construct pWM91-flgG. We thank Bingi Wen and Zhuo Li for initial cryotomographic studies. This work was supported by NIAID grants AI-018045 and AI-26289 to J.J.M. and NIGMS grant GM094800B to G.J.J.
Footnotes
Author Contributions:
All authors helped design and analyze experiments; M.B., M.P. and G.P.H. performed experiments, and M.B., M.P., G.J., and J.M. wrote the paper.
References
- 1.Pukatzki S, et al. Identification of a conserved bacterial protein secretion system in Vibrio cholerae using the Dictyostelium host model system. Proc Natl Acad Sci USA. 2006;103:1528–1533. doi: 10.1073/pnas.0510322103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ma AT, McAuley S, Pukatzki S, Mekalanos JJ. Translocation of a Vibrio cholerae type VI secretion effector requires bacterial endocytosis by host cells. Cell Host Microbe. 2009;5:234–243. doi: 10.1016/j.chom.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Russell AB, et al. Type VI secretion delivers bacteriolytic effectors to target cells. Nature. 2011;475:343–347. doi: 10.1038/nature10244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.MacIntyre DL, Miyata ST, Kitaoka M, Pukatzki S. The Vibrio cholerae type VI secretion system displays antimicrobial properties. Proc Natl Acad Sci USA. 2010;107:19520–19524. doi: 10.1073/pnas.1012931107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schwarz S, et al. Burkholderia Type VI Secretion Systems Have Distinct Roles in Eukaryotic and Bacterial Cell Interactions. PLoS Pathog. 2010;6:e1001068. doi: 10.1371/journal.ppat.1001068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hood RD, et al. A type VI secretion system of Pseudomonas aeruginosa targets a toxin to bacteria. Cell Host Microbe. 2010;7:25–37. doi: 10.1016/j.chom.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zheng J, Ho B, Mekalanos JJ. Genetic Analysis of Anti-Amoebae and AntiBacterial Activities of the Type VI Secretion System in Vibrio cholerae. PLoS One. 2011;6:e23876. doi: 10.1371/journal.pone.0023876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ma AT, Mekalanos JJ. In vivo actin cross-linking induced by Vibrio cholerae type VI secretion system is associated with intestinal inflammation. Proc Natl Acad Sci USA. 2010;107:4365–4370. doi: 10.1073/pnas.0915156107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pukatzki S, Ma AT, Revel AT, Sturtevant D, Mekalanos JJ. Type VI secretion system translocates a phage tail spike-like protein into target cells where it cross-links actin. Proc Natl Acad Sci USA. 2007;104:15508–15513. doi: 10.1073/pnas.0706532104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leiman PG, et al. Type VI secretion apparatus and phage tail-associated protein complexes share a common evolutionary origin. Proc Natl Acad Sci USA. 2009;106:4154–4159. doi: 10.1073/pnas.0813360106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pell LG, Kanelis V, Donaldson LW, Howell PL, Davidson AR. The phage lambda major tail protein structure reveals a common evolution for long-tailed phages and the type VI bacterial secretion system. Proc Natl Acad Sci USA. 2009;106:4160–4165. doi: 10.1073/pnas.0900044106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leiman PG, Chipman PR, Kostyuchenko VA, Mesyanzhinov VV, Rossmann MG. Three-dimensional rearrangement of proteins in the tail of bacteriophage T4 on infection of its host. Cell. 2004;118:419–429. doi: 10.1016/j.cell.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 13.Lossi NS, Dajani R, Freemont P, Filloux A. Structure-function analysis of HsiF, a gp25-like component of the type VI secretion system in Pseudomonas aeruginosa. Microbiology. 2011;157:3292–3305. doi: 10.1099/mic.0.051987-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bonemann G, Pietrosiuk A, Diemand A, Zentgraf H, Mogk A. Remodelling of VipA/VipB tubules by ClpV-mediated threading is crucial for type VI protein secretion. EMBO J. 2009;28:315–325. doi: 10.1038/emboj.2008.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pietrosiuk A, et al. Molecular Basis for the Unique Role of the AAA+ Chaperone ClpV in Type VI Protein Secretion. J Biol Chem. 2011;286:30010–30021. doi: 10.1074/jbc.M111.253377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mougous JD, et al. A virulence locus of Pseudomonas aeruginosa encodes a protein secretion apparatus. Science. 2006;312:1526–1530. doi: 10.1126/science.1128393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pedelacq JD, Cabantous S, Tran T, Terwilliger TC, Waldo GS. Engineering and characterization of a superfolder green fluorescent protein. Nat Biotechnol. 2006;24:79–88. doi: 10.1038/nbt1172. [DOI] [PubMed] [Google Scholar]
- 18.Cho H, McManus HR, Dove SL, Bernhardt TG. Nucleoid occlusion factor SlmA is a DNA-activated FtsZ polymerization antagonist. Proc Natl Acad Sci USA. 2011;108:3773–3778. doi: 10.1073/pnas.1018674108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pilhofer M, Ladinsky MS, McDowall AW, Jensen GJ. Bacterial TEM: new insights from cryo-microscopy. Methods Cell Biol. 2010;96:21–45. doi: 10.1016/S0091-679X(10)96002-0. [DOI] [PubMed] [Google Scholar]
- 20.Aschtgen MS, Bernard CS, De Bentzmann S, Lloubes R, Cascales E. SciN is an outer membrane lipoprotein required for type VI secretion in enteroaggregative Escherichia coli. J Bacteriol. 2008;190:7523–7531. doi: 10.1128/JB.00945-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aschtgen MS, Thomas MS, Cascales E. Anchoring the type VI secretion system to the peptidoglycan: TssL, TagL, TagP … what else? Virulence. 2010;1:535–540. doi: 10.4161/viru.1.6.13732. [DOI] [PubMed] [Google Scholar]
- 22.Aschtgen MS, Gavioli M, Dessen A, Lloubes R, Cascales E. The SciZ protein anchors the enteroaggregative Escherichia coli Type VI secretion system to the cell wall. Mol Microbiol. 2010;75:886–899. doi: 10.1111/j.1365-2958.2009.07028.x. [DOI] [PubMed] [Google Scholar]
- 23.Kostyuchenko VA, et al. The tail structure of bacteriophage T4 and its mechanism of contraction. Nat Struct Mol Biol. 2005;12:810–813. doi: 10.1038/nsmb975. [DOI] [PubMed] [Google Scholar]
- 24.Ballister ER, Lai AH, Zuckermann RN, Cheng Y, Mougous JD. In vitro self-assembly of tailorable nanotubes from a simple protein building block. Proc Natl Acad Sci USA. 2008;105:3733–3738. doi: 10.1073/pnas.0712247105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goldberg S, Murphy JR. Molecular epidemiological studies of United States Gulf Coast Vibrio cholerae strains: integration site of mutator vibriophage VcA-3. Infect Immun. 1983;42:224–230. doi: 10.1128/iai.42.1.224-230.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bina JE, Mekalanos JJ. Vibrio cholerae tolC is required for bile resistance and colonization. Infect Immun. 2001;69:4681–4685. doi: 10.1128/IAI.69.7.4681-4685.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guzman LM, Belin D, Carson MJ, Beckwith J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol. 1995;177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tivol WF, Briegel A, Jensen GJ. An improved cryogen for plunge freezing. Microsc Microanal. 2008;14:375–379. doi: 10.1017/S1431927608080781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Suloway C, et al. Fully automated, sequential tilt-series acquisition with Leginon. J Struct Biol. 2009;167:11–18. doi: 10.1016/j.jsb.2009.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zheng SQ, et al. UCSF tomography: an integrated software suite for real-time electron microscopic tomographic data collection, alignment, and reconstruction. J Struct Biol. 2007;157:138–147. doi: 10.1016/j.jsb.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 31.Mastronarde DN. Correction for non-perpendicularity of beam and tilt axis in tomographic reconstructions with the IMOD package. J Microsc. 2008;230:212–217. doi: 10.1111/j.1365-2818.2008.01977.x. [DOI] [PubMed] [Google Scholar]
- 32.Amat F, et al. Markov random field based automatic image alignment for electron tomography. J Struct Biol. 2008;161:260–275. doi: 10.1016/j.jsb.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 33.Nicastro D, et al. The molecular architecture of axonemes revealed by cryoelectron tomography. Science. 2006;313:944–948. doi: 10.1126/science.1128618. [DOI] [PubMed] [Google Scholar]
- 34.Pettersen EF, et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 35.Metcalf WW, et al. Conditionally replicative and conjugative plasmids carrying lacZ alpha for cloning, mutagenesis, and allele replacement in bacteria. Plasmid. 1996;35:1–13. doi: 10.1006/plas.1996.0001. [DOI] [PubMed] [Google Scholar]
- 36.Iancu CV, et al. Electron cryotomography sample preparation using the Vitrobot. Nat Protoc. 2006;1:2813–2819. doi: 10.1038/nprot.2006.432. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.