

Abstract
Despite extensive understanding of sleep regulation, the molecular-level cause and function of sleep are unknown. I suggest that they originate in individual neurons and stem from increased production of protein fragments during wakefulness. These fragments are transient parts of protein complexes in which the fragments were generated. Neuronal Ca2+ fluxes are higher during wakefulness than during sleep. Subunits of transmembrane channels and other proteins are cleaved by Ca2+-activated calpains and by other nonprocessive proteases, including caspases and secretases. In the proposed concept, termed the fragment generation (FG) hypothesis, sleep is a state during which the production of fragments is decreased (owing to lower Ca2+ transients) while fragment-destroying pathways are upregulated. These changes facilitate the elimination of fragments and the remodeling of protein complexes in which the fragments resided. The FG hypothesis posits that a proteolytic cleavage, which produces two fragments, can have both deleterious effects and fitness-increasing functions. This (previously not considered) dichotomy can explain both the conservation of cleavage sites in proteins and the evolutionary persistence of sleep, because sleep would counteract deleterious aspects of protein fragments. The FG hypothesis leads to new explanations of sleep phenomena, including a longer sleep after sleep deprivation. Studies in the 1970s showed that ethanol-induced sleep in mice can be strikingly prolonged by intracerebroventricular injections of either Ca2+ alone or Ca2+ and its ionophore (Erickson et al., Science 1978;199:1219–1221; Harris, Pharmacol Biochem Behav 1979;10:527–534; Erickson et al., Pharmacol Biochem Behav 1980;12:651–656). These results, which were never interpreted in connection to protein fragments or the function of sleep, may be accounted for by the FG hypothesis about molecular causation of sleep.
Keywords: neurons, proteolysis, N-end rule, remodeling, memory, muscle
INTRODUCTION
The modern era of sleep research began with the 1949 discovery of the reticular activating system in the brainstem that controls wakefulness and sleep.1 Over the last six decades, great strides have been made in studies of sleep regulation. However, the molecular-level function of sleep remains unknown. This lack of understanding is particularly remarkable vis-à-vis the broad knowledge about neuronal circuits that control sleep in animals, including humans. Sleep appears to be universal among vertebrates, from fishes to mammals. Sleep-like states are also present in invertebrates such as the fly Drosophila melanogaster (∼105 neurons) and even the nematode Caenorhabditis elegans (∼300 neurons). Although it is likely that all vertebrates sleep and that sleep in Drosophila and other invertebrates is fundamentally similar to mammalian sleep, these issues remain to be settled definitively. Neuronal pathways that regulate sleep are linked to circadian circuits that control daily rhythms. However, circadian aspects of sleep do not define it entirely, because sleep has, in addition, specific homeostatic properties, described below. The evident complexity of sleep, including its distinct stages in mammals, such as rapid eye movement (REM) and nonrapid eye movement (NREM) sleep, suggests that sleep may have several causes and functions. A new hypothesis described below considers, in the spirit of Occam's razor, one molecular function of sleep.
In mammals, sleep can be initiated in small networks of neurons in the cortex and other regions of the brain.2–6 Despite profound anatomical differences and size disparities between, for example, the human and Drosophila brains, the molecular designs of neurons are highly similar in these divergent organisms. Given this experimental background, I suggest that the fundamental cause and function of sleep may reside in individual neurons, as distinguished from neuronal networks. There is no strong evidence either for or against this conjecture, in part because sleep has been defined, thus far, either behaviorally or at the level of neuronal assemblies. The probability of awake-to-sleep transition in a neuronal network (including, possibly, single neurons of that network) is increased by the preceding electrochemical activity of these neurons. In this view, sleep that is evident behaviorally is an emergent state that coalesces from local sleep foci that might be, initially, single neurons.
Sleep is either counteracted or facilitated by specific neuronal circuits that employ wakefulness-enhancing neurotransmitters such as orexins and somnogenic (sleep-promoting) compounds such as extracellular adenosine. Viewing sleep as a process initiated in small neuronal networks and/or individual neurons suggests that sleep-regulating circuits have been sculpted, on evolutionary timescales, by selection pressures to mitigate cognition-perturbing cellular stress, i.e., the molecular cause of sleep. Discovery of this cause is expected to account, among other things, for sleep's homeostatic character. Animals deprived of sleep exhibit impaired cognition, increased propensity to sleep (sleep drive), and respond to the incurred sleep “debt” through a longer and deeper subsequent sleep. For example, humans not only sleep longer after sleep deprivation but also spend more time in NREM sleep, particularly in a part of it called deep sleep, characterized by slow-wave activity in the electroencephalogram (EEG), by the lowest mean frequency of action potentials, and by the slowest brain metabolism, in comparison with other stages of sleep, and with wakefulness as well. For reviews of sleep, see Refs. 3,4,7–40.
Previously suggested functions of sleep include the null hypothesis,41,42 i.e., the conjecture that sleep is a peculiar form of indolence that has little adaptive value. Cogent arguments against this possibility were discussed previously.18,43,44 Other ideas about the functions of sleep are of two kinds: (i) molecular-level and metabolic conjectures; (ii) hypotheses with key assumptions at higher than molecular levels, for example, specific alterations in the strength of synaptic connections or other modifications of neuronal circuits.
One molecular-level hypothesis about the function of sleep is that during wakefulness the energy charge (a measure of ATP concentration) in the brain becomes sufficiently low to require a functionally distinct period, sleep, that restores the energy charge.45 This conjecture is extant in the field, in the absence of a definitive positive or negative evidence. For example, glucose metabolism in the brain cortex (but not, for example, in the hippocampus or hypothalamus) is decreased during NREM sleep.46 On the other hand, oxidative phosphorylation is upregulated during sleep deprivation, suggesting an increase in ATP production.47 In addition, there is no convincing evidence that neuronal energy gains during sleep are significant enough to be functionally relevant (for a recent discussion, see Refs. 48–51).
It was also suggested that extracellular adenosine is a natural somnogenic compound that contributes to the initiation and maintenance of sleep.11,21,45,52,53 Extracellular adenosine is derived in part from intracellular adenosine via equilibrative adenosine transporters. Adenosine is also secreted through regulated vesicular exocytosis.54 Another source of extracellular adenosine is ATP, which can be released from cells either by vesicular exocytosis or through specific transmembrane channels, followed by conversion of ATP to adenosine in the extracellular space.54–59 Adenosine facilitates sleep in part through binding to the neuronal G protein-coupled adenosine receptors. Recent evidence strongly suggests a role for extracellular adenosine as an endogenous somnogen.53,57,60–64
ATP can act as a somnogen not only through its conversion to adenosine but also through the binding of extracellular ATP to P2-type transmembrane receptors, a step that leads to the release, largely from microglial cells (brain phagocytes), of cytokines that include interleukin-1β (IL1β) and tumor necrosis factor (TNF)-α.56,65,66 As described below, these and other inflammatory cytokines can act as somnogens, indicating a major but incompletely understood connection between the immune system and sleep.4,16,29,31,36,67–79 Another potent endogenous somnogen is the PGD2 prostaglandin, which preferentially induces NREM sleep.11,28,80,81 The somnogenic activity of PGD2 is mediated at least in part by the ability of brain-produced PGD2 to increase the concentration of extracellular adenosine.11,28,81
Small compounds such as adenosine, ATP, and PGD2, proteins such as inflammatory cytokines, and several other endogenous effectors that act as somnogens are a part of the growing understanding of specific circuits that initiate, maintain, and regulate sleep. However, none of this understanding addresses, by itself, the underlying molecular cause and molecular-level function of sleep, which remain unknown.
Nonmolecular conjectures about the functions of sleep have in common the concept of sleep as a means of modifying synaptic connections in ways that optimize their activities during wakefulness. One example of these hypotheses is the suggestion that the function of REM sleep is to minimize deleterious modes of interaction among neurons in the cerebral cortex.82,83 It was also proposed that sleep is influenced by prior synaptic use, and functions to alter synaptic connections, in part through the local production of somnogenic compounds that include cytokines.84,85 Another conjecture is that sleep acts to downscale the strengths of synaptic connections, so that increases in synaptic strength during wakefulness stay below mechanistically infeasible limits.8,26,86–89 This hypothesis is related to the earlier demonstration that the firing by neurons and the strengths of their synapses are homeostatically increased or decreased to maintain firing rates within certain boundaries.90 The synaptic downscaling hypothesis posits, in effect, that the previously characterized recalibration of synaptic strengths90 does not suffice during wakefulness, and that sleep is required for a further downscaling.26 Studies on a role of sleep in memory and the evidence for such a role91–95 are also based on nonmolecular models, in part because we still do not know what exactly a long-term memory trace is. In sum, even if these plausible nonmolecular conjectures about sleep are eventually found to be correct in their domains, they would not, by themselves, identify the fundamental molecular problem that the function of sleep may have to address. “May” in the preceding sentence refers to the still extant possibility that the cause of sleep is not reducible to molecular perturbations in individual neurons and may be, instead, an evolved response of neuronal assemblies to “high-level” parameters of such networks, as distinguished from changes in individual neurons that result from molecular stress during wakefulness.
In contrast, the (unproven) premise of the present paper is that the fundamental cause of sleep originates in individual neurons. Described below is a molecular-level hypothesis about the causation and function of sleep, specific predictions of this hypothesis, and new explanations of the properties of sleep, including its evolutionary persistence and homeostatic character.
THE FRAGMENT GENERATION HYPOTHESIS
In considering causes of sleep, it might be helpful to view it as a metacellular (organismal) adaptation to a specific cellular stress during wakefulness. Responses to sleep deprivation suggest that sleep functions to counteract a cognition-perturbing molecular process that occurs in neurons, “builds up” during wakefulness, causes the initiation of sleep, can prolong its duration and intensity, and is downregulated during sleep. If so, and if sleep is an adaptive response to a cellular stress that impairs brain circuits, what is the molecular nature of a key perturbation that renders sleep necessary? Furthermore, how does sleep deal with this perturbation? There is also an evolutionary problem: given the maladaptive dimension of sleep, why was it impossible to eliminate or bypass the (unknown) molecular cause of sleep in the course of evolution? Deleterious aspects of sleep include a prolonged and fitness-reducing neglect of mating opportunities, and also the combination of immobility, eye closure, and higher arousal thresholds that results in a decreased vigilance. Save for a minority of animals such as adult top predators, a diminished vigilance of a sleeping animal usually signifies its increased vulnerability to predation. Because suppression of consciousness during sleep contributes to decreased vigilance, this suppression, too, is maladaptive at least behaviorally. Given these downsides of sleep, why not, for example, a quiet wakefulness, instead of sleep?
I suggest that the molecular cause and function of sleep originate in individual neurons and stem from increased production of protein fragments during wakefulness. These fragments, a multitude of them, are produced in all cells, including neurons, and are transient parts of protein complexes that initially contain full-length precursors of fragments (Fig. 1). Specific cleavage sites in proteins that give rise to fragments are maintained by positive selection during evolution, because many fragments have adaptive (fitness-increasing) functions. At the same time, at least some fragments can also perturb brain circuits. As described below, this (previously not considered) dichotomy between adaptive and maladaptive aspects of protein fragments can account for the evolutionary persistence of sleep.
Figure 1.

Generation, targeting and degradation of protein fragments, and remodeling of cleaved protein complexes. This diagram illustrates a multistage process that is expected to occur in a great variety of structural contexts and is a part of reactions encompassed by the fragment generation (FG) hypothesis. The process begins with a proteolytic cleavage (lightning arrow) of an oligomeric (in this example, heterodimeric) protein by a nonprocessive protease such as, for example, calpain or caspase. The process continues with the subunit-selective degradation of a resulting C-terminal fragment either by the Arg/N-end rule pathway (in this example) or by another proteolytic pathway, and ends with reconstitution of the initial heterodimer. Such processes can involve both “soluble” and transmembrane proteins (Figs. 3–6). The arrangement of two subunits in the heterodimer is (arbitrarily) antiparallel, with N-termini and C-termini denoted by “N” and “C,” respectively. The rate constants k1–k6 illustrate the possibility of independent regulation of specific reaction steps. In this example, the N-terminal residue of the C-terminal fragment (it remains associated with the other subunit of heterodimer) is Gln (Q), a tertiary destabilizing residue [Fig. 2(A)]. The C-terminal fragment is sequentially modified by the Ntaq1 NQ-amidase and the Ate1 R-transferase (specific enzymes of the Arg/N-end rule pathway) [Fig. 2(A)], followed by polyubiquitylation of the fragment by N-recognins (ubiquitin ligases of the Arg/N-end rule pathway) such as the UBR1 E3 (only E3 component of the holoenzyme ligase is shown) and the proteasome-mediated degradation of the targeted C-terminal fragment. For illustrative purposes, each of two subunits is shown to consist of two domains. The N-terminal fragment of the cleaved subunit may also be targeted for degradation, either by the Ac/N-end rule pathway (via an Ac/N-degron of the fragment) in this example, [Fig. 2(B)], or by another proteolytic pathway. See Fig. 2 and the main text for additional details. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Although this article considers largely intracellular protein fragments, essentially the same logic and analogous physiological ramifications also apply to extracellular protein fragments that are secreted from cells or produced from extracellular proteins (including extracellular domains of transmembrane proteins) by proteases in the extracellular space.
In comparison with sleep, the state of wakefulness entails, on average, significantly higher Ca2+ influxes into neurons. These transient fluxes (Ca2+ transients) are mediated by Ca2+ channels.96–98 Activation of specific transmembrane channels increases the levels of Ca2+ in the nucleus and the cytosol, including the cytosol's dendritic and axonal spaces.99–104 Ca2+ channels reside in the neuron's plasma membrane, particularly its synaptic regions, and also in intracellular membranes of the endoplasmic reticulum (ER), which is present in axons and dendrites as well.96,97 The ER lumen is one site of Ca2+ storage, in addition to extracellular space. The plumes (microdomains105,106) of Ca2+ ions entering specific regions of the cytosol are transient under normal conditions, owing to the activity of transmembrane pumps/exchangers that return Ca2+ to the extracellular space and the ER. Despite the transiency of Ca2+ microdomains and despite the buffering of Ca2+ by Ca2+-binding proteins such as calmodulin,107 increased concentrations of Ca2+ inside microdomains suffice for a local activation of calpains. While the cleavages of proteins by calpains are the chief proteolytic consequence of Ca2+ transients, these fluxes are also capable of activating some secretases,108–110 and caspases as well, in part through activation of the apoptosome and inflammasome.111,112 Many, possibly most neurons contain activated caspases such as caspase-3 (an effector caspase) under conditions in which caspase activation and caspase-mediated protein cuts are relatively constrained and do not cause cell death. The resulting protein fragments can be involved in neuronal remodeling, including changes that underlie memory.113–115 As described below, these fragments may also contribute to the causation of sleep.
An individual mammalian genome encodes ∼15 distinct calpains, including two major ones, calpain-1 (μ-calpain) and calpain-2 (m-calpain). Both neurons and glial cells contain these and other calpains. To simplify discussion, only Ca2+ transients and the resulting local increases of Ca2+ are cited below as the cause of calpain activation. Note, however, that other calpain regulators, such as phospholipids, protein kinases, calpain-binding proteins, and the ability of some calpains to activate themselves and other calpains through specific cuts, are also involved in controlling calpains,116–120 thereby shaping the in vivo repertoires of protein fragments generated by these proteases.
Production of protein fragments takes place on both presynaptic and postsynaptic sides of active synapses, throughout axons and dendrites, in the neuronal soma, in glial cells, and in most other cells of an organism. Moreover, the maintenance of brain anatomy, including the suppression of sprouting in a stable neurite shaft, has been shown to be an active process mediated by the proteolytic activity of calpains (particularly calpain-2) throughout dendrites and axons, even in the absence of action potentials and accompanying Ca2+ transients.121 In other words, the preservation of normal morphologies and connections throughout the nervous system involves a constant generation of protein fragments, above and beyond their production during Ca2+ transients at active synapses.
Subunits of transmembrane channels and many other proteins are cleaved by Ca2+-activated calpains and by other nonprocessive proteases. The resulting C-terminal fragments often have N-terminal residues that are “destabilizing” in that they can be recognized by the Arg/N-end rule pathway, a processive proteolytic system that targets proteins bearing specific N-terminal residues122,123 [Figs. 1 and 2(A)]. (Ramifications of this fact for the in vivo dynamics of protein fragments are discussed below.) Activated calpains have been shown to cleave approximately 100 different proteins, most of which are present in neurons (Fig. 3). Because calpain substrates were identified by nonsystematic means, the number of different proteins in mammalian neurons that are cleaved by calpains is certain to be significantly larger than 100. The known substrates of activated mammalian caspases are nearly 1000 different proteins177–180 (Fig. 4). Other proteases, including secretases, separases, and aminopeptidases, also produce intracellular protein fragments, including fragments that bear destabilizing N-terminal residues (Fig. 5). In addition, proteins that reside in or move through secretory compartments such as the ER can be cleaved by compartment-specific proteases. Most fragments generated by nonprocessive proteases are protein-size molecules (Figs. 3–6). They are degraded processively to short peptides at varying rates, often after delays, by specific branches of the N-end rule pathway (Figs. 1 and 2), by other pathways of the ubiquitin system, and through the autophagy-mediated lysosomal proteolysis as well.
Figure 2.
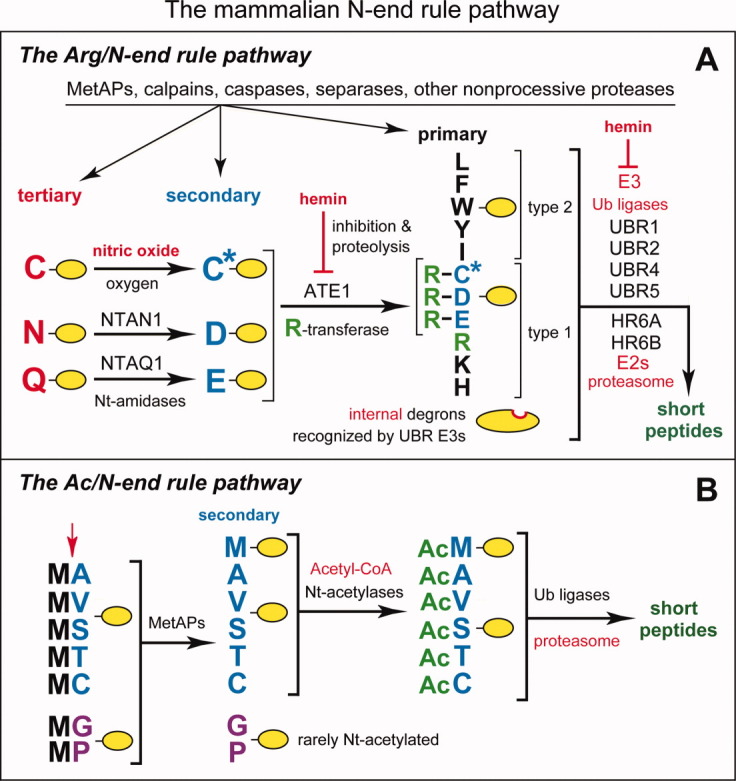
The mammalian N-end rule pathway.122,124,125,126 N-terminal residues are indicated by single-letter abbreviations for amino acids. A yellow oval denotes the rest of a protein substrate. E3 ubiquitin ligases of the N-end rule pathway are called N-recognins. A: The Arg/N-end rule pathway. “Primary,” “secondary,” and “tertiary” denote mechanistically distinct subsets of destabilizing N-terminal residues. C* denotes oxidized N-terminal Cys, either Cys-sulfinate or Cys-sulfonate, produced in vivo by reactions that require nitric oxide (NO) and oxygen. Oxidized N-terminal Cys is arginylated by ATE1-encoded isoforms of arginyl-tRNA-protein transferase (R-transferase), which also arginylates N-terminal Asp (D) and Glu (E). N-terminal Asn (N) and Gln (Q) are deamidated by the NTAN1-encoded NtN-amidase and the NTAQ1-encoded NtQ-amidase, respectively. In addition to the binding sites that recognize primary destabilizing N-terminal residues, the UBR1, UBR2, UBR4 and UBR5 (EDD) N-recognins contain binding sites for substrates (denoted by a larger oval) that lack N-degrons and have internal (non-N-terminal) degradation signals.122,126 Polyubiquitylated Arg/N-end rule substrates are degraded to short peptides by the 26S proteasome. Hemin (Fe3+-heme) binds to R-transferase, inhibits its arginylation activity and accelerates its in vivo degradation.122,126,127 Hemin also binds to UBR-type N-recognins.127 Regulated degradation of specific proteins by the Arg/N-end rule pathway mediates the sensing of heme, NO, and oxygen; the elimination of misfolded proteins; the regulation of DNA repair; the fidelity of chromosome cohesion/segregation; the signaling by G proteins; the control of peptide import; the regulation of apoptosis, meiosis, viral infections, fat metabolism, cell migration, actin filaments, cardiovascular development, spermatogenesis, neurogenesis and memory; the functioning of adult organs, including the brain, muscle, testis and pancreas; and many functions in plants (Refs. 122–124,126,128,129,130,131,132, and references therein). B: The Ac/N-end rule pathway. Although it is clear that this pathway is present in all or most eukaryotes,122 it has been characterized, thus far, only in yeast.125 This diagram illustrates the mammalian Ac/N-end rule pathway through an extrapolation from its S. cerevisiae version. Red arrow on the left indicates the removal of N-terminal Met by Met-aminopeptidases (MetAPs). This Met residue is retained if a residue at position 2 is nonpermissive (too large) for MetAPs. If the retained N-terminal Met or N-terminal Ala, Val, Ser, Thr, and Cys are followed by acetylation-permissive residues, the above N-terminal residues are Nt-acetylated by ribosome-associated Nt-acetylases.133 The resulting N-degrons are called Ac/N-degrons.125 The term “secondary” refers to the necessity of modification (Nt-acetylation) of a destabilizing N-terminal residue before a protein can be recognized by a cognate N-recognin. Although the second-position Gly or Pro residues can be made N-terminal by MetAPs, few proteins with N-terminal Gly or Pro are Nt-acetylated.133 [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Figure 3.

Calpain-generated C-terminal fragments of mammalian proteins that are either identified or predicted substrates of the Arg/N-end rule pathway. Each entry cites, on the left, a C-terminal (Ct) fragment and its N-terminal (Nt) residue (in red, using three-letter abbreviations for amino acids), followed by a brief description of the full-length (uncleaved) precursor protein. The right side of each entry shows the cleavage site of a full-length protein, using single-letter abbreviations for amino acids. An enlarged residue name, in red (preceded by an arrowhead denoting the cleavage site), indicates the P1′ residue, that is, the residue that becomes Nt upon the cleavage. Unless stated otherwise, the residue numbers are of human proteins. Two indicated residue numbers are the number of the first shown residue of a full-length protein and the number of its last residue, respectively. Most proteins on the list are present at least in neurons. These 34 proteins encompass the bulk of the previously characterized calpain substrates whose cleavage sites have been mapped and whose Ct fragments bear Nt residues that can be recognized by the Arg/N-end rule pathway [Fig. 2(A)]. Not shown are ∼20 calpain-generated, previously mapped Ct fragments bearing Nt residues that are not recognized by the Arg/N-end rule pathway. The Nt residues of this class, including Ala, Ser, and Thr, can be recognized by the Ac/N-end rule pathway [Fig. 2(B)] if these residues can be Nt-acetylated after having become Nt through a calpain-mediated cleavage. Whether these post-translationally generated N-terminal residues can be efficaciously Nt-acetylated in vivo remains to be determined. In addition, there are ∼40 other identified mammalian calpain substrates in which the exact locations of cleavage sites are unknown. Two calpain-generated Ct fragments, Asp-BCLXL (#16) and Arg-BID (#17), have been shown to be short-lived substrates of the Arg/N-end rule pathway.123 Other Ct fragments on this list are predicted Arg/N-end rule substrates. The recent finding that 10 out of 10 Ct fragments (generated by caspases or calpains) that were predicted to be Arg/N-rule substrates were actually found to be such123 suggests that most Ct fragments on the present list are also degraded by the Arg/N-end rule pathway. Calpain-generated fragments: #1. Tyr-mGluR1α is the Ct fragment of the mGluR1α subunit of the transmembrane metabotropic glutamate receptor.134 Receptors containing the calpain-truncated mGluR1α subunit could elevate cytosolic Ca2+ but could not activate PI3K-Akt signaling pathways, in contrast to uncleaved receptors.134,135 #2. Leu-NR2A is the Ct-fragment of the NR2A subunit of the transmembrane ionotropic glutamate receptor (NMDAR).136 The NR2B subunit of NMDAR can also be cleaved by calpains.137 Ct fragments of NR2A and NR2B contain domains required for the association of these subunits with synaptic proteins. NMDAR receptors lacking the Ct region of NR2A could function as glutamate-gated Ca2+ channels but the intracellular traffic of cleaved receptors and their electrophysiological properties were altered.138 #3. Lys-ATP2B2 is the Ct fragment of the transmembrane ATP2B2 plasma membrane Ca2+ pump (PMCA) that ejects Ca2+ from cells. This pump is activated either by the binding of Ca2+/calmodulin or by the calpain-mediated truncation of ATP2B2 that generates the Lys-ATP2B2 fragment and thereby activates the pump.139 #4. Gln-RYR1 is the Ct fragment of the RYR1 ryanodine receptor, a Ca2+ channel in the ER140 that mediates the efflux of Ca2+ from the ER into the cytosol. Calpain-mediated cleavage of RYR1 increases Ca2+ efflux.141 #5. Gln-EGFR is one of the calpain-generated Ct fragments of the transmembrane epidermal growth factor (EGF) receptor protein kinase.142 Remarkably, all seven calpain cleavage sites in the cytosol-exposed domain of the 170-kDa EGFR contain P1′ residues (which become Nt upon a cleavage)142 that are destabilizing in the Arg/N-end rule [Fig. 2(A)]. #6. Asn-Cav1.1 is the Ct fragment of the voltage-gated transmembrane Ca2+ channel. This (apparently) calpain-generated fragment is noncovalently associated with the rest of the channel and can inhibit its activity. Upon dissociation from the channel, the Asn-Cav1.1 fragment migrates to the nucleus and functions as a transcriptional regulator.97,143,144,145 #7. Arg-GlyT1A is the Ct fragment of the transmembrane GlyT1A glycine transporter.146 Another Gly transporter, GlyT1B, is also cleaved by calpains, yielding the Arg-GlyT1B fragment.146 These Ct fragments are still active as transporters but are impaired in their ability to remove Gly (an inhibitory neurotransmitter) from synaptic clefts.146 #8. Leu-RAD21 is the Ct-fragment of the SCC1/RAD21 subunit of the chromosome-associated cohesin complex.147 Calpain-mediated generation of Leu-RAD21 contributes to the control of chromosome cohesion/segregation, together with processes that include the separase-mediated cleavage of the same RAD21 subunit147,148,149,150 [see also Fig. 6(F)]. #9. Lys-cortactin is the Ct fragment of cortactin, an actin-binding protein that regulates actin polymerization.151 #10. Leu-vimentin is the Ct fragment of vimentin, a component of intermediate filaments.152 #11. Arg-dystrophin is the Ct fragment of a major cytoskeletal protein in the skeletal muscle.153 #12. Gln-talin is the Ct fragment of talin, an adaptor protein that interacts with the integrin family of cell adhesion transmembrane proteins.139,154,155 #13. Leu-NF2 is the Ct fragment of NF2 (merlin), a tumor suppressor and cytoskeletal protein. Loss-of-function NF2 mutants result in autosomal-dominant neurofibromatosis, a predisposition to specific kinds of brain tumors156 [see also Fig. 6(E)]. #14. Leu-troponin T2 is the Ct fragment of the cardiac troponin T that is produced by calpain-1 from the troponin-containing cardiac myofibril complex.157 #15. Glu-BAK is the Ct fragment of the proapoptotic regulator BAK. Glu-BAK is generated by calpain-1 in vitro and may be formed in vivo as well.158 #16. Asp-BCLXL is the Ct fragment of the BCLXL antiapoptotic protein. In contrast to its full-length precursor, the Asp-BCLXL fragment is proapoptotic, and has been shown to be a short-lived substrate of the Arg/N-end rule pathway.156 #17. Arg-BID is the Ct fragment of the proapoptotic BID regulator. The Arg-BID fragment is also proapoptotic, and in addition a short-lived Arg/N-end rule substrate.156 #18. Asn-DSCR1 (RCAN1) is the Ct fragment of the Down syndrome critical region 1 protein DSCR1, which binds to Raf1, inhibits the phosphatase activity of calcineurin, and enhances its degradation. Calpain-generated Asn-DSCR1 does not bind to the Raf1 kinase.159 #19. Arg-c-FOS is the Ct fragment of the c-FOS transcriptional regulator. c-FOS is targeted for degradation through more than one degron, including the path that includes the cleavage by calpains160 and predicted degradation of Arg-c-FOS by the Arg/N-end rule pathway. #20. Arg-MEF2D is the Ct fragment of the MEF2D myocyte enhancer factor 2D, a transcriptional regulator that contributes to neuronal survival, development, and synaptic plasticity.161 #21. Leu-STEP33 is the Ct fragment of the striatal-enriched STEP61 phosphatase, a brain-specific Tyr-phosphatase whose substrates include the MAPK-family kinases ERK1/2 and p38. Calpain-generated Leu-STEP33 fragment lacks phosphatase activity.162 #22. Leu-β-catenin is the Ct-fragment of β-catenin, a conditionally short-lived cytoskeletal protein and transcriptional regulator. The Leu-β-catenin fragment is a nuclear protein that activates specific genes in conjunction with other transcription factors.163 #23. Arg-IGFBP2 is the Ct fragment of the insulin-like growth factor binding protein-2 that is cleaved by calpain-2 at least in vitro.164 #24. Glu-Iκ-Bα is the Ct fragment of the Iκ-Bα subunit of the autoinhibited NF-κB–Iκ-Bα complex in which the NF-κB transcriptional regulator is inhibited by Iκ-Bα. The Iκ-Bα subunit is targeted for degradation either through a conditional phosphodegron or through the calpain-mediated cleavage165 which produces Glu-Iκ-Bα, predicted to be an Arg/N-end rule substrate. #25. Phe-PKCγ is the Ct fragment of PKCγ, a Ser/Thr kinase of the PKC family.166 The Phe-PKCγ fragment is constitutively active as a kinase, because it lacks the regulatory Nt domain of the full-length PKCγ kinase.166 #26. Leu-CAMK-IV is the Ct fragment of the Ca2+/calmodulin-dependent kinase-IV. This fragment lacks kinase activity.167 #27. Lys-PKCα is the Ct fragment of PKCα, a broadly expressed Ser/Thr kinase of the PKC family.166 Being catalytically active but no longer controlled by the regulatory Nt domain of the full-length PKCα, the Lys-PKCα fragment can be toxic, for example, upon its formation in an ischemic heart.168 #28. Arg-p39 is the Ct fragment of the p39 activator of the Cdk5 protein kinase.169 The indicated cleavage site is located immediately downstream of two other closely spaced (and strongly conserved) calpain cleavage sites in p39. A cleavage at any one of these sites yields a predicted Arg/N-end rule substrate [see Fig. 6(H)]. #29. Lys-GAD65-2 is the Ct fragment of the glutamic acid decarboxylase-65-2 (GAD65-2), which is bound to membranes of synaptic vesicles and mediates the synthesis of the inhibitory neurotransmitter γ-aminobutyric acid (GABA) from the excitatory neurotransmitter glutamate. Calpain-generated Lys-GAD65-2 retains the enzymatic activity of uncleaved GAD65-2 but is no longer associated with synaptic vesicles170–172 [see also Fig. 6(G)]. #30. Arg-caspase-9 is the Ct fragment of caspase-9, which can be inactivated by calpains,173 followed by the (predicted) degradation of the Arg-caspase-9 fragment by the Arg/N-end rule pathway. #31. Leu-calpain-1 is the Ct fragment of human calpain-1. The Leu-calpain-1 fragment is an activated form of this calpain.174,175 #32. Asp-calpain (reg. subunit) is the Ct fragment of the calpain regulatory subunit that is cleaved by activated calpains.116,120 #33. Lys-calpain-2 is the Ct fragment of human calpain-2, an activated form of this calpain.116–120 #34. Asn-calpain-B is the calpain-generated Ct fragment of one of two major D. melanogaster calpains and an activated form of this calpain.176 [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Figure 4.

Caspase-generated C-terminal fragments of mammalian proteins that are either identified or predicted substrates of the Arg/N-end rule pathway. The 30 caspase substrates cited in this list are a small fraction of ∼1000 known and mapped mammalian caspase substrates (see the main text). For designations, including residue numbering, see the legend of Fig. 3. The first eight caspase-generated, proapoptotic Ct fragments are the recently examined and confirmed short-lived substrates of the Arg/N-end rule pathway.123 #1. Cys-RIPK1 is the proapoptotic Ct fragment of the RIPK1 kinase, a regulator of apoptosis, necroptosis, and other processes, including antiviral responses that do not involve cell death178,181–183 [see also Fig. 6(A)]. #2. Cys-TRAF1 is the proapoptotic Ct fragment of TRAF1, which functions to minimize the activation of caspase-8 and other proapoptotic reactions, in part by contributing to upregulation of the antiapoptotic NF-κB regulon.178,184 #3. Asp-BRCA1 is the proapoptotic Ct fragment of BRCA1, a RING-type E3 ubiquitin ligase that functions as a tumor suppressor and participates in DNA repair, cell-cycle regulation, transcriptional control, and other processes185,186 [see also Fig. 6(B)]. #4. Leu-LIMK1 is the proapoptotic Ct fragment of LIMK1, a Ser/Thr kinase that functions, in particular, as a downstream effector of Rho signaling pathways and regulator of actin dynamic.187 #5. Tyr-NEDD9 is the proapoptotic Ct fragment of NEDD9, a scaffolding protein whose functions include cell attachment, migration, and mitotic control.188 #6. Arg-BIMEL is the proapoptotic Ct fragment of BIMEL a regulator of apoptosis.178 #7. Asp-EPHA4 is the proapoptotic Ct fragment of EPHA4, a member of the family of more than 10 mammalian “dependence” receptors (DpRs). These structurally distinct receptors are functionally analogous because of their ability to mediate two opposite physiological outcomes. In the presence of its cognate ligand, a DpR receptor activates signaling pathways that mediate cell survival, migration, proliferation, or differentiation. In the absence of its ligand, a dependence receptor produces a proapoptotic signal, often through the formation, by caspases or other nonprocessive proteases, of a C-terminal proapoptotic fragment that functions in the cytosol and/or the nucleus.189,190 #8. Tyr-MET is the proapoptotic Ct fragment of MET, another dependence receptor, with functions in embryonic development and organ formation.189,190 The next six caspase-generated proapoptotic Ct fragments are predicted substrates of the Arg/N-end rule pathway.123 #9. Asn-PKCδ is the proapoptotic Ct fragment of the PKCδ protein kinase.191 #10. Lys-PKCδ is the proapoptotic Ct fragment of the PKCδ protein kinase.192 #11. Trp-ETK is the proapoptotic Ct fragment of the ETK/BMC tyrosine kinase, a member of the Btk/Tek kinase family.193 #12. Gln-SLK is the proapoptotic Ct fragment of SLK, a STE20-related protein kinase that plays a role in regulation of actin fibers.194 #13. Ile-HPK1 is the proapoptotic Ct fragment of HPK1, the a STE20-related protein kinase whose functions include stimulation of the stress-activated protein kinases SAPKs/JNKs and the NF-κB transcriptional regulon.195 #14. Ile-MLH1 is the proapoptotic Ct fragment of the MLH1 DNA mismatch repair protein.196 The next 16 caspase-generated Ct fragments and predicted substrates of the Arg/N-end rule pathway that are not necessarily proapoptotic. These likely Arg/N-end rule substrates are cited to illustrate the remarkable diversity of their precursor proteins. #15. Tyr-CYLD is the Ct fragment of a deubiquitylase that regulates apoptosis and necroptosis197 [see also Fig. 6(D)[. #16. Leu-p21Cip1/Waf1 is the Ct fragment of p21Cip1/Waf, an inhibitor of cell division.198 #17. Arg-IP3R is the Ct fragment of the inositol 1,4,5-triphosphate receptor.199 #18. Asn-LMN1 is the Ct fragment of lamin-A, a component of nuclear lamina.200 #19. Arg-ETS-1 is the Ct fragment of a transcription factor.201 #20. Tyr-TOP1 is the Ct fragment of type I DNA topoisomerase.202 #21. Leu-MEFD2 is the Ct fragment of a transcription factor.203 #22. Asn-DNA-PK is the Ct fragment of the DNA-dependent protein kinase.204 #23. Asn-CAD1 is the Ct fragment of E-cadherin, an adhesion receptor.205 #24. Gln-synphilin-1 is the Ct fragment of synphilin-1, a ligand of α-synuclein206 [see also Fig. 6(C)]. #25. Tyr-ACINUS is the Ct fragment of a mediator of apoptotic chromatin condensation.207 #26. Lys-PLECTIN is the Ct fragment of a cytoskeletal protein.208 #27. Cys-CCNE1 is the Ct fragment of a specific G1/S cyclin.209 #28. His-PMCA4b is the Ct fragment of a Ca2+ extrusion pump.210 #29. Asp-CDC42 is the Ct fragment of CDC42, a RAS superfamily member.211 #30. Tyr-iPLA2 is the Ct fragment of the phospholipase A2 (Ref. 212). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Figure 5.
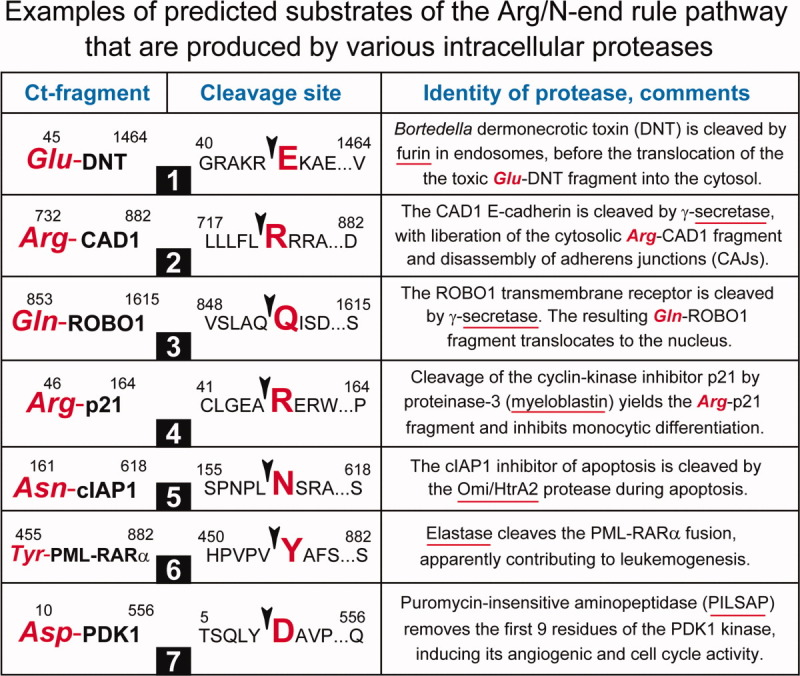
Examples of predicted substrates of the Arg/N-end rule pathway that are produced by nonprocessive proteases other than Met-aminopeptidases, calpains, or caspases. For designations, see the legend of Fig. 3. Although some of the proteases mentioned below (e.g., furin) are usually localized outside the nucleus and cytosol, the cited articles indicate that these proteases can also generate cytosolic or nuclear Ct protein fragments. #1. Furin-mediated cleavage of the bacterial (Bortedella) DNT toxin produces the Glu-DNT fragment, which translocates into the cytosol.213 #2. Cleavage by γ-secretase generates the Arg-CAD1 fragment of E-cadherin.205 #3. Another predicted Arg/N-end rule substrate that can be produced by γ-secretase is Gln-ROBO1, the Ct fragment of the transmembrane ROBO1 receptor.214 #4. Proteinase-3 (myeloblastin), a cytosolic/nuclear protease, produces Arg-p21, the Ct fragment of p21, a cell division inhibitor.215 #5. The Omi/Htr2 protease can cleave the cIAP1 protein (an antiapoptotic regulator and inhibitor of caspases), yielding the Asn-cIAP1 Ct fragment.216 #6. An intracellular form of elastase cleaves the PML-RARα oncoprotein fusion, yielding the Tyr-PML-RARα Ct fragment.217 #7. The aminopeptidase PILSAP removes the first nine residues of the kinase PDK1, yielding the enzymatically active Asp-PDK1 Ct fragment.218 [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Figure 6.
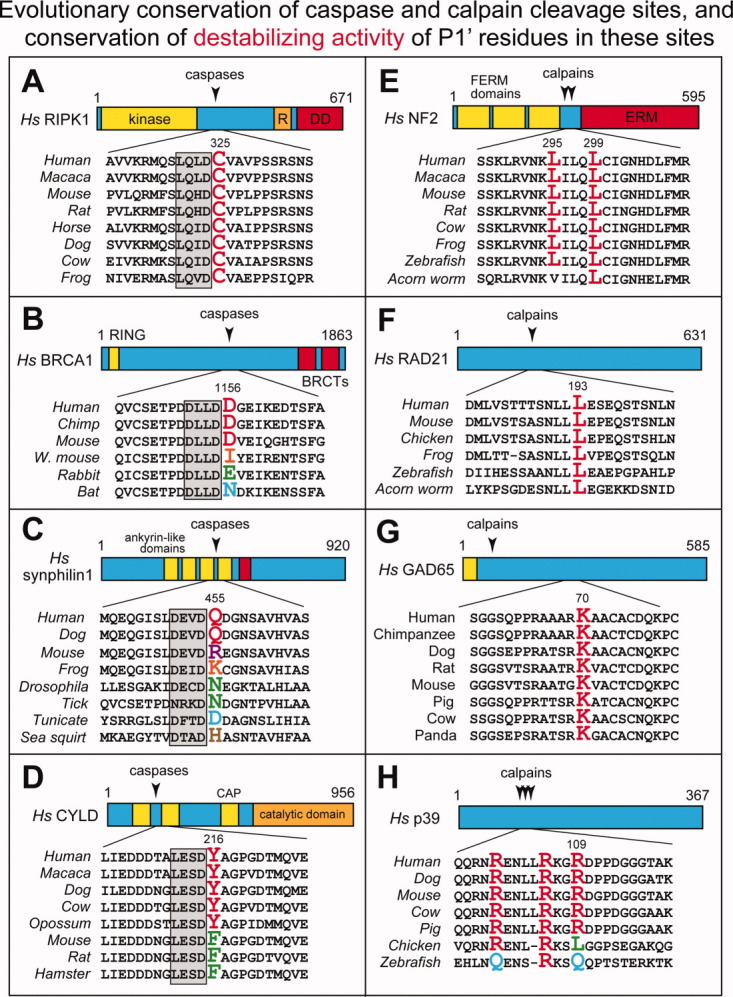
Evolutionary conservation of caspase (A–D) and calpain (E–H) cleavage sites, and the conservation of destabilizing activity of P1′ residues in these sites. The caspase cleavage sites in A–D (positions P4–P1) are framed by gray rectangles. The indicated residue numbers, including those of P1′ residues (in color), are of human versions of the full-length proteins. In many caspase and calpain cleavage sites (including those shown in Figs. 3 and 4), P1′ residues are completely conserved at least among vertebrates. By contrast, in some cleavage sites, for example those of BRCA1 in B (Fig. 4 (#3)), synphilin-1 in C (Fig. 4(#24)), CYLD in D (Fig. 4(#15)), and p39 in H (Fig. 3(#28)), the P1′ residues (they are shown in different colors, with predominant identities in red) are not conserved among vertebrates. Remarkably, however, their destabilizing activity in the Arg/N-end rule pathway [Fig. 2(A)] is invariably conserved (see the main text). For brief descriptions of the other cited caspase- or calpain-cleaved proteins RIPK1, NF2, RAD21, and GAD65, see Fig. 4(#1), Fig. 3(#13), Fig. 3(#8), and Fig. 3(#29), respectively. See also the Protein Fragments, Their Generation In Spite of Deleterious Effects, and the Evolutionary Persistence of Sleep section. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
In the proposed concept, termed the fragment generation (FG) hypothesis, sleep is a state that originates in individual neurons and involves a decreased production of protein fragments (owing in part to lower Ca2+ transients) as well as increased activities of fragment-destroying proteolytic pathways. These changes would facilitate the elimination of fragments and the remodeling of protein complexes in which the fragments were produced and transiently resided. The FG hypothesis leads to mechanistic explanations of specific properties of sleep, including its evolutionary persistence and homeostatic character (a longer sleep after sleep deprivation), as well as a broad variation of sleep duration and other features among different species.
MONITORING PROTEIN FRAGMENTS AND REACTING TO THEIR ACCUMULATION
If the increased generation of protein fragments during wakefulness and specific cognition-perturbing effects of this process underlie the causation of sleep, the individual neurons and circuits that regulate sleep must be able to “sense” either changes in the concentrations of fragments or consequences of these changes. One idea is that some proteins have acquired, during evolution, specific cleavage sites for yielding protease-generated fragments that function as somnogenic proteins. Such sensor-effector proteins and their fragments would allow systems that regulate sleep to gauge the increased production of many different fragments during wakefulness and to react by increasing the levels of somnogenic compounds, for example, extracellular adenosine, ATP or PGD2. These compounds, acting autocrinally and paracrinally (on cells that release them and on nearby cells), would promote a switch to a distinct state, sleep, whose salient features, according to the FG hypothesis, include lower Ca2+ transients, a slower production of fragments, and upregulated pathways that destroy fragments and remodel protein complexes in which the fragments resided.
PANX1, a transmembrane protein and a conditional ATP release channel, is one plausible example of what the postulated sensor-effector protein may be, in the framework of the FG hypothesis. The level of extracellular ATP (a somnogenic compound; see Introduction) could be upregulated, for example, through the activity of the caspase-generated fragment of PANX1 (Fig. 7). Specifically, the ability of PANX1 to function as an ATP release channel has been shown to be greatly increased by its caspase-mediated truncation.219,220 As mentioned above, caspases, including effector caspases, can be active in nonapoptotic neurons.113–115,221,222 It is unknown, at present, whether activated calpains can also cleave and activate PANX1 and/or several other, related transmembrane proteins that function as ATP release channels. For a further discussion of connections between ATP/cytokine pathways and protein fragments, see the FG Hypothesis and the Somnogenic Activity of Cytokines section below.
Figure 7.
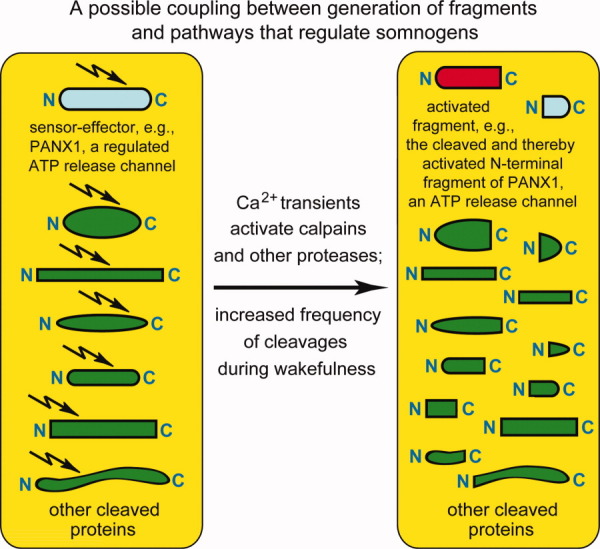
Monitoring protein fragments and reacting to their accumulation. This diagram mentions PANX1, a transmembrane protein and conditional ATP release channel (light blue oval near the top on the left) as a possible example of a sensor-effector protein. A cleavage of PANX1 is depicted to induce its activity as an ATP release channel (red semi-oval near the top on the right). By linking fragment formation to upregulation of somnogenic pathways that involve extracellular ATP and cytokines (see the FG Hypothesis and Somnogenic Activity of Cytokines section), this arrangement would make it possible for sleep-regulating pathways to gauge an increased production of many different fragments (green ovals and rectangles) during wakefulness, and to react by increasing the levels of somnogenic compounds, for example, extracellular adenosine, ATP or PGD2. The PANX1 channel was used to illustrate this regulatory arrangement because PANX1 has been shown to be strongly activated upon its cleavage by caspases.219,220 See also the Monitoring Protein Fragments and Reacting to Their Accumulation section. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Another possible mechanism for coupling a rising concentration of fragments to the activity of sleep-regulating circuits involves E3 ubiquitin ligases of the Arg/N-end rule pathway, called N-recognins. They bind to destabilizing N-terminal residues of protein fragments [Fig. 2(A)]. A fragment of a cleaved subunit of a protein complex may remain associated with the rest of a complex, owing to partially retained interactions within the complex. These interactions may include an association between the N-terminal and C-terminal fragments of a cleaved, complex-bound protein subunit. Examples of nondissociating or slowly dissociating cleaved protein complexes, including transmembrane channels, specific enzymes, and transcriptional regulators, are described in Refs. 143,223–226. The ability of the Arg/N-end rule pathway to destroy a C-terminal fragment that is embedded in a protein complex would be constrained by the fragment's noncovalent interactions within the complex. If so, a pool of slowly dissociating fragments that bear sterically accessible destabilizing N-terminal residues may act as a physical sink for N-recognins, thereby decreasing the ability of the Arg/N-end rule pathway to target other Arg/N-end rule substrates.
Now suppose that a positive regulator of somnogens is a naturally short-lived protein substrate of the Arg/N-end rule pathway. If so, the rise of protease-generated fragments that can interact with N-recognins but cannot be eliminated by them efficaciously enough would titrate available N-recognins. As a result, the normally short-lived regulator would be partially stabilized, increasing in steady-state levels and thereby upregulating the levels of a cognate somnogen as well. Given large distances between the soma and distal regions of axons and dendrites, a transient inhibition of the Arg/N-end rule pathway by this mechanism may occur locally rather than cell-wide. NTAN1 mRNA, which encodes NtN-amidase, a component of the Arg/N-end rule pathway [Fig. 2(A)], has been detected in axons,127 suggesting that both NtN-amidase and other components of the pathway can be produced not only in the neuronal soma but in axons and dendrites as well, through spatially localized translation of specific mRNAs.
A transient trapping/inhibition of specific ubiquitin ligases by protein fragments that are embedded in protein complexes may involve other proteolytic pathways as well, for instance those that can target N-terminal (as distinguished from C-terminal) protein fragments. One example of such systems is the Ac/N-end rule pathway, which targets Nα-terminally acetylated (Nt-acetylated) proteins [Fig. 2(B)]. To address experimentally these, already expected complexities, it would be essential to develop methods for measuring the in vivo activity of the Arg/N-end rule pathway and analogous proteolytic systems specifically in axons and dendrites.
There is also another, non-alternative and mechanistically feasible link through which an increase, during wakefulness, in the levels of either neuronal protein fragments or un-remodeled protein complexes (in which the fragments transiently reside) may upregulate the sleep drive. Either the known (e.g., Fig. 3) or still to be identified protease-mediated cleavages of specific transmembrane ion channels may alter properties of these channels in ways that would favor the signaling by inhibitory interneurons, as distinguished from excitatory neurons. Because small numbers of inhibitory interneurons tend to control significantly larger numbers of excitatory neurons, the resulting alteration (mediated by cleaved ion channels) of neuronal firing patterns in sleep-regulating circuits would favor the transition to sleep.
PROTEIN FRAGMENTS, THEIR GENERATION DESPITE DELETERIOUS EFFECTS, AND THE EVOLUTIONARY PERSISTENCE OF SLEEP
The FG hypothesis posits that proteolytic cleavages and the resultant protein fragments have both deleterious effects and adaptive (fitness-increasing) functions. For example, a specific cleavage-generated protein fragment can be deleterious while the other fragment, produced by the same cut, can have an adaptive function. Thus, remarkably, a cleavage of a protein may have both adaptive and harmful consequences at the same time. This is an important but previously not considered feature of in vivo proteolytic cuts. This property of cleavages, in addition to other dichotomies that involve protein fragments, can account for the observed evolutionary stability of specific cleavage sites in proteins (Fig. 6), because at least one of two fragments formed by a cleavage would have an adaptive function.
For example, calpain-mediated cleavage of a protein may act as a timing device that “tags” a transient event such as a Ca2+ microdomain by generating a cleaved protein complex that may be spatially confined to a specific dendritic spine. Such events were proposed to involve spectrin as a cytoskeletal protein whose cleavage by calpains at active synapses might contribute to memory processes.228 Many other proteins can also be cleaved by activated calpains (Fig. 3). Some of these cleavages and the resulting protein fragments may be a part of memory processes. According to the FG hypothesis, deleterious aspects of these and other protein fragments underlie the causation of sleep.
The necessity of apoptosis and other forms of programmed cell death in multicellular eukaryotes is one reason for the acquisition and retention of caspase cleavage sites (and of some calpain cleavage sites) in proteins during evolution. An example of fitness costs imposed by calpain cleavage sites are excessive calpain-mediated cleavages of neuronal proteins under conditions of excitotoxicity, for example, upon sustained increases of extracellular glutamate (which induces Ca2+ transients) in brain regions affected by stroke. Note, however, that similar cleavages of neuronal and glial intracellular proteins take place during normal wakefulness as well (Fig. 3), but at frequencies that are too low for irreversible cytotoxic effects. Nevertheless, these lower frequencies of cleavages are still high enough, according to the FG hypothesis, for a gradual perturbation of cognition and for eliciting responses by circuits that regulate sleep. With most calpain substrates cited in Figure 3, a rough estimate is that less than 20% of a substrate is cleaved at steady-state during normal wakefulness. Given low percentages of cleaved protein complexes, their (postulated) ability to perturb cognition may stem from gain-of-function effects, either by an initially cleaved complex that retains both products of a cut, or by an unremodeled complex, after the elimination of at least one fragment.
A protein fragment may be deleterious by not dissociating rapidly enough from a protein complex in which the fragment had been produced, or by acting as a perturbant in its free (dissociated) state. For example, a free fragment may form toxic oligomers or larger aggregates. A deleterious effect of a fragment may be accompanied by an independently present adaptive function of the same fragment, for example, by its necessity for a role that cannot be performed by the full-length precursor protein. Another possibility, mentioned above, is that one of two cleavage-generated protein fragments is deleterious while the other one has an adaptive function.
If a maladaptive aspect of a protein cleavage is not accompanied by a different, fitness-increasing effect of this event, there would be a selection pressure to eliminate the cleavage site. Calpain cleavage sites encompass ∼ 10 residues, involve both conformational and sequence determinants, and can be impaired or inactivated by missense mutations.116–120,229 Caspase cleavage sites are simpler and can be readily inactivated by missense mutations.179,230 Given the ease of elimination, on evolutionary timescales, of deleterious cleavage sites in neuronal and other proteins, it is striking, and most telling, that both calpain and caspase cleavage sites are highly conserved at least among vertebrate proteins (Fig. 6).
Also telling, in a different and independent way, is the evolutionary pattern of P1′ residues in these cleavage sites (P1′ residues become N-terminal after a cleavage). Specifically, the exact identity of a P1′ residue is not always conserved during evolution. Remarkably, however, the destabilizing nature of a P1′ residue, i.e., its ability, once it becomes N-terminal, to be recognized by the Arg/N-end rule pathway [Fig. 2(A)], is virtually always conserved (Fig. 6). This evolutionary pattern of P1′ residues in the precursors of natural protein fragments strongly suggests that the ability of C-terminal fragments to be targeted as substrates of the Arg/N-end rule pathway is a positively selected property of these fragments.
A caveat to this view of evolution of the P1′ residues is that their conservation in a specific cleavage site often occurs in a sequence context that is also conserved, at least among vertebrates [Fig. 6(A,E,F), and data not shown]. An alternative interpretation, in such cases, is that the observed conservation of a P1′ residue stems from a low tolerance for sequence alterations throughout the cleavage site (e.g., because this site is a critical part of a structure required for a protein's function), as distinguished from selection that acts to retain destabilizing activity of a P1′ residue. In such cases (and also, strictly speaking, in all cases that involve P1′ residues), the hypothesis of positive selection for destabilizing P1′ residues must be verified by causative (genetic) tests that examine fitness changes of organisms in which a wild-type destabilizing P1′ residue in a precursor of C-terminal fragment has been replaced by a residue that is not recognized by the Arg/N-end rule pathway [Fig. 2(A)].
Based on this experimental background (Figs. 1–6), the FG hypothesis posits a specific explanation for the observed evolutionary persistence of sleep, despite its maladaptive properties cited above. Specifically, the retention, through positive selection, of cleavage sites in proteins and the resulting evolutionary persistence of protease-generated fragments compel the existence of sleep as well, because sleep counteracts deleterious aspects of protein fragments. As discussed below, the FG hypothesis also leads to new explanations of the documented malleability of sleep, including its striking variations among different animals. In the framework of this hypothesis, the rates of generation of specific protein fragments by nonprocessive proteases, the rates of subsequent degradation of fragments to short peptides, and the rates of remodeling of protein complexes in which these fragments initially resided (Fig. 1) are independently tunable parameters that can be altered in individual neurons and other cells.
Consequently, variations in the rates of initial protein cleavages, in the rates of fragments' processive degradation, and in the rates of subsequent protein remodeling that would be favored by the ecology of an organismal lineage can be selected and optimized on evolutionary timescales. The results of such adjustments may underlie and explain many differences in homeostatic aspects and other properties of sleep among different animal species. For example, the duration of sleep in mammals varies from ∼4 hr in mountain goats to ∼16 hr in hedgehogs. The fact that sleep is universal at least among vertebrates and takes hours rather than minutes per day even among shortest-sleeping animals implies that a massive decrease or elimination of sleep during evolution was impossible cost-wise. According to the FG hypothesis, the cost of sleep reduction stems from the necessity of counteracting, through sleep, specific deleterious aspects of (positively selected) protein fragments. If so, one task at hand is to identify protein fragments that are particularly relevant to sleep causation, as well as specific mechanisms of fragment-mediated deleterious effects that occur in the absence of sleep.
EXAMPLES OF PROTEOLYTIC CUTS THAT CAN BE BOTH BENEFICIAL AND DELETERIOUS
Proapoptotic protein fragments
A recent study by our laboratory described the discovery that the Arg/N-end rule pathway is a repressor of apoptosis, through the ability of this pathway to destroy caspase- or calpain-generated proapoptotic protein fragments.123 Such fragments are defined, operationally, as those that increase the probability of apoptosis compared with full-length precursors of fragments. Metabolic stabilization of the proapoptotic, normally short-lived Cys-RIPK1 fragment of the RIPK1 kinase [Fig. 4(#1) and Fig. 6(A)] through the mutational conversion of its destabilizing N-terminal Cys to Val (which is not recognized by the Arg/N-end rule pathway) was found to elevate the fragment's in vivo levels and thereby to greatly increase its (intrinsic) proapoptotic activity.123 Abnormally high proapoptotic activity of a naturally produced proapoptotic fragment such as Cys-RIPK1 would be expected to be maladaptive, strongly suggesting (though not proving) that the presence and evolutionary conservation of a destabilizing N-terminal residue in Cys-RIPK1 [Fig. 4(#1) and Fig. 6(A)] stems from a positive selection for such a residue.
Ten proapoptotic fragments characterized as short-lived N-end rule substrates in the cited study123 [Fig. 3(#16, #17) and Fig. 4(#1–8)] are likely to be produced, at low but nonzero yields, in nonapoptotic cells as well. It remains to be determined whether any one among these C-terminal fragments (and/or their N-terminal counterparts) can be beneficial under conditions distinct from apoptosis. These 10 fragments (at least 20 with N-terminal fragments as well) are the tip of the iceberg vis-à-vis the total number of different fragments (possibly hundreds of them) that are likely to be relevant to the FG hypothesis (Figs. 3–6).
Effects of inflammatory cytokines such as TNF-α include the activation of caspases in target cells (Ref. 123 and references therein). I suggest that the somnogenic activity of TNF-α and analogous cytokines (see Introduction) is mediated in part by activation of caspases in neurons. This activation would yield a variety of protein fragments, including the previously characterized proapoptotic fragments that are targeted for degradation by the Arg/N-end rule pathway123 [Fig. 4(#1–8)]. In this view, increasing concentrations of extracellular TNF-α and other somnogenic cytokines in the brain during wakefulness generate activated caspases and specific intracellular protein fragments at levels that do not cause apoptosis but result, instead, in higher probabilities of awake-to-sleep transitions in individual neurons and neuronal networks (see the FG Hypothesis and the Somnogenic Activity of Cytokines section below).
A fragment of cohesin
Another example of functional dichotomies in proteolytic cuts is a subunit of cohesin.148 Cohesin rings encircle two replicated daughter chromatids and thereby maintain, through a topological confinement, the cohesion of chromatids until mitosis.231–233 An endoprotease called separase is activated during mitosis and cleaves the Scc1/Rad21 (kleisin) subunit, thereby opening the ring of cohesin and making chromatid separation possible. In the yeast Saccharomyces cerevisiae, the C-terminal fragment of separase-cleaved Scc1/Rad21 bears N-terminal Arg, a destabilizing residue [Fig. 2(A)]. This fragment, which remains associated with cohesin complex, is dislodged and degraded by the Arg/N-end rule pathway.148 The rest of cohesin complex is apparently spared, owing to the subunit selectivity of the Arg/N-end rule pathway.122,234–236
The identity of a residue at the N-terminus of a C-terminal fragment of Scc1/Rad21 varies in a tellingly constrained manner. Specifically, this residue is Arg in the budding yeast S. cerevisiae, Asn in the fission yeast Schizosaccharomyces pombe, Cys in D. melanogaster, and Glu in mammals. All these N-terminal residues are destabilizing in the Arg/N-end rule pathway148,149 [Fig. 2(A)]. This evolutionary constraint indicates the necessity of “proactive” elimination of the C-terminal fragment of Scc1/Rad21. The removal of this fragment leads to the eventual reconstitution (remodeling) of cohesin through incorporation of the uncleaved Scc1/Rad21 subunit.148
In ubr1Δ S. cerevisiae, which lack the Arg/N-end rule pathway, the normally short-lived C-terminal fragment of Scc1/Rad21 is a long-lived protein.148 In this setting, the C-terminal fragment can apparently still dissociate (or is displaced) from the rest of cleaved oligomeric cohesin in vivo at a rate that is compatible with the functioning of cohesin pathways. However, this degradation-independent route is inefficient enough to cause a striking (∼1000-fold) increase in the frequency of chromosome loss in ubr1Δ cells.148 Similar genomic instability is observed in ate1Δ mouse cells, which lack R-transferase and therefore lack the arginylation branch of the Arg/N-end rule pathway [Fig. 2(A)]. R-transferase is required for the degradation of the C-terminal fragment of the mammalian Scc1/Rad21 subunit, as this fragment bears N-terminal Glu, a secondary destabilizing residue122,149 [Fig. 2(A)] (Zhou et al., unpublished data).
Viewed together, the separase-mediated cleavage of Scc1/Rad21, the necessity of this cleavage for chromosome segregation, and the overtly deleterious effect of the resulting (cohesin-bound) C-terminal Scc1/Rad21 fragment in the absence of the Arg/N-end rule pathway122,148 are an illustration of some concepts that underlie the FG hypothesis.
The amyloid-β (Aβ) protein fragment
Remarkably, an example of a deleterious protein fragment that accumulates in the brain, is down-regulated during sleep, and is augmented during sleep deprivation, is already known.237 The 42-residue amyloid-β (Aβ) protein fragment is produced through cleavages, by proteases of the secretase family, from the transmembrane amyloid precursor protein (APP). Upon its formation at the cell's plasma membrane, Aβ becomes largely extracellular but can also be found in the cytoplasm. Aβ forms toxic oligomers and larger aggregates. Its accumulation in the brain is among the causes of Alzheimer's disease. At the same time, significantly lower concentrations of Aβ might have beneficial effects. For example, Aβ may function as an antimicrobial peptide.238 The largely extracellular localization of Aβ is consistent with the FG hypothesis, because essentially the same logic and analogous physiological ramifications would apply to cognition-perturbing intracellular and extracellular protein fragments. It should be noted that the study that described the circadian and sleep-deprivation pattern of Aβ expression237 did not consider the possibility, proposed by the FG hypothesis, that such a pattern may be a manifestation of the far more general phenomenon in which a multitude of different protein fragments are generated in the brain during wakefulness and are, collectively, the molecular cause of sleep.
FRAGMENTS OF NEURONAL PROTEINS AS N-END RULE SUBSTRATES
More than 50% of the previously mapped C-terminal fragments of calpain-cleaved proteins bear N-terminal residues that can be targeted by the Arg/N-end rule pathway (Fig. 3). The N-end rule relates the in vivo half-life of a protein to the identity of its N-terminal residue. Regulated degradation of specific proteins by the N-end rule pathway mediates a multitude of biological functions (Fig. 2 and its legend). The N-end rule pathway polyubiquitylates proteins that contain specific degradation signals (degrons), either internal or N-terminal (N-degrons). The main determinant of an N-degron is a destabilizing N-terminal residue of a protein (Fig. 2). The N-end rule pathway consists of two branches, the Ac/N-end rule and the Arg/N-end rule pathways (for reviews, see Refs. 122,124,128,129,239). The Ac/N-end rule pathway, described below, recognizes proteins with Nt-acetylated N-terminal residues.122,125 The Arg/N-end rule pathway targets specific unacetylated N-terminal residues. The primary destabilizing N-terminal residues Arg, Lys, His, Leu, Phe, Tyr, Trp, and Ile are directly recognized by N-recognins (E3 ubiquitin ligases of the N-end rule pathway), whereas N-terminal Asp, Glu, Asn, Gln, and Cys function as destabilizing residues through their preliminary enzymatic modifications [Figs. 1 and 2(A)].
Altogether, 71 C-terminal fragments of specific cleaved proteins that are either identified or predicted Arg/N-end rule substrates are described in Figures 3–6. This list, based on extensive literature searches, contains a majority of mapped calpain substrates (34 proteins; Fig. 3); a small subset of mapped caspase substrates (30 proteins; Fig. 4); and a few examples of proteins that are cleaved in vivo by other nonprocessive proteases (Fig. 5). The total number of fragments is more than twice the cited number, given the production of N-terminal/C-terminal pairs of fragments and frequent multiple cleavages of a full-length protein. Among the proteins that are cleaved by calpains and yield predicted Arg/N-end rule substrates are ligand- or voltage-regulated transmembrane proteins that mediate or control Ca2+ transients [Fig. 3(#1–4, #6)]; (#6)]; specific enzymes [Fig. 3(#25, #29)]; transcriptional regulators [Fig. 3(#19)]; and many other proteins (Fig. 3).
Although the discussion was confined, thus far, to C-terminal fragments of protein precursors, many N-terminal fragments are also likely to be N-end rule substrates, owing to the Ac/N-end rule pathway, the second branch of the N-end rule pathway [Fig. 2(B)]. More than 80% of human proteins are Nt-acetylated as nascent polypeptide chains by ribosome-associated Nt-acetylases.240 In 2010, it was discovered that the Nt-acetylated residues of cellular proteins act as specific degradation signals, termed Ac/N-degrons, to distinguish them from N-degrons of the Arg/N-end rule pathway, which recognizes specific unacetylated N-terminal residues125 (Fig. 2). The new field of the Ac/N-end rule pathway is just beginning to be explored.
As suggested previously,125 a key feature of Ac/N-degrons is their conditionality, in that usually a protein would not be targeted for degradation via its Ac/N-degron, save for a brief interval between the cotranslational formation of an Ac/N-degron in a nascent protein and the subsequent incorporation (sequestration) of this protein in a cognate oligomeric complex. What might be the consequence of a proteolytic cut in a protein subunit for the metabolic fate of a resulting N-terminal fragment? Some N-terminal fragments may be sufficiently destabilized conformationally to expose, and thereby to reactivate, their Ac/N-degrons (Fig. 1). Other N-terminal fragments of cleaved proteins may remain associated with their protein complexes and remain either inaccessible or at most intermittently (stochastically) accessible to the Ac/N-end rule pathway. In sum, the functional effects and metabolic fates of N-terminal fragments in cleaved protein complexes are at least as relevant to the FG hypothesis as the effects and fates of C-terminal fragments.
Either dissociation of fragments from protein complexes or the subsequent processive degradation of fragments may be slow enough to result in accumulation of specific fragments during wakefulness. (For many fragments, the dissociation and degradation steps would be mechanistically coupled.) According to the FG hypothesis, at least some of these fragments and/or unremodeled protein complexes in which the fragments transiently resided can perturb cognition. As mentioned above, the rate at which a newly generated fragment becomes susceptible to the targeting and processive degradation may be limited by the rate of its dissociation from a protein complex, owing to noncovalent interactions within the complex and also, for example, to an intermittent (stochastic) steric accessibility of a fragment's degradation signal.
RELATIONSHIP OF PROTEIN FRAGMENTS TO OTHER SOMNOGENS
The FG hypothesis and somnogenic activity of cytokines
Positive regulation of sleep involves both small compounds (adenosine, ATP, PGD2, nitric oxide, oleamide) and protein-size effectors, including inflammatory cytokines such as TNF-α and IL-1β, and other hormones/growth factors such as epidermal growth factor (EGF), growth hormone releasing hormone (GHRH), prolactin, nerve growth factor (NGF), and brain-derived neurotrophic factor (BDNF) (reviewed in Refs. 7,19,70, and 71). Inflammatory cytokines of the immune system are produced both peripherally and in the brain, and have somnogenic activity. (Hence the common observation of increased sleepiness during a bout of infectious disease.) Bacterial lipopolysaccharide (LPS) and other components of microorganisms that activate the innate immune system are also somnogenic.31,72 LPS and analogous bacterial compounds act as somnogens owing largely to their ability to upregulate somnogenic cytokines. A prolonged sleep deprivation increases susceptibility to infection in both humans and rodents.29,36,73–75 In healthy humans, the levels of IL-1β in blood plasma are highest at the onset of sleep.16,76
This and other evidence, including circadian control of the production of somnogenic cytokines, indicates their involvement in physiological sleep regulation. Activated processing and release of cytokines by microglia (brain phagocytes) is caused in part by rising levels of extracellular ATP during wakefulness. ATP is released from cells by vesicular exocytosis and through transmembrane channels (ATP release channels) such as PANX1, which can be activated by Ca2+ transients and by other effectors. Acting autocrinally and paracrinally, the extracellular ATP binds to P2-type receptors such as P2X7, which are present largely on microglial cells. Through mechanisms that remain to be clearly understood, this binding results in proteolytic processing and release of inflammatory cytokines.4,16,56,67,68 Given this understanding, how might an increased generation of protein fragments during wakefulness—the proposed molecular cause of sleep—interact with ATP/cytokine pathways?
First, as suggested above (Fig. 7) (see Monitoring Protein Fragments and Reacting to Their Accumulation section), an increased generation of protein fragments during wakefulness may be conveyed to sleep-regulating circuits through cleavages of (postulated) sensor-effector proteins. PANX1, a conditional ATP release channel, might be one such protein. PANX1 can be cleaved by caspases, a truncation that has been shown to activate PANX1 as an ATP release channel.219,220 The proposed PANX1-mediated mechanism is but one of several possible ways to link augmented generation of protein fragments during wakefulness to the ATP/cytokine system that contributes to sleep initiation. Note that the effects of inflammatory cytokines such as TNF-α include the activation of caspases in target cells.123 As suggested above (see the Proapoptotic Protein Fragments section), the somnogenic activity of TNF-α and analogous cytokines may be mediated in part by caspases. Their activation in neurons, while remaining below apoptotic levels, results in augmented generation of specific protein fragments and therefore, according to the FG hypothesis, in higher probabilities of awake-to-sleep transitions in individual neurons and neuronal networks.
Second, a somnogenic response of neurons to cytokines may be modulated by the levels of specific protein fragments that neurons contain at the time of their exposure to cytokines. As a result, at a given concentration of extracellular cytokines some neurons may undergo awake-to-sleep transitions, whereas other nearby neurons may not. In this model, the levels of specific protein fragments in a set of neurons would be increased by their recent electrochemical activity, owing to Ca2+ transients. The resulting concentrations of fragments would determine the probability of awake-to-sleep transitions, in a neuron or a neuronal assembly, in response to a given level of extracellular cytokines. Being both the molecular cause of sleep and a function of Ca2+ transients, the generation of protein fragments can track electrochemical activity of neurons and convey the level of activity-generated stress to ATP/cytokine pathways and other somnogenic systems.
Third, it has not been considered so far that although inflammatory cytokines and other protein-size somnogens signal through distinct cell surface receptors, these diverse effectors have a feature in common: they augment, directly or indirectly, calpain-activating Ca2+ transients in both neurons and glial cells.241–248 In addition, some somnogenic effectors can activate calpains independently of Ca2+ transients. For example, EGF can activate calpain-2 by upregulating kinases that phosphorylate and thereby activate this calpain.249 I suggest that these properties of protein-size somnogens and the resulting activation of calpains and other nonprocessive proteases such as caspases, comprise a specific somnogenic mechanism. In other words, it is proposed that inflammatory cytokines and analogous extracellular effectors act as somnogens at least in part because their binding to cognate receptors accelerates calpain-mediated and caspase-mediated generation of protein fragments. As suggested above, it is the concentrations of specific protein fragments in neurons that may determine the probabilities of awake-to-sleep transitions in response to a given level of extracellular cytokines. If so, the ability of cytokines, in this model, to upregulate the very process (generation of fragments) that contributes to their somnogenic activity would result in a positive feedback loop, a verifiable possibility.
Although the overall model is sufficiently specific to allow experimental testing, it is underdetermined, for example, in regard to identities of protein fragments that are functionally critical for responses to specific cytokines or other somnogenic effectors. On the (unproven) assumption that the FG hypothesis is correct at least in outline, one can ask, for instance, whether calpain-mediated cleavages of specific Ca2+ channels [Fig. 3(#2, #4, #6)] are more important for determining the probability of awake-to-sleep transition in response to a somnogen than the cleavages of other neuronal proteins, such as those described in Figure 3. Is there, in fact, a specific subset of protease-generated fragments (and their parental protein complexes) that largely determines the sensitivity of a neuron (or a neuronal assembly) to a given somnogen? These are some of experimentally addressable questions in this domain of the FG hypothesis.
Ethanol-induced sleep and Ca2+ transients
Ethanol readily crosses the blood-brain barrier and at sufficiently high doses acts as a somnogen. The main molecular mechanism through which ethanol exerts its somnogenic effect is unclear. Although biochemical impacts of ethanol on the brain are many and varied, the available evidence indicates that one major effect of ethanol is to increase Ca2+ transients in neurons and astrocytes.250–250 Higher Ca2+ transients promote the activation of calpains and other nonprocessive proteases. Hence the prediction, by the FG hypothesis, that a major somnogenic effect of ethanol may be its ability to increase the generation of protein fragments in neurons and other brain cells. In the 1970s, several studies showed that ethanol-induced sleep in mice can be strikingly prolonged by injections of either Ca2+ alone or Ca2+ and its ionophore.253–255 (Sleep was measured by the duration of unconsciousness and the loss of righting reflex after intraperitoneal injection of ethanol.) For example, at a dose of ethanol that resulted in a sleep for ∼ 68 min, an intracerebroventricular injection of CaCl2 (20 μmoles/kg) 30 min before ethanol injection resulted in a continuous sleep for ∼ 234 min, a nearly 3.5-fold increase compared with the control injection of buffer alone.253 Similar effects were observed upon injections of a lower amount of CaCl2 in the presence of a Ca2+ ionophore.253–255 These results were never interpreted in connection to protein fragments or the function of sleep. Moreover, these studies from three decades ago seem to be no longer discussed in the current literature about mechanisms of sleep. Note, however, that these results, obtained for unrelated reasons,253–255 may be accounted for by the FG hypothesis, in that an increase in Ca2+ transients (which augment the levels of activated calpains and other nonprocessive proteases) is predicted to be somnogenic. Further exploration of ethanol/calcium-induced sleep and extensions of this approach to mutants in the mouse Arg/N-end rule pathway,123,130 with measurements of specific protein fragments in the brain, are a promising way to explore the cause of sleep vis-á-vis the FG hypothesis.
PROTEIN FRAGMENTS AND SPECIFIC PROPERTIES OF SLEEP
Sleep deprivation and homeostatic aspects of sleep
The FG hypothesis can account, in general terms so far, for homeostatic aspects of sleep. Specifically, a prolongation of wakefulness, i.e., sleep deprivation would be expected to generate a larger load of undestroyed protein fragments and unremodeled protein complexes. The result, according to the FG hypothesis, would be increasingly perturbed cognition, in agreement with experimental evidence about sleep deprivation. Given higher than normal levels of protein fragments and unremodeled protein complexes by the end of sleep deprivation, adjustments of those levels during subsequent sleep would be expected to take a longer time, resulting in a longer sleep, in agreement with known effects of sleep deprivation.
Suppression of sleep and unihemispheric sleep
Described below are variations of sleep that offer additional opportunities to address predictions of the FG hypothesis. Studies of Danio rerio fishes (zebrafishes) identified a specific dichotomy in their responses to sleep deprivation. Adult zebrafishes are diurnal, being active during the day and less active at night, with prolonged episodes of sleep, defined behaviorally.12,256,257 If adult zebrafishes were sleep-deprived, in darkness, through repetitive electric stimulation and thereafter observed during an extended period of darkness, they exhibited prolonged sleep, as would be expected of responses to sleep deprivation in mammals. However, this compensatory prolongation of sleep after sleep deprivation was not observed if sleep-deprived zebrafishes were released into light.256 Moreover, sleep-deprived zebrafishes that were kept under constant light conditions exhibited little sleep to begin with, despite the preceding sleep deprivation in darkness.256 A similar and nearly complete suppression of sleep was also observed with zebrafishes kept under constant light for at least 3 days. Sleep reappeared and progressively increased by 8 days in the presence of light. No significant rebound sleep was observed afterwards in darkness, despite the (formally defined) sleep deprivation under constant-light conditions.256
In the framework of the FG hypothesis, these, currently unexplained, sleep patterns in zebrafishes may be accounted for by different rates of reactions (under different conditions of sleep deprivation described above) that mediate the generation or degradation of specific protein fragments and the remodeling of protein complexes that initially contained these fragments. Addressing these issues experimentally with zebrafishes can involve, at first, quantitative comparisons of the levels of protease-generated protein fragments, either of a kind described in Figures 3 to 6 or of any kind, using proteome-scale, mass spectrometry-based techniques. For example, one verifiable possibility is that long-term illumination of zebrafishes may upregulate their intracellular fragment-degrading pathways, including, possibly, the N-end rule pathway, so that specific (relevant to sleep causation) protein fragments are destroyed faster than in the dark. As a result, the levels of these fragments under constant light would be predicted to be, at least initially, lower than they would be during the diurnal pattern of alternating light and darkness, resulting in a low sleep drive in the light, despite the lack of sleep. Specific causative studies, including genetic approaches (they are feasible with zebrafishes), would be essential as well, to verify and deepen FG-based mechanistic interpretations.
Sleep in cetaceans (dolphins and whales) and in some pinnipeds (walrus, sea lions, fur seals, eared seals, and earless seals) has a property called the unihemispheric slow wave (USW) sleep. This is a form of NREM sleep in which the state of being asleep alternates between two brain hemispheres and is accompanied by only one closed eye at a time, the one that is contralateral to (and is innervated by) the sleeping hemisphere.24 The likely adaptive nature of USW sleep stems from the ability of, for example, a fur seal to monitor its immediate environment for predators and conspecifics using a single open eye, to continue to swim, and to continue staying “halfway” alert most of the time. Among terrestrial animals, only birds exhibit aspects of unihemispheric sleep, but not as pronounced as in cetaceans.24,258 The USW sleep has evolved in marine mammals presumably because it is particularly adaptive in aquatic settings, as the terrestrial environment can provide an animal with a measure of protection during sleep, for example, in burrows and trees. In addition, vulnerable grazing ungulates, such as antelopes, tend to sleep in herds, in which there are always some animals (sentinels) that are not asleep.258 Strikingly, the NREM sleep in fur seals can be of two kinds. On land, seals exhibit, most of the time, a bilaterally symmetrical slow wave (BSW) sleep, similar to NREM sleep in terrestrial mammals. In contrast, when a fur seal sleeps in water, its EEG patterns indicate a unihemispheric slow wave (USW) sleep, as in whales and dolphins.24,259
This switch from BSW sleep on land to USW sleep in water may be an opportunity to address a prediction of the FG hypothesis. In the BSW sleep (on land), when both hemispheres sleep at the same time, specific (e.g., relevant to sleep causation) protein fragments in the two hemispheres of the seal's brain would be expected to be present at equal levels, irrespective of the timing of brain analyses during sleep–wakefulness cycles. In contrast, if it is the destruction of protein fragments that does not keep pace with fragments' accumulation during wakefulness, the FG hypothesis predicts that during the USW sleep (in water), the sleeping brain hemisphere of a fur seal would contain lower levels of these fragments than the awake hemisphere of the same brain. This is a verifiable proposition. Its upside is that a negative result (the absence of significant differences between the levels of assayed protein fragments in a sleeping vs. awake hemisphere of a seal or a bird) would make the FG hypothesis less likely to be correct. A downside of such comparisons is their correlational (noncausative) nature.
Protein fragments and the size of a nervous system
Generation of protein fragments and the accompanying production of protein complexes that have to be remodeled after fragments' removal are cell-autonomous processes. Therefore there is no obvious lower boundary, in the current FG hypothesis, to the size of a nervous system that may exhibit a sleep-like behavior as an adaptive response to protease-generated fragments and modified protein complexes in which these fragments resided. C. elegans (∼300 neurons) has a quiescent behavioral state during a period called lethargus.260–262 It occurs before each of the four molts and exhibits some aspects of sleep, including higher arousal thresholds and a homeostatic response to mechanical stimulation during lethargus (specifically, an augmented quiescence after stimulation).260 The periods of lethargus involve alterations of the worm's nervous system, suggesting that lethargus evolved to accommodate specific requirements of such alterations.
Although it remains to be determined whether lethargus in C. elegans is fundamentally similar to sleep-like states in Drosophila, let alone to sleep in mammals, there are analogies between lethargus circuits and sleep regulation in larger animals, in that both settings involve proteins that control the epidermal growth factor (EGF) pathway, an ancient system whose functions encompass quiescence.260–264 The FG hypothesis predicts higher levels of specific protease-generated protein fragments in some or most C. elegans neurons (and possibly in other cell types as well) shortly before lethargus. If the FG hypothesis proves relevant to the causation and function of sleep in mammals, the resulting understanding could be “turned around” by asking whether the lethargus in C. elegans involves the generation and degradation of specific fragments and the remodeling of protein complexes after fragments' removal, and whether these processes are the molecular cause of lethargus.
It is also unknown whether FG concepts might be relevant to organisms such as protists or other single-cell eukaryotes, which exhibit cycles of metabolic transitions under normal growth conditions.265 Fundamentally, the FG hypothesis posits that a multitude of diverse protein fragments generated by nonprocessive proteases under conditions of daily life, as well as the necessity to remodel cleaved protein complexes can perturb the functioning of a cell. Being a challenge to stress response systems, these perturbations might require, at least in some cells, a quasi-periodic change of regimen that in more complex cellular assemblies would be called sleep. If the FG hypothesis proves germane to animals with elaborate nervous systems, this advance might also clarify whether FG concepts are also relevant to organisms where “sleep” is not an applicable term.
THE FG HYPOTHESIS AND SETTINGS OTHER THAN SLEEP
Although the FG hypothesis was described here in the context of sleep, the relevance of proposed mechanisms is not confined to neurons. Resetting of a circuit that involves a protease-generated protein fragment embedded in a multisubunit complex may involve the subunit-selective degradation of this fragment in any cell type and for reasons unrelated to sleep. Some of these reasons, such as the cycles of cleavage, subunit-selective degradation, and remodeling of cohesin complexes,122 were described above (see a Fragment of Cohesin section).
Viewed generally, the concepts of the FG hypothesis should be relevant to any in vivo setting that involves accumulation of either a protease-generated fragment or a cleaved, unremodeled protein complex, owing to insufficiently high rates of either degradation or remodeling, respectively. Increased levels of protein fragments and unremodeled complexes in such cases may be accompanied by an (evolved) cellular adaptation that amounts to a period of quiescence for the cell type involved. “Quiescence” is defined here as the time during which the generation of fragments slows down, the activity of fragment-destroying pathways goes up, and the remodeling of cleaved complexes can take place after the fragments' removal. Just two examples of the likely relevance of FG concepts outside sleep—muscle fatigue and mechanisms of memory—are briefly mentioned below, but many such settings may exist in all organisms, including prokaryotes, which contain both nonprocessive proteases and specific versions of the N-end rule pathway.122,129,266,267
The roles of calpains in mammalian cells of different types are broadly similar to calpain functions in neurons. In myotubes of the skeletal muscle and in cardiomyocytes of the heart, calpains can cleave cytoskeletal proteins such as cortactin, vimentin, dystrophin, talin, troponin, actin, and titin [Fig. 3(#9, #10, #11, #12, #14)], and many other proteins as well. A detailed discussion of this subject is beyond the scope of the present article, but it should be mentioned that the role of calpains in the cleavage of muscle proteins (with generation of protein fragments) and processive degradation of fragments under normal conditions or upon a strenuous exercise is a significant theme in muscle research.268–270 The existing evidence is consistent with the relevance of FG concepts to muscle physiology.
Another FG-related subject that will be mentioned but not discussed in detail are mechanisms of long-term memory. The previously proposed notion that calpain-mediated cleavages of specific cytoskeletal proteins such as spectrin may be relevant to memory processes228 remains unproven but likely, especially in view of many other calpain and caspase substrates in neurons that may be components of memory circuits (Figs. 3 and 4). There is growing evidence that cleavages of specific neuronal proteins by calpains and caspases play a role in long-term memory.114,271 Studies of Ntan1−/− mice, which lacked the Asn-specific Ntan1 NtN-amidase and therefore lacked the Asn-deamidation branch of the Arg/N-end rule pathway [Fig. 2(A)], showed that these mice had significant defects in learning and long-term memory.272 The recent discovery and molecular characterization of the Gln-specific Ntaq1 NtQ-amidase, another component of the Arg/N-end rule pathway [Fig. 2(A)], has revealed that the previously identified fly mutant termed tungus, which had impaired long-term memory,273 was apparently a null mutant in the Drosophila Ntaq1 NtQ-amidase.274 In addition, physiological substrates of calpains and caspases can yield C-terminal fragments with N-terminal Asn or Gln. Such fragments are predicted substrates of NtN-amidase and NtQ-amidase, respectively [Fig. 3(#4, #5, #6, #12, #18, #34), Fig. 4(#9, #12, #18, #22, #23, #24), and data not shown]. This fact and the above genetic results270–274 strongly suggest a role of the Arg/N-end rule pathway in memory processes.
CONCLUDING REMARKS
The motivation to describe these linked ideas and their ramifications before experimental testing was fourfold. First, although the proposed model differs from previous thinking in research on sleep, the FG hypothesis is not inconsistent, to my knowledge, with any solid data about sleep. Second, in addition to possibly identifying the molecular cause and function of sleep, the FG hypothesis leads to new explanations of specific properties of sleep, including its evolutionary persistence and homeostatic character. Third, the concepts about protein fragments and their dynamics that underlie the FG hypothesis are likely to be relevant to other biological problems, including mechanisms of memory and the functioning of cells other than neurons. Fourth, the complexity of pathways encompassed by the FG hypothesis suggests that its verification won't be a short affair. Experiments to address specific predictions of this hypothesis were recently initiated in our laboratory.
Acknowledgments
I thank Christopher Brower, Raymond Deshaies, William Dunphy, Konstantin Piatkov, Jevgenij Raskatov, Connor Rosen, Brenda Schulman, Anna Shemorry, and Brandon Wadas for helpful discussions and comments on the manuscript. I am particularly grateful to Roger Kornberg and William Tansey for their detailed suggestions. Our studies of the ubiquitin system and the N-end rule pathway are supported by grants from the National Institutes of Health.
References
- 1.Moruzzi G, Magoun HW. Brain stem reticular formation and activation of the EEG. Electroencephalogr Clin Neurophysiol. 1949;1:455–473. [PubMed] [Google Scholar]
- 2.Vyazovskiy VV, Olcese U, Hanlon EC, Nir Y, Cirelli C, Tononi G. Local sleep in awake rats. Nature. 2011;472:443–447. doi: 10.1038/nature10009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Krueger JM, Rector DM, Roy S, Van Dongen HPA, Belenky G, Panksepp J. Sleep as a fundamental property of neuronal assemblies. Nat Rev Neurosci. 2008;9:910–919. doi: 10.1038/nrn2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sengupta P, Roy S, Krueger JM. The ATP-cytokine-adenosine hypothesis: how the brain translates past activity into sleep. Sleep Biol Rhythms. 2011;9:29–33. [Google Scholar]
- 5.Kattler H, Dijk DJ, Borbely A. Effect of unilateral somatosensory stimulation prior to sleep on the sleep EEG in humans. J Sleep Res. 1994;3:159–164. doi: 10.1111/j.1365-2869.1994.tb00123.x. [DOI] [PubMed] [Google Scholar]
- 6.Vyazovskiy VV, Welker E, Fritschy JM, Tobler I. Regional pattern of metabolic activation is reflected in the sleep EEG after sleep deprivation combined with unilateral whisker stimulation in mice. Eur J Neurosci. 2004;20:1363–1370. doi: 10.1111/j.1460-9568.2004.03583.x. [DOI] [PubMed] [Google Scholar]
- 7.Rattenborg NC, Lima SL, Lesku JA. Sleep locally, act globally. Neuroscientist. 18:308. doi: 10.1177/1073858412441086. (in press) [DOI] [PubMed] [Google Scholar]
- 8.Tononi G, Cirelli C. Time to be SHY? Some comments on sleep and synaptic homeostasis. Neural Plast. 2012 doi: 10.1155/2012/415250. (in press) 415250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frank MG. Erasing synapses in sleep: is it time to be SHY? Neural Plast. 2012 doi: 10.1155/2012/264378. (in press) 264378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Urade Y, Hayaishi O. Prostaglandin D2 and sleep/wake regulation. Sleep Med Rev. 2011;15:411–418. doi: 10.1016/j.smrv.2011.08.003. [DOI] [PubMed] [Google Scholar]
- 11.Chikahisa S, Séi H. The role of ATP in sleep regulation. Front Neurol. 2011;2:1–5. doi: 10.3389/fneur.2011.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhdanova IV. Sleep and its regulation in zebrafish. Rev Neurosci. 2011;22:27–36. doi: 10.1515/RNS.2011.005. [DOI] [PubMed] [Google Scholar]
- 13.Saper CB, Fuller PM, Pedersen NP, Lu JV, Scammell TE. Sleep state switching. Neuron. 2010;68:1023–1042. doi: 10.1016/j.neuron.2010.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kilgore WD. Effects of sleep deprivation on cognition. Prog Brain Res. 2010;185:105–129. doi: 10.1016/B978-0-444-53702-7.00007-5. [DOI] [PubMed] [Google Scholar]
- 15.Siegel JM. Sleep viewed as a state of adaptive inactivity. Nat Rev Neurosci. 2009;10:747–753. doi: 10.1038/nrn2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Imeri L, Opp MR. How (and why) the immune system makes us sleep. Nat Rev Neurosci. 2009;10:199–210. doi: 10.1038/nrn2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hartse KM. Sleep in insects. In: McNamara P, Barton RA, Nunn CL, editors. Evolution of sleep. Cambridge, UK: Cambridge University Press; 2009. [Google Scholar]
- 18.Cirelli C, Tononi G. Is sleep essential? PLoS Biol. 2008;6:e216. doi: 10.1371/journal.pbio.0060216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Allada R, Siegel JS. Unearting the phylogenetic roots of sleep. Curr Biol. 2008;18:R670–R679. doi: 10.1016/j.cub.2008.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mignot E. Why we sleep: the temporal organization of recovery. PLoS Biol. 2008;6:e106. doi: 10.1371/journal.pbio.0060106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scharf MT, Naidoo N, Zimmerman JE, Pack AI. The energy hypothesis of sleep revisited. Prog Neurobiol. 2008;86:264–280. doi: 10.1016/j.pneurobio.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zimmerman JE, Naidoo N, Raizen DM, Pack AI. Conservation of sleep: insights from non-mammalian model systems. Trends Neurosci. 2008;31:371–376. doi: 10.1016/j.tins.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Andretic R, Franken P, Tafti M. Genetics of sleep. Annu Rev Genet. 2008;42:361–388. doi: 10.1146/annurev.genet.42.110807.091541. [DOI] [PubMed] [Google Scholar]
- 24.Lyamin OI, Manger PR, Ridgway SH, Mukhametov LM, Siegel JM. Cetacean sleep: an unusual form of mammalian sleep. Neurosci Biobehav Rev. 2008;32:1451–1484. doi: 10.1016/j.neubiorev.2008.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walker MP, Stickgold R. Sleep, memory, and plasticity. Annu Rev Psychol. 2006;57:139–166. doi: 10.1146/annurev.psych.56.091103.070307. [DOI] [PubMed] [Google Scholar]
- 26.Tononi G, Cirelli C. Sleep function and synaptic homeostasis. Sleep Med Rev. 2006;10:49–62. doi: 10.1016/j.smrv.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 27.Hobson JA. Sleep is of the brain, by the brain and for the brain. Nature. 2005;437:1254–1256. doi: 10.1038/nature04283. [DOI] [PubMed] [Google Scholar]
- 28.Hayaishi O. Molecular mechanisms of sleep-wake regulation: a role of prostaglandin D2. Philos Trans R Soc Lond Biol Sci. 2005;355:275–280. doi: 10.1098/rstb.2000.0564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Everson CA. Animal models of sleep deprivation/restriction. Basics of sleep guide. Westchester, IL: Sleep Research Society; 2005. pp. 31–36. [Google Scholar]
- 30.Basheer R, Strecker RE, Thakkar MM, McCarley RW. Adenosine and sleep-wake regulation. Prog Neurobiol. 2004;73:379–396. doi: 10.1016/j.pneurobio.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 31.Bryant PA, Trinder J, Curtis N. Sick and tired: does sleep have a vital role in the immune system? Nat Rev Immunol. 2004;4:457–467. doi: 10.1038/nri1369. [DOI] [PubMed] [Google Scholar]
- 32.Pace-Schott EF, Hobson JA. The neurobiology of sleep: genetics, cellular physiology and subcortical networks. Nat Rev Neurosci. 2002;3:591–605. doi: 10.1038/nrn895. [DOI] [PubMed] [Google Scholar]
- 33.Shaw PJ, Franken P. Perchance to dream: solving the mystery of sleep through genetic analysis. J Neurobiol. 2002;54:179–202. doi: 10.1002/neu.10167. [DOI] [PubMed] [Google Scholar]
- 34.Benington JH. Sleep homeostasis and the function of sleep. Sleep. 2000;23:959–966. [PubMed] [Google Scholar]
- 35.Borbely AA, Acherman P. Sleep homeostasis and models of sleep regulation. J Biol Rhythms. 1999;14:557–568. doi: 10.1177/074873099129000894. [DOI] [PubMed] [Google Scholar]
- 36.Rechtschafen A. Current perspectives on the function of sleep. Persp Biol Med. 1998;41:359–380. doi: 10.1353/pbm.1998.0051. [DOI] [PubMed] [Google Scholar]
- 37.Kavanau JL. Origin and evolution of sleep: roles of vision and endothermy. Brain Res Bull. 1997;42:245–264. doi: 10.1016/s0361-9230(96)00331-0. [DOI] [PubMed] [Google Scholar]
- 38.Berger RJ, Philips NH. Energy conservation and sleep. Behav Brain Res. 1995;69:65–73. doi: 10.1016/0166-4328(95)00002-b. [DOI] [PubMed] [Google Scholar]
- 39.Allison T, Cicchetti DV. Sleep in mammals: ecological and constitutional correlates. Science. 1976;194:732–734. doi: 10.1126/science.982039. [DOI] [PubMed] [Google Scholar]
- 40.Zepelin H, Rechtschaffen A. Mammalian sleep, longevity, and energy metabolism. Brain Behav Evol. 1974;10:425–470. doi: 10.1159/000124330. [DOI] [PubMed] [Google Scholar]
- 41.Rial RV, Nicolau MC, Gamundí A, Akaârir M, Aparicio S, Garau CT, S, Roca C, Gené L, Moranta D, Esteban S. The trivial function of sleep. Sleep Med Rev. 2007;11:311–325. doi: 10.1016/j.smrv.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 42.Rial RV, Nicolau MC, Gamundí A, Akaârir M, Aparicio S, Garau C, Tejada S, Roca C, Gené L, Moranta D, Esteban S. Sleep and wakefulness, trivial and non-trivial: which is which? Sleep Med Rev. 2007;11:411–417. doi: 10.1016/j.smrv.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 43.Siegel JM. REM sleep must have an adaptive value. Sleep Med Rev. 2011;15:111. [Google Scholar]
- 44.Rattenborg NC, Lesku JA, Martinez-Gonzalez D, Lima SL. The non-trivial functions of sleep. Sleep Med Rev. 2007;11:405–409. doi: 10.1016/j.smrv.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 45.Benington JH, Heller HC. Restoration of brain energy metabolism as the function of sleep. Prog Neurobiol. 1995;45:347–360. doi: 10.1016/0301-0082(94)00057-o. [DOI] [PubMed] [Google Scholar]
- 46.Nofzinger EA, Buysse DJ, Miewald JM, Meltzer CC, Price JC, Sembrat RC, Ombao H, Reynolds CF, Monk TH, Hall M, Kupfer DJ, Moore RY. Human regional cerebral glucose metabolism during nonrapid eye movement sleep in relation to waking. Brain. 2002;125:1105–1115. doi: 10.1093/brain/awf103. [DOI] [PubMed] [Google Scholar]
- 47.Nikonova EV, Naidoo N, Zhang L, Romer M, Cater JR, Scharf MT, Galante RJ, Pack AI. Changes in components of energy regulation in mouse cortex with increases in wakefulness. Sleep. 2010;33:889–900. doi: 10.1093/sleep/33.7.889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Haddad GG. Does the brain gain back energy during sleep? But what does it mean? Sleep. 2011;34:835–836. doi: 10.5665/SLEEP.1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dworak M, McCarley RW, Kim T, Kalinchuk AV, Basheer R. Sleep and brain energy levels: ATP changes during sleep. J Neurosci. 2010;30:9007–9016. doi: 10.1523/JNEUROSCI.1423-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wilson DF. Measuring in vivo metabolite levels in brain. Sleep. 2011;34:837. doi: 10.5665/SLEEP.1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dworak M, McCarley RW, Kim T, Kalinchuk AV, Basheer R. Replies to commentaries on ATP changes during sleep. Sleep. 2011;34:841–843. doi: 10.5665/SLEEP.1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Porkka-Heiskanena T, Kalinchuk AV. Adenosine, energy metabolism and sleep homeostasis. Sleep Med Rev. 2011;15:123–135. doi: 10.1016/j.smrv.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 53.Huang ZL, Urade Y, Hayaishi O. The role of adenosine in the regulation of sleep. Curr Top Med Chem. 2011;11:1047–1057. doi: 10.2174/156802611795347654. [DOI] [PubMed] [Google Scholar]
- 54.Klyuch BP, Dale N, Wall MJ. Deletion of ecto-5′-nucleotidase (CD73) reveals direct action potential-dependent adenosine release. J Neurosci. 2012;32:3842–3847. doi: 10.1523/JNEUROSCI.6052-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Trautmann A. Extracellular ATP in the immune system: more than just a “danger signal”. Sci Signal. 2009;2:pe6. doi: 10.1126/scisignal.256pe6. [DOI] [PubMed] [Google Scholar]
- 56.Krueger JM, Taishi P, De A, Davis CJ, Winters BD, Clinton J, Szentirmai E, Zielinski MR. ATP and the purine type 2 X7 receptor affect sleep. J Appl Physiol. 2010;109:1318–1327. doi: 10.1152/japplphysiol.00586.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Halassa MM, Haydon PG. Integrated brain circuits: astrocytic networks modulate neuronal activity and behavior. Annu Rev Physiol. 2010;72:335–355. doi: 10.1146/annurev-physiol-021909-135843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Qiu F, Dahl G. A permeant regulating its permeation pore: inhibition of pannexin 1 channels by ATP. Am J Physiol Cell Physiol. 2009;296:C250–C255. doi: 10.1152/ajpcell.00433.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Corriden R, Insel PA. Basal release of ATP: an autocrine-paracrine mechanism for cell regulation. Sci Signal. 2010;3:re1. doi: 10.1126/scisignal.3104re1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rétey JV, Adam M, Honegger E, Khatami R, Luhmann UF, Jung HH, Berger W, Landolt HP. A functional genetic variation of adenosine deaminase affects the duration and intensity of deep sleep in humans. Proc Natl Acad Sci USA. 2005;102:15676–15681. doi: 10.1073/pnas.0505414102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Halassa MM, Florian C, Fellin T, Munoz JR, Lee SY, Abel T, Haydon PG, Frank MG. Astrocytic modulation of sleep homeostasis and cognitive consequences of sleep loss. Neuron. 2009;61:213–219. doi: 10.1016/j.neuron.2008.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jones B. Glia, adenosine, and sleep. Neuron. 2009;61:156–157. doi: 10.1016/j.neuron.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 63.Florian C, Vecsey CG, Halassa MM, Haydon PG, Abel T. Astrocyte-derived adenosine and A1 receptor activity contribute to sleep loss-induced deficits in hippocampal synaptic plasticity and memory in mice. J Neurosci. 2011;31:6956–6962. doi: 10.1523/JNEUROSCI.5761-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schmitt LI, Sims RE, Dale N, Haydon PG. Wakefulness affects synaptic and network activity by increasing extracellular astrocyte-derived adenosine. J Neurosci. 2012;32:4417–4425. doi: 10.1523/JNEUROSCI.5689-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ferrari D, Pizzirani C, Adinolfi E, Lemoli RM, Curti A, Idzko M, Panther E, Di Virgilio F. The P2X7 receptor: a key player in IL-1 processing and release. J Immunol. 2006;176:3877–3883. doi: 10.4049/jimmunol.176.7.3877. [DOI] [PubMed] [Google Scholar]
- 66.Bianco F, Pravettoni E, Colombo A, Schenk U, Möller T, Matteoli M, Verderio C. Astrocyte-derived ATP induces vesicle shedding and IL-1 beta release from microglia. J Immunol. 2005;174:7268–7277. doi: 10.4049/jimmunol.174.11.7268. [DOI] [PubMed] [Google Scholar]
- 67.Krueger JM, Majde JA, Rector DM. Cytokines in immune function and sleep regulation. Handb Clin Neurol. 2011;98:229–240. doi: 10.1016/B978-0-444-52006-7.00015-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zielinski MR, Krueger JM. Sleep and innate immunity. Front Biosci. 2011;3:632–642. doi: 10.2741/s176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Krueger JM. The role of cytokines in sleep regulation. Curr Pharm Des. 2008;14:3408–3416. doi: 10.2174/138161208786549281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Taishi P, Davis CJ, Bayomy O, Zielinski MR, Liao F, Clinton JM, Smith DE, Krueger JM. Brain-specific interleukin-1 receptor accessory protein in sleep regulation. J Appl Physiol. 2012;112:1015–1022. doi: 10.1152/japplphysiol.01307.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kushikata T, Fang J, Chen Z, Wang Y, Krueger JM. Epidermal growth factor enhances spontaneous sleep in rabbits. Am J Physiol. 1998;275:R509–R514. doi: 10.1152/ajpregu.1998.275.2.R509. [DOI] [PubMed] [Google Scholar]
- 72.Mullington J, Korth C, Hermann DM, Orth A, Galanos C, Holsboer F, Pollmächer T. Dose-dependent effects of endotoxin on human sleep. Am J Physiol Regul Integr Comp Physiol. 2000;278:R947–R955. doi: 10.1152/ajpregu.2000.278.4.R947. [DOI] [PubMed] [Google Scholar]
- 73.Everson CA, Szabo A. Repeated exposure to severely limited sleep results in distinctive and persistent physiological imbalances in rats. PLoS One. 2011;6:e22987. doi: 10.1371/journal.pone.0022987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Spiegel K, Sheridann JF, Van Cauter E. Effect of sleep deprivation on response to immunization. JAMA. 2002;288:1471–1472. doi: 10.1001/jama.288.12.1471-a. [DOI] [PubMed] [Google Scholar]
- 75.Cohen S, Doyle WJ, Alper CM, Janicki-Deverrts D, Turner RB. Sleep habits and susceptibility to the common cold. Arch Intern Med. 2009;169:62–67. doi: 10.1001/archinternmed.2008.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Moldofsky H, Lue FA, Eisen J, Keystone E, Gorzynski RM. The relationship of interleukin-1 and immune function to sleep in humans. Psychosom Med. 1986;48:309–318. doi: 10.1097/00006842-198605000-00001. [DOI] [PubMed] [Google Scholar]
- 77.Luk WP, Zhang Y, White TD, Lue FA, Wu C, Jiang CG, Zhang L, Moldofsky H. Adenosine: a mediator of interleukin-1beta-induced hippocampal synaptic inhibition. J Neurosci. 1999;19:4238–4244. doi: 10.1523/JNEUROSCI.19-11-04238.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sperlágh B, Baranyi M, Haskó G, Vizi ES. Potent effect of interleukin-1 beta to evoke ATP and adenosine release from rat hippocampal slices. J Neuroimmunol. 2004;151:33–39. doi: 10.1016/j.jneuroim.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 79.Zhu G, Okada M, Yoshida S, Mori F, Hirose S, Wakabayashi K, Kaneko S. Involvement of Ca(2+)-induced Ca2+ releasing system in interleukin-1beta-associated adenosine release. Eur J Pharmacol. 2006;532:246–252. doi: 10.1016/j.ejphar.2005.12.085. [DOI] [PubMed] [Google Scholar]
- 80.Ueno R, Honda K, Inoué S, Hayaishi O. Prostaglandin D2, a cerebral sleep-inducing substance in rats. Proc Natl Acad Sci USA. 1983;80:1735–1737. doi: 10.1073/pnas.80.6.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pinzar E, Kanaoka Y, Inui T, Eguchi N, Urade Y, Hayashi O. Prostaglandin D synthase gene is involved in the regulation of non-rapid eye movement sleep. Proc Natl Acad Sci USA. 2000;97:4903–4907. doi: 10.1073/pnas.090093997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Crick FHC, Mitchison G. The function of dream sleep. Nature. 1983;304:111–113. doi: 10.1038/304111a0. [DOI] [PubMed] [Google Scholar]
- 83.Crick FHC, Mitchison G. REM sleep and neural nets. Behav Brain Res. 1995;69:147–155. doi: 10.1016/0166-4328(95)00006-f. [DOI] [PubMed] [Google Scholar]
- 84.Krueger JM, Obál FJ. A neuronal group theory of sleep function. Sleep Res. 1993;2:63–69. doi: 10.1111/j.1365-2869.1993.tb00064.x. [DOI] [PubMed] [Google Scholar]
- 85.Krueger JM, Obál F, Fang J. Why we sleep: a theoretical view of sleep function. Sleep Med Rev. 1999;3:119–129. doi: 10.1016/s1087-0792(99)90019-9. [DOI] [PubMed] [Google Scholar]
- 86.Tononi G, Cirelli C. Sleep and synaptic homeostasis: a hypothesis. Brain Res Bull. 2003;62:143–150. doi: 10.1016/j.brainresbull.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 87.Maret S, Faraguna U, Nelson AB, Cirelli C, Tononi G. Sleep and waking modulate spine turnover in the adolescent mouse cortex. Nat Neurosci. 2011;14:1418–1420. doi: 10.1038/nn.2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bushey D, Tononi G, Cirelli C. Sleep and synaptic homeostasis: structural evidence in Drosophila. Science. 2011;332:1576–1582. doi: 10.1126/science.1202839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hanlon EC, Vyazovskiy VV, Faraguna U, Tononi G, Cirelli C. Synaptic potentiation and sleep need: clues from molecular and electrophysiological studies. Curr Top Med Chem. 2011;11:2472–2482. doi: 10.2174/156802611797470312. [DOI] [PubMed] [Google Scholar]
- 90.Turigiano G. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell. 2008;135:422–435. doi: 10.1016/j.cell.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Girardeau G, Zugaro M. Hippocampal ripples and memory consolidation. Curr Opin Neurobiol. 2011;21:452–459. doi: 10.1016/j.conb.2011.02.005. [DOI] [PubMed] [Google Scholar]
- 92.Mednick SC, Cai DJ, Shuman T, Anagnostaras S, Wixted JT. An opportunistic theory of cellular and systems consolidation. Trends Neurosci. 2011;34:504–514. doi: 10.1016/j.tins.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Diekelman S, Born J. The memory function of sleep. Nat Rev Neurosci. 2010;11:114–126. doi: 10.1038/nrn2762. [DOI] [PubMed] [Google Scholar]
- 94.Wang G, Grone B, Colas D, Appelbaum L, Mourrain P. Synaptic plasticity and sleep: learning, homeostasis and disease. Trends Neurosci. 2011;34:452–463. doi: 10.1016/j.tins.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Seibt J, Dumoulin MC, Aton SJ, Coleman T, Watson A, Naidoo N, Frank MG. Protein synthesis during sleep consolidates cortical plasticity in vivo. Curr Biol. 2012;22:1–7. doi: 10.1016/j.cub.2012.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Berridge MJ. Calcium signalling remodelling and disease. Biochem Soc Trans. 2012;40:297–309. doi: 10.1042/BST20110766. [DOI] [PubMed] [Google Scholar]
- 97.Catterall WA. Voltage-gated calcium channels. Cold Spring Harb Perspect Biol. 2011;3:a003947. doi: 10.1101/cshperspect.a003947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Higley MJ, Sabatini BL. Calcium signaling in dendritic spines. Cold Spring Harb Perspect Biol. 2012;4:a005686. doi: 10.1101/cshperspect.a005686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yuste R. Dendritic spines. Cambridge, MA: MIT Press; 2010. [Google Scholar]
- 100.Lisman J, Yasuda R, Raghavachari S. Mechanisms of CaMKII action in long-term potentiation. Nat Rev Neurosci. 2012;13:169–182. doi: 10.1038/nrn3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Südhof TC. Calcium control of neurotransmitter release. Cold Spring Harb Perspect Biol. 2012;4:a011353. doi: 10.1101/cshperspect.a011353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sheng M, Kim E. The postsynaptic organization of synapses. Cold Spring Harb Perspect Biol. 2011;3:a005678. doi: 10.1101/cshperspect.a005678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Greer PL, Greenberg ME. From synapse to nucleus: calcium-dependent gene transcription in the control of synapse development and function. Neuron. 2008;59:846–860. doi: 10.1016/j.neuron.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 104.Kennedy MB, Beale HC, Carlisle HJ, Washburn LR. Integration of biochemical signalling in spines. Nat Rev Neurosci. 2005;6:423–434. doi: 10.1038/nrn1685. [DOI] [PubMed] [Google Scholar]
- 105.Berridge MJ. Calcium microdomains: organization and function. Cell Calcium. 2006;40:405–412. doi: 10.1016/j.ceca.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 106.Nakamura H, Bannai H, Inoue T, Michikawa T, Sano M, Mikoshiba K. Cooperative and stochastic calcium releases from multiple calcium puff sites generate calcium microdomains in intact HeLa cells. J Biol Chem. 2012;287:24563–24572. doi: 10.1074/jbc.M111.311399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Faas GC, Raghavachari S, Lisman JE, Mody I. Calmodulin as a direct detector of Ca2+ signals. Nat Neurosci. 2011;14:301–304. doi: 10.1038/nn.2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chen M. Alzheimer's alpha-secretase may be a calcium-dependent protease. FEBS Lett. 1997;417:163–167. doi: 10.1016/s0014-5793(97)01214-3. [DOI] [PubMed] [Google Scholar]
- 109.Litterst C, Georgakopoulos A, Shioi J, Ghersi E, Wisniewski T, Wang R, Ludwig A, Robakis NK. Ligand binding and calcium influx induce distinct ectodomain/gamma-secretase-processing pathways of EphB2 receptor. J Biol Chem. 2007;282:16155–16163. doi: 10.1074/jbc.M611449200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ho M, Hoke DE, Chua YJ, Li QX, Culvenor JG, Masters C, White AR, Evin G. Effect of metal chelators on γ-secretase indicates that calcium and magnesium ions facilitate cleavage of Alzheimer amyloid precursor substrate. Int J Alzheimers Dis. 2011;2011:950932. doi: 10.4061/2011/950932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Pinan-Lucarre B, Gabel CV, Reina CP, Hulme SE, Shevkoplyas SS, Slone RD, Xue J, Qiao Y, Weisberg S, Roodhouse K, Sun L, Whitesides GM, Samuel A, Driscoll M. The core apoptotic executioner proteins CED-3 and CED-4 promote initiation of neuronal regeneration in Caenorhabditis elegans. PLoS Biol. 2012;10:e1001331. doi: 10.1371/journal.pbio.1001331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Murakamia T, Ockingera J, Yua J, Bylesa V, A M, Hoferb AM, Hornga T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc Natl Acad Sci USA. 2012;109:11282–11287. doi: 10.1073/pnas.1117765109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Williams DW, Kondo S, Krzyzanowska A, Hiromi Y, Truman JW. Local caspase activity directs engulfment of dendrites during pruning. Nat Neurosci. 2011;9:1234–1236. doi: 10.1038/nn1774. [DOI] [PubMed] [Google Scholar]
- 114.Li Z, Jo J, Jia J-M, Lo S-C, Whitcomb DJ, Jiao S, Cho K, Sheng M. Caspase-3 activation via mitochondria is required for long-term depression and AMPA receptor internalization. Cell. 2010;141:859–871. doi: 10.1016/j.cell.2010.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Galluzzi L, Kepp O, Trojel-Hansen C, Kroemer G. Non-apoptotic functions of apoptosis-regulatory proteins. EMBO Rep. 2012;13:322–330. doi: 10.1038/embor.2012.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ono Y, Sorimachi H. Calpains: an elaborate proteolytic system. Biochim Biophys Acta. 2012;1824:224–236. doi: 10.1016/j.bbapap.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 117.Storr SJ, Carragher NO, Frame MC, Parr T, Martin SG. The calpain system and cancer. Nat Rev Cancer. 2011;11:364–374. doi: 10.1038/nrc3050. [DOI] [PubMed] [Google Scholar]
- 118.Vosler PS, Brennan CS, Chen J. Calpain-mediated signaling mechanisms in neuronal injury and neurodegeneration. Mol Neurobiol. 2008;38:78–100. doi: 10.1007/s12035-008-8036-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Croall DE, Ersfeld K. The calpains: modular designs and functional diversity. Genome Biol. 2007;8:218. doi: 10.1186/gb-2007-8-6-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Goll DE, Thompson VF, Li H, Wei W, Cong J. The calpain system. Physiol Rev. 2003;83:731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- 121.Mingorance-Le Meur A, O'Connor TP. Neurite consolidation is an active process requiring constant repression of protrusive activity. EMBO J. 2009;28:248–260. doi: 10.1038/emboj.2008.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Varshavsky A. The N-end rule pathway and regulation by proteolysis. Prot Sci. 2011;20:1298–1345. doi: 10.1002/pro.666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Piatkov KI, Brower CS, Varshavsky A. The N-end rule pathway counteracts cell death by destroying proapoptotic protein fragments. Proc Natl Acad Sci USA. 2012;109:E1839–E1847. doi: 10.1073/pnas.1207786109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Dougan DAM, D, Truscott KN. The N-end rule pathway: from recognition by N-recognins to destruction by AAA+ proteases. Biochim Biophys Acta. 2011;1823:83–91. doi: 10.1016/j.bbamcr.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 125.Hwang C-S, Shemorry A, Varshavsky A. N-terminal acetylation of cellular proteins creates specific degradation signals. Science. 2010;327:973–977. doi: 10.1126/science.1183147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tasaki TS, Sriram SM, Park KS, Kwon YT. Regulation of protein function by N-terminal degradation signal. Annu Rev Biochem. 2012;81:261–289. doi: 10.1146/annurev-biochem-051710-093308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hu R-G, Wang H, Xia Z, Varshavsky A. The N-end rule pathway is a sensor of heme. Proc Natl Acad Sci USA. 2008;105:76–81. doi: 10.1073/pnas.0710568105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Graciet E, Wellmer F. The plant N-end rule pathway: structure and functions. Trends Plant Sci. 2010;15:447–453. doi: 10.1016/j.tplants.2010.04.011. [DOI] [PubMed] [Google Scholar]
- 129.Mogk A, Schmidt R, Bukau B. The N-end rule pathway of regulated proteolysis: prokaryotic and eukaryotic strategies. Trends Cell Biol. 2007;17:165–172. doi: 10.1016/j.tcb.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 130.Brower CS, Varshavsky A. Ablation of arginylation in the mouse N-end rule pathway: loss of fat, higher metabolic rate, damaged spermatogenesis, and neurological perturbations. PLoS One. 2009;4:e7757. doi: 10.1371/journal.pone.0007757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Eisele F, Wolf DH. Degradation of misfolded proteins in the cytoplasm by the ubiquitin ligase Ubr1. FEBS Lett. 2008;582:4143–4146. doi: 10.1016/j.febslet.2008.11.015. [DOI] [PubMed] [Google Scholar]
- 132.Heck JW, Cheung SK, Hampton RY. Cytoplasmic protein quality control degradation mediated by parallel actions of the E3 ubiquitin ligases Ubr1 and San1. Proc Natl Acad Sci USA. 2010;107:1106–1111. doi: 10.1073/pnas.0910591107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Arnesen T, Van Damme P, Polevoda B, Helsens K, Evjenth R, Colaert N, Varhaug JE, Vandekerckhove J, Lillehaug JR, Sherman F, Gevaert K. Proteomics analyses reveal the evolutionary conservation and divergence of N-terminal acetyltransferases from yeast to humans. Proc Natl Acad Sci USA. 2009;106:8157–8162. doi: 10.1073/pnas.0901931106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Xu W, Wong TP, Chery N, Gaertner T, Wang YT, Baudry M. Calpain-mediated mGluR1alpha truncation: a key step in excitotoxicity. Neuron. 2007;53:399–412. doi: 10.1016/j.neuron.2006.12.020. [DOI] [PubMed] [Google Scholar]
- 135.Niswender CM, Conn PJ. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol. 2010;50:295–322. doi: 10.1146/annurev.pharmtox.011008.145533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bi R, Rong Y, Bernard A, Khrestchatisky M, Baudry M. Src-mediated tyrosine phosphorylation of NR2 subunits of N-methyl-d-aspartate receptors protects from calpain-mediated truncation of their C-terminal domains. J Biol Chem. 2000;275:26477–26483. doi: 10.1074/jbc.M003763200. [DOI] [PubMed] [Google Scholar]
- 137.Simpkins KL, Guttmann RP, Dong Y, Chen Z, Sokol S, Neumar RW, Lynch1 DR. Selective activation induced cleavage of the NR2B subunit by calpain. J Neurosci. 2003;23:11322–11331. doi: 10.1523/JNEUROSCI.23-36-11322.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Steigerwald F, Schulz TW, Schenker LT, Kennedy MB, Seeburg PH, Köhr G. C-Terminal truncation of NR2A subunits impairs synaptic but not extrasynaptic localization of NMDA receptors. J Neurosci. 2000;20:4573–4581. doi: 10.1523/JNEUROSCI.20-12-04573.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Di Leva F, Domi T, Fedrizzi L, Lim d, Carafoli E. The plasma membrane Ca2+ ATPase of animal cells: structure, function and regulation. Arch Biochem Biophys. 2008;476:65–74. doi: 10.1016/j.abb.2008.02.026. [DOI] [PubMed] [Google Scholar]
- 140.Wu Y, Aghdasi B, Dou SJ, Zhang JZ, Liu SQ, Hamilton SL. Functional interactions between cytoplasmic domains of the skeletal muscle Ca2+ release channel. J Biol Chem. 1997;272:25051–25061. doi: 10.1074/jbc.272.40.25051. [DOI] [PubMed] [Google Scholar]
- 141.Shoshan-Barmatz V, Weil S, Meyer H, Varsanyi M, Heilmeyer LM. Endogenous, Ca(2+)-dependent cysteine-protease cleaves specifically the ryanodine receptor/Ca2+ release channel in skeletal muscle. J Membr Biol. 1994;142:281–288. doi: 10.1007/BF00233435. [DOI] [PubMed] [Google Scholar]
- 142.Gregoriou M, Willis AC, Pearson MA, Crawford C. The calpain cleavage sites in the epidermal growth factor receptor kinase domain. Eur J Biochem. 1994;223:455–464. doi: 10.1111/j.1432-1033.1994.tb19013.x. [DOI] [PubMed] [Google Scholar]
- 143.Hulme JT, Konoki K, Lin TW-C, Gritsenko MA, Camp DG, Bigelow DJ, Catterall WA. Sites of proteolytic processing and noncovalent association of the distal C-terminal domain of CaV1.1 channels in skeletal muscle. Proc Natl Acad Sci USA. 2005;102:5274–5279. doi: 10.1073/pnas.0409885102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hulme JT, Yarov-Yarovoy V, Lin TWC, Scheuer T, Catterall WA. Autoinhibitory control of the CaV1.2 channel by its proteolytically processed distal C-terminal domain. J Physiol. 2006;576:87–102. doi: 10.1113/jphysiol.2006.111799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gomez-Ospina N, Tsuruta F, Barreto-Chang O, Hu L, Dolmetsch R. The C terminus of the L-type voltage-gated calcium channel Ca(V)1.2 encodes a transcription factor. Cell. 2006;127:591–606. doi: 10.1016/j.cell.2006.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Baliova M, Jursky F. Calpain sensitive regions in the N-terminal cytoplasmic domains of glycine transporters GlyT1A and GlyT1B. Neurochem Res. 2005;30:1093–1100. doi: 10.1007/s11064-005-7520-1. [DOI] [PubMed] [Google Scholar]
- 147.Panigrahi AK, Zhang N, Mao Q, Pati D. Calpain-1 cleaves Rad21 to promote sister chromatid separation. Mol Cell Biol. 2011;31:4335–4447. doi: 10.1128/MCB.06075-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Rao H, Uhlmann F, Nasmyth K, Varshavsky A. Degradation of a cohesin subunit by the N-end rule pathway is essential for chromosome stability. Nature. 2001;410:955–960. doi: 10.1038/35073627. [DOI] [PubMed] [Google Scholar]
- 149.Hauf S, Waizenegger IC, Peters J-M. Cohesin cleavage by separase required for anaphase and cytokinesis in human cells. Science. 2001;293:1320–1323. doi: 10.1126/science.1061376. [DOI] [PubMed] [Google Scholar]
- 150.Varshavsky A. Discovery of cellular regulation by protein degradation. J Biol Chem. 2008;283:34469–34489. doi: 10.1074/jbc.X800009200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Perrin BJ, Amann KJ, Huttenlocher A. Proteolysis of cortactin by calpain regulates membrane protrusion during cell migration. Mol Biol Cell. 2006;17:239–250. doi: 10.1091/mbc.E05-06-0488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Fischer S, Vandekerckhove J, Ampe C, Traub P, Weber K. Protein-chemical identification of the major cleavage sites of the Ca2+ proteinase on murine vimentin, the mesenchymal intermediate filament protein. Biol Chem Hoppe Seyler. 1986;367:1147–1152. doi: 10.1515/bchm3.1986.367.2.1147. [DOI] [PubMed] [Google Scholar]
- 153.Yoshida M, Suzuki A, Shimizu T, Ozawa E. Proteinase-sensitive sites on isolated rabbit dystrophin. J Biochem. 1992;112:433–439. doi: 10.1093/oxfordjournals.jbchem.a123918. [DOI] [PubMed] [Google Scholar]
- 154.Rees DJG, Ades SE, Singer SJ, Hynes RO. Sequence and domain structure of talin. Nature. 1990;347:685–689. doi: 10.1038/347685a0. [DOI] [PubMed] [Google Scholar]
- 155.Bate N, Gingras AR, Bachir A, Horwitz R, Ye F, Patel B, Goult BT, Critchley DR. Talin contains a C-terminal calpain-2 cleavage site important in focal adhesion dynamics. PLoS One. 2012;7:e34461. doi: 10.1371/journal.pone.0034461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kimura Y, Koga H, Araki N, Mugita N, Fujita N, Takeshima H, Nishi T, Yamashima T, Saido TC, Yamasaki T, Moritake K, Saya H, Nakao M. The involvement of calpain-dependent proteolysis of the tumor suppressor NF2 (merlin) in schwannomas and meningiomas. Nat Med. 1998;4:915–922. doi: 10.1038/nm0898-915. [DOI] [PubMed] [Google Scholar]
- 157.Zhang Z, Biesiadecki BJ, Jin JP. Selective deletion of the NH2-terminal variable region of cardiac troponin T in ischemia reperfusion by myofibril-associated mu-calpain cleavage. Biochemistry. 2006;45:11681–11694. doi: 10.1021/bi060273s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Moldoveanu T, Liu Q, Tocilj A, Watson M, Shore G, Gehring K. The X-ray structure of a BAK homodimer reveals an inhibitory zinc binding site. Mol Cell. 2006;24:677–688. doi: 10.1016/j.molcel.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 159.Cho Y-J, Abe M, Kim SY, Sato Y. Raf-1 is a binding partner of DSCR1. Arch Biochem Biophys. 2005;439:121–128. doi: 10.1016/j.abb.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 160.Pariat M, Salvat C, Bébien M, Brockly F, Altieri E, Carillo S, Jariel-Encontre I, Piechaczyk M. The sensitivity of c-Jun and c-Fos proteins to calpains depends on conformational determinants of the monomers and not on formation of dimers. Biochem J. 2000;345:129–138. [PMC free article] [PubMed] [Google Scholar]
- 161.Wei G, Yin Y, Li W, Bito H, She H, Mao Z. Calpain-mediated degradation of myocyte enhancer factor 2D contributes to excitotoxicity by activation of extrasynaptic NMDA receptors. J Biol Chem. 2012;287:5797–5805. doi: 10.1074/jbc.M111.260109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Xu J, Kurup P, Zhang Y, Goebel-Goody SM, Wu PH, Hawasli AH, Baum ML, Bibb JA, Lombroso PJ. Extrasynaptic NMDA receptors couple preferentially to excitotoxicity via calpain-mediated cleavage of STEP. J Neurosci. 2009;29:9330–9343. doi: 10.1523/JNEUROSCI.2212-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Abe K, Takeichi M. NMDA-receptor activation induces calpain-mediated beta-catenin cleavages for triggering gene expression. Neuron. 2007;53:387–397. doi: 10.1016/j.neuron.2007.01.016. [DOI] [PubMed] [Google Scholar]
- 164.Berg U, Bang P, Carlsson-Skwirut C. Calpain proteolysis of insulin-like growth factor binding protein (IGFBP) -2 and -3, but not of IGFBP-1. Biol Chem. 2007;388:859–863. doi: 10.1515/BC.2007.098. [DOI] [PubMed] [Google Scholar]
- 165.Schaecher K, Goust JM, Banik NL. The effects of calpain inhibition on IkB alpha degradation after activation of PBMCs: identification of the calpain cleavage sites. Neurochem Res. 2004;29:1443–1451. doi: 10.1023/b:nere.0000026410.56000.dd. [DOI] [PubMed] [Google Scholar]
- 166.Kishimoto A, Mikawa K, Hashimoto K, Yasuda I, Tanaka S, Tominaga M, Kuroda T, Nishizuka Y. Limited proteolysis of protein kinase C subspecies by calcium-dependent neutral protease (calpain) J Biol Chem. 1989;264:4088–4092. [PubMed] [Google Scholar]
- 167.McGinnis KM, Whitton MM, Gnegy ME, Wang KK. Calcium/calmodulin-dependent protein kinase IV is cleaved by caspase-3 and calpain in SH-SY5Y human neuroblastoma cells undergoing apoptosis. J Biol Chem. 1998;273:19993–20000. doi: 10.1074/jbc.273.32.19993. [DOI] [PubMed] [Google Scholar]
- 168.Kang MY, Zhang Y, Matkovich SJ, Diwan A, Chishti AH, Dorn GW., 2nd Receptor-independent cardiac protein kinase Calpha activation by calpain-mediated truncation of regulatory domains. Circ Res. 2010;107:903–912. doi: 10.1161/CIRCRESAHA.110.220772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Patzke H, Tsai LH. Calpain-mediated cleavage of the cyclin-dependent kinase-5 activator p39 to p29. J Biol Chem. 2002;277:8054–8060. doi: 10.1074/jbc.M109645200. [DOI] [PubMed] [Google Scholar]
- 170.Wei J, Lin CH, Wu H, Jin Y, Lee YH, Wu JY. Activity-dependent cleavage of brain glutamic acid decarboxylase 65 by calpain. J Neurochem. 2006;98:1688–1695. doi: 10.1111/j.1471-4159.2006.04074.x. [DOI] [PubMed] [Google Scholar]
- 171.Buddhala C, Suarez M, Modi J, Prentice H, Ma Z, Tao R, Wu JY. Calpain cleavage of brain glutamic acid decarboxylase 65 is pathological and impairs GABA neurotransmission. PLoS One. 2012;7:e33002. doi: 10.1371/journal.pone.0033002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Sha D, Jin Y, Wu H, Wei J, Lin CH, Lee YH, Buddhala C, Kuchay S, Chishti AH, Wu JY. Role of mu-calpain in proteolytic cleavage of brain l-glutamic acid decarboxylase. Brain Res. 2008;1207:9–18. doi: 10.1016/j.brainres.2008.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Chua BT, Guo K, Li P. Direct cleavage by the calcium-activated protease calpain can lead to inactivation of caspases. J Biol Chem. 2000;275:5131–5135. doi: 10.1074/jbc.275.7.5131. [DOI] [PubMed] [Google Scholar]
- 174.Brown N, Crawford C. Structural modifications associated with the change in Ca2+ sensitivity on activation of m-calpain. FEBS Lett. 1993;322:65–68. doi: 10.1016/0014-5793(93)81112-d. [DOI] [PubMed] [Google Scholar]
- 175.Molinari M, Anagli J, Carafoli E. Ca2+-activated neutral protease is active in the erythrocyte membrane in its noautolized 80-kDa form. J Biol Chem. 1994;269:27992–27995. [PubMed] [Google Scholar]
- 176.Farkas A, Tompa P, Schád E, Sinka R, Jekely G, Friedrich P. Autolitic activation and localization in Schneider cells (S2) of calpain B from Drosophila. Biochem J. 2004;378:299–305. doi: 10.1042/BJ20031310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Agard NJ, Mahrus S, Trinidad JC, Lynn A, Burlingame AL, Wells JA. Global kinetic analysis of proteolysis via quantitative targeted proteomics. Proc Natl Acad Sci USA. 2012;109:1913–1918. doi: 10.1073/pnas.1117158109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Green DR. Means to an end: apoptosis and other cell death mechanisms. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2011. [Google Scholar]
- 179.Pop C, Salvesen GS. Human caspases: activation, specificity, and regulation. J Biol Chem. 2009;284:21777–21781. doi: 10.1074/jbc.R800084200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Dix MM, Simon GM, Cravatt BF. Global mapping of the topography and magnitude of proteolytic events in apoptosis. Cell. 2008;134:679–691. doi: 10.1016/j.cell.2008.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Lin Y, Devin A, Rodriguez Y, Liu Z-G. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev. 1999;13:2514–2526. doi: 10.1101/gad.13.19.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Kim JW, Choi E-J, Joe CO. Activation of death-inducing signaling complex (DISC) by pro-apoptotic C-terminal fragment of RIP. Oncogene. 2000;19:4491–4499. doi: 10.1038/sj.onc.1203796. [DOI] [PubMed] [Google Scholar]
- 183.Rajput A, Kovalenko A, Bogdanov K, Yang S-H, Kang T-B, Kim J-CDJ, Wallach D. RIG-I RNA helicase activation of IRF3 transcription factor is negatively regulated by caspase-8-mediated cleavage of the RIP1 protein. Immunity. 2011;34:340–351. doi: 10.1016/j.immuni.2010.12.018. [DOI] [PubMed] [Google Scholar]
- 184.Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008;9:231–241. doi: 10.1038/nrm2312. [DOI] [PubMed] [Google Scholar]
- 185.O'Donovan PJ, Livingston DM. BRCA1 and BRCA2: breast/ovarian cancer susceptibility gene products and participants in DNA double-strand break repair. Carcinogenesis. 2010;31:961–967. doi: 10.1093/carcin/bgq069. [DOI] [PubMed] [Google Scholar]
- 186.Pathania S, Nguyen J, Hill SJ, Scully R, Adelmant GO, Marto JA, Feunteun J, Livingston DM. BRCA1 is required for postreplication repair after UV-induced DNA damage. Mol Cell. 2011;44:235–251. doi: 10.1016/j.molcel.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Tomioshi G, Horita Y, Nishita M, Ohashi K, Mizuno K. Caspase-mediated cleavage and activation of LIM-kinase 1 and its role in apoptotic membrane blebbing. Genes Cells. 2004;9:591–600. doi: 10.1111/j.1356-9597.2004.00745.x. [DOI] [PubMed] [Google Scholar]
- 188.Tikhmyanova N, Golemis EA. NEDD9 and BCAR1 negatively regulate E-cadherin membrane localization, and promote E-cadherin degradation. PLoS One. 2011;6:e22102. doi: 10.1371/journal.pone.0022102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Mehlen P, Thibert C. Dependence receptors: between life and death. Cell Mol Life Sci. 2004;61:1854–1866. doi: 10.1007/s00018-004-3467-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Goldschneider D, Mehlen P. Dependence receptors: a new paradigm in cell signaling and cancer. Oncogene. 2010;29:1865–1882. doi: 10.1038/onc.2010.13. [DOI] [PubMed] [Google Scholar]
- 191.LaGory EL, Sitailo LA, Denning MF. The protein kinase Cdelta catalytic fragment is critical for maintenance of the G2/M DNA damage checkpoint. J Biol Chem. 2010;285:1879–1887. doi: 10.1074/jbc.M109.055392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Datta R, Kojima H, Yoshida K, Kufe D. Caspase-3-mediated cleavage of protein kinase C-theta in induction of apoptosis. J Biol Chem. 1997;272:20317–20320. doi: 10.1074/jbc.272.33.20317. [DOI] [PubMed] [Google Scholar]
- 193.Wu YM, Huang CL, Kung HJ, Huang CYF. Proteolytic activation of Etk/Bmx tyrosine kinase by caspases. J Biol Chem. 2001;276:17672–17678. doi: 10.1074/jbc.M010964200. [DOI] [PubMed] [Google Scholar]
- 194.Sabourin LA, Seale P, Wagner J, Rudnicki MA. Caspase 3 cleavage of the Ste20-related kinase SLK releases and activates an apoptosis-inducing kinase domain and an actin-disassembling region. Mol Cell Biol. 2000;20:684–696. doi: 10.1128/mcb.20.2.684-696.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Arnold R, Liou J, Drexler HCA, Weiss A, Friedemann K. Caspase-mediated cleavage of hematopoietic progenitor kinase 1 (HPK1) converts an activator of NF-kappaB into an inhibitor of NF-kappaB. J Biol Chem. 2001;276:14675–14684. doi: 10.1074/jbc.M008343200. [DOI] [PubMed] [Google Scholar]
- 196.Chen F, Arseven OK, Cryns VL. Proteolysis of the mismatch repair protein MLH1 by caspase-3 promotes DNA damage-induced apoptosis. J Biol Chem. 2004;279:27542–27548. doi: 10.1074/jbc.M400971200. [DOI] [PubMed] [Google Scholar]
- 197.O'Donnell MA, Perez-Jimenez E, Oberst A, Ng A, Massoumi R, Xavier R, Green DR, Ting AT. Caspase 8 inhibits programmed necrosis by processing CYLD. Nat Cell Biol. 2011;13:1437–1442. doi: 10.1038/ncb2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Gervais JLM, Seth P, Zhang H. Cleavage of CDK inhibitor p21Cip1/Waf1 by caspases is an early event during DNA damage-induced apoptosis. J Biol Chem. 1998;273:19207–11912. doi: 10.1074/jbc.273.30.19207. [DOI] [PubMed] [Google Scholar]
- 199.Hirota J, Furuichi T, Mikoshiba K. Inositol 1,4,5-trisphosphate receptor type 1 is a substrate for caspase-3 and is cleaved during apoptosis in a caspase-3-dependent manner. J Biol Chem. 1999;274:34433–34437. doi: 10.1074/jbc.274.48.34433. [DOI] [PubMed] [Google Scholar]
- 200.Takahashi A, Alnemri ES, Lazebnik Y, Fernandes-Alnemri T, Litwack G, Moir RD, Goldman RD, Poirier GG, Kaufmann SH, Earnshaw WC. Cleavage of lamin A by Mch2-alpha but CPP32: multiple interleukin 1-beta-converting enzyme-related proteases with distinct substrate recognition properties are active in apoptosis. Proc Natl Acad Sci USA. 1996;93:8395–8400. doi: 10.1073/pnas.93.16.8395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Choul-Li S, Leroy C, Leprivier G, Laitem C, Tulasne D, Aumercier M. Caspase cleavage of Ets-1 p51 generates fragments with transcriptional dominant-negative function. Biochem J. 2010;426:229–241. doi: 10.1042/BJ20090877. [DOI] [PubMed] [Google Scholar]
- 202.Samedjima K, Svingen PA, Basi GS, Kottke T, Mesner PW, Jr, Stewart L, Durrieu F, Poirier GG, Alnemri ES, Champoux JJ, Kauffmann SH, Earnshaw WC. Caspase-mediated cleavage of DNA topoisomerase I at unconventional sites during apoptosis. J Biol Chem. 1999;274:4335–4340. doi: 10.1074/jbc.274.7.4335. [DOI] [PubMed] [Google Scholar]
- 203.Okamoto S-I, Li Z, Ju C, Schölzke MN, Mathews e, Cui J, Salvesen GS, Bossy-Wetzel e, Lipton SA. Dominant-interfering forms of MEF2 generated by caspase cleavage contribute to NMDA-induced neuronal apoptosis. Proc Natl Acad Sci USA. 2002;99:3974–3979. doi: 10.1073/pnas.022036399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Song Q, Lees-Miller SP, Kumar S, Zhang N, Chan DW, Smith GCM, Jackson SP, Alnemri ES, Litwack G, Khanna KK, Lavin MF. DNA-dependent protein kinase catalytic subunit: a target for an ICE-like protease in apoptosis. EMBO J. 1996;13:3238–3246. [PMC free article] [PubMed] [Google Scholar]
- 205.Marambaud P, Shioi J, Serban G, Georgakopoulos A, Sarner S, Nagy V, Baki L, Wen P, Efthimiopoulos S, Shao Z, Wisniewski T, Robakis NK. A presenilin-1/gamma-secretase cleavage releases the E-cadherin intracellular domain and regulates disassembly of adherens junctions. EMBO J. 2002;21:1948–1956. doi: 10.1093/emboj/21.8.1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Giaime E, Sunyach C, Herrant M, Grosso S, Auberger P, McLean PJ, Checler F, da Costa CA. Caspase-3-derived C-terminal product of synphilin-1 displays antiapoptotic function via modulation of the p53-dependent cell death pathway. J Biol Chem. 2006;281:11515–11522. doi: 10.1074/jbc.M508619200. [DOI] [PubMed] [Google Scholar]
- 207.Sahara S, Aoto M, Eguchi Y, Imamoto N, Yoneda Y, Tsujimoto Y. Acinus is a caspase-3-activated protein required for apoptotic chromatin condensation. Nature. 1999;401:168–173. doi: 10.1038/43678. [DOI] [PubMed] [Google Scholar]
- 208.Stegh AH, Herrmann H, Lampel S, Weisenberger D, Andrä K, Seper M, Wiche G, Krammer PH, Peter ME. Identification of the cytolinker plectin as a major early in vivo substrate for caspase 8 during CD95- and tumor necrosis factor receptor-mediated apoptosis. Mol Cell Biol. 2000;20:5665–5679. doi: 10.1128/mcb.20.15.5665-5679.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Mazumder S, Gong B, Chen Q, Drazba JA, Bushcsbaum JC, Almasan A. Proteolytic cleavage of cyclin E leads to inactivation of associated kinase activity and amplification of apoptosis in hemetopoietic cells. Mol Cell Biol. 2002;22:2398–2409. doi: 10.1128/MCB.22.7.2398-2409.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Pászty K, Antalffy G, Penheiter AR, Homolya L, Padányi R, Iliás A, Filoteo AG, Penniston JT, Enyedi A. The caspase-3 cleavage product of the plasma membrane Ca2+-ATPase 4b is activated and appropriately targeted. Biochem J. 2005;391:687–692. doi: 10.1042/BJ20051012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Tu S, Cerione RA. Cdc42 is a substrate for caspase and influences Fas-induced apoptosis. J Biol Chem. 2001;276:19656–19663. doi: 10.1074/jbc.M009838200. [DOI] [PubMed] [Google Scholar]
- 212.Zhao X, Wang D, Zhao Z, Xiao Y, Sengupta S, Xiao Y, Zhang R, Lauber K, Wesselborg S, Feng L, Rose TM, Shen YZ, J, Prestwich G, Xu Y. Caspase-3-dependent activation of calcium-independent phospholipase A2 enhances cell migration in non-apoptotic ovarian cancer cells. J Biol Chem. 2006;281:29357–29368. doi: 10.1074/jbc.M513105200. [DOI] [PubMed] [Google Scholar]
- 213.Matsuzawa T, Fukui A, Kashimoto T, Nagao K, Oka K, Miyake M, Horiguchi Y. Bordetella dermonecrotic toxin undergoes proteolytic processing to be translocated from a dynamin-related endosome into the cytoplasm in an acidification-independent manner. J Biol Chem. 2004;279:2866–2872. doi: 10.1074/jbc.M310340200. [DOI] [PubMed] [Google Scholar]
- 214.Seki M, Watanabe A, Enomoto S, Kawamura T, Ito H, Kodama T, Hamakubo T, Aburatani H. Human ROBO1 is cleaved by metalloproteinases and gamma-secretase and migrates to the nucleus in cancer cells. FEBS Lett. 2010;584:2909–2915. doi: 10.1016/j.febslet.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 215.Dublet B, Ruello A, Pederzoli M, Hajjar E, Courbebaisse M, Canteloup S, Reuter N, Witko-Sarsat V. Cleavage of p21/WAF1/CIP1 by proteinase 3 modulates differentiation of a monocytic cell line. Molecular analysis of the cleavage site. J Biol Chem. 2005;280:30242–30253. doi: 10.1074/jbc.M414609200. [DOI] [PubMed] [Google Scholar]
- 216.Yang Q-H, Church-Hajduk R, Ren J, Newton ML, Du C. Omi/HtrA2 catalytic cleavage of inhibitor of apoptosis (IAP) irreversibly inactivates IAPs and facilitates caspase activity in apoptosis. Genes Dev. 2003;17:1487–1496. doi: 10.1101/gad.1097903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Lane AA, Ley TJ. Neutrophil elastase cleaves PML-RARalpha and is important for the development of acute promyelocytic leukemia in mice. Cell. 2003;115:305–318. doi: 10.1016/s0092-8674(03)00852-3. [DOI] [PubMed] [Google Scholar]
- 218.Yamazaki T, Akada T, Niizeki O, Suzuki T, Miyashita H, Sato Y. Puromycin-insensitive leucyl-specific aminopeptidase (PILSAP) binds and catalyzes PDK1, allowing VEGF-stimulated activation of S6K for endothelial cell proliferation and angiogenesis. Blood. 2011;104:2345–2352. doi: 10.1182/blood-2003-12-4260. [DOI] [PubMed] [Google Scholar]
- 219.Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, Armstrong AJ, Penuela S, Laird DW, Salvesen GS, Isakson BE, Bayliss DA, Ravichandran KS. Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature. 2010;467:863–867. doi: 10.1038/nature09413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Sandilos JK, Chiu YHC, Chekeni FB, Armstrong AJ, Walk SF, Ravichandran KS, Bayliss DA. Pannexin 1, an ATP release channel, is activated by caspase cleavage of its pore-associated C-terminal autoinhibitory region. J Biol Chem. 2012;287:11303–11311. doi: 10.1074/jbc.M111.323378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Ruiz-Vela A, de Buitrago GG, Martínez C. Implication of calpain in caspase activation during B cell clonal deletion. EMBO J. 1999;18:4988–4998. doi: 10.1093/emboj/18.18.4988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Nakagawa T, Yuana J. Activation of caspase-12 by calpain in apoptosis. J Cell Biol. 2000;150:887–894. doi: 10.1083/jcb.150.4.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Vogel JL, Kristie TM. Autocatalytic proteolysis of the transcription factor-coactivator C1 (HCF): a potential role for proteolytic regulation of coactivator function. Proc Natl Acad Sci USA. 2000;97:9425–9430. doi: 10.1073/pnas.160266697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Butkinaree C, Cheung WD, Park S, Park K, Barber M, Hart GW. Characterization of beta-N-acetylglucosaminidase cleavage by caspase-3 during apoptosis. J Biol Chem. 2008;283:23557–23566. doi: 10.1074/jbc.M804116200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Hsieh JJ, Ernst P, Erdjument-Bromage H, Tempst P, Korsmeyer SJ. Proteolytic cleavage of MLL generates a complex of N- and C-terminal fragments that confers protein stability and subnuclear localization. Mol Cell Biol. 2003;23:186–194. doi: 10.1128/MCB.23.1.186-194.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Yokoyama A, Ficara F, Murphy MJ, Meisel C, Naresh A, Kitabayashi I, Cleary ML. Proteolytically cleaved MLL subunits are susceptible to distinct degradation pathways. J Cell Sci. 2011;124:2208–2219. doi: 10.1242/jcs.080523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Gumy LF, Yeo GS, Tung YCZ KH, Willis D, Coppola G, Lam BY, Twiss JL, Holt CE, Fawcett JW. Transcriptome analysis of embryonic and adult sensory axons reveals changes in mRNA repertoire localization. RNA. 2011;17:85–98. doi: 10.1261/rna.2386111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Baudry M, Lynch G. Remembrance of arguments past: how well is the glutamate receptor hypothesis of LTP is holding up after 20 years? Neurobiol Learn Mem. 2001;76:284–297. doi: 10.1006/nlme.2001.4023. [DOI] [PubMed] [Google Scholar]
- 229.Friedrich P, Bozóky Z. Digestive versus regulatory proteases: on calpain action in vivo. Biol chem. 2005;386:609–612. doi: 10.1515/BC.2005.071. [DOI] [PubMed] [Google Scholar]
- 230.Crawford ED, Wells JA. Caspase substrates and cellular remodeling. Annu Rev Biochem. 2011;80:1055–1087. doi: 10.1146/annurev-biochem-061809-121639. [DOI] [PubMed] [Google Scholar]
- 231.Onn I, Heidinger-Pauli JM, Guacci V, Unal E, Koshland DE. Sister chromatid cohesion: a simple concept with a complex reality. Annu Rev Cell Dev Biol. 2008;24:105–129. doi: 10.1146/annurev.cellbio.24.110707.175350. [DOI] [PubMed] [Google Scholar]
- 232.Ocampo-Hafalla MT, Uhlmann F. Cohesin loading and sliding. J Cell Sci. 2011;124:685–691. doi: 10.1242/jcs.073866. [DOI] [PubMed] [Google Scholar]
- 233.Nasmyth K. Cohesin: a catenase with separate entry and exit gates? Nat Cell Biol. 2011;13:1170–1177. doi: 10.1038/ncb2349. [DOI] [PubMed] [Google Scholar]
- 234.Johnson ES, Gonda DK, Varshavsky A. Cis-trans recognition and subunit-specific degradation of short-lived proteins. Nature. 1990;346:287–291. doi: 10.1038/346287a0. [DOI] [PubMed] [Google Scholar]
- 235.Hochstrasser M, Varshavsky A. In vivo degradation of a transcriptional regulator: the yeast MATalpha2 repressor. Cell. 1990;61:697–708. doi: 10.1016/0092-8674(90)90481-s. [DOI] [PubMed] [Google Scholar]
- 236.Schrader EK, Harstad KG, Matouschek A. Targeting proteins for degradation. Nat Chem Biol. 2009;5:815–822. doi: 10.1038/nchembio.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Kang J-E, Lim MM, Bateman RJ, Lee JJ, Smyth LP, Cirrito JR, Fujiki N, Nishino S, Holtzman DM. Amyloid-β dynamics are regulated by orexin and the sleep-wake cycle. Science. 2009;326:1005–1007. doi: 10.1126/science.1180962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Soscia SJ, Kirby JE, Washicosky KJ, Tucker SM, Ingelsson M, Hyman B, Burton MA, Goldstein LE, Duong S, Tanzi RE, Moir RD. The Alzheimer's disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One. 2010;5:e9505. doi: 10.1371/journal.pone.0009505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Tasaki T, Kwon YT. The mammalian N-end rule pathway: new insights into its components and physiological roles. Trends Biochem Sci. 2007;32:520–528. doi: 10.1016/j.tibs.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 240.Starheim KK, Gevaert K, Arnesen T. Protein N-terminal acetyltransferases: when the start matters. Trends Biochem Sci. 2012;37:152–161. doi: 10.1016/j.tibs.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 241.White TA, Xue A, Chini EN, Thompson M, Sieck GC, Wylam ME. Role of transient receptor potential C3 in TNF-alpha-enhanced calcium influx in human airway myocytes. Am J Respir Cell Mol Biol. 2006;35:243–251. doi: 10.1165/rcmb.2006-0003OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Fu T, Xu Y, Jiang W, Zhang H, Zhu P, Wu J. EGF receptor-mediated intracellular calcium increase in human hepatoma BEL-7404 cells. Cell Res. 1994;4:145–153. [Google Scholar]
- 243.Størling J, Zaitsev SV, Kapelioukh IL, Karlsen AE, Billestrup N, Berggren PO, Mandrup-Poulsen T. Calcium has a permissive role in interleukin-1beta-induced c-jun N-terminal kinase activation in insulin-secreting cells. Endocrinology. 2005;146:3026–3036. doi: 10.1210/en.2005-0036. [DOI] [PubMed] [Google Scholar]
- 244.Pritchard S, Guilak F. Effects of interleukin-1 on calcium signaling and the increase of filamentous actin in isolated and in situ articular chondrocytes. Arthritis Rheum. 2006;54:2164–2174. doi: 10.1002/art.21941. [DOI] [PubMed] [Google Scholar]
- 245.Arora PD, Ma J, Min W, Cruz T, McCulloch CA. Interleukin-1-induced calcium flux in human fibroblasts is mediated through focal adhesions. J Biol Chem. 1995;270:6042–6049. doi: 10.1074/jbc.270.11.6042. [DOI] [PubMed] [Google Scholar]
- 246.Rozanski GJ, Witt RC. Interleukin-1 enhances beta-responsiveness of cardiac L-type calcium current suppressed by acidosis. Am J Physiol. 1994;267:H1361–H1367. doi: 10.1152/ajpheart.1994.267.4.H1361. [DOI] [PubMed] [Google Scholar]
- 247.Viviani B, Bartesaghi S, Gardoni F, Vezzani A, Behrens MM, Bartfai T, Binaglia M, Corsini E, Di Luca M, Galli CL, Marinovich M. Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J Neurosci. 2003;23:8692–8700. doi: 10.1523/JNEUROSCI.23-25-08692.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Tuschil A, Lam C, Haslberger A, Lindley I. Interleukin-8 stimulates calcium transients and promotes epidermal cell proliferation. J Invest Dermatol. 1992;99:294–298. doi: 10.1111/1523-1747.ep12616634. [DOI] [PubMed] [Google Scholar]
- 249.Glading A, Bodnar RJ, Reynolds I, Shiraha H, Satish L, Potter DA, Blair HC, Wells A. Epidermal growth factor activates m-calpain (calpain II), at least in part, by extracellular signal-regulated kinase-mediated phosphorylation. Mol Cell Biol. 2004;24:2499–2512. doi: 10.1128/MCB.24.6.2499-2512.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Catlin MC, Guitzetti M, Costa LG. Effects of ethanol on calcium homeostasis in the nervous system. Mol Neurobiol. 1999;19:1–24. doi: 10.1007/BF02741375. [DOI] [PubMed] [Google Scholar]
- 251.Simasko SM, Boyadjieva N, De A, Sarkar DK. Effect of ethanol on calcium regulation in rat fetal hypothalamic cells in culture. Brain Res. 1999;824:89–96. doi: 10.1016/s0006-8993(99)01188-9. [DOI] [PubMed] [Google Scholar]
- 252.Debelak-Kragtorp KA, Armant DR, Smith SM. Ethanol-induced cephalic apoptosis requires phospholipase C-dependent intracellular calcium signaling. Alcohol Clin Exp Res. 2003;27:515–523. doi: 10.1097/01.ALC.0000056615.34253.A8. [DOI] [PubMed] [Google Scholar]
- 253.Erickson CK, Tyler TD, Harris RA. Ethanol: modification of acute intoxication by divalent cations. Science. 1978;199:1219–1221. doi: 10.1126/science.343251. [DOI] [PubMed] [Google Scholar]
- 254.Harris RA. Alteration of alcohol effects by calcium and other inorganic ions. Pharmacol Biochem Behav. 1979;10:527–534. doi: 10.1016/0091-3057(79)90228-4. [DOI] [PubMed] [Google Scholar]
- 255.Erickson CK, Tyler TD, Beck LK, Duensing KL. Calcium enhancement of alcohol and drug-induced sleeping time in mice and rats. Pharmacol Biochem Behav. 1980;12:651–656. doi: 10.1016/0091-3057(80)90143-4. [DOI] [PubMed] [Google Scholar]
- 256.Yokogawa T, Marin W, Faraco M, Pézeron G, Appelbaum L, Zhang J, Rosa F, Mourrain P, Mignot E. Characterization of sleep in zebrafish and insomnia in hypocretin receptor mutants. PLoS Biol. 2007;5:2379–2397. doi: 10.1371/journal.pbio.0050277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Rihel J, Prober DA, Schier AF. Monitoring sleep and arousal in zebrafish. Meth Cell Biol. 2010;100:281–294. doi: 10.1016/B978-0-12-384892-5.00011-6. [DOI] [PubMed] [Google Scholar]
- 258.Lima SL, Rattenborg NC, Lesku JA, Amlaner CJ. Sleeping under the risk of predation. Anim Behav. 2005;70:723–736. [Google Scholar]
- 259.Lyamin OI, Kosenko PO, Lapierre JL, Mukhametov LM, Siegel JM. Fur seals display a strong drive for bilateral slow-wave sleep while on land. J Neurosci. 2008;28:12614–12621. doi: 10.1523/JNEUROSCI.2306-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Raizen DM, Zimmerman JE, Maycock MH, Ta UD, You YJ, Sundaram MV, Pack AI. Lethargus is a Caenorhabditis elegans sleep-like state. Nature. 2008;451:569–572. doi: 10.1038/nature06535. [DOI] [PubMed] [Google Scholar]
- 261.Olofsson B, de Bono M. Sleep: dozy worms and sleepy flies. Curr Biol. 2008;18:R204–R206. doi: 10.1016/j.cub.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 262.Buskirk CV, Sternberg PW. Epidermal growth factor signaling induces behavioral quiescence in Caenorhabditis elegans. Nat Neurosci. 2007;10:1300–1307. doi: 10.1038/nn1981. [DOI] [PubMed] [Google Scholar]
- 263.You Y-J, Kim J, Raizen D, Avery L. Insulin and TGF-beta act through a PKG to control satiety behavior in C. elegans. Cell Met. 2008;7:247–257. [Google Scholar]
- 264.Buskirk CV, Sternberg PW. Paired and LIM class homeodomain proteins corrdinate differentiation of the C. elegans ALA neuron. Development. 2010;137:2065–2074. doi: 10.1242/dev.040881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Tu BP, McKnight SL. Evidence of carbon monoxide-mediated phase advancement of the yeast metabolic cycle. Proc Natl Acad Sci USA. 2009;106:14293–14296. doi: 10.1073/pnas.0907786106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Tobias JW, Shrader TE, Rocap G, Varshavsky A. The N-end rule in bacteria. Science. 1991;254:1374–1377. doi: 10.1126/science.1962196. [DOI] [PubMed] [Google Scholar]
- 267.Román-Hernández G, Hou JY, Grant RA, Sauer RT, Baker TA. The ClpS adaptor mediates staged delivery of N-end rule substrates to the AAA+ ClpAP protease. Mol Cell. 2011;43:217–228. doi: 10.1016/j.molcel.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Dargelos E, Poussard S, Brulé C, Daury L, Cottin P. Calcium-dependent proteolytic system and muscle dysfunctions: a possible role of calpains in sarcopenia. Biochimie. 2008;90:359–368. doi: 10.1016/j.biochi.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 269.Belcastro AN, Shewchuk LD, Raj DA. Exercise-induced muscle injury: a calpain hypothesis. Mol Cell Biochem. 1998;179:135–145. doi: 10.1023/a:1006816123601. [DOI] [PubMed] [Google Scholar]
- 270.Patterson C, Portbury AL, Schisler JC, Willis MS. Tear me down: role of calpain in the development of cardiac ventricular hypertrophy. Circ Res. 2011;109:453–462. doi: 10.1161/CIRCRESAHA.110.239749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Shimizu K, Phan T, Mansuy IM, Storm DR. Proteolytic degradation of SCOP in the hippocampus contributes to activation of MAP kinase and memory. Cell. 2007;128:1219–1229. doi: 10.1016/j.cell.2006.12.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Kwon YT, Balogh SA, Davydov IV, Kashina AS, Yoon JK, Xie Y, Gaur A, Hyde L, Denenberg VH, Varshavsky A. Altered activity, social behavior, and spatial memory in mice lacking the NTAN1 amidase and the asparagine branch of the N-end rule pathway. Mol Cell Biol. 2000;20:4135–4148. doi: 10.1128/mcb.20.11.4135-4148.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Dubnau J, Chinag AS, Grady L, Barditch J, Gossweiler S, McNeil J, Smith P, Buldoc F, Scott R, Certa U, Broger C, Tully T. The staufen/pumilio pathway is involved in Drosophila long-term memory. Curr Biol. 2003;13:286–296. doi: 10.1016/s0960-9822(03)00064-2. [DOI] [PubMed] [Google Scholar]
- 274.Wang H, Piatkov KI, Brower CS, Varshavsky A. Glutamine-specific N-terminal amidase, a component of the N-end rule pathway. Mol Cell. 2009;34:686–695. doi: 10.1016/j.molcel.2009.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]