

Abstract
Despite existing aggressive treatment modalities, the prognosis for advanced stage neuroblastoma remains poor with significant long-term illness in disease survivors. Advance stage disease features are associated with tumor vascularity, and as such, angiogenesis inhibitors may prove useful along with current therapies. The matricellular protein, secreted protein acidic and rich in cysteine (SPARC), is known to inhibit proliferation and migration of endothelial cells stimulated by growth factors. Here, we sought to determine the effect of SPARC on neuroblastoma tumor cell-induced angiogenesis and to decipher the molecular mechanisms involved in angiogenesis inhibition. Conditioned medium from SPARC-overexpressed neuroblastoma cells (pSPARC-CM) inhibited endothelial tube formation, cell proliferation, induced programmed cell death and suppressed expression of pro-angiogenic molecules such as VEGF, FGF, PDGF, and MMP-9 in endothelial cells. Further analyses revealed that pSPARC-CM-suppressed expression of growth factors was mediated by inhibition of the Notch signaling pathway, and cells cultured on conditioned medium from tumor cells that overexpress both Notch intracellular domain (NICD-CM) and SPARC resumed the pSPARC-CM-suppressed capillary tube formation and growth factor expression in vitro. Further, SPARC overexpression in neuroblastoma cells inhibited neo-vascularization in vivo in a mouse dorsal air sac model. Furthermore, SPARC overexpression-induced endothelial cell death was observed by co-localization studies with TUNEL assay and an endothelial marker, CD31, in xenograft tumor sections from SPARC-overexpressed mice. Our data collectively suggest that SPARC overexpression induces endothelial cell apoptosis and inhibits angiogenesis both in vitro and in vivo.
Keywords: Neuroblastoma, SPARC, Angiogenesis, Apoptosis
Introduction
Neuroblastoma is a common pediatric solid tumor of the central nervous system. Prognosis for neuroblastoma is variable and highly dependent on measurable tumor biologic features [1]. Neuroblastoma has the highest rate of spontaneous regression of any human cancer, and patients with localized disease may be successfully treated with surgery alone or with minimal therapy; however, more than 50 percent of neuroblastoma patients present with metastatic disease and/or adverse tumor-specific biologic features [1]. Regardless of rigorous multimodal therapy, these neuroblastoma high-risk patients have a poor prognosis, with a long-term survival rate of less than 40 percent (compared with 90% for low-risk patients) [2]. The patients who survive frequently experience significant long-term morbidity as a result of receiving dose-intensive therapy at a young age [3]. Novel therapeutic approaches that will both improve survival and decrease treatment-related toxicity are clearly needed.
Angiogenesis is a biological process by which new capillaries are formed from pre-existing vessels [2]. It plays an important role in the neoplastic process and is important for the local progression and metastatic spread of solid and hematologic tumors [2]. Tumor vascularity is associated with an aggressive phenotype in neuroblastoma, thereby signifying that anti-angiogenic therapy might be a useful addition to current high-risk neuroblastoma treatment approaches. High-risk neuroblastoma is highly vascular, and that tumor vascularity is strongly correlated with adverse prognostic features, including the presence of metastases, unfavorable histopathology and poor survival probability [4]. In contrast, localized tumors with promising biological features are often less vascular and have a rich stromal component composed largely of nonmalignant cells. Thus, neuroblastoma vascularity is intrinsically related to the underlying tumor biology as well as tumor-host interactions.
Cell–extracellular matrix (ECM) and cell–cell interactions influence cell proliferation and survival. By their ability to modify the nature and structure of ECM proteins, matricellular proteins modulate ECM–cell interactions and subsequently, cell proliferation [5]. Secreted protein acidic and rich in cysteine (SPARC/osteonectin/BM-40) belongs to the family of non-structural components of the extracellular matrix (ECM) called matricellular proteins that modulate interactions between cells and their environment [6]. SPARC functions as a tumor suppressor in neuroblastoma, breast, pancreatic, lung and ovarian cancers [7]. In addition, SPARC modulates angiogenesis and regulates the production, assembly and organization of the ECM [8;9]. SPARC is highly expressed in a variety of cell types associated with tissue remodeling [10]. The mechanism for SPARC’s anti-angiogenic activity is not well understood; SPARC is known to interfere with the binding of pro-angiogenic factors [vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF) and basic fibroblast growth factor (bFGF)] to their receptors in endothelial cells, resulting in inhibited proliferation and angiogenesis [10]. SPARC has also been shown to downregulate VEGF in glioma cells [11]. The role of SPARC in cancer seems to be cell-type specific due to its diverse functions. In some types of cancers, SPARC expression has been shown to correlate with disease progression and poor outcome.
Usually adult solid tumor malignancies arise in fully differentiated tissues, whereas embryonal malignancies (e.g., neuroblastoma and other childhood tumors) arise during normal tissue development. Therefore, the methods of providing a blood supply for the growing primary tumor mass may be characteristically different. A strong correlation between high-risk tumors and angiogenesis in neuroblastoma has been reported by various investigators [4;10;12;13] suggesting that vascularature may be clinically relevant to therapeutic targets. To prioritize development of anti-angiogenic compounds, delineation of the most clinically relevant angiogenic signaling pathways intrinsic to neuroblastoma and the most efficient ways to inhibit them is needed. The most appropriate use of anti-angiogenic strategies as potential adjuncts to current treatment regimens can then be determined.
Radiotherapy is considered to be a vital component of the therapeutic regimen for advanced neuroblastoma. However, recent studies have shown that radiotherapy can promote malignant behavior by activating several genes involved in tumor invasiveness and metastasis [14;15]. It is generally assumed that tumor progression towards metastasis, during or after therapy, is due to the appearance of resistant tumor cells. Further, several studies indicate that radiotherapy also rapidly and persistently alters the tumor microenvironment [16;17]. Radiation (IR) induces the production of various tumor-inducing proteins including pro-angiogenic molecules [18–23] which may activate the microenvironment, including the vasculature [16;17]. On the other hand, while adjuvant radiotherapy significantly improves local tumor control, recurrence within a pre-irradiated field is associated with a higher risk of local invasion and metastasis and with poor prognosis [17;24–27]. In accordance, anti-angiogenic approaches enhance the anti-tumor effects of IR [22;27;28].
Previously, we demonstrated that SPARC could inhibit medulloblastoma-induced angiogenesis by inhibiting MMP-9 levels [29]. In this study, we sought to determine the underlying molecular mechanisms involved in inhibition of endothelial cell proliferation and apoptosis, leading to decreased angiogenesis in neuroblastoma upon SPARC overexpression in tumor cells.
Materials and methods
Cells and reagents
SK-N-AS cells were obtained from ATCC (Manassas, VA) and NB-1691 cells were obtained from Dr. Houghton of St. Jude Children’s Research Hospital (Memphis, TN). SK-N-AS and NB-1691 cells were cultured on RPMI-1640 (ATCC, Manassas, VA) supplemented with 10% fetal bovine serum (FBS), 50 units/mL penicillin, and 50 μg/mL streptomycin (Life Technologies Inc., Frederick, MD). Human dermal microvascular endothelial cells (HMEC) provided by Dr. Francisco J Candal (Centers for Disease Control and Prevention, Atlanta, GA, USA) were cultured on Advanced MEM medium (Invitrogen, Carlsbad, CA) and supplemented with 10% FBS, penicillin and streptomycin, glutamine, EGF (San Jose, CA) and hydrocortisone (Stem Cell Technologies, British Columbia, Canada). Cells were incubated in a humidified 5% CO2 atmosphere at 37°C. The antibodies specific for SPARC,VEGF, VEGFR2, phospho-VEGFR2, ERK, phospho-ERK, PI3K, Akt, phospho-Akt, Notch1, Notch2, Notch3, Noth4, Hes1, Hey1, Stat3, phospho-Stat3 (Tyr 705), phospho-Stat3 (Ser 727), Jagged, Ki-67, PECAM1 (CD31), Bcl-XL/S, MMP-9, Caspase-3, Fak, Phospho-Fak, FGF, uPA, Delta, HRP-conjugated secondary antibodies (Santa Cruz Biotechnology, Santa Cruz, CA), GAPDH (Novus Biologicals, Inc., Littleton, CO), mTOR, Caspase-8, p70S6K (Cell Signaling Technology Inc, Danvers, MA) PARP (EMD Biosciences, Rockland, MA), PDGFA (Abcam, Cambridge, MA) and were used in this study. The other materials used in this study were Transcriptor First Strand cDNA Synthesis Kit, In Situ Cell Death Detection Kit, Fluorescein (Roche Applied Science, Indianapolis, IN), MTT cell growth assay kit (Millipore Corporation, Billerica, MA), and Stattic, a small molecule inhibitor of Stat3 (TOCRIS Bioscience, Ellisville, MO). pStat3-C (constructs of constitutively activated Stat3) was purchased from Addgene Inc. (Cambridge, MA) (Addgene plasmid 8722).
Construction of pcDNA3.1-SPARC and transfection of neuroblastoma cells
An 1100-bp cDNA fragment of human SPARC was cloned into a pcDNA3.1 vector (Invitrogen, San Diego, CA, USA) in sense orientation as described previously [29]. Neuroblastoma cells were transfected with full-length cDNA of SPARC containing vector (pSPARC) or empty vector (pEV) using FuGene HD (Roche, Indianapolis, IN, USA) according to the manufacturer’s instructions.
Preparation of tumor conditioned media (CM)
Tumor cell conditioned medium was prepared as described previously [30]. Briefly, 1.5 × 106 neuroblastoma cells (SK-N-AS and NB-1691) were seeded in 100-mm Petri dishes and left overnight. Cells were transfected with either mock (PBS), pEV or pSPARC, and cultured for 24 hrs. The medium was then replaced with serum-free DMEM/F-12 50/50 medium, and cells were irradiated with 8Gy. Sixteen hrs after irradiation, the conditioned medium was collected and used to grow endothelial cells. Conditioned media collected from mock, pEV and pSPARC transfected neuroblastoma cells were designated as mock-CM, pEV-CM and pSPARC-CM, respectively. All the experiments were performed in the presence of serum-free endothelial culture media to see if conditioned medium by itself had any effect on endothelial cells.
In vitro angiogenic assay
Angiogenesis was performed as described earlier [29] with minor modifications. HMECs (2×104 cells/well) were grown in the presence of CM in 96-well plates coated with growth factor reduced Matrigel and incubated for 16 hrs at 37°C. The formation of capillary-like structures was captured. The degree of angiogenesis was quantified for the number of branch points per view and cumulative tube length.
Mouse dorsal air sac model
Athymic nude mice were maintained within a specific-pathogen, germ-free environment. The implantation technique of the dorsal skin-fold chamber model has been described previously [29]. Briefly, diffusion chambers with mock, pEV- or pSPARC-transfected neuroblastoma cells (2×106) were placed underneath the skin into the superficial incision made horizontally along the edge of the dorsal air sac. After 10 days, the mice were carefully skinned around the implanted chambers, and the skin fold covering the chambers was photographed under a visible light microscope. The number of blood vessels within the chamber area of the air sac fascia was counted and quantitated.
Western blotting
Western blot analysis was performed as described previously [31]. Briefly, SPARC-overexpressed and irradiated neuroblastoma cells, Stattic treated (10nM for 30 minutes) or HMECs grown in tumor conditioned medium were lysed in radioimmunoprecipitation assay (RIPA) lysis buffer containing 1 mM sodium orthovanadate, 0.5 mM PMSF, 10 μg/mL aprotinin, and 10 μg/mL leupeptin. Equal amounts of total protein fractions of lysates were resolved by SDS-PAGE and transferred to PVDF membrane. The blot was blocked with 5% non-fat dry milk and probed overnight with primary antibodies followed by HRP-conjugated secondary antibodies. An ECL system was used to detect chemiluminescent signals. All blots were re-probed with GAPDH antibody to confirm equal loading.
RT-PCR and RT2 Profiler™ PCR Array
Neuroblastoma cells were transfected with mock, pEV or pSPARC and irradiated as described above. Total RNA was extracted from these cells and cDNA synthesized using poly-dT primers as described earlier [30]. PCR was performed using the following primers: SPARC: 5′-GGAAGAAACTGTGGCAGAGG-3′ (sense), and 5′-ATTGCTGCACACCTTCTCAA-3′ (antisense); GAPDH: 5′-TGAAGGTCGGAGTCAACGGATTTGGT-3′ (sense), and 5′-CATGTGGGCCATGAGGTCCACCAC-3′ (antisense). Quantification of SPARC mRNA levels was determined based on densitometry. For the human angiogenesis signaling pathway RT2 Profiler™ PCR Array, the above synthesized cDNA was used according to the manufacturer’s instructions.
Immunocytochemical analysis for Ki-67 index
Immunocytochemical analysis was done as described previously [30]. Briefly, HMECs (5×103 cells/well) were seeded in 8-well chamber slides and cultured for 72 hrs in tumor cell conditioned medium from neuroblastoma cells transfected with either mock, pEV or pSPARC. The effects of the conditioned medium on HMEC cellular proliferation were measured by analysis for Ki-67 immunoreactivity. Cells were fixed in cold methanol and permeabilized in 0.1% Triton X-100 in PBS. After blocking with 1% BSA in PBS for 1 h at room temperature (RT), cells were incubated overnight with anti-Ki-67 (1:100 dilution). Mouse IgG was used as a negative control. After incubation with HRP-conjugated secondary antibody (1:200 dilution) for 1 hr, 3,3-diaminobenzidine solution (Sigma, St. Louis, MO) was used for developing chromogen and counterstained with hematoxylin and mounted. The bright field images were captured with an Olympus BX-60 research fluorescence microscope attached to a CCD camera.
Cell proliferation assays
Cell growth rate was determined using a modified 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay as a measurement of mitochondrial metabolic activity as described earlier [32]. Cells were transfected with indicated plasmids and incubated at 37°C. After 0 to 96 hrs, MTT reagent was added, and cells were incubated for 4 hrs at 37°C. After removing the medium, formazan crystals were dissolved in DMSO, the absorbance at 550 nm was read using a microplate spectrophotometer and the results were expressed graphically.
Terminal deoxy nucleotidyl transferase-mediated nick labeling (TUNEL) assay and immunohistochemistry
Induction of apoptosis in HMECs cultured on conditioned medium as well as in the xenograft tumor tissue sections of pSPARC-treated mice was detected using TUNEL enzyme reagent (Roche) following the manufacturer’s instructions and as described previously [30]. Briefly, 5 × 103 HMECs were cultured in CM for 72 hrs (fresh CM was replaced every 24 hrs), fixed in 4% paraformaldehyde in PBS for 1 hr at room temperature (RT), and permeabilized with 0.1% Triton-X100 in 0.1% sodium citrate in PBS for 2 min (for cells) or 10 min (for tissue sections) on ice. The samples were incubated in TUNEL reaction mixture in a humidified atmosphere at 37°C for 1 hr in the dark. After TUNEL, immunohistochemical analysis was performed for tissue sections using anti-CD31 antibody as described previously [30]. Images were captured with an Olympus BX 60 research fluorescence microscope attached to a CCD camera and cells were counted. The apoptotic index was defined as follows: apoptotic index (%) = 100 × (apoptotic cells/total cells).
Intra-adrenal tumor model and immunohistochemistry
The Institutional Animal Care and Use Committee at the University of Illinois College of Medicine at Peoria approved all experimental procedures involving the use of animals. Orthotopic, localized neuroblastoma tumors were established in C.B-17 SCID mice by injection of 1×106 NB-1691 cells in 100 μL of PBS intra adrenally as described earlier [33]. After 2 weeks of tumor cell implantation, the mice were separated into six groups containing 6 animals per group, and each group was injected intravenously with PBS (mock) or pEV or pSPARC (3–6mg/kg body weight; 100 μL volume) in three doses on alternate days. Between the first and the second injections, and the second and the third injections, one group was radiated with a dose of 5 Gy each time. Mice were euthanized when they had lost >20% of body weight or had trouble ambulating, feeding, or grooming. One week following the last treatment, tumors were removed and either fixed in 10% phosphate-buffered formaldehyde or snap frozen and maintained at −70 °C until sectioning. Briefly, all tumors were serially sectioned and tissue sections (5 μm thick) obtained from the paraffin blocks were stained with hematoxylin and eosin (H&E) using standard histologic techniques. For immunohistochemical analysis, sections were incubated with primary antibody for 1 hr at room temperature followed by the appropriate secondary antibody. For HRP-conjugated secondary antibodies, we used DAB solution as the chromogen. Negative control slides were obtained by nonspecific IgG. Sections were mounted with mounting solution and analyzed with an inverted microscope. The microvessel density (CD31 positive area) was quantified as described previously [34].
Data and Statistical analysis
Quantification of band intensities and in vitro capillary tube formation were done by using ImageJ software (NIH). All data are presented as means ± standard deviation (SD) of at least three independent experiments, each performed at least in triplicate. One way analysis of variance (ANOVA) combined with the Tukey post-hoc test of means was used for multiple comparisons. Statistical differences are presented at probability levels of p < 0.05, p < 0.01 and p < 0.001.
Results
SPARC overexpression inhibits tumor-induced angiogenesis
We have shown previously that the overexpression of SPARC in medulloblastoma inhibited tumor growth and tumor-induced angiogenesis in a mouse model [29]. Previous studies demonstrated that a sub-lethal dose of radiation-treatment induces pro-angiogenic molecules in tumors [18–23] and radiation inhibits expression levels of SPARC in neuroblastoma cells [35]. In the present study, we measured the SPARC expression in SK-N-AS and NB-1691 neuroblastoma cells transfected with either mock, pEV, or pSPARC and irradiated. The SPARC protein and mRNA levels were increased in pSPARC-transfected neuroblastoma cells when compared to mock or pEV-transfected cells. We observed a ~3- to 4-fold increase in SPARC protein and mRNA levels in pSPARC-transfected cells compared to controls (Supplementary Fig. 1a). However, SPARC expression was inhibited significantly by IR (8Gy) compared to un-treated controls (Supplementary Fig. 1a). Next, tumor conditioned medium (CM) from SK-N-AS and NB-1691 neuroblastoma cells transfected with either mock (Mock-CM), pEV (pEV-CM) or pSPARC (pSPARC-CM) and irradiated (IR-CM, pEV-IR-CM or pSPARC-IR-CM) were used to induce a capillary-like network formation in an in vitro angiogenic assay. Mock-CM and pEV-CM elicited a strong angiogenic response and induced HMECs to differentiate into capillary-like structures within 16 hrs (Figs. 1a and 1b). In contrast, pSPARC-CM inhibited microvessel morphogenesis (Figs. 1a and 1b). Cells grown in endothelial culture medium without serum (SFM) were just beginning to differentiate into capillaries at the 16-hrs time point (inset in Fig. 1a). Quantification indicated about 60–75% decrease in branch points and a 70–80% decrease in tube length in HMECs cultured on pSPARC-CM in combination with radiation when compared with HMECs cultured on mock-CM or pEV-CM with or without radiation (Fig. 1b). Further, we measured secreted SPARC in conditioned medium by western blot analysis. Figure 1c shows that SPARC-overexpressed neuroblastoma cells led to a 2–3 fold increase in the secretion of SPARC into the CM compared to mock and pEV-overexpressed cells. In contrast, the secretion of SPARC into CM was decreased by 30–40% in IR-treated neuroblastoma cells compared to untreated control cells (Fig. 1c).
Figure 1. SPARC overexpression inhibits tumor-induced angiogenesis.

Neuroblastoma cells (SK-N-AS and NB-1691) were seeded in 100mm Petri dishes and left overnight. Cells were transfected with mock (PBS), pEV or pSPARC and cultured for 24 hrs. Later the cells were irradiated with 8Gy and incubated for further 16 hrs in serum-free DMEM/F-12 50/50 medium. The tumor cell conditioned medium (CM) was aseptically collected, centrifuged for 5 min, and used to grow human dermal microvascular endothelial cells (HMECs). Inset: HMECs grown on normal serum free medium. (a) In vitro angiogenesis assay: CM was added into 96-well plates, which were coated with matrigel and pre-seeded with HMEC (2 × 104 cells/well). After overnight incubation at 37°C, cells were observed under the bright field microscope for formation of capillary-like structures. (b) The degree of angiogenic induction by Mock-CM, pEV-CM and pSPARC-CM alone or in combination with radiation was quantified for the numerical value of the product of the relative tube length and number of branch points per field. Columns, mean of three experiments; bars, SD. * p < 0.01 vs pEV-CM; ** p < 0.01 vs IR+pEV-CM. (c) Western blot analysis for SPARC in the CM from SK-N-AS and NB-1691cells. (d) In vivo angiogenic assay using the dorsal skin fold model as described in Materials and Methods. Ten days after implantation, the animals were sacrificed and the skin fold covering the diffusion chamber was observed under a bright field light microscope for the presence of tumor-induced neovasculature (indicated by arrows).
We also examined whether pSPARC could inhibit tumor angiogenesis in vivo as assessed by the dorsal window model. Implantation of a chamber containing mock or pEV–transfected neuroblastoma cells in the dorsal air sac chamber resulted in the development of microvessels (indicated by red arrows in Fig. 1d) with curved thin structures and many tiny bleeding spots. Further, the implantation of irradiated neuroblastoma cells significantly enhanced the number of microvessels with curved thin structures compared to untreated control cell implantations. In contrast, implantation of SPARC-overexpressed neuroblastoma cells alone or in combination with radiation treatment resulted in the development of a fewer microvessels when compared to EV-overexpressed cells alone or in combination with radiation (Fig. 1d).
pSPARC-CM inhibits proliferation and induces apoptosis in endothelial cells
To elucidate the effect of SPARC-overexpression on tumor induced angiogenesis, we investigated the effect of CM on the proliferation and survival of HMECs. We examined cell proliferation indices in HMECs grown on CM from mock, pEV, pSPARC overexpressed cells with or without IR-treatment. We assayed for the presence of the Ki-67 molecular marker (expressed in the G1, S, and G2 phases of the cell cycle), and following immunocytochemical analysis, we quantified the number of positive cells/microscopic field in all treatment groups at 400X magnification. Figure 2a indicates that pSPARC-IR-CM caused a decrease in the fraction of cells expressing the proliferation marker Ki-67 at 72 hrs. HMECs cultured on pSPARC-CM or pSPARC+IR-CM showed a significantly lower Ki-67 index at 50–60% when compared to HMECs grown on mock-CM or pEV-CM alone or in combination with radiation (Fig. 2a). We also confirmed Ki-67 data by performing MTT assay for cell proliferation. MTT proliferation assay revealed 40–60% reduced growth rate of HMECs cultured on pSPARC-CM when compared to HMECs cultured on Mock-CM or pEV-CM (Fig. 2b). These observations led us to check pSPARC-CM effect on HMECs death, for which we determined the presence of apoptosis by TUNEL assay and FACS analysis (Fig. 2c). TUNEL labeling indices in HMECs grown on mock-CM and pEV-CM showed barely any apoptosis for up to 72 hrs (Supplementary Fig. 1b). However, endothelial cells grown on pSPARC-CM displayed a time-dependent increase in apoptosis. At the 24 hrs time point, 5–7% of the cells were TUNEL-positive; 11–14% and 27–30% of the cells were TUNEL-positive at 48 hrs and 72 hrs time point, respectively (Supplementary Fig. 1b). Further, the SPARC overexpression-induced apoptotic population was increased significantly in combination with irradiation. The quantification of TUNEL data revealed that the induction of apoptosis in SPARC-overexpressed and irradiated cells was ~10% at the 24 hrs time point, ~20% and ~40% at 48 hrs and 72 hrs time points, respectively (Supplementary Fig. 1b). In addition, FACS analysis also revealed that at 72 hrs time point, about 35% HMECs grown on pSPARC-IR-CM were in the sub-G1 phase when compared to HMECs grown on mock-CM or pEV-CM. (Fig. 2c). We further confirmed endothelial cell apoptosis by western blot analysis for expression of Bcl-XL/S, and cleavage of caspases-3, -8 and PARP. CM from either pSPARC alone or in combination with radiation showed increased levels of Bcl-XS expression and decreased levels of Bcl-XL expression, an indication of induction of apoptosis. Further, pSPARC-CM or pSPARC+IR-CM showed significant levels of cleaved caspases-3, -8 and PARP in HMECs when compared to mock-CM or pEV-CM alone or in combination with radiation (Fig. 2d).
Figure 2. Conditioned medium from SPARC overexpressed neuroblastoma cells inhibits endothelial cell survival and proliferation.

Tumor conditioned medium from SPARC-overexpressed and irradiated neuroblastoma cells was prepared as described in Materials and Methods. (a) Human dermal microvascular endothelial cells (HMECs) were grown on tumor cell conditioned medium from SPARC and radiation-treated neuroblastoma cells for 72 hrs. Proliferating HMECs were identified by Ki-67 indirect immunofluorescence. Values were calculated as the number of Ki67-positive cells/number of cells with intact nuclei (hematoxylin staining) at 400X magnification. Columns: mean of quadruplicate experiments; bars: SD. * p < 0.01 vs pEV-CM; ** p < 0.01 vs IR+pEV-CM. (b) HMEC (5 × 103 cells/well) were plated in 96-well plates and cultured in tumor cell conditioned medium for 0–96 hrs, and 20μL of 0.5 mg/mL MTT (3-4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium) reagent in PBS were added to cells. The cells were incubated for another 4 hrs. Next, the medium was removed from each well and DMSO (100μL) was added to each well to dissolve the formazan crystals. Absorbance values at 550 nm were measured with a microplate reader and the results were presented with the comparison of cells treated with vehicle. Points, mean of triplicate experiments. p < 0.05 pSPARC-CM vs pEV-CM; p < 0.01 pSPARC+IR-CM vs IR+pEV-CM. (c) HMECs were cultured in the presence of tumor cell conditioned medium for 72 hrs. The degree of apoptosis was determined by FACS analysis using propidium iodide staining. (d) HMECs were cultured in the presence of tumor cell conditioned medium for 72 hrs. The levels Bcl-XL/S, and cleavage of caspases-3, -8 and PARP were assessed by western blot analysis. Results are representative of three independent experiments. GAPDH served as a loading control.
pSPARC-CM suppresses PI3K/AKT signaling and inhibits VEGFR2 phosphorylation in HMECs
To understand the molecular basis of SPARC-mediated anti-angiogenic effects, we next examined the signaling pathways in HMECs cultured on CM. Accumulating evidence shows that VEGFR2 is the crucial and main receptor mediating angiogenic and vascular permeability activity [36]. Thus, we examined the action of pSPARC-CM on the phosphorylation of VEGFR2. The results showed that pSPARC-CM effectively inhibited the activity of VEGFR2 (Figs. 3a and 3b). We also found that pSPARC-CM significantly suppressed the activation of PI3K and Akt signaling (Figs. 3a and 3b), but not extracellular signal-regulated kinase (ERK) and focal adhesion kinase (FAK) (Fig. 3a). As a result of PI3K/Akt inhibition, the activation of mTOR and p70S6K kinase was assessed and found to be blocked. These data indicated that pSPARC-CM exerted its anti-angiogenic function primarily through blockade of the VEGFR2-mediated PI3K/Akt/mTOR pathway (Figs. 3a and 3b).
Figure 3. pSPARC-CM suppresses PI3K/AKT signaling in HMECs.

HMECs were cultured in the presence of tumor cell conditioned medium for 72 hrs. (a) Cell lysates were used to perform western blot analysis for VEGFR2, pVEGFR2, PI3K, pAKT, AKT, mTOR, p70S6K, pERK, ERK, pFAK and FAK. GAPDH served as a loading control. (b) Protein levels were quantified by densitometry as shown in the corresponding bar graph. All experiments were performed in triplicate with consistent results. Columns, mean of triplicate experiments; bars, SD. * p < 0.01 vs pEV; ** p < 0.01 vs IR+pEV.
pSPARC led to Notch-mediated inhibition of Stat3 signaling in neuroblastoma cells
General blockade of Notch signaling in tumor-bearing mice could lead to decreased angiogenesis in tumors; however, this is dependent on tumor cell type since general inhibition of Notch signaling might result in tumor regression, progression, or metastasis [37]. Our earlier studies in medulloblastoma showed inhibition of Notch signaling in SPARC-overexpressed cells [38]. Here, we sought to determine the effect of SPARC overexpression on Notch signaling and its role in neuroblastoma-induced angiogenesis. We therefore examined the effects of SPARC overexpression on the expression of Notch family members (Notch 1–4) and their downstream molecules Jagged, Delta, Hes1 and Hey1 in neuroblastoma cells. Western blot analysis revealed that Notch 1,2 and 4 were suppressed more than 55% in SPARC-overexpressed SK-N-AS and NB-1691 cells when compared to mock or pEV-transfected cells (Fig. 4a). It was shown that Stat3 was activated in the presence of active Notch, as well as the Notch effectors Hes1, Hes5, Hey, Hrt, Jagged and Delta [39–42]. We evaluated the levels of total Stat3 and its phosphorylation status in SPARC-overexpressed neuroblastoma cells. Stat3 phosphorylation was decreased more than 60% in SPARC-overexpressed neuroblastoma cells when compared to mock or pEV-transfected cells (Fig. 4b). Further, phosphorylation of Stat3 increased significantly by IR treatment; while, SPARC-overexpression inhibited radiation induced Stat3 phosphorylation remarkably (Fig. 4b). To further confirm the role of Notch signaling in the regulation of Stat3 and angiogenesis, we overexpressed Notch intra cellular domain (NICD) in the SPARC-overexpressed neuroblastoma cells. NICD overexpression in SPARC-overexpressed cells led to ~ 2–3 fold increase in active Notch levels and its downstream targets Hes1, Hey1 and Jagged. Further, since Stat3 is regulated by Notch downstream molecules Hes1 and Hey1, we analyzed Stat3 levels and its phosphorylation status. NICD and SPARC-overexpression in tumor cells showed 2 to 2.5 fold increase in Stat3 phosphorylation (Fig. 4c). Further, the CM collected from NICD and SPARC-overexpressed neuroblastoma cells increased angiogenic response and tube formation in HMECs when compared to SPARC-overexpression alone. The angiogenic response was comparable with the mock or empty vector-transfected cell conditioned medium-treated HMECs network formation (data not shown).
Figure 4. SPARC overexpression inhibits Notch-mediated Stat3 signaling in neuroblastoma cells.

Neuroblastoma cells (SK-N-AS and NB-1691) were transfected with mock (PBS), pEV or pSPARC and cultured for 24 hrs. Cells were irradiated with 8Gy and incubated for another 16 hrs. (a) Cell lysates were used to perform western blot analysis for Notch1, Notch2, Notch3, Notch4, Jagged, Delta, Hes1 and Hey1. GAPDH served as a loading control. Protein levels were quantified by densitometry as shown in the corresponding bar graph. Columns, mean of triplicate experiments; bars, SD. * p < 0.01 vs pEV; ** p < 0.01 vs IR+pEV. (b) Cell lysates were used to perform western blot analysis for phospho-Stat3 (Tyr-705), phospho-Stat3 (Ser-727) and total Stat3. GAPDH served as a loading control. Protein levels were quantified by densitometry as shown in the corresponding bar graph. Columns, mean of triplicate experiments; bars, SD. * p < 0.01 vs pEV; ** p < 0.01 vs IR+pEV. (c) Neuroblastoma cells (SK-N-AS and NB-1691) were transfected with either mock (PBS), pEV or pSPARC alone or in combination with pNICD and cultured for 24 hrs. The medium was then replaced with serum-free DMEM/F-12 50/50 medium, irradiated with 8Gy and incubated for another 16 hrs. Cell lysates were used to perform western blot analysis for Notch1 (120kDa endogenous; 80 kDa NICD fragment), Notch2, Hes1, Hey1, Jagged, phospho-Stat3 (Tyr 705), phospho-Stat3 (Ser 727) and Stat3. GAPDH served as a loading control. Protein levels were quantified by densitometry as shown in the corresponding bar graph. All experiments were performed in triplicate with consistent results. Columns, mean of triplicate experiments; bars, SD. * p < 0.01 vs pEV; ** p < 0.01 vs IR+pNICD; @ p < 0.01 vs pEV+IR; @@ p < 0.01 vs IR+pNICD.
pSPARC suppresses Stat3 signaling in neuroblastoma cells leading to decreased VEGF
Recent studies have also shown that Stat3 acts as a direct transcription activator of the VEGF gene [43;44]. Activation of Stat3 leads to tumor angiogenesis in vivo [36] and blocking Stat3 signaling in tumors can reduce tumor angiogenesis [43]. Our earlier publications also demonstrated that SPARC overexpression inhibited Stat3 signaling in medulloblastoma [38]. We therefore investigated whether interrupting Stat3 signaling by SPARC overexpression could block VEGF expression in neuroblastoma. To confirm these results, we overexpressed Stat3 after SPARC overexpression in neuroblastoma cells by using constitutively active Stat3 (pStat3). Stat3 overexpression in neuroblastoma cells resulted in the increase of VEGF otherwise suppressed by SPARC overexpression (Fig. 5a). Further, the CM collected from Stat3- and SPARC-overexpressed neuroblastoma cells showed increased angiogenic response and tube formation in HMECs when compared with pSPARC alone (Figs. 5b and 5c). The anti-angiogenic response of pSPARC was nullified by constitutively active Stat3 overexpression and is comparable to mock or empty vector-treated cell conditioned medium-treated HMECs network formation (Figs. 5b and 5c). We next examined whether Stat3 phophorylation (Tyr-705) has any influence on SPARC expression in neuroblastoma cell lines using a small molecule inhibitor (Stattic). Stattic inhibits Stat3 activation, dimerization and nuclear translocation [45]. Stat3 inhibitor did not affect the endogenous levels of SPARC in either SK-N-AS or NB-1691 neuroblastoma cell lines, indicating the downstream activity of Stat3 to SPARC (Fig. 5d). Taken together, these results show that Stat3 plays an important role in angiogenesis to downstream of SPARC in neuroblastoma cells.
Figure 5. SPARC overexpression inhibits Stat3-mediated angiogenesis in neuroblastoma cells.

(a) Neuroblastoma cells (SK-N-AS and NB-1691) were transfected with either with mock (PBS), pEV or pSPARC alone or in combination with pStat3 and cultured for 24 hrs. Cells were irradiated with 8Gy and incubated for another 16 hrs. Cell lysates were used for western blot analysis for phospho-Stat3 (Tyr 705), Stat3 and VEGF. GAPDH served as a loading control. (b) In vitro angiogenesis: neuroblastoma cells (SK-N-AS and NB-1691) were transfected with either mock (PBS), pEV or pSPARC alone or in combination with pStat3 and cultured for 24 hrs. The medium was then replaced with serum-free DMEM/F-12 50/50 medium, irradiated with 8Gy and incubated for another 16 hrs. The conditioned medium (CM) was collected and added into 96-well plates, which were coated with matrigel and pre-seeded with HMECs (2 × 104 cells/well). After overnight incubation (16 hrs) at 37°C, cells were observed under the bright field microscope for formation of capillary-like structures. (c) The degree of angiogenic induction by Mock-CM, pEV-CM and pSPARC-CM alone or in combination with radiation was quantified for the numerical value of the product of the relative tube length and number of branch points per field. The capillary length and number of branch points were reduced drastically in HMECs grown in conditioned medium from pSPARC-transfected neuroblastoma cells. Columns, mean of three experiments; bars, SD. * p < 0.01 vs pEV-CM; ** p < 0.01 vs pStat3; # p < 0.05 vs IR+pEV-CM. ## p < 0.01 vs IR+pStat3; @ p <0.01 vs pEV-CM and not significant difference from IR+pEV and pStat3. (d) Neuroblastoma cells (SK-N-AS and NB-1691) were treated with 10nM Stattic for 30 minutes and incubated overnight in serum-free medium. Western blot analysis was performed on cell lysates and CM for pStat-3 and SPARC, respectively.
SPARC overexpression inhibits pro-angiogenic molecules and induces anti-angiogenic molecules in neuroblastoma cells
To better determine the mechanisms underlying SPARC-mediated inhibition of angiogenesis, we used the human angiogenesis signaling pathway RT2 Profiler PCR array to profile the expression of 84 genes related to the angiogenesis signaling pathway. Total RNA from mock, pEV- or pSPARC-transfected neuroblastoma cells were used to synthesize cDNA. Real-time PCR was performed following the manufacturer’s instructions and fold change of mRNA expression was calculated on the basis of Ct values. It was evident from the results that SPARC overexpression led to decreased expression of pro-angiogenic factors (e.g., VEGF, FGF, EGF and MMP-9), as well as increased expression of anti-angiogenic factors (e.g., TIMP-3 and transforming growth factor-β; Fig 6a). To confirm the PCR array results, protein levels of pro-angiogenic molecules in cell lysates from mock, pEV- and pSPARC-transfected cells were assessed using western blotting (Fig. 6b). When normalized to controls, densitometry analysis revealed about 60–70% decrease in VEGF, MMP-9 in SPARC-overexpressed neuroblastoma cells when compared to controls (Fig. 6b).
Figure 6. SPARC-overexpression inhibits expression of pro-angiogenic molecules and inhibits anti-angiogenic molecules in neuroblastoma cells.
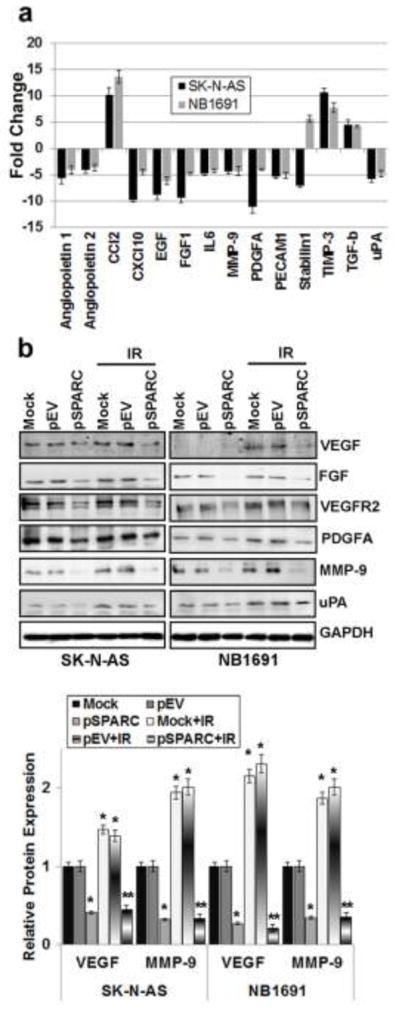
(a) Neuroblastoma cells (SK-N-AS and NB-1691) were transfected with pEV and pSPARC. cDNA was synthesized using a commercial kit and used in the human angiogenesis signaling pathway RT2 Profiler™ PCR Array to profile the expression of 84 genes related to the angiogenesis signaling pathway. Graph showing the effect of SPARC overexpression on increased/decreased expression of several angiogenic factors compared to pEV transfected cells. All experiments were performed in triplicate with consistent results. Columns, mean of triplicate experiments; bars, SD (p < 0.01 vs pEV). (b) Neuroblastoma cells (SK-N-AS and NB-1691) were transfected with mock (PBS), pEV or pSPARC and cultured for 24 hrs. Cells were irradiated with 8Gy and incubated for another 16 hrs. Total cell lysates were used to perform western blot analysis for the angiogenic molecules: VEGF, FGF, VEGFR2, PDGFA, MMP-9, and uPA. GAPDH served as a loading control. VEGF and MMP-9 protein levels were quantified by densitometry as shown in the corresponding bar graph. All experiments were performed in triplicate with consistent results. Columns, mean of triplicate experiments; bars, SE. * p < 0.01 vs pEV; ** p < 0.01 vs IR+pEV.
SPARC overexpression in orthotopic neuroblastoma model suppresses angiogenesis and tumor growth in vivo
We showed that the SPARC overexpression in neuroblastoma cells inhibited angiogenesis using capillary formation assay in vitro and an in vivo dorsal air sac assay. We next sought to determine whether the tumor growth inhibition observed in an orthotopic model of neuroblastoma after pSPARC treatment was due to the extension of anti-angiogenic activity. To evaluate this phenomenon in vivo, we stereotactically implanted NB-1691 cells intra-adrenally in SCID mice. The tumors that arose were challenged with intravenous injections of pSPARC along with or without irradiation (2 × 5 Gy). There was a significant decrease in the tumor volume in mice treated with pSPARC when compared with mice treated with mock (PBS) and pEV. We observed decreased tumor growth in the pSPARC-treated mice compared to mock or pEV-treated mice. To determine SPARC overexpression in vivo, tumor sections were stained with human anti-SPARC anti-body. The tumor sections from pSPARC alone or in combination with radiation treatment showed intense staining for SPARC as compared to radiation alone or in combination with mock or the pEV treatment. Further, to examine whether SPARC caused Notch-mediated inhibition of angiogenesis, tumor sections were subjected to immunohistochemical analysis for Notch1, phospho-Stat3 (Tyr-705), and VEGF. Notch1, phospho-Stat3 (Tyr-705), and VEGF staining was significantly high in mock or pEV treatment and remarkably increased with irradiation treatment (Fig. 7a). However, a drastic reduction was observed in immunoreactivity of anti-Notch1, anti-phospho-Stat3 (Tyr-705), and anti-VEGF in tumor section from mice that received SPARC-treatment alone or in combination with irradiation compared to tumor sections from mice that received mock and pEV-treatment. Next, we analyzed immunoreactivity of anti-CD31 in the tumor sections from mock, pEV-, and pSPARC-treated SCID mice. The immunoreactivity of CD31 in the tissue sections from the tumors derived from mice that received each treatment provided some measure of vascularity. Compared with mock and pEV-treated mice, tumor sections from pSPARC-treated mice showed decreased CD31 (Fig. 7b). The quantification of the CD31 positive area shows a significant increase in tumor vasculature with radiation treatment compared to untreated controls. However, tumor vasculature in tumor sections from mice that received pSPARC-treatment was diminished significantly when compared to tumor sections from mice that received mock and pEV treatments (Fig. 7c). To confirm endothelial cell apoptosis, we performed TUNEL staining on the same sections. Endothelial cell apoptosis as determined by TUNEL and CD31 double staining was significantly higher in the tumor sections from mice treated with pSPARC when compared with the sections from mock and pEV-treated mice (Figs. 7b and 7c). These results suggest that overexpression of SPARC in a neuroblastoma orthotopic tumor model inhibited angiogenesis through induction of apoptosis in endothelial cells.
Figure 7. SPARC overexpression alone or in combination with radiation decreased angiogenesis and induced endothelial cell apoptosis in neuroblastoma tumors.

Representative sections (400X magnification) from mock and pEV-treated tumors and those treated with pSPARC alone or in combination with radiation. (a) Paraffin-embedded sections of tumors were used for Hematoxylin and Eosin (H&E) staining and immunohistochemical analysis for SPARC, VEGF, phospho-Stat3 (Tyr-705), Notch1 and Hes1. (Inset: Negative control.) (b) Dual staining for TUNEL-positive and CD31 positive cells in xenograft tissue sections from mock, pEV, pSPARC alone or in combination with irradiation (2 × 5Gy)-treated mice was performed as described in Materials and Methods. (Inset: Negative control.) (c) CD31 positive blood vessels were quantified as vessel area per optical field and data represent mean of 9–10 fields from 6 tumor sections. Columns, mean of 9–10 optical fields; bars, SD. * p < 0.01 vs pEV; ** p < 0.01 vs IR+pEV.
Discussion
Since tumors require blood supply for nutrition and growth, management of cancer is focused on developing treatments based on inhibiting tumor angiogenesis. This “anti-angiogenic” therapy has now been documented to prolong the survival of patients with several types of cancer in large randomized clinical trials [46–49]. In this study, we report that SPARC is a strong inhibitor of tumor-induced angiogenesis, and SPARC overexpression in tumors lead to endothelial cell apoptosis, which resulted in decreased angiogenesis. These two processes can lead to decreased tumor growth in neuroblastoma upon overexpression of SPARC. Our study shows that SPARC overexpression in neuroblastoma cells develop a primary response to angiogenesis by inhibiting the angiogenic growth factors and this eventually triggers endothelial cell apoptosis. Angiogenesis is mediated by multiple regulatory factors, such as growth factors, adhesion molecules, and matrix-degrading enzymes [50]. Earlier publications suggest that activation of endothelial cell proliferation and migration is mainly regulated by receptor tyrosine kinase ligands such as vascular endothelial growth factor (VEGF), fibroblast growth factor-2 (FGF-2), platelet derived growth factor (PDGF), epidermal growth factor (EGF), transforming growth factor-alpha (TGF-α), and angiopoietins (Ang-1 and Ang-2) [50]. In this study, we observed that the tumor conditioned medium (CM) from irradiated neuroblastoma cells enhanced the degree of capillary tube formation in endothelial cells compared to CM from non-irradiated control cells. In contrast, the CM from SPARC-overexpressed neuroblastoma cells with or without radiation inhibited capillary tube formation in endothelial cells as compared to the controls. We also observed decreased proliferative index Ki-67 and increased apoptosis in endothelial cells grown in the presence of CM from neuroblastoma cells with SPARC-overexpression alone or in combination of radiation.
The PI3K/Akt pathway is one of the central pathways involved in survival signaling [51]. Several receptors, including those for VEGF [52] and IGF-1 [53], transmit survival signals through these pathways. To elucidate the molecular basis of SPARC-mediated anti-angiogenic effects and endothelial cell death, we next examined the signaling pathways in treated HMECs. CM from SPARC-overexpressed neuroblastoma cells with or without irradiation led to an increased endothelial cell death when compared to CM from cells treated with mock or empty vector-transfected cells alone or in combination with radiation. We observed that irradiation significantly induced expression of PI3K/Akt pathway molecules in neuroblastoma cells and overexpression of SPARC in these cells significantly inhibited expression of PI3K/Akt pathway molecules. The downstream targets of PI3K/Akt pathway, mTOR and p70S6K, were also inhibited by overexpression of SPARC in neuroblastoma cells. Several reports suggest that the serine/threonine kinase mTOR is an important downstream target of PI3K/Akt, and mTOR signaling has been shown to be involved in the control of cell growth and proliferation [54]. Recent publications have demonstrated that 15(S)-hydroxyeicosatetraenoic acid activates the PI3K/Akt/mTOR /p70S6K pathway and activation of this pathway induces tube formation and migration of human dermal microvascular endothelial cells in vitro and Matrigel plug angiogenesis in vivo [55].
The Notch pathway is involved in numerous aspects of vascular development and angiogenesis [43;44;56]. Recent evidences indicate that Notch signaling from tumor cells is able to activate endothelial cells and trigger tumor angiogenesis in vitro and in a xenograft mouse tumor model [56]. Selective interruption of Notch signaling within tumors may provide an anti-angiogenic strategy [56]. In this study, we observed that irradiation induced Notch and its downstream targets in neuroblastoma cells, and overexpression of SPARC alone or in combination with radiation suppressed Notch signaling molecules and their downstream targets, thereby leading to inhibition of tumor-induced angiogenesis. Further, as Stat3 is regulated by Notch downstream molecules Hes1, Delta, Jagged and Hey1, and recent studies have also shown that Stat3 is a direct transcription activator of the VEGF gene [43;44], we analyzed Stat3 phosphorylation status. Our results show that SPARC overexpression in neuroblastoma cells inhibited the Stat3 phosphorylation at Tyr-705 and Ser-727. To further confirm the role of Notch and Stat3 in neuroblastoma-induced angiogenesis, we overexpressed Notch intracellular domain (NICD) or constitutively active Stat3 (pStat3) in neuroblastoma cells after SPARC overexpression. These studies show that Notch signaling regulates Stat3-mediated VEGF expression in neuroblastoma.
We next carried out experiments designed to identify signaling molecules secreted by cancer cells that might be responsible for the observed effect on endothelial cells. It is known that endothelial cell rearrangement leading to tubule formation is primarily established by activation of VEGF and its receptor [30]. VEGFR2 is the main human receptor responsible for both physiological and pathological vascular development, and the VEGF-VEGFR2 signaling pathway has become an important target for the development of anti-angiogenic agents [57]. To evaluate this hypothesis, we performed angiogenesis PCR array analysis for pro- and anti-angiogenesis molecules in SPARC-overexpressed neuroblastoma cells and compared it with pEV-overexpressed cells. The PCR array analysis showed that SPARC overexpression alone led to decreased expression of pro-angiogenic factors (e.g., VEGF, FGFR, ECGF and MMP-9) as well as increased expression of anti-angiogenic factors (e.g., TIMP-3 and TGF-β). Similarly, results from the western blot analysis also showed the downregulation of some potent angiogenic molecules, including MMP-9, VEGF, PDGFR and FGF. These results suggest that SPARC expression alters the angiogenic balance in the tumor microenvironment by altering the expression of a complex array of inhibitors and stimuli. Growth factors, such as VEGF or PDGF, act directly on endothelial cells and/or activate inflammatory cells (monocytes and T lymphocytes), which in turn synthesize angiogenic factors [58]. Our previous studies demonstrated that SPARC regulates additional components and coordinates the activity of growth factors on endothelial cell proliferation and migration [29]. Other studies demonstrated that SPARC regulates additional components and coordinates the activity of growth factors on endothelial cell proliferation and migration [59]. SPARC antagonised the migratory response of endothelial cells to bFGF without the binding of SPARC to bFGF or the blocking of the ligand–receptor interaction [59]. SPARC modulates glioma growth by altering the tumor microenvironment and suppressing tumor vascularity through suppression of VEGF expression and secretion. [11]. In this context, the suppression of angiogenesis-mediated tumor growth by SPARC in neuroblastoma seems to be the consequence of its ability to inhibit the expression of angiogenic factors in tumor tissues, which may in turn inhibit capillary infiltration into tumors.
Our in vivo results demonstrated that Notch signaling regulated Stat3 and its downstream pro-angiogenic molecules were inhibited by SPARC overexpression. The expression of phosphorylated Stat3 and VEGF and density of vasculature were increased in tumor sections from mice treated with irradiation compared to tumor sections from mock and pEV-treated controls. SPARC-overexpression alone or in combination with radiation suppressed the expression levels of phospho-Stat3 and VEGF and vascular density in tumor sections. Further, we demonstrated that the SPARC overexpression induced endothelial cell death in the tumor core, which is an indication of the destruction of blood vessels in tumors. Taken together, these results suggest that, upon SPARC overexpression in neuroblastoma cells, Notch signaling is inhibited and leads to decreased Stat3 activation, which results in inhibition of tumor-induced angiogenesis.
Although, radiotherapy is considered to be a vital component of the therapeutic regimen for advanced neuroblastoma it can induce the expression of various tumor promoting proteins including pro-angiogenic molecules [18–23]. Our work provided insights on molecular mechanisms and evidence that SPARC-overexpression in neuroblastoma tumor cells inhibits radiation-induced, Notch-mediated Stat3 activation in tumors cells through decreased PI3K/Akt/mTOR/P70S6K pathway in endothelial cells. This in turn, results in decreased endothelial cell proliferation and increased endothelial cell apoptosis. We have also established the importance of these pathways in the induction of endothelial cell apoptosis and tube formation. These data provide insights about the mechanisms of SPARC overexpression in tumor cells, which led to decreased angiogenesis and resulted in tumor growth inhibition. This study provides insights into the possible functional roles of SPARC in neuroblastoma angiogenesis, and the data demonstrate that SPARC expression is inversely correlated to neuroblastoma tumor angiogenesis and growth in vivo.
Supplementary Material
Acknowledgments
The authors thank Alicia Woodworth for technical assistance and Diana Meister and Sushma Jasti for manuscript review. We also thank Dr. P. Houghton (St. Jude Children’s Research Hospital, Memphis, TN) for providing NB-1691 neuroblastoma cell line and Dr. Francisco J Candal (Centers for Disease Control and Prevention, Atlanta, GA, USA) for providing HMEC cells.
This project was supported by award number CA147792 (to J.S.R.) from the National Institutes of Health (NIH). Contents are solely the responsibility of the authors and do not necessarily represent the official views of NIH. The funding agency had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Ethical Standards
The experiments comply with the current laws of the United States of America.
Conflict of interest
The authors disclose no conflict of interest.
Reference List
- 1.Morowitz MJ, Barr R, Wang Q, King R, Rhodin N, Pawel B, et al. Methionine aminopeptidase 2 inhibition is an effective treatment strategy for neuroblastoma in preclinical models. Clin Cancer Res. 2005;11(7):2680–2685. doi: 10.1158/1078-0432.CCR-04-1917. [DOI] [PubMed] [Google Scholar]
- 2.Matthay KK, Villablanca JG, Seeger RC, Stram DO, Harris RE, Ramsay NK, et al. Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. Children’s Cancer Group. N Engl J Med. 1999;341(16):1165–1173. doi: 10.1056/NEJM199910143411601. [DOI] [PubMed] [Google Scholar]
- 3.Meadows AT, Tsunematsu Y. Late effects of treatment for neuroblastoma. In: Brodeur GM, Sawada T, Tsuchida Y, Voute PA, editors. Neuroblastoma. Amsterdam: Elsevier Science B.V; 2000. pp. 561–570. [Google Scholar]
- 4.Meitar D, Crawford SE, Rademaker AW, Cohn SL. Tumor angiogenesis correlates with metastatic disease, N-myc amplification, and poor outcome in human neuroblastoma. J Clin Oncol. 1996;14(2):405–414. doi: 10.1200/JCO.1996.14.2.405. [DOI] [PubMed] [Google Scholar]
- 5.Brekken RA, Sage EH. SPARC, a matricellular protein: at the crossroads of cell-matrix. Matrix Biol. 2000;19(7):569–580. doi: 10.1016/s0945-053x(00)00105-0. [DOI] [PubMed] [Google Scholar]
- 6.Martinek N, Shahab J, Sodek J, Ringuette M. Is SPARC an evolutionarily conserved collagen chaperone? J Dent Res. 2007;86(4):296–305. doi: 10.1177/154405910708600402. [DOI] [PubMed] [Google Scholar]
- 7.Framson PE, Sage EH. SPARC and tumor growth: where the seed meets the soil? J Cell Biochem. 2004;92(4):679–690. doi: 10.1002/jcb.20091. [DOI] [PubMed] [Google Scholar]
- 8.Bradshaw AD, Sage EH. SPARC, a matricellular protein that functions in cellular differentiation and tissue response to injury. J Clin Invest. 2001;107(9):1049–1054. doi: 10.1172/JCI12939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sage H, Johnson C, Bornstein P. Characterization of a novel serum albumin-binding glycoprotein secreted by endothelial cells in culture. J Biol Chem. 1984;259(6):3993–4007. [PubMed] [Google Scholar]
- 10.Chlenski A, Guerrero LJ, Peddinti R, Spitz JA, Leonhardt PT, Yang Q, et al. Anti-angiogenic SPARC peptides inhibit progression of neuroblastoma tumors. Mol Cancer. 2010;9:138. doi: 10.1186/1476-4598-9-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yunker CK, Golembieski W, Lemke N, Schultz CR, Cazacu S, Brodie C, et al. SPARC-induced increase in glioma matrix and decrease in vascularity are associated with reduced VEGF expression and secretion. Int J Cancer. 2008;122(12):2735–2743. doi: 10.1002/ijc.23450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rossler J, Taylor M, Geoerger B, Farace F, Lagodny J, Peschka-Suss R, et al. Angiogenesis as a target in neuroblastoma. Eur J Cancer. 2008;44(12):1645–1656. doi: 10.1016/j.ejca.2008.05.015. [DOI] [PubMed] [Google Scholar]
- 13.Eggert A, Ikegaki N, Kwiatkowski J, Zhao H, Brodeur GM, Himelstein BP. High-level expression of angiogenic factors is associated with advanced tumor stage in human neuroblastomas. Clin Cancer Res. 2000;6(5):1900–1908. [PubMed] [Google Scholar]
- 14.Engebraaten O, Schwachenwald R, Valen H, Bjerkvig R, Laerum OD, Backlund EO. Effects of high and low single dose irradiation on glioma spheroid invasion into normal rat brain tissue in vitro. Anticancer Res. 1992;12(5):1501–1506. [PubMed] [Google Scholar]
- 15.Li T, Zeng ZC, Wang L, Qiu SJ, Zhou JW, Zhi XT, et al. Radiation enhances long-term metastasis potential of residual hepatocellular carcinoma in nude mice through TMPRSS4-induced epithelial-mesenchymal transition. Cancer Gene Ther. 2011 doi: 10.1038/cgt.2011.29. [DOI] [PubMed] [Google Scholar]
- 16.Karar J, Maity A. Modulating the tumor microenvironment to increase radiation responsiveness. Cancer Biol Ther. 2009;8(21):1994–2001. doi: 10.4161/cbt.8.21.9988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lorusso G, Ruegg C. The tumor microenvironment and its contribution to tumor evolution toward metastasis. Histochem Cell Biol. 2008;130(6):1091–1103. doi: 10.1007/s00418-008-0530-8. [DOI] [PubMed] [Google Scholar]
- 18.Adair JC, Baldwin N, Kornfeld M, Rosenberg GA. Radiation-induced blood-brain barrier damage in astrocytoma: relation to elevated gelatinase B and urokinase. J Neurooncol. 1999;44(3):283–289. doi: 10.1023/a:1006337912345. [DOI] [PubMed] [Google Scholar]
- 19.Bivik CA, Larsson PK, Kagedal KM, Rosdahl IK, Ollinger KM. UVA/B-induced apoptosis in human melanocytes involves translocation of cathepsins and Bcl-2 family members. J Invest Dermatol. 2006;126(5):1119–1127. doi: 10.1038/sj.jid.5700124. [DOI] [PubMed] [Google Scholar]
- 20.Heissig B, Rafii S, Akiyama H, Ohki Y, Sato Y, Rafael T, et al. Low-dose irradiation promotes tissue revascularization through VEGF release from mast cells and MMP-9-mediated progenitor cell mobilization. J Exp Med. 2005;202(6):739–750. doi: 10.1084/jem.20050959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hovdenak N, Karlsdottir A, Sorbye H, Dahl O. Profiles and time course of acute radiation toxicity symptoms during conformal radiotherapy for cancer of the prostate. Acta Oncol. 2003;42(7):741–748. doi: 10.1080/02841860310011302. [DOI] [PubMed] [Google Scholar]
- 22.Trog D, Yeghiazaryan K, Fountoulakis M, Friedlein A, Moenkemann H, Haertel N, et al. Pro-invasive gene regulating effect of irradiation and combined temozolomide-radiation treatment on surviving human malignant glioma cells. Eur J Pharmacol. 2006;542(1–3):8–15. doi: 10.1016/j.ejphar.2006.05.026. [DOI] [PubMed] [Google Scholar]
- 23.Zhai GG, Malhotra R, Delaney M, Latham D, Nestler U, Zhang M, et al. Radiation enhances the invasive potential of primary glioblastoma cells via activation of the Rho signaling pathway. J Neurooncol. 2006;76(3):227–237. doi: 10.1007/s11060-005-6499-4. [DOI] [PubMed] [Google Scholar]
- 24.Knizetova P, Ehrmann J, Hlobilkova A, Vancova I, Kalita O, Kolar Z, et al. Autocrine regulation of glioblastoma cell cycle progression, viability and radioresistance through the VEGF-VEGFR2 (KDR) interplay. Cell Cycle. 2008;7(16):2553–2561. doi: 10.4161/cc.7.16.6442. [DOI] [PubMed] [Google Scholar]
- 25.Zheng M, Morgan-Lappe SE, Yang J, Bockbrader KM, Pamarthy D, Thomas D, et al. Growth inhibition and radiosensitization of glioblastoma and lung cancer cells by small interfering RNA silencing of tumor necrosis factor receptor-associated factor 2. Cancer Res. 2008;68(18):7570–7578. doi: 10.1158/0008-5472.CAN-08-0632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vicini FA, Kestin L, Huang R, Martinez A. Does local recurrence affect the rate of distant metastases and survival in patients with early-stage breast carcinoma treated with breast-conserving therapy? Cancer. 2003;97(4):910–919. doi: 10.1002/cncr.11143. [DOI] [PubMed] [Google Scholar]
- 27.Lee CG, Heijn M, diTomaso E, Griffon-Etienne G, Ancukiewicz M, Koike C, et al. Anti-Vascular endothelial growth factor treatment augments tumor radiation response under normoxic or hypoxic conditions. Cancer Res. 2000;60(19):5565–5570. [PubMed] [Google Scholar]
- 28.Sofia VI, Martins LR, Imaizumi N, Nunes RJ, Rino J, Kuonen F, et al. Low doses of ionizing radiation promote tumor growth and metastasis by enhancing angiogenesis. PLoS One. 2010;5(6):e11222. doi: 10.1371/journal.pone.0011222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bhoopathi P, Chetty C, Gujrati M, Dinh DH, Rao JS, Lakka SS. The role of MMP-9 in the anti-angiogenic effect of secreted protein acidic and rich in cysteine. Br J Cancer. 2010;102(3):530–540. doi: 10.1038/sj.bjc.6605538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chetty C, Lakka SS, Bhoopathi P, Kunigal S, Geiss R, Rao JS. Tissue inhibitor of metalloproteinase 3 suppresses tumor angiogenesis in matrix metalloproteinase 2-down-regulated lung cancer. Cancer Res. 2008;68(12):4736–4745. doi: 10.1158/0008-5472.CAN-07-6612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bhoopathi P, Chetty C, Kunigal S, Vanamala SK, Rao JS, Lakka SS. Blockade of tumor growth due to matrix metalloproteinase-9 inhibition is mediated by sequential activation of beta1-integrin, ERK, and NF-kappaB. J Biol Chem. 2008;283(3):1545–1552. doi: 10.1074/jbc.M707931200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Chetty C, Bhoopathi P, Rao JS, Lakka SS. Inhibition of matrix metalloproteinase-2 enhances radiosensitivity by abrogating radiation-induced FoxM1-mediated G2/M arrest in A549 lung cancer cells. Int J Cancer. 2009;124:2468–2477. doi: 10.1002/ijc.24209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tivnan A, Tracey L, Buckley PG, Alcock LC, Davidoff AM, Stallings RL. MicroRNA-34a is a potent tumor suppressor molecule in vivo in neuroblastoma. BMC Cancer. 2011;11:33. doi: 10.1186/1471-2407-11-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xue Y, Cao R, Nilsson D, Chen S, Westergren R, Hedlund EM, et al. FOXC2 controls Ang-2 expression and modulates angiogenesis, vascular patterning, remodeling, and functions in adipose tissue. Proc Natl Acad Sci U S A. 2008;105(29):10167–10172. doi: 10.1073/pnas.0802486105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bhoopathi P, Gorantla B, Sailaja GS, Gondi CS, Gujrati M, Klopfenstein JD, et al. SPARC overexpression inhibits cell proliferation in neuroblastoma and is partly mediated by tumor suppressor protein PTEN and AKT. PLoS One. 2012;7(5):e36093. doi: 10.1371/journal.pone.0036093. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 36.Xue Y, Chen F, Zhang D, Lim S, Cao Y. Tumor-derived VEGF modulates hematopoiesis. J Angiogenes Res. 2009;1:9. doi: 10.1186/2040-2384-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu XB, Feng F, Wang YC, Wang L, He F, Dou GR, et al. Blockade of Notch signaling in tumor-bearing mice may lead to tumor regression, progression, or metastasis, depending on tumor cell types. Neoplasia. 2009;11(1):32–38. doi: 10.1593/neo.81008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bhoopathi P, Chetty C, Dontula R, Gujrati M, Dinh DH, Rao JS, et al. SPARC Stimulates Neuronal Differentiation of Medulloblastoma Cells via the Notch1/STAT3 Pathway. Cancer Res. 2011;71(14):4908–4919. doi: 10.1158/0008-5472.CAN-10-3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kamakura S, Oishi K, Yoshimatsu T, Nakafuku M, Masuyama N, Gotoh Y. Hes binding to STAT3 mediates crosstalk between Notch and JAK-STAT signalling. Nat Cell Biol. 2004;6(6):547–554. doi: 10.1038/ncb1138. [DOI] [PubMed] [Google Scholar]
- 40.Kluppel M, Wrana JL. Turning it up a Notch: cross-talk between TGF beta and Notch signaling. Bioessays. 2005;27(2):115–118. doi: 10.1002/bies.20187. [DOI] [PubMed] [Google Scholar]
- 41.Sakurai T, Kudo M. Signaling pathways governing tumor angiogenesis. Oncology. 2011;81 (Suppl 1):24–29. doi: 10.1159/000333256. [DOI] [PubMed] [Google Scholar]
- 42.Shi W, Harris AL. Notch signaling in breast cancer and tumor angiogenesis: cross-talk and therapeutic potentials. J Mammary Gland Biol Neoplasia. 2006;11(1):41–52. doi: 10.1007/s10911-006-9011-7. [DOI] [PubMed] [Google Scholar]
- 43.Niu G, Wright KL, Huang M, Song L, Haura E, Turkson J, et al. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene. 2002;21(13):2000–2008. doi: 10.1038/sj.onc.1205260. [DOI] [PubMed] [Google Scholar]
- 44.Wei D, Le X, Zheng L, Wang L, Frey JA, Gao AC, et al. Stat3 activation regulates the expression of vascular endothelial growth factor and human pancreatic cancer angiogenesis and metastasis. Oncogene. 2003;22(3):319–329. doi: 10.1038/sj.onc.1206122. [DOI] [PubMed] [Google Scholar]
- 45.Schust J, Sperl B, Hollis A, Mayer TU, Berg T. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol. 2006;13(11):1235–1242. doi: 10.1016/j.chembiol.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 46.Assifi MM, Hines OJ. Anti-angiogenic agents in pancreatic cancer: a review. Anticancer Agents Med Chem. 2011;11(5):464–469. doi: 10.2174/187152011795677463. [DOI] [PubMed] [Google Scholar]
- 47.Jain RK, diTomaso E, Duda DG, Loeffler JS, Sorensen AG, Batchelor TT. Angiogenesis in brain tumours. Nat Rev Neurosci. 2007;8(8):610–622. doi: 10.1038/nrn2175. [DOI] [PubMed] [Google Scholar]
- 48.Sharma PS, Sharma R, Tyagi T. VEGF/VEGFR pathway inhibitors as anti-angiogenic agents: present and future. Curr Cancer Drug Targets. 2011;11(5):624–653. doi: 10.2174/156800911795655985. [DOI] [PubMed] [Google Scholar]
- 49.Tugues S, Koch S, Gualandi L, Li X, Claesson-Welsh L. Vascular endothelial growth factors and receptors: anti-angiogenic therapy in the treatment of cancer. Mol Aspects Med. 2011;32(2):88–111. doi: 10.1016/j.mam.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 50.Roy CS, Karmakar S, Banik NL, Ray SK. Targeting angiogenesis for controlling neuroblastoma. J Oncol. 2012;2012:782020. doi: 10.1155/2012/782020. Epub;%2011 Aug 25.:782020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Talapatra S, Thompson CB. Growth factor signaling in cell survival: implications for cancer treatment. J Pharmacol Exp Ther. 2001;298(3):873–878. [PubMed] [Google Scholar]
- 52.Gerber HP, McMurtrey A, Kowalski J, Yan M, Keyt BA, Dixit V, et al. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J Biol Chem. 1998;273(46):30336–30343. doi: 10.1074/jbc.273.46.30336. [DOI] [PubMed] [Google Scholar]
- 53.Wu W, Lee WL, Wu YY, Chen D, Liu TJ, Jang A, et al. Expression of constitutively active phosphatidylinositol 3-kinase inhibits activation of caspase 3 and apoptosis of cardiac muscle cells. J Biol Chem. 2000;275(51):40113–40119. doi: 10.1074/jbc.M004108200. [DOI] [PubMed] [Google Scholar]
- 54.Bhatt AP, Bhende PM, Sin SH, Roy D, Dittmer DP, Damania B. Dual inhibition of PI3K and mTOR inhibits autocrine and paracrine proliferative loops in PI3K/Akt/mTOR-addicted lymphomas. Blood. 2010;115(22):4455–4463. doi: 10.1182/blood-2009-10-251082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang B, Cao H, Rao GN. 15(S)-hydroxyeicosatetraenoic acid induces angiogenesis via activation of PI3K-Akt-mTOR-S6K1 signaling. Cancer Res. 2005;65(16):7283–7291. doi: 10.1158/0008-5472.CAN-05-0633. [DOI] [PubMed] [Google Scholar]
- 56.Li JL, Harris AL. Notch signaling from tumor cells: a new mechanism of angiogenesis. Cancer Cell. 2005;8(1):1–3. doi: 10.1016/j.ccr.2005.06.013. [DOI] [PubMed] [Google Scholar]
- 57.Lynn KD, Udugamasooriya DG, Roland CL, Castrillon DH, Kodadek TJ, Brekken RA. GU81, a VEGFR2 antagonist peptoid, enhances the anti-tumor activity of doxorubicin in the murine MMTV-PyMT transgenic model of breast cancer. BMC Cancer. 2010;10:397. doi: 10.1186/1471-2407-10-397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jendraschak E, Sage EH. Regulation of angiogenesis by SPARC and angiostatin: implications for tumor cell biology. Semin Cancer Biol. 1996;7(3):139–146. doi: 10.1006/scbi.1996.0019. [DOI] [PubMed] [Google Scholar]
- 59.Hasselaar P, Sage EH. SPARC antagonizes the effect of basic fibroblast growth factor on the migration of bovine aortic endothelial cells. J Cell Biochem. 1992;49(3):272–283. doi: 10.1002/jcb.240490310. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.