

Proteases are a major class of enzymes that catalyze the hydrolysis of peptide bonds to break down proteins into smaller pieces in a process known as proteolysis.[1] Ubiquitous in nature, proteases are present in all living cells and organisms, and are known to play a pivotal role in the development and control of many biological processes.[2] The importance of proteases is also manifested by a large number of pathological conditions that involve alteration of protease levels, including cancer, arthritis, as well as neurodegenerative and cardiovascular diseases.[3]
In view of the essential role played by proteases, there is a strong need to develop sensitive, accurate, and convenient methods for the detection of proteases in trace amounts. To this end, a large number of qualitative and quantitative methods involving organic chromopores have been developed. The main-stream detection mechanism is based on fluorescence resonance energy transfer (FRET), where a donor and an acceptor are attached to a protease substrate (a peptide or a protein) and the emission from the donor is suppressed due to energy transfer to the acceptor.[4] The fluorescence will be recovered whenever the donor and acceptor are separated from each other due to enzymatic cleavage of the substrate by a protease.[4] Alternatively, various methods based on the conventional bioassays and immunoassays have also been demonstrated with high sensitivity, albeit these methods are typically laborious, costly, and time-consuming.[5] A third approach involves the precipitation of an undigested substrate to measure the hydrolyzed portion by mass spectrometry or inductively coupled plasma mass spectrometry (ICP-MS).[6] Despite the great performance of all these methods, there is a strong need to develop a nanoscale platform that could potentially eliminate the requirement of complicate synthesis (e.g., conjugation of two chromophores to different sites of the same peptide), the limitation on sensitivity and diversity with regard to the enzymatic substrates, and involvement of multiple processing steps such as centrifugation and purification.
Herein we demonstrate a new class of nanoscale biosensors for fluorescence detection of proteases with high sensitivity. The detection mechanism is based on the change in fluorescence intensity for protein-protected Au clusters (AuCs) when a protease is introduced. Fluorescent AuCs, composed of a few to roughly a hundred Au atoms, are novel fluorescent nanomaterials that have continuously drawn research interest in the fields of chemistry, biology, and materials science owing to their ultrasmall size (a few nanometers), facile synthesis, good biocompatibility, and bright fluorescence in the visible and near-infrared regions.[7] Recently, fluorescent AuCs have been used as nanoscale probes to detect analytes such as Hg2+, Cu2+, and CN− by utilizing the fluorescence quenching effect derived from the interactions between AuCs and an analyte.[8] In addition, the fluorescence intensity from AuCs has been reported to depend on the surface ligand or environment, providing another mechanism for sensing.[9] As schematically illustrated in Figure 1, the emission from a protein-protected AuC will drop in intensity when the protein shell is degraded due to protease-catalyzed hydrolysis. We hypothesize that the fluorescence will be effectively quenched by the oxygen (O2) from ambient air once the protein shell is destructed by a protease. As a result, the presence of protease can be detected and quantified by monitoring the intensity of fluorescence from the AuCs. This one-step method is simple, fast, highly selective, and ultrasensitive. Owning to the good bio-inertness of Au, the fluorescent AuCs can be further developed into nanoscale probes for both in vitro and in vivo detection of proteases.[10]
Figure 1.
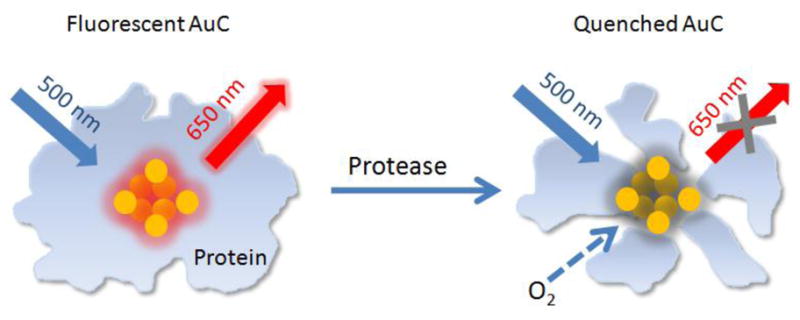
A schematic illustrating fluorescence detection of proteases using protein-protected AuCs. After degradation of the protein shell by a protease, the oxygen molecules (O2) from ambient air can penetrate through the shell and quench the fluorescence from AuC in the core.
We initially focused on fluorescent AuCs protected by bovine serum albumin (BSA-AuCs), which were prepared from chloroauric acid (HAuCl4) and BSA using a procedure previously reported by other group (Fig. S1).[11] Figure S2 shows the fluorescence spectrum of BSA-protected AuCs, presenting a broad emission peak at 650 nm. We demonstrated the concept by incubating BSA-AuCs with proteinase K (1 ×105 ng mL−1), a broad-spectrum serine protease, at 37 °C and pH 7.5 for 4 h. As shown by the fluorescence spectrum in Figure 2a, the emission peak at 650 nm completely disappeared after incubation for 4 h. To further evaluate the sensitivity of this new assay based on the AuCs, proteinase K solutions with designated concentrations from 0.1 ng mL−1 to 1 ×106 ng mL−1 were incubated with the same amount of BSA-AuCs for 4 h under identical conditions. As shown in Figure 2a, the intensity of fluorescence from BSA-AuCs proportionally dropped as the concentration of proteinase K increased. It is worth mentioning that an incubation time of 4 h was sufficient to completely degrade the BSA shells on AuCs, as indicated by a plateau in the fluorescence intensity for samples incubated for different periods of time with proteinase K at different concentrations (Fig. 2b). To ensure that the drop in fluorescence intensity was caused by degradation of the BSA shells by proteinase K, we performed a control experiment with deactivated proteinase K obtained by heating the enzyme at 95 °C for 20 min. As shown in Figure 2c, there was essentially no change to the fluorescence intensity when the BSA-AuCs were incubated with the deactivated proteinase K at concentrations up to 104 ng mL−1, confirming that the drop in fluorescence intensity was associated with the degradation of BSA shells. The linearity of this detection method was confirmed by plotting the peak intensity at 650 nm against log of proteinase K concentration. The linear relation over the range of 5 to 5 × 103 ng mL−1 (R2 = 0.994, Fig. S3) could be described by an equation of I/I0 = Alog(C) + B, where I and I0 denote the fluorescence intensity at 650 nm with and without proteinase K, respectively, and C corresponds to the concentration of proteinase K. Using this assay, the lowest concentration of proteinase K that could be detected was ca. 1 ng mL−1, the same level as what has been reported in literature for other techniques.[12] However, the new assay is able to cover a broader (at least two orders of magnitude) range of protease concentration than those of other conventional assays and the commercially available kits.[6a, 12, 13]
Figure 2.

(a) Fluorescence spectra taken from aqueous suspensions of BSA-AuCs after incubation with proteinase K at different concentrations for 4 h. (b) Plots of I/I0 as a function of incubation time after the introduction of proteinase K at different concentrations. (c) Plots of I/I0 as a function of the concentration of proteinase K or deactivated proteinase K, which went through heating at 95 °C for 20 min. Here I0 and I represent intensities of fluorescence from BSA-AuCs in the absence and presence of proteinase K, respectively. (d) Fluorescence decay curves for BSA-AuCs after incubation with proteinase K at different concentrations. Aqueous suspensions of BSA-AuCs were excited at 500 nm.
In addition to the intensity, the fluorescence lifetime also changed upon the introduction of proteinase K. Figure 2d shows the fluorescence (λem = 650 nm) decay curves of BSA-AuCs after incubation with different concentrations of proteinase K. With the introduction of proteinase K at a concentration of 1 ×104 ng mL−1, the fluorescence decayed faster as the intensity-weighted average lifetime dropped from 498 ns to 338 ns (Fig. S4). This change of fluorescence lifetime also reflects the variation in microenvironment that surrounded the AuCs due to the destruction of the BSA shells.
We next performed a series of experiments to validate the proposed fluorescence quenching mechanism. Previous studies have proven that O2, which is abundant in the ambient air, can lead to fluorescence quenching of many fluorescent or luminescent nanoparticles and dyes.[14] To examine whether the O2 indeed played an essential role in the fluorescence quenching for the AuCs-based system, we saturated the aqueous solution of BSA-AuCs with O2 or argon (Ar) before incubation with proteinase K. Figure 3a shows fluorescence intensities in reference to that of BSA-AuCs incubated in ambient air without proteinase K. In the O2-saturated solution, the drop of fluorescence intensity was much more pronounced. On the contrary, in the Ar-saturated solution, the fluorescence intensities of BSA-AuCs incubated with proteinase K at the same concentrations were much higher than those incubated with both O2- and air-saturated solutions. Even in the presence of proteinase K at high concentrations up to 1 ×104 ng mL−1, the degradation of BSA did not result in significant fluorescence quenching for the Ar-saturated solution. These observations were consistent with the mechanism described in Figure 1. The as-prepared AuC was a core-shell structure comprised of an AuC core and a BSA shell. The protein shell served as a protective layer preventing the fluorescent AuC core from being quenched by the O2 from ambient air. When the protein shell was destructed by a protease, the O2 molecules were able to penetrate through the protein shell and interact with the AuC directly and thus quench its fluorescence. In this case, the fluorescence quenching likely involved an energy transfer from the excited AuC to the triplet O2 (3O2) to generate the singlet O2 (1O2), a mechanism similar to what has been observed for quantum dots and Au nanoparticles.[15]
Figure 3.

(a) Dependence of the intensity of fluorescence from BSA-AuCs on the concentration of proteinase K. The aqueous suspensions of BSA-AuCs were saturated with oxygen (O2), air or argon before and during fluorescence measurements. (b) Dependence of the size of BSA-AuCs on the concentration of proteinase K. (c) Comparison of the intensity of fluorescence for aqueous suspensions of AuCs conjugated with amino acids or methoxy group using PEG as the linker. The amino acids (alanine, tryptophan, and phenylalanine) were first conjugated with succinimidyl propionyl PEG disulfide (Mw ≈ 2,000) and then reacted with BSA-AuCs. The fluorescence intensities were in reference to that of BSA-AuCs incubated in ambient air without proteinase K.
We further investigated whether other mechanisms were also involved in this AuC-based fluorescence quenching process. For the core-shell structure of BSA-AuCs, the presence of a hydrophilic BSA shell helped stabilize the AuC core by forming Au-S bonds between Au atoms and cysteine residues of BSA.[16] It was thus proposed that when the BSA shells were destructed by the protease, the exposed AuCs would quickly aggregate into larger particles, leading to a drop in fluorescence intensity.[17] This proposed mechanism, however, was contradictory to the results of our dynamic light scattering (DLS) measurements. As shown in Figure 3b, the hydrodynamic diameter of the BSA-AuCs decreased from 6.4 nm to 3.2 nm as the concentration of proteinase K increased from 0.1 to 1×107 ng mL−1. No aggregation was observed for all these samples. It can thus be concluded that the reduction in fluorescence intensity was not caused by aggregation of AuCs into larger particles. In addition, since the value of 3.2 nm was larger than the size of the Au25 core (less than 1 nm), there were likely cysteine-containing residues on the surface of the protease-treated AuCs.
It has also been reported that the ligands on the surface of AuCs played an important role in determining their fluorescence efficiency: the fluorescence can be greatly enhanced via charge transfer from the ligand to the Au core.[9] Since BSA contains a number of electron-rich groups, one would expect that the red fluorescence emission from BSA-AuCs arises from the charge transfer from BSA to the AuC core. When the BSA shell is degraded, the electron-donor groups will be detached from the AuCs and the fluorescence will thus disappear. If this is the case, replacing BSA with other ligands having a higher density of electron-donating groups should result in enhancement of fluorescence and vice versa. To test this mechanism, we performed a set of experiments by conjugating amino acids bearing a neutral group (i.e., methoxy group) or an electron-rich group (i.e., alanine phenylalanine or tryptophan) to the surface of the AuCs via a thiolate poly(ethyleneglycol) linker (Mw≈2,000). As shown in Figure 3c, we observed significant reduction (ca. 70–80%) in fluorescence intensity in all cases. However, we found no significant difference between the AuCs conjugated with electron-rich groups and those with neutral groups, weakening the hypothesized mechanism based on charge transfer. Therefore, we can conclude that the fluorescence quenching was exclusively caused by ambient O2 once the protein shell has been destructed by the protease.
A general assay for detecting proteases needs to be sensitive and capable of detecting a variety of proteases. To define its scope of utility, the AuC-based assay was applied to a number of different protein substrates and proteases. Thanks to the facile chemistry of such a detection system, protein-protected AuCs with different shells (e.g., insulin, lysozome, and transferrin) could all be prepared using a protocol similar to that of BSA-AuCs. As shown in Figure 4a, both lysozome- and transferrin-protected AuCs worked well for the assay, with detection limits of 103 and 10 ng mL−1, respectively. In comparison, reduction in fluorescence intensity was only observed for the insulin-protected AuCs when proteinase K was added at concentrations higher than 1 ×104 ng mL−1. Table S1 summarizes the detection ranges of all the protein-protected AuCs synthesized with different proteins. The detection limit seems to decrease with increasing protein molecular weight. The insensitive detection for the insulin-AuCs might be due to the fact that it has a relatively low molecular and loose structure, allowing for the O2 molecules to easily penetrate through the shell and interact with AuCs.
Figure 4.

a) Plots of I/I0 for different types of protein protected AuCs as a function of the concentration of proteinase K. Proteins with different molecular weights were used to prepare the AuCs. The detection sensitivity increased as the molecular weight of protein increased. b) Plots of I/I0 for BSA-AuCs as a function of the concentration of protease for different types of proteases. I0 and I represent intensities of fluorescence from protein protected AuCs in the absence and presence of proteases, respectively.
The application of this assay is not solely limited to a specific protease, but can be easily adapted for other proteases by simply changing the protein substrates surrounding AuCs. To demonstrate the utility of this system for the detection of other proteases, we also examined α-chymotrypsin, trypsin, and papain. As shown in Figure 4b, all of them exhibited similar trends for the reduction of fluorescence intensity although they exhibited different detection ranges due to their different abilities to degrade BSA (Table S1). Along with the requirement on a broad range of detection, an ability to discriminate between active and inactive proteases is also crucial in some applications. We demonstrated such a capability for BSA-AuCs by conducting a control experiment with protease XIII to treat BSA-AuCs. Protease XIII was reported to be unable to degrade BSA.[18] When treated with protease XIII, we did not observe any drop in fluorescence intensity in the entire concentration range up to 1 ×106 ng mL−1, implying that this assay was specifically sensitive to a protease that could degrade the protein shell. Therefore, this assay allows for the use of a rich variety of different protein substrates and thus selective detection of the corresponding proteases.
In summary, we have demonstrated a new nanoscale platform based on protein-protected AuCs for the fluorescence detection of proteases with high sensitivity and selectivity. The detection is based on the concept that the protein shell can serve as a protective layer to prevent the fluorescent AuCs from being exposed to the O2 from ambient air. When the protein shell is degraded by a protease, the O2 molecules can quickly access the AuCs and thus quench the fluorescence. The advantages of this new detection system lies in its simplicity, easiness, low cost, “one-step” of preparation, as well as the potential use with a wide range of protein substrates. We are currently designing and synthesizing new protein-protected AuCs with the protein substrates sensitive to disease-related proteases in an effort to expand its scope of applications.
Experimental Section
Chemicals
Proteinase K from Tritirachium album, α-chymotrypsin from bovine pancreas, papain from papaya latex, trypsin from bovine pancrease, protease XIII from aspergillus saitoi, albumin from bovine serum, insulin from bovine pancreas, apo-transferrin human, lysozome chloride form from chicken egg white, alanine, tryptophan, phenylalanine, hydrochloric acid (HCl, 37%), nitric acid (HNO3, 70%), chloroauric aicd (HAuCl4 3H2O) and poly(ethylene glycol) methyl ether thiol (PEG-SH, Mw ≈ 2000) were all obtained from Sigma-Aldrich (St. Louis, MO) and used as received. Ultrapurified water with a resistivity of 18 MΩ.cm was prepared using a E-Pure filtration system from Barnstead International (Dubuque, IA). Succinimidyl propionyl PEG disulfide (SVA-PEG-OPSS, Mw ≈ 2000) was purchased from Laysan Bio (Arab, AL) and used as received.
Synthesis of Fluorescent Protein-AuCs Conjugates
All glassware was washed with aqua regia (HCl/HNO3 = 3/1, v/v), and rinsed with ultrapurified water before use. In a typical experiment, aqueous HAuCl4 solution (5 mL, 10 mM) was added to BSA solution (5 mL, 50 mg mL−1, 37 °C) under magnetic stirring at 37 °C. Aqueous NaOH solution (0.5 mL, 1 M) was introduced 2 min later, and the reaction was allowed to proceed under magnetic stirring at 37 °C for 12 h. The as-synthesized protein-AuCs conjugates were purified by ultrafiltration at 3000 rpm for 3 times. Fluorescence spectra were recorded from aqueous suspensions of protein-AuCs using a Cary Eclipse fluorescence spectrophotometer (Varian, Palo Alto, CA).
Degradation of Protein-AuCs Conjugates by Proteases
The protein-AuCs conjugate was mixed with phosphate buffer (pH 7.5). The protease assay was initiated by the addition of a protease, with a final concentration ranging from 0.1 ng mL−1 to 1×106 ng mL−1. The reaction mixture was incubated at 37 °C for 4 h and fluorescence emission spectra were collected with a fluorescence spectrometer at an excitation of 500 nm. The control experiments were done without proteases following the same procedures. The size of AuCs during degradation was measured using dynamic light scattering (NanoZS, Malvern, Worcestershire, UK).
Conjugation of Amino Acids with AuCs
In a typical procedure, alanine and SVA-PEG-OPSS were dissolved in aqueous solution and stirred at room temperature overnight to generate conjugate alanine-PEG-OPSS, which was then mixed with BSA-AuCs and reacted at 4°C overnight. The final solution was ultrafiltered (Mw cut off = 3000) at 3000 rpm for 5 times to remove unconjugated alanine-PEG-OPSS and obtain AuCs conjugated with amino acids.
Supplementary Material
Footnotes
This work was supported in part by an NIH Director’s Pioneer Award (DP1 OD000798), a research grant from the NCI (R01 CA13852701), and startup funds from Georgia Institute of Technology.
Supporting Information is available on the WWW under http://www.small-journal.com or from the author.
Contributor Information
Yucai Wang, The Wallace H. Coulter Department of Biomedical Engineering, Georgia Institute of Technology and Emory University, Atlanta, GA 30332 (USA).
Dr. Yi Wang, The Wallace H. Coulter Department of Biomedical Engineering, Georgia Institute of Technology and Emory University, Atlanta, GA 30332 (USA)
Fengbo Zhou, Department of Biomedical Engineering, Washington University, St. Louis, MO 63130 (USA).
Paul Kim, Department of Biomedical Engineering, Washington University, St. Louis, MO 63130 (USA).
Prof. Younan Xia, Email: younan.xia@bme.gatech.edu, The Wallace H. Coulter Department of Biomedical Engineering, Georgia Institute of Technology and Emory University, Atlanta, GA 30332 (USA)
References
- 1.Wolfe MS. Chem Rev. 2009;109:1599. doi: 10.1021/cr8004197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Taylor RC, Cullen SP, Martin SJ. Nat Rev Mol Cell Bio. 2008;9:231. doi: 10.1038/nrm2312. [DOI] [PubMed] [Google Scholar]; b) Page-McCaw A, Ewald AJ, Werb Z. Nat Rev Mol Cell Bio. 2007;8:221. doi: 10.1038/nrm2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Drag M, Salvesen GS. Nat Rev Drug Discov. 2010;9:690. doi: 10.1038/nrd3053. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Friedl P, Wolf K. Cancer Res. 2008;68:7247. doi: 10.1158/0008-5472.CAN-08-0784. [DOI] [PubMed] [Google Scholar]
- 4.a) Ghadiali JE, Lowe SB, Stevens MM. Angew Chem Int Ed. 2011;50:3417. doi: 10.1002/anie.201008263. [DOI] [PubMed] [Google Scholar]; b) Shi LF, De Paoli V, Rosenzweig N, Rosenzweig Z. J Am Chem Soc. 2006;128:10378. doi: 10.1021/ja063509o. [DOI] [PubMed] [Google Scholar]
- 5.Capek P, Kirkconnell KS, Dickerson TJ. J Am Chem Soc. 2010;132:13126. doi: 10.1021/ja104572f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Yan XW, Yang LM, Wang QQ. Angew Chem Int Ed. 2011;50:5130. doi: 10.1002/anie.201101087. [DOI] [PubMed] [Google Scholar]; b) Kaman WE, Hulst AG, van Alphen PTW, Roffel S, van der Schans MJ, Merkel T, van Belkum A, Bikker FJ. Anal Chem. 2011;83:2511. doi: 10.1021/ac102764v. [DOI] [PubMed] [Google Scholar]
- 7.a) Lin CAJ, Yang TY, Lee CH, Huang SH, Sperling RA, Zanella M, Li JK, Shen JL, Wang HH, Yeh HI, Parak WJ, Chang WH. ACS Nano. 2009;3:395. doi: 10.1021/nn800632j. [DOI] [PubMed] [Google Scholar]; b) Jin RC, Zhu Y, Qian HF. Chem Eur J. 2011;17:6584. doi: 10.1002/chem.201002390. [DOI] [PubMed] [Google Scholar]; c) Gao Y, Shao N, Pei Y, Zeng XC. Nano Lett. 2010;10:1055. doi: 10.1021/nl100017u. [DOI] [PubMed] [Google Scholar]; d) Jin RC. Nanoscale. 2010;2:343. [Google Scholar]; e) Herzing AA, Kiely CJ, Carley AF, Landon P, Hutchings GJ. Science. 2008;321:1331. doi: 10.1126/science.1159639. [DOI] [PubMed] [Google Scholar]
- 8.a) Liu YL, Ai KL, Cheng XL, Huo LH, Lu LH. Adv Funct Mater. 2010;20:951. [Google Scholar]; b) Xie J, Zheng Y, Ying JY. Chem Commun. 2010;46:961. doi: 10.1039/b920748a. [DOI] [PubMed] [Google Scholar]; c) Lin YH, Tseng WL. Anal Chem. 2010;82:9194. doi: 10.1021/ac101427y. [DOI] [PubMed] [Google Scholar]
- 9.Wu ZK, Jin RC. Nano Lett. 2010;10:2568. doi: 10.1021/nl101225f. [DOI] [PubMed] [Google Scholar]
- 10.a) Wang HH, Lin CAJ, Lee CH, Lin YC, Tseng YM, Hsieh CL, Chen CH, Tsai CH, Hsieh CT, Shen JL, Chan WH, Chang WH, Yeh HI. ACS Nano. 2011;5:4337. doi: 10.1021/nn102752a. [DOI] [PubMed] [Google Scholar]; b) Liu CL, Wu HT, Hsiao YH, Lai CW, Shih CW, Peng YK, Tang KC, Chang HW, Chien YC, Hsiao JK, Cheng JT, Chou PT. Angew Chem Int Ed. 2011;50:7056. doi: 10.1002/anie.201100299. [DOI] [PubMed] [Google Scholar]; c) Wu X, He X, Wang K, Xie C, Zhou B, Qing Z. Nanoscale. 2010;2:2244. doi: 10.1039/c0nr00359j. [DOI] [PubMed] [Google Scholar]
- 11.Xie J, Zheng Y, Ying JY. J Am Chem Soc. 2009;131:888. doi: 10.1021/ja806804u. [DOI] [PubMed] [Google Scholar]
- 12.Zauner T, Berger-Hoffmann R, Muller K, Hoffmann R, Zuchner T. Anal Chem. 2011;83:7356. doi: 10.1021/ac201274f. [DOI] [PubMed] [Google Scholar]
- 13.Shi LF, De Paoli V, Rosenzweig N, Rosenzweig Z. J Am Chem Soc. 2006;128:10378. doi: 10.1021/ja063509o. [DOI] [PubMed] [Google Scholar]
- 14.a) Achatz DE, Meier RJ, Fischer LH, Wolfbeis OS. Angew Chem Int Ed. 2011;50:260. doi: 10.1002/anie.201004902. [DOI] [PubMed] [Google Scholar]; b) Zhang Y, He J, Wang PN, Chen JY, Lu ZJ, Lu DR, Guo J, Wang CC, Yang WL. J Am Chem Soc. 2006;128:13396. doi: 10.1021/ja061225y. [DOI] [PubMed] [Google Scholar]; c) Borisov SM, Nuss G, Klimant I. Anal Chem. 2008;80:9435. doi: 10.1021/ac801521v. [DOI] [PubMed] [Google Scholar]
- 15.a) Samia ACS, Chen XB, Burda C. J Am Chem Soc. 2003;125:15736. doi: 10.1021/ja0386905. [DOI] [PubMed] [Google Scholar]; b) Zhang YX, Aslan K, Previte MJR, Geddes CD. J Fluoresc. 2007;17:345. doi: 10.1007/s10895-007-0196-y. [DOI] [PubMed] [Google Scholar]
- 16.a) Wei H, Wang ZD, Zhang JO, House S, Gao YG, Yang LM, Robinson H, Tan LH, Xing H, Hou CJ, Robertson IM, Zuo JM, Lu Y. Nat Nanotechnol. 2011;6:92. doi: 10.1038/nnano.2010.280. [DOI] [PubMed] [Google Scholar]; b) Xavier PL, Chaudhari K, Verma PK, Pal SK, Pradeep T. Nanoscale. 2010;2:2769. doi: 10.1039/c0nr00377h. [DOI] [PubMed] [Google Scholar]
- 17.Wen F, Dong YH, Feng L, Wang S, Zhang SC, Zhang XR. Anal Chem. 2011;83:1193. doi: 10.1021/ac1031447. [DOI] [PubMed] [Google Scholar]
- 18.Davies KJA, Lin SW, Pacifici RE. J Biol Chem. 1987;262:9914. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.