

Abstract
We conducted a genome-wide association (GWA) meta-analysis of 4,604 endometriosis cases and 9,393 controls of Japanese1 and European2 ancestry. We show that rs12700667 on chromosome 7p15.2, previously found in Europeans, replicates in Japanese (P = 3.6 × 10−3), and confirm association of rs7521902 on 1p36.12 near WNT4. In addition, we establish association of rs13394619 in GREB1 on 2p25.1 and identify a novel locus on 12q22 near VEZT (rs10859871). Excluding European cases with minimal or unknown severity, we identified additional novel loci on 2p14 (rs4141819), 6p22.3 (rs7739264) and 9p21.3 (rs1537377). All seven SNP effects were replicated in an independent cohort and produced P < 5 × 10−8 in a combined analysis. Finally, we found a significant overlap in polygenic risk for endometriosis between the European and Japanese GWA cohorts (P = 8.8 × 10−11), indicating that many weakly associated SNPs represent true endometriosis risk loci and risk prediction and future targeted disease therapy may be transferred across these populations.
Endometriosis (MIM131200) is a common gynecological disease associated with severe pelvic pain, affecting 6-10% of women in their reproductive years3,4 and 20-50% of women with infertility5. Endometriosis risk is influenced by genetic factors and has an estimated heritability of around 51%3.
Two large endometriosis GWA studies1,2 have reported genome-wide significant associations. The first, in a Japanese sample of 1,423 cases and 1,318 controls obtained from the BioBank Japan (BBJ), with 484 cases and 3,974 controls for replication, implicated a SNP (rs10965235) in the CDKN2BAS gene on chromosome 9p21.3 (overall odds ratio (OR) = 1.44, 95% CI 1.30–1.59; P = 5.57 × 10−12)1. The second, by the International Endogene Consortium (IEC) in a sample of European ancestry from Australia (2,270 cases and 1,870 controls) and the UK (924 cases and 5,190 controls), with 2,392 cases and 2,271 controls from the US for replication, identified an intergenic SNP (rs12700667) on 7p15.2 (overall OR = 1.20, 95% CI 1.13–1.27; P = 1.4 × 10−9)2. These two studies did not report replication of each other’s top locus, partly because rs10965235 is monomorphic in Caucasian populations. The European study did find association with rs7521902 (OR = 1.16, 95% CI 1.08–1.25, P = 9.0 × 10−5) near the WNT4 gene on 1p36.12, that was reported to be suggestively associated in the Japanese (OR = 1.20, 95% CI 1.11–1.29, P = 2.2 × 10−6).
Encouraged by the WNT4 association and with accumulating evidence for many complex traits that the number of discovered variants is strongly correlated with experimental sample size6, we sought to increase the ratio of controls to cases in the Australian GWA cohort and to perform a formal meta-analysis of the Australian (QIMR), UK (OX) and Japanese (BBJ) GWA data.
To increase the power of the Australian GWA dataset we matched the existing QIMR cases and controls2 on ancestry to individuals from the Hunter Community Study (HCS)7. After stringent quality control (QC), the combined QIMRHCS GWA cohort consisted of 2,262 endometriosis cases and 2,924 controls, increasing the number of controls by 1,054 and the Australian effective sample size by 24%. We also performed more stringent QC incorporating the OX dataset, resulting in a revised OX GWA cohort of 919 endometriosis cases and 5,151 controls. All cases in the QIMRHCS and OX studies have surgically confirmed endometriosis and disease stage from surgical records using the rAFS classification system8, subjects are grouped into stage A (stage I or II disease or some ovarian disease with a few adhesions; n = 1,680, 52.8%) or stage B (stage III or IV disease; n = 1,357, 42.7%), or unknown (n = 144, 4.5%). Details of the final GWA and independent replication case-control cohorts are summarized in Table 1 and a schematic of our study design is provided in Fig. 1.
Table 1.
Summary of the endometriosis case-control cohorts
| Cohort | Ancestry | No. of cases (stage B) | No. of controls |
|---|---|---|---|
| QIMRHCS GWA | European | 2,262 (905) | 2,924 |
| OX GWA | European | 919 (452) | 5,151 |
| BBJ GWA | Japanese | 1,423 | 1,318 |
| GWA meta-analysis | 4,604 | 9,393 | |
| Replication | Japanese | 1,044 | 4,017 |
| Total | 5,648 | 13,410 |
Figure 1.

Study design.
Meta-analysis of all endometriosis 4,604 cases and 9,393 controls for the 407,632 SNPs overlapping in the QIMRHCS, OX and BBJ GWA data, showed that the A allele of rs12700667 at the European 7p15.2 locus (OR = 1.22, 95% CI 1.13–1.31, P = 7.2 × 10−8) also replicates in the Japanese GWA data (OR = 1.22, 95% CI 1.07–1.39, P = 3.6 × 10−3), producing an overall OR of 1.22 (95% CI 1.14–1.30) and P = 9.3 × 10−10 in the GWA meta-analysis; we also confirmed association with allele A of rs7521902 at the 1p36.12 WNT4 locus (OR = 1.18, 95% CI 1.11–1.25, P = 4.6 × 10−8) (Table 2).
Table 2.
Summary of the GWA and replication study results for the seven genome-wide significant loci
| GWA | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| QIMRHCS |
OX |
BBJ |
Meta-analysis |
Replication |
Total |
||||||||||||
| Chr | SNP | Position | RA | OA | RAFcase | RAFcontrol | RAFcase | RAFcontrol | RAFcase | RAFcontrol | Pall | PstageB | RAFcase | RAFcontrol | P | Pall | PstageB |
| 1 | rs7521902 | 22490724 | A | C | 0.265 | 0.236 | 0.259 | 0.238 | 0.570 | 0.514 | 4.6 × 10−8 | 2.3 × 10−9 | 0.568 | 0.521 | 6.5 × 10−5 | 3.2 × 10−11 | 7.6 × 10−13 |
| 2 | rs13394619* | 11727507 | G | A | 0.538 | 0.514 | 0.551 | 0.521 | 0.485 | 0.449 | 6.1 × 10−8 | 7.0 × 10−8 | 0.489 | 0.455 | 3.5 × 10−2 | 6.1 × 10−9 | 6.7 × 10−9 |
| 2 | rs4141819 | 67864675 | C | T | 0.331 | 0.298 | 0.343 | 0.309 | 0.226 | 0.203 | 4.0 × 10−7 | 6.5 × 10−8 | 0.220 | 0.203 | 5.1 × 10−2 | 8.5 × 10−8 | 4.1 × 10−8 |
| 6 | rs7739264 | 19785588 | T | C | 0.545 | 0.512 | 0.556 | 0.515 | 0.772 | 0.742 | 1.3 × 10−7 | 5.8 × 10−8 | 0.778 | 0.744 | 6.9 × 10−4 | 3.6 × 10−10 | 2.1 × 10−10 |
| 7 | rs12700667 | 25901639 | A | G | 0.769 | 0.730 | 0.776 | 0.744 | 0.221 | 0.189 | 9.3 × 10−10 | 3.8 × 10−11 | 0.197 | 0.191 | 2.6 × 10−1 | 3.6 × 10−9 | 1.1 × 10−9 |
| 9 | rs1537377 | 22169700 | C | T | 0.424 | 0.395 | 0.436 | 0.401 | 0.410 | 0.379 | 2.5 × 10−6 | 1.0 × 10−8 | 0.402 | 0.359 | 1.3 × 10−4 | 2.4 × 10−9 | 5.8 × 10−12 |
| 12 | rs10859871 | 95711876 | C | T | 0.332 | 0.299 | 0.332 | 0.295 | 0.373 | 0.328 | 5.5 × 10−9 | 3.7 × 10−7 | 0.377 | 0.328 | 1.1 × 10−5 | 5.1 × 10−13 | 2.6 × 10−11 |
Chr = Chromosome, Position = GRCh37 (hg19) bp position, RA = risk allele, OA = other allele, RAF = risk allele frequency.
= GWA meta-analysis and total P values for rs13394619 include results published in Adachi et al. (2010), consisting of P = 6.1 × 10−4 (RAFcase = 0.517, RAFcontrol = 0.414) and P = 1.0 × 10-2 (RAFcase = 0.488, RAFcontrol = 0.429) obtained in their 500K and 6.0 case-control cohorts, respectively. Pall includes all available endometriosis cases. PstageB excludes unknown and minimal (rAFS I-II) endometriosis stage cases where detailed stage data was available.
The GWA meta-analysis identified a novel locus on 12q22 near the VEZT gene (allele C of rs10859871 OR = 1.18, 95% CI 1.12–1.25, P = 5.5 × 10−9). We also established association with allele G of rs13394619 in the GREB1 gene on 2p25.1 (OR = 1.12, 95% CI 1.06–1.18, P = 2.1 × 10−5), previously reported (OR = 1.35, 95% CI 1.17–1.56, P = 3.8 × 10−5) in a small independent Japanese GWA study of 696 cases and 825 controls by Adachi et al (2010)9. The G allele of rs13394619 approached conventional genome-wide significance (P ≤ 5 × 10−8) in combined analysis of the QIMRHCS, OX, BBJ, Adachi500K and Adachi6.0 GWA data (OR = 1.15, 95% CI 1.09–1.20, P = 6.1 × 10−8) (Table 2). In addition to the three genome-wide significant SNPs on chromosomes 1, 7 and 12 (rs7521902, rs12700667, rs10859871), the Manhattan plot of the all endometriosis GWA meta-analysis results (Supplementary Fig. 1) showed 34 SNPs reached genome-wide suggestive association (P ≤ 10−5).
Given the substantially greater genetic loading of moderate to severe (Stage B) endometriosis (rAFS stage III or IV disease) compared to minimal (Stage A) endometriosis (rAFS stage I or II disease)2, a secondary analysis was performed for the SNPs reaching genome-wide suggestive association, where the association results from QIMRHCS and OX Stage B cases versus controls, were meta-analyzed with the BBJ association results (stage information not available).
After excluding endometriosis cases with minimal (rAFS stage I-II) or unknown severity in the QIMRHCS and OX cohorts, GWA meta-analysis implicated novel loci on 2p14 (allele C of rs4141819 OR = 1.22, 95% CI 1.14–1.32, P = 6.5 × 10−8), 6p22.3 (allele T of rs7739264 OR = 1.21, 95% CI 1.13–1.30, P = 5.8 × 10−8) and 9p21.3 (allele C of rs1537377 OR = 1.22, 95% CI 1.14–1.30, P = 1.0 × 10−8) (Table 2, Supplementary Fig. 2, Supplementary Table 1-2 and Supplementary Note).
Annotated plots showing evidence for association in the combined QIMRHCS, OX and BBJ GWA data of genotyped SNPs across the seven implicated loci from the analysis of all cases and of stage B cases only are provided in Supplementary Figs. 3-9. Imputation up to the 1000 Genomes reference panel produced more significant P values and helped resolve the associated region at the 1p36.12 (rs56318008, Pall = 1.3 × 10−10), 2p25.1 (rs77294520, PstageB = 8.6 × 10−8), 2p14 (rs2861694, PstageB = 7.9 × 10−9), 6p22.3 (rs6901079, Pall = 1.9 × 10−8), 9p21.3 (rs7041895, PstageB = 5.1 × 10−10) and 12q22 (rs11107968, Pall = 3.9 × 10−9) loci (Fig. 2 and Supplementary Figs. 10-16). Of particular note, the most significant imputed SNPs on 1p36.12, rs56318008 and rs3820282 (Pall = 1.6 × 10−10), are located 22 bp 5′ and within the WNT4 gene, respectively.
Figure 2.

Evidence for association with endometriosis from the QIMRHCS+OX+BBJ GWA meta-analysis across the 1p36.12 (a), 6p22.3 (b), 9p21.3 (c) and 12q22 (d) regions following imputation using the 1000 Genomes Project reference panel. Diamond and circle symbols represent genotyped and imputed SNPs, respectively. The most significant genotyped SNP is represented by a purple diamond. All other SNPs are color coded according to the strength of LD with the top genotyped SNP (as measured by r2 in the European 1000 Genomes data).
Interestingly, the most associated genotyped SNP at 9p21.3 (rs1537377) is 55 kb centromeric to the genome-wide significant SNP reported in the original BBJ GWA1 (rs10965235) located in the CDKN2BAS gene, and 49 kb 3′ to the transcription end site of CDKN2BAS. SNP rs10965235 is monomorphic in Caucasian populations and we investigated the independence of rs10965235 and rs1537377 in the BBJ GWA data. Firstly, in the BBJ GWA data, alleles of rs10965235 and rs1537377 are very weakly correlated, with linkage disequilibrium (LD) metrics of r2 = 0.028 and D′ = 0.461. Secondly, the allelic association P values for rs10965235 and rs1537377 are P = 1.6 × 10−4 and P = 1.8 × 10−2, respectively. After conditioning on rs10965235, weak residual association remains at rs1537377 (P = 9.0 × 10−2). Consequently, the data suggest there may be two independent genetic risk factors near the CDKN2BAS locus on 9p21.3. CDKN2BAS is a long non-coding RNA adjacent to and transcribed from the opposite strand to CDKN2B (p15), CDKN2A (p16) and ARF (p14). Loss of heterozygosity of CDKN2A and hypermethylation of the CDKN2A promoter have been reported in endometriosis10,11.
To further validate the seven SNPs implicated by the meta-analysis, we carried out a replication study using a cohort of 1,044 cases and 4,017 controls obtained from the BioBank Japan independent of the BBJ GWA cohort. As shown in the forest plots of risk allele effects estimated using all cases versus controls (Fig. 3), the effects (ORs) were in the same direction for all seven implicated SNPs across the GWA and replication cohorts. With the exception of rs12700667, which was previously replicated (P = 1.2 × 10−3) in 2,392 cases and 2,271 controls from the US2, and rs4141819 (with a marginal P = 5.1× 10−2), all SNPs were replicated at the nominal P < 0.05 threshold (Table 2). All seven SNPs surpass the conventional genome-wide significant threshold of P ≤ 5 × 10−8 after combined analysis of the GWA and replication cases and controls (Table 2). A conservative adjustment of the rs4141819 total P values (Pall = 8.5 × 10−8; PstageB = 4.1 × 10−8) for performing two independent GWA studies (all and stage B endometriosis cases versus controls) would produce P > 5 × 10−8 (Pall-adjusted = 1.7 × 10−7; PstageB-adjusted = 8.2 × 10−8). However, the accurately imputed (Rsq > 0.95) SNP rs2861694 (PstageB = 7.9 × 10−9), in strong LD with rs4141819 (r2 = 0.981, D′ = 1.0; and r2 = 0.867, D′ = 1.0, in the 379 European and 286 Asian 1000 Genomes reference samples, respectively), would remain genome-wide significant (PstageB-adjusted = 1.6 × 10−8).
Figure 3.
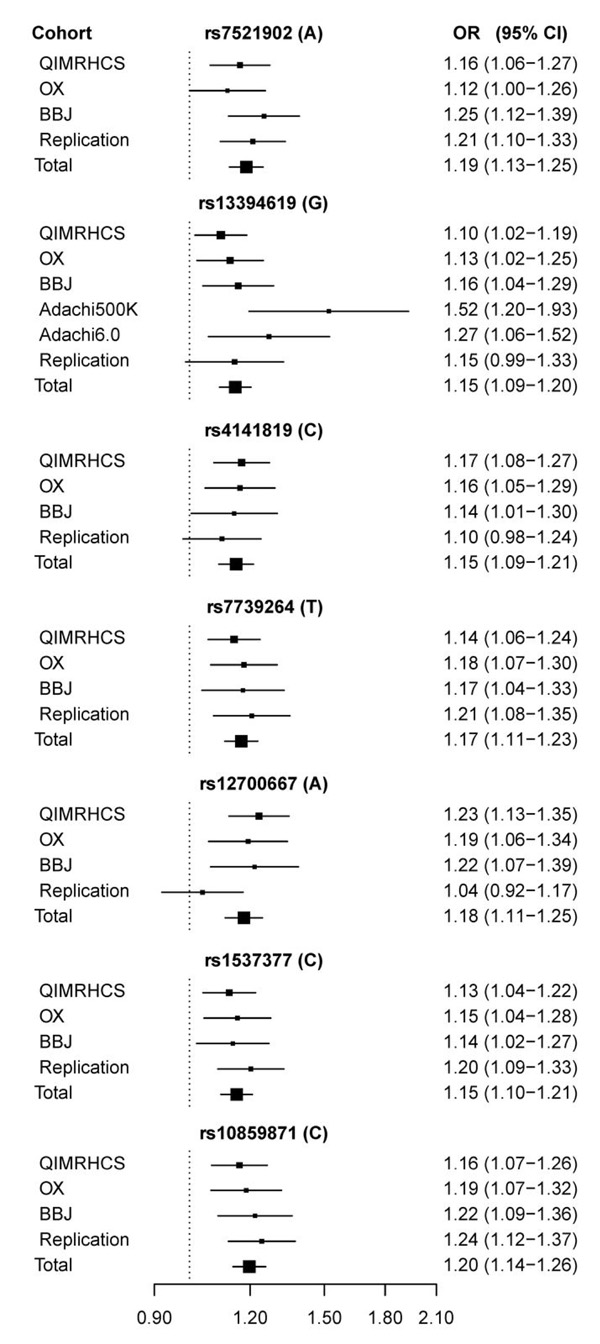
Forest plots of risk allele effects for the seven genome-wide significant SNP loci in the individual and total endometriosis case-control cohorts.
The Q-Q plots for the QIMRHCS, OX and BBJ GWA data (Supplementary Fig. 17a-c) reflect our stringent quality control, while the GWA meta-analysis Q-Q plot (Supplementary Fig. 17d), reveals a significant preponderance of small P values <10−3, suggesting many of these nominally significant SNPs likely represent true signals12. To further examine the shared genetic risk across our European and Japanese populations we performed polygenic prediction analysis13 to evaluate whether the aggregate effects of many variants of small effect in the BBJ GWA cohort, could predict affection status in the European GWA cohorts. The BBJ-derived risk scores significantly predicted affection status in the QIMRHCS (R2 = 0.0064; P = 6.9 × 10−7), OX (R2 = 0.0057; P = 9.6 × 10−6) and combined QIMRHCS+OX all endometriosis case-control sets (R2 = 0.0054; P = 8.8 × 10−11). For the individual and combined QIMRHCS and OX case-control sets, the variance explained peaked in the SNP sets with BBJ GWA P < 0.1, using all GWA meta-analysis SNPs (Fig. 4a) and after excluding all SNPs within ±2500 kb of the seven implicated SNPs listed in Table 1 (Fig. 4b). Analogously, performing the prediction in reverse, the QIMRHCS+OX-derived risk scores significantly predicted affection status in the BBJ case-control set (R2 = 0.0106; P = 3.3 × 10−6) (Supplementary Fig. 18 and Supplementary Note).
Figure 4.

Allele-specific score prediction for endometriosis, using the BBJ population as the discovery dataset and the QIMRHCS+OX population as the target dataset. The variance explained in the target dataset on the basis of allele-specific scores derived in the discovery dataset for twelve significance thresholds (P < 0.01, P < 0.05, P < 0.1, P < 0.2, P < 0.3, P < 0.4, P < 0.5, P < 0.6, P < 0.7, P < 0.8, P < 0.9, P < 1.0, plotted left to right). The y-axis indicates Nagelkerke’s pseudo R2 representing the proportion of variance explained. The number above each bar is the P value for the target dataset prediction analysis (i.e. R2 significance). Prediction was performed using all GWA meta-analysis SNPs (a) and after excluding all SNPs within ±2500 kb of the seven implicated SNPs listed in Table 1 (b). These figures show that the results were not driven by a few highly associated regions, indicating a substantial number of common variants underlie endometriosis risk.
A gene-based GWA analysis using VEGAS14, which accounts for gene size and LD between SNPs, revealed 1,184 genes with a combined P ≤ 0.05 and the top three ranked genes associated with endometriosis to be WNT4 on 1p36.12 (P = 5.0 × 10−9), VEZT on 12q22 (P = 5.7 × 10−7) and GREB1 on 2p25.1 (P = 2.5 × 10−5) (Supplementary Table 3). In addition to having genome-wide significant SNPs near these three genes, the WNT4 and VEZT genes easily surpass our conservative gene-based significant association threshold of P ≤ 2.85 × 10−6 (calculated as P = 0.05 / 17,538 independent genes). WNT4 encodes for wingless-type MMTV integration site family, member 4 and is important for the development of the female reproductive tract15 and steroidogenesis16. VEZT encodes vezatin, an adherens junction transmembrane protein that is down regulated in gastric cancer17. GREB1 encodes growth regulation by estrogen in breast cancer 1, an early response gene in the estrogen regulation pathway involved in hormone dependent breast cancer cell growth18. For the four remaining implicated regions on 2p14, 6p22.3, 7p15.2 and 9p21.3, no genes were significant (P ≤ 1.3 × 10−3) after adjusting VEGAS results for testing 37 genes across all seven regions, see Table 2, Supplementary Figs. 3-9 and Supplementary Table 4.
In conclusion, given their high gene-based ranking, proximity to genome-wide significant SNPs, known pathophysiology and reported gene expression (Supplementary Note and Supplementary Fig. 19), the WNT4, VEZT and GREB1 genes are strong targets for further studies aimed at understanding the molecular pathogenesis of endometriosis. Our results also suggest that a considerable number of SNPs nominally implicated (e.g. P < 0.1) in the European and Japanese GWA cohorts represent true endometriosis risk loci. Moreover, the significant overlap in common polygenic risk for endometriosis indicates genetic risk prediction and future targeted disease therapy may be transferred across these populations.
ONLINE METHODS
GWA samples and phenotyping
Initially, 2,351 surgically-confirmed endometriosis cases were drawn from women recruited by The Queensland Institute of Medical Research (QIMR) study19 and a further 1,030 cases were obtained from women recruited by the Oxford Endometriosis Gene (OXEGENE) study. Australian controls consisted of 1,870 individuals recruited by QIMR2 and 1,244 individuals recruited by the Hunter Community Study (HCS)7. UK controls encompassed 6,000 individuals provided by the Wellcome Trust Case Control Consortium 2 (WTCCC2). Approval for the studies was obtained from the QIMR Human Ethics Research Committee, the University of Newcastle and Hunter New England Population Health Human Research Ethics Committees, and the Oxford regional multi-centre and local research ethics committees. Informed consent was obtained from all participants prior to testing2.
All Japanese GWA case and control samples were obtained from the BioBank Japan (BBJ) at the Institute of Medical Science, the University of Tokyo. A total of 1,423 cases were diagnosed with endometriosis by the following one or more examinations: multiple clinical symptoms, physical examinations, and laparoscopy or imaging tests. We utilized 1,318 female control samples consisting of healthy volunteers from Osaka-Midosuji Rotary Club, Osaka, Japan and women in the Biobank Japan who were registered to have no history of endometriosis. All participants provided written informed consent to this study. This study was approved by the ethical committees at the Institute of Medical Science, the University of Tokyo and Center for Genomic Medicine, RIKEN Yokohama Institute.
GWA genotyping and quality control (QC)
QIMR and OX cases, and QIMR controls were genotyped at deCODE Genetics on Illumina 670-Quad (cases) and 610-Quad (controls) BeadChips (Illumina Inc), respectively. HCS controls were genotyped at the University of Newcastle on 610-Quad BeadChips (Illumina Inc). The WTCCC2 controls were genotyped at the Wellcome Trust Sanger Institute using Illumina HumanHap1M BeadChips. Genotypes for QIMR cases and controls were called with the Illumina BeadStudio software. Standard quality control procedures were applied as outlined previously20. Briefly, individuals with call rates <0.95 then SNPs with a mean BeadStudio GenCall score < 0.7, call rates < 0.95, Hardy-Weinberg equilibrium P < 10−6, and minor allele frequency (MAF) < 0.01 were excluded. Cryptic relatedness between individuals was identified through a full identity-by-state matrix. Ancestry outliers were identified using data from 11 populations of the HapMap 3 and five Northern European populations genotyped by the GenomeEUtwin consortium, using EIGENSOFT21,22. To increase the power of the Australian GWA dataset we ancestrally matched the existing QIMR cases and controls2 to individuals from the Hunter Community Study (HCS)7 genotyped on Illumina 610 chips. After stringent quality control, the resulting QIMRHCS GWA cohort consists of 2,262 endometriosis cases and 2,924 controls, increasing the Australian effective sample size by 24%.2
Quality control procedures for the OX genotype data resulted in the removal of SNPs with a genotype call rate < 0.99 and/or heterozygosity < 0.31 or > 0.33. Genome-wide IBS was estimated for each pair of individuals and one individual from each duplicate or related pair (IBS > 0.82) was removed. Genotype data were combined with CEU, CHB&JPT and YRI genotype data from HapMap 3 and individuals of non Northern European ancestry were identified using EIGENSOFT and subsequently removed. SNPs with a genotype call rate < 0.95 were removed, and this threshold was increased to 0.99 for SNPs with MAF < 0.05. In addition, SNPs showing a significant a) deviation from HWE (P < 1 × 10−6); b) difference in call rate between 58BC and NBS control groups (P < 1 × 10−4); c) difference in allele/genotype frequency between control groups (P < 1 × 10−4); d) difference in call rate between cases and controls (P < 1 × 10−4) and e) a MAF < 0.01 were removed.2
The BBJ cases and controls were genotyped using the Illumina HumanHap550v3 Genotyping BeadChip. Quality control included sample call rate ≥ 0.98, identity-by-state to exclude close relatedness samples and principal component analysis to exclude non-Asian samples. We also performed SNP quality control (call rate of ≥ 0.99 in both cases and controls and Hardy-Weinberg equilibrium test P ≥ 1.0 × 10−6 in controls); 460,945 SNPs on all chromosomes passed the quality control filters and were further analyzed.1
GWA meta-analysis
For SNPs passing QC, tests of allelic association (--assoc) were performed using PLINK23 in the separate QIMRHCS, OX and BBJ GWA datasets. The primary meta-analysis of all endometriosis cases versus controls in the QIMRHCS, OX and BBJ GWA data was performed using a fixed-effect (inverse variance-weighted) model, where the effect size estimates, or β-coefficients, are weighted by their estimated standard errors, utilizing the GWAMA software24.
The threshold of 7.2 × 10−8 for GWA studies of dense SNPs and resequence data25 proposed by Dudbridge and Gusnanto26 was utilized to indicate genome-wide significant association, while SNPs with P ≤ 10−5 were considered to show a suggestive association [as used in the online ‘Catalog of Published Genome-Wide Association Studies’].
Also, given the substantially greater genetic loading of moderate to severe (stage B) endometriosis (rAFS stage III or IV disease) compared to minimal (stage A) endometriosis (rAFS stage I or II disease)2, a secondary analysis was performed for suggestive SNPs (P ≤ 10−5); where the association results from QIMRHCS and OX stage B cases versus controls, were meta-analyzed with the BBJ association results. As previously demonstrated2, the exclusion of minimal endometriosis cases has the potential to enrich true genetic risk effects, even taking into account the reduced sample size.
Consistency of allelic effects across studies was examined utilizing the Cochran’s Q test27. Between-study (effect) heterogeneity was indicated by Q statistic P values < 0.128. Meta-analysis of SNPs associated with fixed-effect P ≤ 10−5 and showing evidence of effect heterogeneity were also analyzed using the recently developed Han and Eskin’s random effects model (RE2) implemented in the Metasoft software29. In contrast to the conventional DerSimonian-Laird random effects (RE) model30, the RE2 model increases power under heterogeneity29.
Genotype imputation analysis
In order to assess the impact of variants not present on the Illumina BeadChips and better define the associated regions, we imputed genotypes within ±2500 kb of the most significant genotyped SNP using the full reference panel from the 1000 Genomes project Interim Phase I Haplotypes (2010-11 data freeze, 2011-06 haplotypes). Imputation was performed separately for the QIMRHCS, OX and BBJ GWA datasets with only the overlapping genotyped SNPs within ±2500kb of the most significant genotyped SNP, using the MaCH and minimac programs31,32 and following the two-step approach outlined in the online ‘Minimac: 1000 Genomes Imputation Cookbook’. Analysis of imputed genotype dosage scores was performed using mach2dat31,32 and PLINK. The quality of imputation was assessed by means of the Rsq statistic. Results for poorly imputed SNPs, defined as having an Rsq < 0.3, were subsequently removed. The results from association analysis of imputed data in the QIMRHCS, OX and BBJ datasets were then combined via meta-analysis of the β-coefficients weighted by their estimated standard errors using GWAMA.
Replication samples and genotyping
Independent of the BBJ GWA case-control cohort, a total of 1,044 cases and 4,017 controls were obtained from the BioBank Japan and utilized for replication. We note that 653 of these 1,044 cases were also utilized in a small GWA study (Adachi et al. 2010) of 696 cases and 825 controls9. To utilize all available association data for rs13394619 maximally, given there is incomplete overlap between the Adachi and our replication cases and zero overlap between the controls, we worked with the published results for rs13394619 in Adachi et al (2010) and the results from comparing our non-overlapping 391 replication cases to our 4,017 replication controls.
The seven SNPs (rs7521902, rs13394619, rs4141819, rs7739264, rs12700667, rs1537377 and rs10859871) reaching genome-wide significance in the GWA meta-analysis were genotyped in the independent Japanese replication cohort using the multiplex PCR-based Invader assay (Third Wave Technologies), as previously described1.
Replication and total association analyses
Tests of allelic association were performed using PLINK in the independent Japanese replication cohort. Because only the associations in the same direction would be considered as replicated, one-sided P values were obtained by halving the standard (two-sided) PLINK P values. To determine the total evidence for association, the one-sided replication P values were meta-analyzed with the QIMRHCS, OX, BBJ [and Adachi9 500K (290 cases and 262 controls) and 6.0 (406 cases and 563 controls) for rs13394619] GWA P values using METAL33. The P values observed in each case-control cohort were converted into a signed Z-score. Z-scores for each allele were combined across samples in a weighted sum, with weights proportional to the square-root of the sample size for each cohort34. Given that our cohorts have unequal numbers of cases and controls, we utilized the effective sample size, where Neff = 4 / (1 / Ncases + 1 / Ncontrols)33. We also performed meta-analysis of the β-coefficients weighted by their estimated standard errors using GWAMA to estimate the overall odds ratio and 95% CI for the genome-wide significant SNPs.
Polygenic prediction
The aim of the prediction analysis was to evaluate the aggregate effects of many variants of small effect. We summarized variation across nominally associated loci into quantitative scores and related the scores to disease state in independent samples. Although variants of small effect (e.g., genotype relative risk of 1.05) are unlikely to achieve even nominal significance, increasing proportions of “true” effects will be detected at increasingly liberal P value thresholds, e.g. P < 0.1 (i.e., ~10% of all SNPs), P < 0.2, etc. Using such thresholds, we defined large sets of “allele specific scores” in the “discovery” sample of the Japanese BioBank (BBJ) endometriosis case-control set (1,423 cases, 1,318 controls) to generate risk scores for individuals in the “target” sample of the QIMRHCS (2,262 cases, 2,924 controls), OX (919 cases, 5,151 controls) and combined European (QIMRHCS+OX) endometriosis case-control sets (3,181 cases, 8,075 controls). The term risk score is used instead of risk, as it is impossible to differentiate the minority of true risk alleles from the non-associated variants. In the discovery sample, we selected sets of allele specific scores for SNPs with the following levels of significance; P < 0.01, P < 0.05, P < 0.1, P < 0.2, P < 0.3, P < 0.4, P < 0.5, P < 0.6, P < 0.7, P < 0.8, P < 0.9, P < 1.0. For each individual in the target sample, we calculated the number of score alleles that they possessed, each weighted by the log odds ratio from the discovery sample. To assess whether the aggregate scores reflect endometriosis risk, we tested for a higher mean score in cases compared to controls. Logistic regression was used to assess the relationship between target sample disease status and aggregate risk score. Nagelkerke’s pseudo R2 was used to assess the variance explained. Prediction was performed using all 407,632 SNPs overlapping the QIMRHCS, OX and BBJ GWA datasets, and after excluding the 6,163 SNPs within ±2500 kb of the seven implicated SNPs listed in Table 1. We also performed the predictions in reverse, using QIMRHCS+OX-derived risk scores to predict affection status in the BBJ case-control set.
Gene-based association analysis
Gene-based approaches can be more powerful than traditional individual-SNP-based approaches in the presence of allelic heterogeneity. Therefore, utilizing the QIMRHCS, OX and BBJ GWA data, we performed a genome-wide gene-based association study using VEGAS14. Briefly, for the 407,632 overlapping SNPs, the P values from the European GWA study (i.e., FE meta-analysis of QIMRHCS and OX GWA data) and the P values from the Japanese (BBJ) GWA study were analyzed separately using VEGAS. The VEGAS test incorporates evidence for association from all SNPs across a gene and accounts for gene size (number of SNPs) and LD between SNPs by using simulations from the multivariate normal distribution. The resulting European and Japanese gene-based P values were meta-analyzed using Stouffer’s Z-score combined p-value method34. A total of 17,538 genes (including 50 kb 5′ and 3′ of their transcription start and end site, respectively14) contained association results for ≥1 SNP, so a Bonferroni adjusted significance threshold of P ≤ 2.85 × 10−6 (0.05 / 17,538) was utilized to indicate genome-wide gene-based significant association.
Supplementary Material
Supplementary information One file ‘Supplementary Text and Figures.pdf’, containing: Supplementary Figures 1–19, Supplementary Tables 1–4, Supplementary Note and References.
Acknowledgments
We acknowledge with appreciation all the women who participated in the QIMR, OX and BBJ studies. We thank Endometriosis Associations for supporting the study recruitment. We also thank the many hospital directors and staff, gynecologists, general practitioners and pathology services in Australia, the UK and the United States who provided assistance with confirmation of diagnoses. We thank Sullivan and Nicolaides Pathology and the Queensland Medical Laboratory Pathology for pro bono collection and delivery of blood samples and other pathology services for assistance with blood collection. The Hunter Community Study team thanks the men and women of the Hunter region who participated in the study.
The QIMR Study was supported by grants from the National Health and Medical Research Council (NHMRC) of Australia (241944, 339462, 389927, 389875, 389891, 389892, 389938, 443036, 442915, 442981, 496610, 496739, 552485 and 552498), the Cooperative Research Centre for Discovery of Genes for Common Human Diseases (CRC), Cerylid Biosciences (Melbourne) and donations from N. Hawkins and S. Hawkins. D.R.N. was supported by the NHMRC Fellowship (339462 and 613674) and the ARC Future Fellowship (FT0991022) schemes. S.M. was supported by NHMRC Career Development Awards (496674, 613705). E.G.H. (631096) and G.W.M. (339446, 619667) were supported by the NHMRC Fellowships Scheme. We thank B. Haddon, D. Smyth, H. Beeby, O. Zheng, B. Chapman and S. Medland for project and database management, sample processing, genotyping and imputation. We thank Brisbane gynecologist D.T. O’Connor for his important role in initiating the early stages of the project and for confirmation of diagnosis and staging of disease from clinical records of many cases, including 251 in these analyses. We are grateful to the many research assistants and interviewers for assistance with the studies contributing to the QIMR collection. The Hunter Community Study was funded by the University of Newcastle, the Gladys M Brawn Fellowship scheme and the Vincent Fairfax Family Foundation in Australia.
The work presented here was supported by a grant from the Wellcome Trust (WT084766/Z/08/Z) and makes use of WTCCC2 control data generated by the Wellcome Trust Case-Control Consortium. A full list of the investigators who contributed to the generation of these data is available from http://www.wtccc.org.uk. Funding for the WTCCC project was provided by the Wellcome Trust under awards 076113 and 085475. C.A.A. was supported by a grant from the Wellcome Trust (098051). A.P.M. was supported by a Wellcome Trust Senior Research Fellowship. S.H.K. is supported by the Oxford Partnership Comprehensive Biomedical Research Centre with funding from the Department of Health NIHR Biomedical Research Centres funding scheme. K.T.Z. is supported by a Wellcome Trust Research Career Development Fellowship (WT085235/Z/08/Z). We thank L. Cotton, L. Pope, G. Chalk and G. Farmer (University of Oxford). We also thank P. Koninckx (Leuven, Belgium), M. Sillem (Heidelberg, Germany), C. O’Herlihy and M. Wingfield (Dublin, Ireland), M. Moen (Trondheim, Norway), L. Adamyan (Moscow, Russia), E. McVeigh (Oxford, UK), C. Sutton (Guildford, UK), D. Adamson (Palo Alto, California, USA) and R. Batt (Buffalo, New York, USA) for providing diagnostic confirmation.
We thank the members of the Rotary Club of Osaka-Midosuji District 2660 Rotary International in Japan for supporting our study. This work was conducted as a part of the BioBank Japan Project that was supported by the Ministry of Education, Culture, Sports, Science and Technology of the Japanese government.
Footnotes
Author contributions:
Manuscript preparation and final approval: D.R.N., S.-K.L., C.A.A., J.N.P., S.U., A.P.M., S.M., S.D.G., A.K.H., N.G.M., J.A., E.G.H., M.M., R.J.S., S.H.K., S.A.T., S.A.M., S.A., K.T., Y.N., K.T.Z., H.Z. & G.W.M.
Study conception and design: D.R.N., S.M., Y.N., K.T.Z., H.Z. & G.W.M.
GWAS data collection, sample preparation and clinical phenotyping: J.N.P., S.U., A.K.H., N.G.M., J.A., E.G.H., M.M., R.J.S., S.H.K., S.A.T., K.T.Z., H.Z. & G.W.M.
Replication data collection, sample preparation and clinical phenotyping: S.A., K.T. & H.Z.
Replication genotyping: H.Z.
Data analysis: GWA analysis: D.R.N., C.A.A. & S.-K.L.; imputation and replication analysis: D.R.N. & S.-K.L.; polygenic prediction, gene-based and meta-analysis: D.R.N.
Obtaining study funding: D.R.N., S.M., N.G.M., S.H.K., S.A.T., S.A.M., Y.N., K.T.Z. & G.W.M.
Competing financial interests The authors declare no competing financial interests.
URLs.
A Catalog of Published Genome-Wide Association Studies, www.genome.gov/gwastudies;
Gene Expression Omnibus (GEO) database, http://www.ncbi.nlm.nih.gov/gds/;
GENe Expression VARiation (Genevar) database, http://www.sanger.ac.uk/resources/software/genevar/;
GWAMA, http://www.well.ox.ac.uk/gwama/;
MaCH, http://www.sph.umich.edu/csg/abecasis/MaCH/;
Mammalian Gene Expression Uterus database (MGEx-Udb), http://resource.ibab.ac.in/cgi-bin/MGEXdb/microarray/scoring/interface/Homepage.pl;
METAL, http://genome.sph.umich.edu/wiki/METAL_Program;
Metasoft, http://genetics.cs.ucla.edu/meta/index.html; minimac, http://genome.sph.umich.edu/wiki/Minimac;
minimac, http://genome.sph.umich.edu/wiki/Minimac;
Minimac: 1000 Genomes Imputation Cookbook, http://genome.sph.umich.edu/wiki/Minimac:_1000_Genomes_Imputation_Cookbook; 1000 Genomes Project, http://www.1000genomes.org/;
PLINK, http://pngu.mgh.harvard.edu/~purcell/plink/;
SNPSpD, http://genepi.qimr.edu.au/general/daleN/SNPSpD/;
Wellcome Trust Case-Control Consortium, http://www.wtccc.org.uk;
References
- 1.Uno S, et al. A genome-wide association study identifies genetic variants in the CDKN2BAS locus associated with endometriosis in Japanese. Nat Genet. 2010;42:707–10. doi: 10.1038/ng.612. [DOI] [PubMed] [Google Scholar]
- 2.Painter JN, et al. Genome-wide association study identifies a locus at 7p15.2 associated with endometriosis. Nat Genet. 2011;43:51–4. doi: 10.1038/ng.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Treloar SA, O’Connor DT, O’Connor VM, Martin NG. Genetic influences on endometriosis in an Australian twin sample. Fertil Steril. 1999;71:701–710. doi: 10.1016/s0015-0282(98)00540-8. [DOI] [PubMed] [Google Scholar]
- 4.Montgomery GW, et al. The search for genes contributing to endometriosis risk. Hum Reprod Update. 2008;14:447–57. doi: 10.1093/humupd/dmn016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gao X, et al. Economic burden of endometriosis. Fertil Steril. 2006;86:1561–72. doi: 10.1016/j.fertnstert.2006.06.015. [DOI] [PubMed] [Google Scholar]
- 6.Visscher PM, Brown MA, McCarthy MI, Yang J. Five Years of GWAS Discovery. Am J Hum Genet. 2012;90:7–24. doi: 10.1016/j.ajhg.2011.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McEvoy M, et al. Cohort profile: The Hunter Community Study. Int J Epidemiol. 2010;39:1452–63. doi: 10.1093/ije/dyp343. [DOI] [PubMed] [Google Scholar]
- 8.American Society for Reproductive Medicine Revised American Society for Reproductive Medicine classification of endometriosis: 1996. Fertil Steril. 1997;67:817–21. doi: 10.1016/s0015-0282(97)81391-x. [DOI] [PubMed] [Google Scholar]
- 9.Adachi S, et al. Meta-analysis of genome-wide association scans for genetic susceptibility to endometriosis in Japanese population. J Hum Genet. 2010;55:816–21. doi: 10.1038/jhg.2010.118. [DOI] [PubMed] [Google Scholar]
- 10.Goumenou AG, Arvanitis DA, Matalliotakis IM, Koumantakis EE, Spandidos DA. Loss of heterozygosity in adenomyosis on hMSH2, hMLH1, p16Ink4 and GALT loci. Int J Mol Med. 2000;6:667–71. doi: 10.3892/ijmm.6.6.667. [DOI] [PubMed] [Google Scholar]
- 11.Martini M, et al. Possible involvement of hMLH1, p16(INK4a) and PTEN in the malignant transformation of endometriosis. Int J Cancer. 2002;102:398–406. doi: 10.1002/ijc.10715. [DOI] [PubMed] [Google Scholar]
- 12.Yang J, et al. Genomic inflation factors under polygenic inheritance. Eur J Hum Genet. 2011;19:807–12. doi: 10.1038/ejhg.2011.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Purcell SM, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–52. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu JZ, et al. A versatile gene-based test for genome-wide association studies. Am J Hum Genet. 2010;87:139–45. doi: 10.1016/j.ajhg.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vainio S, Heikkila M, Kispert A, Chin N, McMahon AP. Female development in mammals is regulated by Wnt-4 signalling. Nature. 1999;397:405–9. doi: 10.1038/17068. [DOI] [PubMed] [Google Scholar]
- 16.Guo X, et al. Down-regulation of VEZT gene expression in human gastric cancer involves promoter methylation and miR-43c. Biochem Biophys Res Commun. 2011;404:622–7. doi: 10.1016/j.bbrc.2010.12.026. [DOI] [PubMed] [Google Scholar]
- 17.Boyer A, et al. WNT4 is required for normal ovarian follicle development and female fertility. Faseb J. 2010;24:3010–25. doi: 10.1096/fj.09-145789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rae JM, et al. GREB 1 is a critical regulator of hormone dependent breast cancer growth. Breast Cancer Res Treat. 2005;92:141–9. doi: 10.1007/s10549-005-1483-4. [DOI] [PubMed] [Google Scholar]
- 19.Treloar SA, et al. Genomewide linkage study in 1,176 affected sister pair families identifies a significant susceptibility locus for endometriosis on chromosome 10q26. Am J Hum Genet. 2005;77:365–376. doi: 10.1086/432960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Medland SE, et al. Common variants in the trichohyalin gene are associated with straight hair in Europeans. Am J Hum Genet. 2009;85:750–5. doi: 10.1016/j.ajhg.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2:e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Price AL, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–9. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 23.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analysis. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Magi R, Morris AP. GWAMA: software for genome-wide association meta-analysis. BMC bioinformatics. 2010;11:288. doi: 10.1186/1471-2105-11-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bajpai AK, et al. MGEx-Udb: a mammalian uterus database for expression-based cataloguing of genes across conditions, including endometriosis and cervical cancer. PLoS One. 2012;7:e36776. doi: 10.1371/journal.pone.0036776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dudbridge F, Gusnanto A. Estimation of significance thresholds for genomewide association scans. Genet Epidemiol. 2008;32:227–34. doi: 10.1002/gepi.20297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cochran WG. The combination of estimates from different experiments. Biometrics. 1954;10:101–129. [Google Scholar]
- 28.Ioannidis JP, Patsopoulos NA, Evangelou E. Heterogeneity in meta-analyses of genome-wide association investigations. PLoS One. 2007;2:e841. doi: 10.1371/journal.pone.0000841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Han B, Eskin E. Random-effects model aimed at discovering associations in meta-analysis of genome-wide association studies. Am J Hum Genet. 2011;88:586–98. doi: 10.1016/j.ajhg.2011.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DerSimonian R, Laird N. Meta-analysis in clinical trials. Control Clin Trials. 1986;7:177–88. doi: 10.1016/0197-2456(86)90046-2. [DOI] [PubMed] [Google Scholar]
- 31.Li Y, Willer C, Sanna S, Abecasis G. Genotype imputation. Annu Rev Genomics Hum Genet. 2009;10:387–406. doi: 10.1146/annurev.genom.9.081307.164242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol. 2010;34:816–34. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–1. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stouffer SA, Suchman EA, DeVinney LC, Star SA, Williams RMJ. Adjustment During Army Life. Princeton University Press; Princeton, NJ: 1949. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.