

Abstract
Enoyl-acyl carrier protein reductase (ENR) is a crucial enzyme in the type II fatty acid synthesis pathway of many pathogens such as Plasmodium falciparum, the etiological agent of the most severe form of malaria. Because of its essential function of fatty acid double bond reduction and the absence of a human homologue, PfENR is an interesting drug target. Although extensive knowledge of the protein structure has been gathered over the last decade, comparatively little remains known about the dynamics of this crucial enzyme. Here, we perform extensive molecular dynamics simulations of tetrameric PfENR in different states of cofactor and ligand binding, and with a variety of different ligands bound. A pocket-volume analysis is also performed, and virtual screening is used to identify potential druggable hotspots. The implications of the results for future drug-discovery projects are discussed.
Keywords: molecular dynamics, protein–drug interactions, computer-aided drug discovery, enoyl-acyl carrier protein reductase
Introduction
The synthesis of fatty acids is an essential process for all living organisms. Many species rely on converting energy derived from sugars into fats for storage. Fatty acid synthesis, a process conducted through the fatty acid synthase (FAS) system, involves using acetyl-CoA, an intermediate in the metabolism of simple sugars, into fatty acids that are subsequently esterified with glycerol to form lipids. There are two types of FAS pathways in nature.1 Humans, vertebrates, and some bacteria utilize type I FAS. In this associated pathway, all reactions are catalyzed by different domains of a single, multifunctional protein. In contrast, type II FAS, utilized by plants and most prokaryotes, is a dissociated pathway, where each reaction is catalyzed by a different enzyme.
The product of each reaction is carried around by a highly acidic small protein called acyl carrier protein (ACP). The proteins of the fatty acid synthesis pathway are interesting drug targets for several reasons. First, FAS is essential for the survival of mammals, plants, fungi, and bacteria.2 Second, though fatty acids are essential for bacterial growth, they cannot be scavenged from the host and must be synthesized de novo.3 Third, the pathway in most pathogenic prokaryotes is different from that in humans; consequently, drugs targeting the prokaryotic FAS pathway would be less likely to cause side effects by interacting with homologous human proteins. A detailed understanding of the structures and processes involved in type II FAS may therefore offer insight that will be helpful in future drug-discovery projects.
In this study, we focus on the enoyl-ACP reductase (ENR) of Plasmodium falciparum, a protozoan parasite that causes a severe form of malaria in humans.4 ENR catalyzes the final step of the fatty acid elongation cycle, NADH-dependent reduction of the double bond of ACP-bound fatty acids (between C2 and C3 of enoyl-ACP5; see Fig. 1 in Ref. 2). This reduction converts a trans-2-enoyl-ACP to acyl-ACP, with NADH serving as the reductant (sometimes also referred to as a coenzyme or cofactor).2 In the process of elongation, a growing fatty acid generally cycles the pathway multiple times until it reaches its final length. Because ENR has been shown to be the rate-determining step of type II FAS,6 ENR plays a crucial role in regulating the catalysis of fatty acids7 and the synthesis of bacterial cell walls.8 By inhibiting the synthesis of fatty acid by ENR, we envision shutting down Plasmodium falciparum's ability for survival.
Figure 1.
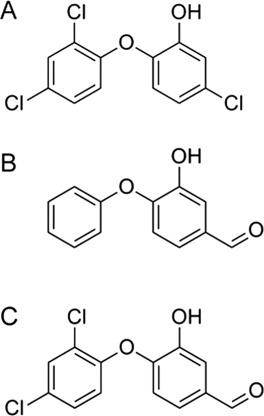
Structures of the ligands used in the simulations: (A) Triclosan, (B) FT0, and (C) FT1.
ENRs are tetrameric α-β proteins with a classic Rossmann fold. They bind the NADH cofactor in an elongated binding pocket reaching from the center of the tetramer back to Trp36. The active site of ENR (with its signature residues tyrosine and lysine) is located in a cleft between the protein core and a protruding helical subdomain.9 Substrate specificity, which varies by species, is determined by the flexible “substrate-binding loop.”9,10 The catalytic reduction mechanism of ENR is depicted in Figure 2 of Ref. 9. Conjugate addition of a NADH hydride ion to the C3 substrate atom causes reduction of the trans-2-acyl C2–C3 double bond, thus completing the synthesis of the acyl chain. An intermediate enolate anion on the substrate C1 carbonyl oxygen atom is formed in the process; the proton is accepted by the key Tyr182 residue.
Figure 2.

RMSF versus residue plots for all nine simulations. (A) 1UH5 apo simulation, (B) 3LT0 apo simulation, (C) 3LSY apo simulation, (D) 1UH5 NAD-bound simulation, (E) 3LT0 NAD-bound simulation, (F) 3LSY NAD-bound simulation, (G) 1UH5 TCL-NAD-bound simulation, (H) 3LT0 FT1-NAD-bound simulation, and (I) 3LSY FT0-NAD-bound simulation. Largest fluctuations are observed in the central part of the substrate-binding loop. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
One of the most studied inhibitors of PfENR is a drug called triclosan (TCL).11 Triclosan is an antibacterial and antifungal compound used in many consumer products such as mouthwashes, soaps, toothpastes, cosmetics, plastics, and textiles.12 Since TCL has been overused commercially, there is resistance and a need for new inhibitors for ENR is imperative. Binding of TCL, a triclorinated biphenyl ether2 [see Fig. 1(A)], is competitive with NADH, but not competitive with NAD+6,13; in fact, inhibition requires that NAD+ be bound.2 TCL targets ENR at the acyl substrate-binding pocket,9 where it forms a stable ternary complex with the protein and the oxidized cofactor (NAD+). A number of binding features explain the high affinity of TCL: extensive van der Waals contacts in the ternary complex facilitated by the movement of the substrate-binding-loop residues 318–324,6,14 a phenol ring that makes π-stacking interactions with the NAD+ nicotinamide ring,2 and hydrogen bonds that form between the phenolic TCL hydroxyl group and both the phenolic oxygen atom of Tyr182 (Tyr156 in FabI) and the 2′-hydroxyl group of the nicotinamide ribose.2,6
Although extensive research has elucidated the structures of ENR in its cofactor and drug bound states, much less is known on the dynamics of this system.8,15–17 Molecular dynamics (MD) simulations can be used to investigate the dynamics of protein–ligand complexes.18 In this study, we describe extensive MD simulations performed on a tetramer complex of PfENR in different states of cofactor and drug binding. Simulations with TCL and two of its variants are performed. In addition to a general analysis of the fluctuations of the system, the evolution of TCL interactions with the protein and cofactor over time is investigated. Additionally, a pocket-volume analysis is performed of each simulation, giving valuable insight into the time evolution of active-site size and PfENR geometry. Finally, an extensive search for alternative druggable ENR sites is performed. These simulations will be helpful for those pursuing further pharmacological studies. Additionally, the structures and active sites of ENR from different species are similar; thus, knowledge gained by studying PfENR may be applicable to the ENR enzymes and its homologues.
Results and Discussion
MD simulations show fluctuations in the substrate-binding loop
A total of 100 ns MD simulations were performed for each of the three structures under investigation (1UH5, 3LSY, and 3LT0), in apo (no ligand, no NAD cofactor), NAD-bound (no ligand, with NAD cofactor), and ligand-NAD-bound (with ligand and NAD cofactor) states. When monomers are considered separately, this amounts to a total of 3.6 μs of MD simulation on a single ENR subunit. Heavy atom RMSD versus time plots (Supporting Information Figure 1) show that the simulations equilibrated after 10–20 ns. General RMSD values of 2.5–3.5 Å with respect to the starting structures are low given the large size of the protein (1316 residues, 329 residues per subunit) and suggest that the crystal structures represent stable, low-energy structures. Furthermore, the fluctuations over the course of the simulations are not distributed evenly over all residues. A root mean square fluctuation analysis (see Fig. 2) demonstrates that the vast majority of the residues (between 85 and 89%) stay well below 2 Å of fluctuations. By far the largest fluctuations are observed for the residues that were originally missing from the crystal structures (325–366). Indeed, it may well be that these residues could not be resolved in the crystal structures because they are so mobile.
The following residues are known to interact with the cofactor: W131, D168, S170, S215, L216, N218, L265, S317, and A319. These residues have been labeled blue in the RMSF plots. Similarly, residues Y267, Y277, K285, and I323 (Fig. 2, in red) interact with the ligand. It is clear from Figure 2 that these residues are among the least flexible residues of the protein, suggesting that tight interactions between the protein and its bound substituents persist throughout the simulations and that the residues in contact with the ligand and cofactor are substantially stabilized. Since these residues are relatively stable even in the apo and cofactor-bound simulations one may speculate that low general fluctuations of binding residues support ligand binding. Another interesting observation evident in the RMSF analysis is that the individual subunits behave independently of each other. While the overall fluctuations are similar across subunits, there are distinct differences (particularly with respect to the amplitude of the fluctuations) between the different subunits. This is particularly important for drug-discovery studies, as it shows that each subunit is independent. Consequently each monomer can provide independent conformations all of which are valid drug targets.
The results of the PCA analysis (see Supporting Information Figure 2) were less conclusive. When projected into the PC1–PC2 space, all the simulations sampled roughly the same area, close to the projections of the crystal structures. There are no obvious differences in terms of principal-component space sampled between the different proteins or ligand binding states.
Pocket-volume analysis
Binding-pocket volumes were monitored over the course of the nine simulations. The volumes of the ligand-NAD-bound pockets were calculated after removal of both the cofactor and ligand. The pocket volumes extracted from the NAD-bound simulations were calculated in the presence of the cofactor. Thus these values describe the volume accessible to the ligand only. Figure 3 displays the calculated pocket volumes as a function of simulation time for all simulations.
Figure 3.

Active-site pocket volumes over the course of the simulations. (A) 1UH5 apo simulation, (B) 3LT0 apo simulation, (C) 3LSY apo simulation, (D) 1UH5 NAD-bound simulation, (E) 3LT0 NAD-bound simulation, (F) 3LSY NAD-bound simulation, (G) 1UH5 TCL-NAD-bound simulation, (H) 3LT0 FT1-NAD-bound simulation, and (I) 3LSY FT0-NAD-bound simulation. Volumes were calculated using POVME and are shown for each of the subunits in the protein (Pockets 1–4). Volumes in the apo and ligand-NAD-bound conformations were calculated without any ligands or cofactors in the pockets. Volumes in the NAD-bound simulations were calculated with the cofactor present. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
The most obvious finding is that the binding pocket is highly dynamic and flexible. In some cases, significant differences in the volumes of the pockets of the same tetramer are even observed. This suggests that there is no unique static protein conformation that defines the ENR binding pocket. Binding pockets can open and close completely within a fraction of the simulation time. This is important as it suggests that docking procedures that account for receptor flexibility, like the relaxed complex scheme, may be a crucial part of drug-discovery studies on the molecule. While a multitude of pocket shapes are seen in the simulations, the bulk of the opening in the widest observed pockets is due to a widening of the part of the pocket accommodating the adenine part of the cofactor. Naturally, the values for the ligand-NAD-bound simulations are slightly larger than those observed in the apo simulations because the presence of the ligands and cofactors prevent the pocket from collapsing beyond a certain point. Large pocket conformations are observed in both the apo (up to ∼800 Å3 in the 3LT0 simulation) and ligand-cofactor-bound (up to ∼1000 Å3 in the 1UH5 simulation) trajectories. Picking conformations based on pocket volume may be an interesting approach to structure-based drug discovery, as has been previously shown by demonstrating better correlation between ligand binding affinities and docking scores for certain wide open pockets.19 In summary, these results suggest that the protein adapts rapidly to accommodate different ligands. We thus speculate that accounting for receptor flexibility is crucial in drug-discovery studies focusing on PfENR.
Role of fluctuations in the substrate-binding loop
The substrate-binding loop is comprised of residues L315 through Y375.20 In crystal structures of the protein, residues 325 through 366 are generally not resolved.1,21 Although partly unresolved, these residues are critical because residues in the N- and C-terminal ten-residue fragments of the loop are known to interact with drugs in the substrate-binding site. Additionally, a conserved salt bridge between K316 and D370 stabilizes the PfENR fold.20 We monitored this conserved salt bridge over the course of the simulations (Fig. 4). Since distances were measured between the nitrogen atom of the lysine ammonium ion and the carbon atom of the aspartate carboxylate ion, values up to about 5 Å are representative of salt bridges. Driven by the large fluctuations in residues 325 through 366, this critical salt bridge is continually broken and reformed. These dynamics not only allow substrate/inhibitor access to the binding site; they also correlate with fluctuations in the pocket volume. A comparison between the pocket volumes in Figure 3 and the salt bridge distances in Figure 4 reveals that many of the largest pocket volumes coincide with large K316–D370 distances.
Figure 4.
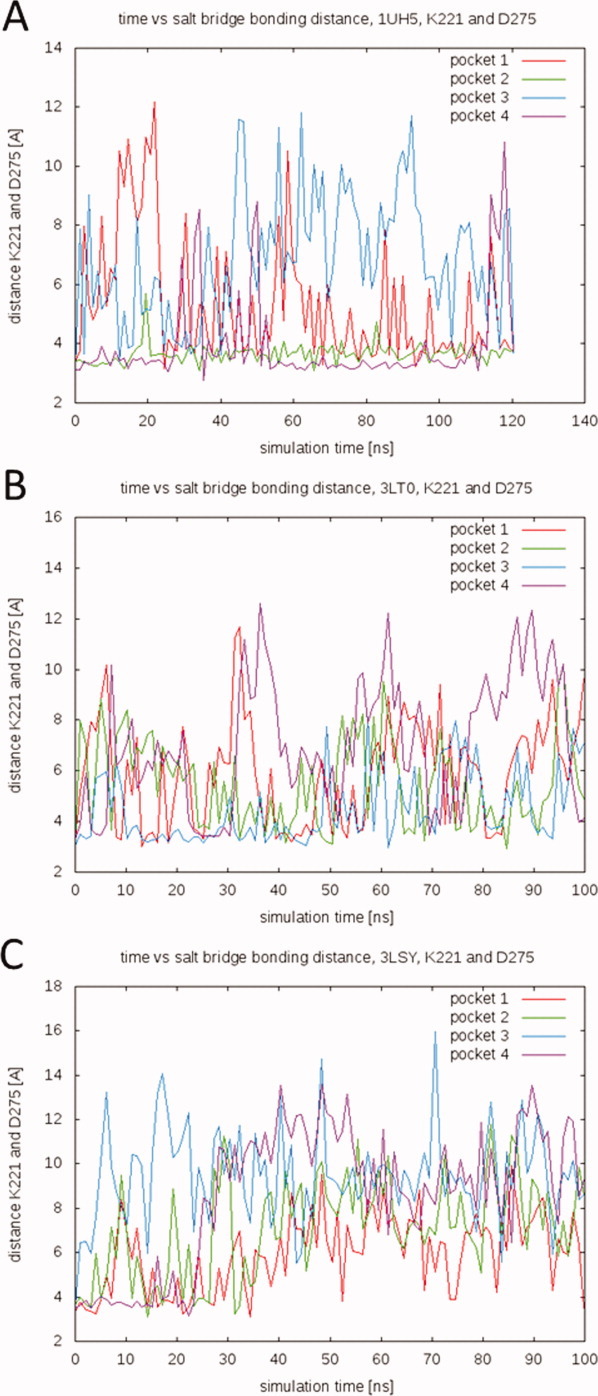
Distances between salt-bridge forming residues K316 and D370 over the course of the ligand-cofactor-bound simulations. Distances are measured between the nitrogen atom of the lysine ammonium ion (NZ) and the carbon atom of the aspartate carboxylate ion (CG). A spline smoothing has been applied for visualization purposes. (A) 1UH5 TCL-NAD-bound simulation, (B) 3LT0 FT1-NAD-bound simulation, and (C) 3LSY FT0-NAD-bound simulation. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
The simulations also demonstrate that the missing crystallographic residues of the substrate-binding loop in fact serve an important function, acting as a gate to open and close access to the active site. This is particularly evident in the 3LSY ligand- and cofactor-bound simulation (Fig. 5), where a flexible-loop conformation that allows access to the active site [Fig. 5(A)] is eventually replaced by a closed conformation that locks FT0 into the active site [Fig. 5(B)]. Knowledge of this gating mechanism has important implications on drug design studies as it allows selecting structures for docking where the binding site is accessible to ligands.
Figure 5.
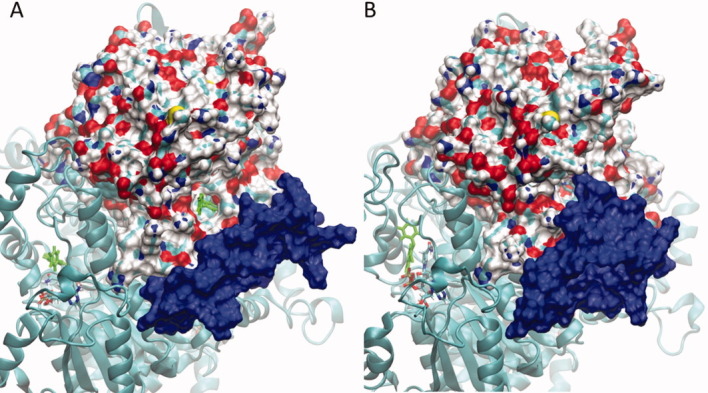
Active-site gating of the substrate-binding loop. Snapshots from the ligand-NAD-bound 3LSY simulation are shown (A) at the beginning of the simulation, and (B) after about 45 ns of simulation time. One subunit of pfENR from the ligand-NAD-bound 3LSY simulation is shown in surface representation. The cofactor and ligand are shown in bond representation with the ligand colored green. The surface from the highly flexible residues of the substrate binding loop is shown in blue. At the beginning of the simulations, (A) access to the active site is open, the ligand is visible. Over the course of the simulation, the conformation of the substrate-binding loop changes to almost completely block access to the ligand active site (B).
Stabilizing interactions between the ligand, cofactor, and protein
TCL and its variants, FT0 and FT1, are characterized by high binding affinities (Ki = 100–200 nM against PfENR), suggesting strong noncovalent interactions between these drugs and the protein-cofactor complex.21 Based on the ligand-NAD-bound simulations, the time evolution of some of the well-known interactions can be elucidated. A double hydrogen bond between the phenolic ligand hydroxyl group and the phenolic oxygen atom of Tyr182 (Tyr156 in FabI), as well as the 2′-hydroxyl group of the nucleotide nicotinamide ribose, is widely considered to be the major contributor to the high affinity of triclosan.2,6 These two distances were monitored over the course of the relevant simulations and are displayed in Figure 6. Distances between 2.0 and 3.5 Å are representative of hydrogen bonds.22
Figure 6.
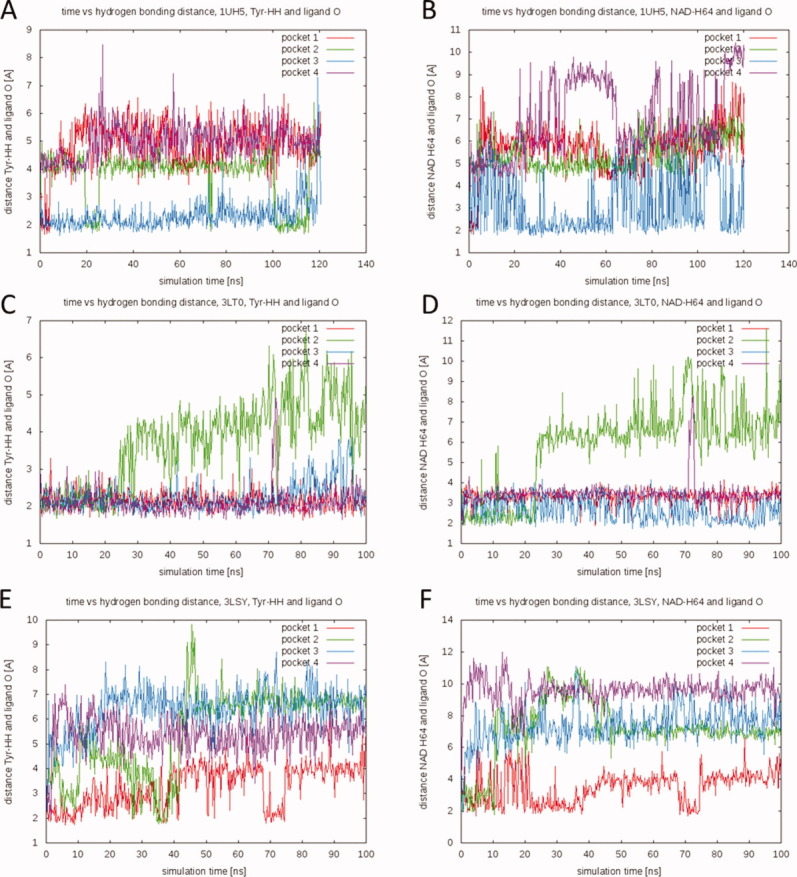
Time evolution of the double hydrogen bond between ligand, NAD, and Tyr182. (A, C, E) The distance between the oxygen atom of the ligand hydroxyl group and the hydrogen atom of the Tyr182 phenolic hydroxyl group over the course of the simulations. (B, D, F) The distance between the oxygen atom of the ligand hydroxyl group and the 2′-hydroxyl group of the nucleotide nicotinamide ribose. (A, B) 1UH5 TCL-NAD-bound simulation, (C, D) 3LT0 FT1-NAD-bound simulation, and (E, F) 3LSY FT0-NAD-bound simulation. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Interestingly, the results were highly ligand specific. FT1 (3LT0) maintains the double hydrogen bond with the protein and cofactor more or less throughout the course of the simulation in three of four binding pockets. TCL maintains the Tyr hydrogen bond in one pocket over the entire simulation, while in other pockets the bond transiently reforms and breaks. In the case of FT0, however, none of the hydrogen bonds remain unbroken over the course of the simulation, in any of the pockets. The most stable bonding occurs in Pocket 1, where the double hydrogen bond is reformed for about 5 ns after ∼70 ns of simulation. In the other three pockets, the hydrogen bonds typically break within the first 15 ns of simulation. Of particular interest is the behavior in Pocket 2, where the ligand transiently reforms the hydrogen bond with the Tyr182 side chain multiple times (between 25 and 40 ns of simulation time), but does not reform the H-bond with the cofactor during that time period. This suggests that the two hydrogen bonds can exist independently even though they are often formed in concert (Fig. 6).
Despite the fact that the hydrogen bonds in one to four of the subunits are broken during the course of the simulation, the ligands remained tightly in the binding pocket. This suggests that other potentially pharmaceutically relevant interactions stabilize the ligand even in the absence of the double hydrogen bond. Close inspection of the trajectory yielded the following previously unreported hydrogen-bond interactions: a hydrogen bond between the phenolic ligand hydroxyl group and the carbonyl oxygen atom of the NAD amide [Fig. 7(A)], a hydrogen bond between the phenolic ligand hydroxyl group and the amine of the NAD amide [Fig. 7(B)], and a hydrogen bond between the phenolic ligand hydroxyl group and a NAD phosphate oxygen atom [Fig. 7(C)]. Computer-aided drug-discovery studies could directly target these interactions by computationally restraining the ligand to form specific hydrogen bonds with the cofactor.
Figure 7.
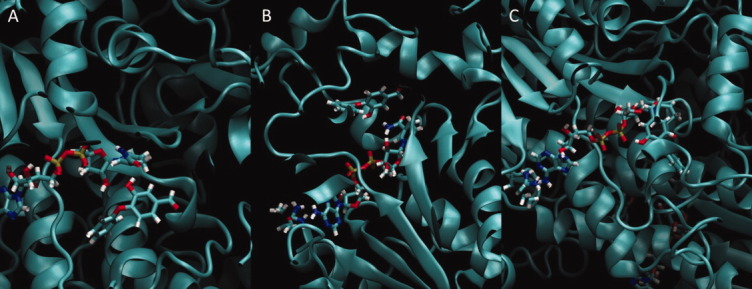
Examples of other hydrogen-bond interactions that stabilize the ligand in the binding pocket. All snapshots are taken from different pockets of the 3LSY FT0-NAD-bound simulation. (A) Hydrogen bond between the phenolic ligand hydroxyl group and the carbonyl oxygen atom of the NAD amide, (B) hydrogen bond between the phenolic ligand hydroxyl group and the amine of the NAD amide, and (C) hydrogen bond between the phenolic ligand hydroxyl group and a NAD phosphate oxygen atom. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Other known stabilizing interactions in the ligand-bound complex include π-stacking interactions between the ligand phenol ring and the NAD nicotinamide ring.2 This interaction in the crystal structures is best characterized as being in the parallel-displaced configuration.23 The evolution of the same stacking interaction over the course of the MD simulations was analyzed by calculating the center-of-mass distances between the rings (see Supporting Information Figure 3). Here again FT1 maintains the stacking interaction with NAD most of the time. TCL is stable in one pocket over the course of the simulation. However, FT0 rapidly breaks the stacking interactions in most pockets.
In the absence of the stabilizing π-stacking interaction between the ligand and the cofactor, aromatic active-site residues can provide π-stacking interactions with the ligand, thereby stabilizing it within the binding pocket. Pocket 2 of the 3LT0 ligand-cofactor-bound simulation illustrates this well [see Fig. 6(C,D) and Supporting Information Figure 3B]. After about 25 ns of simulation, the two hydrogen bonds are broken and the π-stacking rings move apart. The ensuing conformation is stabilized by a hydrogen bond between the phenolic ligand hydroxyl group and the carbonyl oxygen atom of the NAD+ amide [such as in Fig. 7(A)]. Additionally, a hydrogen bond forms between the phenolic oxygen atom of Tyr182 and one of the hydroxyl groups of the cofactor. After about 90 ns, these hydrogen bonds break, and the ligand separates from the cofactor while being stabilized by a π-stacking interaction with the Tyr182 ring (see Supporting Information Figure 4). The ligand forms a π-stacking interaction for 10 ns leading up to the end of the simulation. While this indicates a stable interaction, we cannot exclude the possibility of the ligand movement being a chance occurrence of a rare event. An in-depth investigation of this phenomenon will be the focus of follow-up studies. Conformations like these are particularly interesting from a drug-discovery perspective as they provide structural information beyond what has been known from the crystal structure. The conformation in Supporting Information Figure 4 for example coincides with one of the largest pocket volumes observed. Restraining the ligand to interact with the Tyr182 ring may yield interesting docking results.
Binding-pocket mapping (FTMAP)
The FTMAP server24 was used to perform a fragment-based search for druggable ENR hotspots. Representative structures from all the simulations (members of the respective ensembles) were stripped of any ligands and cofactors and submitted to the server. By using representative conformations from the individual simulations, this approach constitutes a comparative analysis of conformational dynamics of ENR in different states of ligand and cofactor binding. Different dynamics will lead to different identified hotspots. FTMAP identified only two druggable hotspots. The first can be described as the known active site. Depending on the structure submitted, FTMAP recognized between one and three of the four ENR active-site binding pockets. Probes were exclusively placed to occupy space generally filled by the ligands (TCL, FT0, and FT1) or the 1,4-dihydropyridine-3-carboxamide end of the cofactor. We hypothesize that the reason that not every pocket was recognized all the time is that much of the tight interactions contributing to the ligands' high binding affinity are between the ligand and cofactor. The ligand only has modest binding affinity to the binding pocket in the absence of the cofactor. However, more interestingly, FTMAP identified the central cavity of the tetramer as a second hotspot. Dozens of probes were placed in this region, in every representative structure from the simulations. While probes were found in virtually all parts of the cavity, there were two dominant regions wherein the vast majority of the probes interacted. The first was a pocket created by a short helical turn about six residues from the C-terminus of the subunit and a strand from the neighboring subunit comprised of residues 314–318. Supporting Information Figure 5 shows a representative structure of 10 fragments placed in this region of the central cavity. The second notable hot-spot region of the central cavity is the large central helix (approximately residues 185–207). When comparing the number of central cavity hotspots in dependence on the presence of ligand and cofactor, we observe a higher number of identified hotspots in the C-terminal regions for the ligand-bound simulations. Thus it seems that the presence of the ligand modifies the dynamics of the molecule to predispose the C-terminal region for binding of small molecules. This phenomenon will be investigated in much greater detail in the future and may prove very interesting in the search for alternative druggable sites of ENR.
Given the predicted proclivity of these hot spots for fragment binding, it may be interesting to look for novel ligands that target the central cavity in hopes of modifying ENR function allosterically. The two central regions exhibit low fluctuations in the RMSF plots, further supporting druggability; ligand binding could further stabilize and influence both the structure and dynamics of the enzyme. Allosteric modulation of these hot spots might also interrupt or alter inter-subunit signals, given the proximity of these regions to subunit boundaries.
Implications for drug discovery
In the current work, extensive MD simulations of pfENR have been performed. Tetrameric systems from three different ligand-bound structures were simulated in three different states of ligand binding. In total, the simulations add up to more than 3.6 μs of monomer dynamics. To our knowledge, this is the most comprehensive computational study of pfENR dynamics to date. The results further our understanding of the dynamics of pfENR and its interactions with bound inhibitors and cofactors.
The current work also provides valuable information for future drug-discovery studies. There are three areas where we believe these results may be beneficial. First, the pocket-volume analysis suggested that the active-site binding pocket is far from static. Considering receptor flexibility in docking studies has been shown to be valuable in countless cases, one outstanding example being the discovery of a novel binding trench in HIV integrase that eventually lead to the FDA approval of the drug raltegravir.25 Representative structures extracted from these simulations can be used in conjunction with the relaxed complex scheme, a docking technique that accounts for receptor flexibility.26–28 Alternatively, the pocket-volume analysis makes it possible to extract snapshots from the simulations that exhibit particularly open pocket conformations. Many of these pockets are wider in general, allowing the ligand to assume conformations different from those seen in the crystal structures utilizing many more possible alternative stabilizing interaction. In at least one example, this approach has yielded better structures for virtual screening than a simple cluster analysis.19
Second, the dynamics of the ligand inside the binding pocket suggest several stabilizing interactions in addition to the ones known from crystallography. Designing drugs that exploit these additional, critical interactions will certainly be the target of future drug-discovery efforts. Furthermore, MD simulations demonstrate that the ligand can occupy parts of the binding pocket that are not occupied in the crystal structures; indeed, the ligand can even change its entire orientation when binding as seen in Pocket 2 of the 3LSY simulation. The region of the pocket that should be targeted in future virtual screening efforts is therefore larger than what one would expect by examining the crystal structures alone.
Finally, a fragment-based docking scheme identified druggable ENR hotspots in the region of the central cavity at the interface of the subunit C-termini. Despite the fact that this area is far removed from the active site, it may be interesting to screen the region for ligands and to test for allosteric effects on ENR function.
Materials and Methods
System preparation
Systems based on three different crystal structures were prepared for simulations: PDB entries 1UH5,1 3LSY, and 3LT0.21 These structures contain triclosan [TCL; Fig. 1(A)] and two triclosan variants [FT0 and FT1; Fig. 1(B,C)] as ligands, respectively. The NAD+ cofactor is bound in all structures. In all three structures, residues 325–366 of the substrate-binding loop are missing. Swiss model server was used to build a homology model for the missing sequence.29 The crystal structures are dimers, but it is widely known that ENR functions as a tetramer.30 To obtain coordinates for the tetramer structure, PDB entry 2O2Y31 with a sequence similarity of 99% to the reference structures was used. The matchmaker tool in UCSF chimera32 was used to superimpose the dimer coordinates of the three structures onto the tetramer coordinates of 2O2Y to build tetrameric models of 1UH5, 3LSY, and 3LT0. Tleap33 was used to neutralize the systems by adding Cl− counter ions (20 Cl− for 1UH5, 3LSY, and 3LT0) and solvating using a TIP3P water box. For each of the starting structures, three different systems were built: apo (no ligand, no NAD cofactor), NAD-bound (no ligand, with NAD cofactor), and ligand-NAD-bound (with ligand and NAD cofactor). The fully solvated ligand-NAD-bound systems contained 179681 (1UH5), 173710 (3LSY), and 161062 (3LT0) atoms, respectively. Simulations were performed on each of the nine different systems. Minimization using SANDER33 was carried out in two stages: 1000 steps of minimization of solvent and ions with the protein, ligand, and cofactor restrained using a force constant of 500 kcal/mol/Å2, followed by a 2500 step minimization of the entire system. A short initial 20 ps MD simulation with weak restraints (10 kcal/mol/Å2) on the protein, ligand, and cofactor residues was used to heat the system to a temperature of 300 K. Subsequently, 100 ns of MD simulations were performed on each of the nine systems under investigation.
MD simulations
All MD simulations were performed under the NPT ensemble at 300 K using AMBER33 and the ff99SB force field.34 Periodic boundary conditions were used, along with a nonbonded interaction cutoff of 10 Å. Bonds involving hydrogen atoms were constrained using the SHAKE algorithm,35 allowing for a time step of 2 fs. For each system, 100 ns MD trajectories were generated totaling a simulation time of 900 ns.
Principal component analysis
The principal component analysis was performed using the bio3D package in R.36 The PC space was built up using known structures from the protein data bank. A blast profile of the 3LSY sequence revealed 626 hits. The 44 most similar hits were chosen and their experimentally determined structures were obtained from the protein data bank. The 44 structures underwent iterative rounds of structural superposition to determine the invariant core of the protein, a region that exhibits the least structural variance between the protein structures. This core consists of residues 98–151, 159, 161–176, 186–187, and 190–317. Subsequently the experimental structures were superimposed onto this core, and a principal component analysis was employed.37,38 In this process, a covariance matrix from the coordinates of the superimposed structures is diagonalized. The eigenvectors of this matrix represent the principal components of the system (parts of the structure within which there is the most variety among the set of superimposed experimental structures), while the eigenvalues are a measure of the variance within the distribution along the respective eigenvectors. All the experimental structures have been projected into the space spanned by principal components one and two, along which there is the most variance among the structures. The first two principal components account for over 53% of all the variance observed. The principal-component space generated from the experimental structures served as basis for the projection of the MD trajectories.
Pocket-volume calculations
To analyze the geometry of the binding pocket over time, its volume was calculated from the MD trajectories. Snapshots were extracted every 100 frames (200 ps of simulation time) from all nine trajectories. POVME39 was used for pocket-volume calculations. The coordinates of the following atoms were used as centers for POVME inclusion spheres: C1 and C7 of the respective ligands (TCL, FT0, and FT1), and C2, C10, C14, C17, and P2 of the NAD cofactor. Every extracted frame of the apo and NAD-bound simulations was superimposed onto the starting coordinates of the ligand-NAD-bound simulation from which the atom positions were extracted. In the case of the ligand-NAD-bound simulation, the atom coordinates were directly extracted from each individual frame. Points were generated in POVME with a grid spacing of 1 Å using inclusion spheres of 9 Å radius around the atom positions. The volume was calculated using the contiguous option (with contiguous seed spheres of radius 4 Å centered at the same coordinates as the inclusion spheres).
Distance measurements
Several key interactions have been reported to stabilize the binding of TCL and its variants. Among these are hydrogen bonds between the ligand, cofactor, and protein side chains.2 Additionally, the conserved salt bridge between K316 and D370 stabilizes the PfENR fold.20 To elucidate the time evolution of these interactions, distance measurements between key atoms were performed. The double hydrogen bond was investigated by measuring two distances. To monitor the hydrogen bond between the ligand hydroxyl group and the Tyr182, the distance between the ligand O2 (1UH5) or O3 (3LSY and 3LT0) atom and the Tyr182 HH atom was measured. To monitor the hydrogen bond between the ligand hydroxyl group and the cofactor, the distance between the ligand O2 (1UH5) or O3 (3LSY and 3LT0) atom and the cofactor H64 atom was measured. Distances were measured in all four binding pockets individually. To analyze the stacking interaction between the ligand phenol ring and the cofactor nicotinamide ring, the center of mass of each of the rings was calculated (atoms in ligand ring: C1, C2, C3, C4, C5, and C6; atoms in cofactor ring: C16, C17, C19, C20, C21, and N6). The distance between these two centers of mass as a function of simulation time was subsequently measured. Finally, to monitor the salt bridge, the distance between the nitrogen atom of the lysine ammonium ion (NZ) and the carbon atom of the aspartate carboxylate ion (CG) was measured. This approximates the salt-bridge distance, since the exact side chain atoms between which the interaction occurs change over the course of the simulation. All distance measurements were carried out at 200 ps intervals.
Cluster analysis and binding-pocket mapping
Frames were extracted from the MD trajectories every 10 ps, aligned using all protein Cα atoms, and clustered by RMSD using GROMOS++ conformational clustering.40 RMSD cutoffs of 2.0 Å (3LSY-ligand-NAD, 3LT0-apo), 1.9 Å (1UH5-ligand-NAD, 3LT0-NAD), 1.8 Å (3LT0-ligand-NAD, 1UH5-NAD, 3LSY-NAD), and 1.7 Å (1UH5-apo, 3LSY-apo) were chosen, respectively. These cutoffs resulted in three clusters that represented at least 90% of the respective trajectories for all but the 3LT0 NAD-bound simulation, where four clusters were generated. The central members of each of these clusters were chosen as representative. The set of all these representative structures is said to constitute an ensemble. Ligands and cofactors were removed from each ensemble structure when present. A total of 28 structures were submitted to the FTMAP server24 for fragment-based identification of ENR druggable hotspots, areas of the protein that are likely to bind small molecules. Particular focus was given to hits that were not located within the ligand and cofactor binding pockets.
Acknowledgments
The authors thank Dr. Jacob Durrant, Lori Tallorin and Prof. Dr. Michael Burkart for helpful discussions and careful reading of the manuscript.
Supplementary material
Additional Supporting Information may be found in the online version of this article.
Supplemental Figure 1. RMSD vs. time plots for all nine simulations. A) 1UH5 apo simulation, B) 3LT0 apo simulation, C) 3LSY apo simulation, D) 1UH5 NAD-bound simulation, E) 3LT0 NAD-bound simulation, F) 3LSY NAD-bound simulation, G) 1UH5 TCL-NAD-bound simulation, H) 3LT0 FT1-NAD-bound simulation, and I) 3LSY FT0-NAD-bound simulation. All simulations equilibrate within 10-20 ns.
Supplemental Figure 2. Principal-component analysis for all nine simulations. Projections of the MD trajectory into PC1-PC2 space are shown in panels A through I. A) 1UH5 apo simulation, B) 3LT0 apo simulation, C) 3LSY apo simulation, D) 1UH5 NAD-bound simulation, E) 3LT0 NAD-bound simulation, F) 3LSY NAD-bound simulation, G) 1UH5 TCL-NAD-bound simulation, H) 3LT0 FT1-NAD-bound simulation, and I) 3LSY FT0-NAD-bound simulation. Panel I shows the PC1-PC2 space generated from experimental structures. The subunits of the dimer starting structures are labeled red (1UH5), blue (3LT0), and green (3LSY).
Supplemental Figure 3. ϕ-stacking interactions as seen by the center-of-mass distances between a ligand phenol ring and the NAD nicotinamide ring over the course of the simulation. A) 1UH5 TCL-NAD-bound simulation, B) 3LT0 FT1-NAD-bound simulation, and C) 3LSY FT0-NAD-bound simulation.
Supplemental Figure 4. ϕ-stacking interaction between the FT1 ligand and Tyr182 residue. Snapshot taken from pocket 2 of the 3LT0 ligand-cofactor-bound simulation. The coordinates of Tyr182 are colored in red.
Supplemental Figure 5. Ten small organic probes placed by FTMAP into the central cavity of ENR. The ENR subunits are colored in different colors. Probes associated with a beta sheet region at the interface between two subunit C-termini.
References
- 1.Pidugu LS, Kapoor M, Surolia N, Surolia A, Suguna K. Structural basis for the variation in triclosan affinity to enoyl reductases. J Mol Biol. 2004;343:147–155. doi: 10.1016/j.jmb.2004.08.033. [DOI] [PubMed] [Google Scholar]
- 2.Massengo-Tiasse RP, Cronan JE. Diversity in enoyl-acyl carrier protein reductases. Cell Mol Life Sci. 2009;66:1507–1517. doi: 10.1007/s00018-009-8704-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu H, Tonge PJ. Inhibitors of FabI, an enzyme drug target in the bacterial fatty acid biosynthesis pathway. Acc Chem Res. 2008;41:11–20. doi: 10.1021/ar700156e. [DOI] [PubMed] [Google Scholar]
- 4.Rich SM, Leendertz FH, Xu G, LeBreton M, Djoko CF, Aminake MN, Takang EE, Diffo JL, Pike BL, Rosenthal BM, Formenty P, Boesch C, Ayala FJ, Wolfe ND. The origin of malignant malaria. Proc Natl Acad Sci USA. 2009;106:14902–14907. doi: 10.1073/pnas.0907740106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ward WH, Holdgate GA, Rowsell S, McLean EG, Pauptit RA, Clayton E, Nichols WW, Colls JG, Minshull CA, Jude DA, Mistry A, Timms D, Camble R, Hales NJ, Britton CJ, Taylor IW. Kinetic and structural characteristics of the inhibition of enoyl (acyl carrier protein) reductase by triclosan. Biochemistry. 1999;38:12514–12525. doi: 10.1021/bi9907779. [DOI] [PubMed] [Google Scholar]
- 6.Oliveira JS, Vasconcelos IB, Moreira IS, Santos DS, Basso LA. Enoyl reductases as targets for the development of anti-tubercular and anti-malarial agents. Curr Drug Targets. 2007;8:399–411. doi: 10.2174/138945007780058942. [DOI] [PubMed] [Google Scholar]
- 7.Heath RJ, Rock CO. Enoyl-acyl carrier protein reductase (fabI) plays a determinant role in completing cycles of fatty acid elongation in Escherichia coli. J Biol Chem. 1995;270:26538–26542. doi: 10.1074/jbc.270.44.26538. [DOI] [PubMed] [Google Scholar]
- 8.Singh NJ, Shin D, Lee HM, Kim HT, Chang HJ, Cho JM, Kim KS, Ro S. Structural basis of triclosan resistance. J Struct Biol. 2011;174:173–179. doi: 10.1016/j.jsb.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 9.Lu X, Huang K, You Q. Enoyl acyl carrier protein reductase inhibitors: a patent review (2006–2010) Expert Opin Ther Pat. 2011;21:1007–1022. doi: 10.1517/13543776.2011.581227. [DOI] [PubMed] [Google Scholar]
- 10.Qiu X, Janson CA, Court RI, Smyth MG, Payne DJ, Abdel-Meguid SS. Molecular basis for triclosan activity involves a flipping loop in the active site. Prot Sci. 1999;8:2529–2532. doi: 10.1110/ps.8.11.2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heath RJ, Yu YT, Shapiro MA, Olson E, Rock CO. Broad spectrum antimicrobial biocides target the FabI component of fatty acid synthesis. J Biol Chem. 1998;273:30316–30320. doi: 10.1074/jbc.273.46.30316. [DOI] [PubMed] [Google Scholar]
- 12.Bhargava HN, Leonard PA. Triclosan: applications and safety. Am J Infect Control. 1996;24:209–218. doi: 10.1016/s0196-6553(96)90017-6. [DOI] [PubMed] [Google Scholar]
- 13.Kapoor M, Dar MJ, Surolia A, Surolia N. Kinetic determinants of the interaction of enoyl-ACP reductase from Plasmodium falciparum with its substrates and inhibitors. Biochem Biophys Res Commun. 2001;289:832–837. doi: 10.1006/bbrc.2001.6061. [DOI] [PubMed] [Google Scholar]
- 14.Kapoor M, Reddy CC, Krishnasastry MV, Surolia N, Surolia A. Slow-tight-binding inhibition of enoyl-acyl carrier protein reductase from Plasmodium falciparum by triclosan. Biochem J. 2004;381:719–724. doi: 10.1042/BJ20031821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee HM, Singh NJ. Understanding drug-protein interactions in Escherichia coli FabI and various FabI inhibitor complexes. B Korean Chem Soc. 2011;32:162–168. [Google Scholar]
- 16.Pasqualoto KFM, Ferreira MMC. Application of a receptor pruning methodology to the enoyl-ACP reductase from Escherichia coli (Fabl) Qsar Comb Sci. 2006;25:629–636. [Google Scholar]
- 17.Yang L, Liu Y, Sternberg C, Molin S. Evaluation of enoyl-acyl carrier protein reductase inhibitors as Pseudomonas aeruginosa quorum-quenching reagents. Molecules. 2010;15:780–792. doi: 10.3390/molecules15020780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Azevedo WF., Jr Molecular dynamics simulations of protein targets identified in Mycobacterium tuberculosis. Curr Med Chem. 2011;18:1353–1366. doi: 10.2174/092986711795029519. [DOI] [PubMed] [Google Scholar]
- 19.Sinko W, de Oliveira C, Williams S, Van Wynsberghe A, Durrant JD, Cao R, Oldfield E, McCammon JA. Applying molecular dynamics simulations to identify rarely sampled ligand-bound conformational states of undecaprenyl pyrophosphate synthase, an antibacterial target. Chem Biol Drug Des. 2011;77:412–420. doi: 10.1111/j.1747-0285.2011.01101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maity K, Banerjee T, Prabakaran N, Surolia N, Surolia A, Suguna K. Effect of substrate binding loop mutations on the structure, kinetics, and inhibition of enoyl acyl carrier protein reductase from Plasmodium falciparum. IUBMB Life. 2011;63:30–41. doi: 10.1002/iub.412. [DOI] [PubMed] [Google Scholar]
- 21.Maity K, Bhargav SP, Sankaran B, Surolia N, Surolia A, Suguna K. X-ray crystallographic analysis of the complexes of enoyl acyl carrier protein reductase of Plasmodium falciparum with triclosan variants to elucidate the importance of different functional groups in enzyme inhibition. IUBMB Life. 2010;62:467–476. doi: 10.1002/iub.327. [DOI] [PubMed] [Google Scholar]
- 22.Wallwork SC. Hydrogen-bond radii. Acta Cryst. 1962;15:758–759. [Google Scholar]
- 23.Sinnokrot MO, Valeev EF, Sherrill CD. Estimates of the ab initio limit for pi-pi interactions: the benzene dimer. J Am Chem Soc. 2002;124:10887–10893. doi: 10.1021/ja025896h. [DOI] [PubMed] [Google Scholar]
- 24.Brenke R, Kozakov D, Chuang GY, Beglov D, Hall D, Landon MR, Mattos C, Vajda S. Fragment-based identification of druggable 'hot spots' of proteins using Fourier domain correlation techniques. Bioinformatics. 2009;25:621–627. doi: 10.1093/bioinformatics/btp036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schames JR, Henchman RH, Siegel JS, Sotriffer CA, Ni H, McCammon JA. Discovery of a novel binding trench in HIV integrase. J Med Chem. 2004;47:1879–1881. doi: 10.1021/jm0341913. [DOI] [PubMed] [Google Scholar]
- 26.Amaro R, Baron R, McCammon J. An improved relaxed complex scheme for receptor flexibility in computer-aided drug design. J Comp Mol Des. 2008;22:693–705. doi: 10.1007/s10822-007-9159-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin JH, Perryman AL, Schames JR, McCammon JA. Computational drug design accommodating receptor flexibility: the relaxed complex scheme. J Am Chem Soc. 2002;124:5632–5633. doi: 10.1021/ja0260162. [DOI] [PubMed] [Google Scholar]
- 28.Lin JH, Perryman AL, Schames JR, McCammon JA. The relaxed complex method: accommodating receptor flexibility for drug design with an improved scoring scheme. Biopolymers. 2003;68:47–62. doi: 10.1002/bip.10218. [DOI] [PubMed] [Google Scholar]
- 29.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 30.Perozzo R, Kuo M, Sidhu AS, Valiyaveettil JT, Bittman R, Jacobs WR, Jr, Fidock DA, Sacchettini JC. Structural elucidation of the specificity of the antibacterial agent triclosan for malarial enoyl acyl carrier protein reductase. J Biol Chem. 2002;277:13106–13114. doi: 10.1074/jbc.M112000200. [DOI] [PubMed] [Google Scholar]
- 31.Muench SP, Prigge ST, McLeod R, Rafferty JB, Kirisits MJ, Roberts CW, Mui EJ, Rice DW. Studies of Toxoplasma gondii and Plasmodium falciparum enoyl acyl carrier protein reductase and implications for the development of antiparasitic agents. Acta Cryst. 2007;D63:328–338. doi: 10.1107/S0907444906053625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera—a visualization system for exploratory research and analysis. J Comp Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 33.Case DA, Cheatham TE, III, Darden T, Gohlke H, Luo R, Merz KM, Jr, Onufriev A, Simmerling C, Wang B, Woods RJ. The Amber biomolecular simulation programs. J Comp Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ryckaert J-P, Ciccotti G, Berendsen HJC. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comp Phys. 1977;23:327–341. [Google Scholar]
- 36.Grant BJ, Rodrigues AP, ElSawy KM, McCammon JA, Caves LS. Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics. 2006;22:2695–2696. doi: 10.1093/bioinformatics/btl461. [DOI] [PubMed] [Google Scholar]
- 37.Abseher R, Nilges M. Are there non-trivial dynamic cross-correlations in proteins? J Mol Biol. 1998;279:911–920. doi: 10.1006/jmbi.1998.1807. [DOI] [PubMed] [Google Scholar]
- 38.van Aalten DMF, DeGroot BL, Findlay JBC, Berendsen HJC, Amadei A. A comparison of techniques for calculating protein essential dynamics. J Comp Chem. 1997;18:169–181. [Google Scholar]
- 39.Durrant JD, de Oliveira CA, McCammon JA. POVME: an algorithm for measuring binding-pocket volumes. J Mol Graph Model. 2011;29:773–776. doi: 10.1016/j.jmgm.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Christen M, Hunenberger PH, Bakowies D, Baron R, Burgi R, Geerke DP, Heinz TN, Kastenholz MA, Krautler V, Oostenbrink C, Peter C, Trzesniak D, van Gunsteren WF. The GROMOS software for biomolecular simulation: GROMOS05. J Comp Chem. 2005;26:1719–1751. doi: 10.1002/jcc.20303. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.