

Fibroblast growth factor receptor 2 (FGFR2) pre-mRNA is alternatively spliced to form the epithelial-specific and mesenchymal-specific IIIb and IIIc isoforms, respectively, which are useful markers of epithelial–mesenchymal transitions (EMT). The authors developed a fluorescence-based reporter system to visualize exon IIIc regulation in vitro and in vivo. They show the application of this reporter system to the study of EMT in vitro in cell culture and in vivo in transgenic mice harboring these splicing constructs. In explant studies, the reporters revealed that FGFR2 isoform switching is not required for keratinocyte migration during cutaneous wound closure.
Keywords: pre-mRNA splicing, epithelial–mesenchymal transition, wound healing, in vivo imaging, enhanced green fluorescent protein, DsRed
Abstract
Alternative splicing generates a vast diversity of protein isoforms from a limited number of protein-coding genes, with many of the isoforms possessing unique, and even contrasting, functions. Fluorescence-based splicing reporters have the potential to facilitate studies of alternative splicing at the single-cell level and can provide valuable information on phenotypic transitions in almost real time. Fibroblast growth factor receptor 2 (FGFR2) pre-mRNA is alternatively spliced to form the epithelial-specific and mesenchymal-specific IIIb and IIIc isoforms, respectively, which are useful markers of epithelial–mesenchymal transitions (EMT). We have used our knowledge of FGFR2 splicing regulation to develop a fluorescence-based reporter system to visualize exon IIIc regulation in vitro and in vivo. Here we show the application of this reporter system to the study of EMT in vitro in cell culture and in vivo in transgenic mice harboring these splicing constructs. In explant studies, the reporters revealed that FGFR2 isoform switching is not required for keratinocyte migration during cutaneous wound closure. Our results demonstrate the value of the splicing reporters as tools to study phenotypic transitions and cell fates at single cell resolution. Moreover, our data suggest that keratinocytes migrate efficiently in the absence of a complete EMT.
INTRODUCTION
Alternative splicing enables eukaryotic cells to create multiple protein-encoding transcripts from a single pre-mRNA. It has been estimated that as many as 92%–94% of human pre-mRNAs undergo alternative splicing (Wang et al. 2008), and there are several examples in which the protein isoforms resulting from alternative splicing have profoundly different cellular functions (David and Manley 2010).
A well-characterized example of the effect alternative splicing can have on protein function is that observed among fibroblast growth factor receptor 2 (FGFR2) transcripts. FGFR2 pre-mRNAs undergo alternative splicing within sequences encoding the C-terminal half of the FGFR2 third immunoglobulin-like domain. FGFR2 alternative splicing is cell type specific; epithelial cells include exon 8 (IIIb) and skip exon 9 (IIIc), whereas mesenchymal cells skip IIIb and include IIIc (Turner and Grose 2010). The FGFR2 IIIb and IIIc isoforms possess up to a 1000-fold difference in affinity for some fibroblast growth factors (FGFs) (Dell and Williams 1992). For example, stromal-derived FGF-7 specifically activates FGFR2-IIIb (Turner and Grose 2010), while FGF-2 activates the IIIc but not the IIIb isoform (Marek et al. 2009).
FGFR2 isoform switching has been implicated in some types of epithelial–mesenchymal transitions (EMT), a process whereby epithelial cells alter their motility, lose apico-basal polarity and cell-to-cell adhesion molecules, and are able to penetrate the basement membrane (Yang and Weinberg 2008). These phenotypic cellular transitions are observed during development, wound healing, fibrosis, and cancer progression. The in-depth characterization of IIIb and IIIc splicing events has led to the development of a series of fluorescence-based reporters that reproducibly and predictably mimic the splicing pattern of the endogenous FGFR2 pre-mRNA (Bonano et al. 2006, 2007; Oltean 2006; Oltean et al. 2008; Takeuchi et al. 2010). In the present study, we generated an in vivo mouse model of epithelial-specific FGFR2-IIIc skipping using a system of fluorescence-based alternative splicing reporters, which we refer to here as FSRs (fluorescence splicing reporters). These transgenic mice have the benefit of employing allele-specific fluorescence controls and targeted insertion of controls and reporters within the ubiquitously expressed ROSA26 locus. Using these mice, we show that deletion of the stem–loop structure within intron 8 in our in vivo reporters recapitulates in vitro findings in which removal of this element abrogates exon IIIc skipping. We also demonstrate epithelial-specific exon IIIc repression of the FSRs using primary cultures of keratinocytes and dermal fibroblasts and apply this model to ex vivo cutaneous wound healing assays. The explant studies reveal that FGFR2 isoform switching is not required for keratinocyte migration and suggest that keratinocytes do not undergo a complete EMT during wound closure. The use of the FSRs in multiple studies of EMT highlights their utility as tools for the discovery of novel insights into phenotypic transitions in vitro and in vivo at single-cell resolution.
RESULTS
Design rationale for alternative splicing reporters
The rigorous characterization of FGFR2 alternative splicing facilitated the design of splicing reporters that accurately recapitulate splicing of the endogenous gene (Kemp et al. 2005; Bonano et al. 2007; Kuroyanagi et al. 2010). Using our knowledge of exon IIIc splicing control, we generated a series of FSRs that would allow us to interrogate epithelial-specific exon IIIc regulation in vitro and in vivo. The pGIIIcI2 reporter contains the endogenous IIIc exon and flanking intronic segments, including intronic activating sequence 2 (IAS2) and intronic silencing and activating region (ISAR) (indicated by black boxes in Fig. 1), which interrupt the enhanced green fluorescent protein (EGFP) reading frame (Fig. 1). A second reporter, pGIIIcIΔΔ, harbors deletions in IAS2 and ISAR (Fig. 1). IAS2 and ISAR are required for formation of a stem–loop within intron 8 of the FGFR2 pre-mRNA (Muh 2002; Baraniak et al. 2003). The stem–loop is postulated to bring epithelial-specific regulatory factors into close proximity to control proper splicing (Muh 2002; Baraniak et al. 2003). Deletion of IAS2 and ISAR results in loss of epithelial-specific exon IIIc skipping (dotted lines in Fig. 1; Baraniak et al. 2003). The pGIIIcIΔΔ construct allows us to evaluate the importance of IAS2 and ISAR in exon IIIc skipping in normal tissues in vivo. These reporters have the same design as pRIIIcI2 and pRIIIcΔ,Δ previously used to observe epithelial transitions in tumors (Oltean 2006); however, the expression of EGFP permitted us to use them in transgenic mice with the allelic control construct pRint. The pRint reporter, a version of which was described by Bonano et al. (2006), is comprised of the Discosoma red fluorescent protein (DsRed) open reading frame interrupted by a short, constitutively spliced adenoviral intron (Fig. 1). Like the pGIIIc reporters, pRint is driven by the CMV immediate early promoter, so any disruption in CMV-based transcription or widespread alterations in the splicing machinery will result in the loss of DsRed expression. Therefore, we only interrogated the splicing of those cell types that expressed DsRed. This allele-restricted reporter is particularly useful in controlling for methylation and subsequent shutdown of the CMV promoter observed in some transgenic animals (Mehta et al. 2009).
FIGURE 1.

Design strategy for the in vivo splicing reporters. The FGFR2 pre-mRNA is alternatively spliced within the last half of its third immunoglobulin-like domain to produce the IIIb (exon 8) or IIIc (exon 9) isoforms (top). To generate the pGIIIcI2 splicing reporter, the IIIc exon and flanking introns, including intronic activating sequence 2 (IAS2) and the intronic silencing and activating region (ISAR; black boxes), were inserted within the enhanced green fluorescent protein (EGFP) reading frame (green boxes). Skipping of IIIc (bottom lines) leads to fusion of the EGFP reading frame and expression of EGFP, while IIIc inclusion (top lines) interrupts the EGFP reading frame. Another reporter, pGIIIcIΔΔ, harbors deletions in IAS2 and ISAR, which abrogates cell-type-specific exon IIIc skipping (indicated by the dashed lines), resulting in almost complete inclusion of exon IIIc (solid lines). The pRint construct contains the DsRed open reading frame interrupted by a short adenoviral intron with consensus 5′ and 3′ splice sites, a branch point consensus sequence, and a polypyrimidine tract. This reporter controls for CMV-driven transcription and proper constitutive splicing at the ROSA26 locus in a specified cell type or tissue. Using the pRint control allows us to interrogate the splicing pattern of only those cells or tissue types that are expressing DsRed. If a cell or tissue is not expressing DsRed, we assume there is an interruption in transcription via CMV or constitutive splicing and do not interrogate GIIIc splicing regulation in these locations.
FGFR2 splicing constructs enable predictions of epithelial and mesenchymal cell types in vitro
To test the ability of the constructs to report on epithelial-specific splicing in vitro, we stably transfected pGIIIcI2 and pGIIIcIΔΔ into rat DT and AT3 cells. Western blots using lysates from DT and AT3 cells indicate that DT cells express epithelial markers E-cadherin and pan-cytokeratin, while AT3 cells express the mesenchymal markers fibronectin and vimentin (Fig. 2A). Equally, analysis of endogenous FGFR2 RNAs using reverse transcription polymerase chain reaction (RT-PCR) followed by isoform-sensitive restriction endonuclease cleavage revealed that DT cells and AT3 cells express FGFR2-IIIb and -IIIc, respectively (Fig. 2B). RT-PCR of the reporters indicated that both DT and AT3 cells transfected with pGIIIcIΔΔ robustly included exon IIIc (Fig. 2C), while DT cells predominantly skipped exon IIIc and AT3 cells included the IIIc exon when transfected with pGIIIcI2 (Fig. 2C). Most importantly, these molecular events resulted in the expected fluorescence. Epithelial-specific exclusion of exon IIIc was partially abrogated by deletion of IAS2 and ISAR in epithelial DT cells (Fig. 2D, left panels), whereas DT cells transfected with the pGIIIcI2 reporter showed strong expression of EGFP and AT3 cells revealed only background levels of EGFP (Fig. 2D, right panels). Taken together, these data indicate that the pGIIIcI2 reporter can be effectively used to predict epithelial and mesenchymal cell types in vitro.
FIGURE 2.

Validation of GIIIc reporters in vitro. (A) Western blots indicate that rat DT cells differentially express epithelial markers E-cadherin and pan-cytokeratin, and rat AT3 cells express mesenchymal markers fibronectin and vimetin. β-actin was used as a loading control. (B) RT-PCR and restriction digestion reveal that DT and AT3 cells express the endogenous FGFR2 IIIb and IIIc isoforms, respectively. (M) Mock digested RT-PCR amplicon; (AvaI) IIIb-specific restriction digest; (EcoRV) IIIc-specific restriction digest. (C) RT-PCR reveals that both DT and AT3 cells include IIIc in the pGIIIcIΔΔ construct, but only DT cells robustly skip IIIc in the pGIIIcI2 construct. Bars indicate the percent inclusion for each sample, and error bars represent standard deviations of the mean. (D) The GIIIcIΔΔ and GIIIcI2 reporters were stably transfected into DT and AT3 cells. Untransfected cells that did not receive both the reporter and the G418 selection gene were gated away using fluorescence activated cell sorting (FACS). As expected, epithelial-specific IIIc skipping of pGIIIcI2 is observed in DT cells, but deletion of IAS2 and ISAR partially abrogates epithelial-specific control of IIIc.
The GIIIcI2 reporter is a reliable tool for the study of epithelial plasticity in vitro
FSRs have been used in previous studies to observe the splicing of different cell types; however, these constructs have never been employed in EMT studies in which a single cell line is induced to undergo EMT. Considering the numerous investigations of EMT, a simple system for interrogating phenotypic plasticity at single-cell resolution would be of great value to the field. We postulated that the pGIIIcI2 reporter could be uniquely suited for the analysis of EMT and phenotypic transitions. Along these lines, we induced EMT using TGF-β in normal murine mammary gland (NMuMG) epithelial cells stably transfected with pGIIIcI2 and looked for loss of EGFP. Cells were treated for 7 d to account for the >24-h half-life of EGFP (Verkhusha et al. 2003). After 7 d, we observed a clear loss of EGFP in the TGF-β-treated wells when compared with cells incubated with vehicle alone (Fig. 3A). Reproducible reductions in EGFP were noted as early as 72 h. The down-regulation of EGFP was accompanied by a disappearance of cell-to-cell contacts, an increase in cellular elongation, and an increase in actin stress fiber formation in cells exposed to TGF-β (Fig. 3A, F-actin staining). We also noted a reduction in growth in the cells exposed to TGF-β (cf. Hoechst staining in left panels of Fig. 3A), which has been observed in prior studies (Gal et al. 2008). Moreover, E-cadherin was markedly down-regulated following EMT induction (Fig. 3A). We did not observe any changes in EGFP after incubation with TGF-β in NMuMGs transfected with pGint, a reporter that contains a constitutively spliced intron interrupting the EGFP reading frame (data not shown).
FIGURE 3.

Fluorescence-based splicing reporters mark epithelial–mesenchymal transitions (EMT) in vitro. (A) Normal mouse mammary (NMuMG) cells stably transfected with pGIIIcI2 were cultured in the presence of TGF-β or vehicle for 7 d. Addition of TGF-β resulted in a marked decrease in EGFP expression (second column of images), and the loss of EGFP was related to the formation of actin stress fibers (third column, top two rows) and down-regulation of E-cadherin (third column, bottom two rows). (B) NMuMGs reduced EGFP expression by nearly threefold after incubation with TGF-β, as estimated by flow cytometry.
As detected by fluorescence microscopy, the phenotypic changes in NMuMGs treated with TGF-β were marked by a drop in EGFP intensity. Using flow cytometry, we were able to quantify the differences in EGFP intensity in cells exposed to vehicle or TGF-β. On average, the number of EGFP-negative cells shifted by 2.9-fold from 15.05% in the cells exposed to vehicle alone (Fig. 3B, black histogram) to 44.02% in the cells incubated in the presence of TGF-β (Fig. 3B, gray histogram). To determine whether this loss of EGFP reflected an increase in exon IIIc inclusion, the amount of exon IIIc inclusion was ascertained by quantitative polymerase chain reaction (qPCR). To do this, we used a forward primer within the first EGFP exon and a reverse primer spanning the junction between the first EGFP exon and the IIIc exon. In a parallel set of reactions, we used the same forward primer and a reverse primer in the downstream EGFP exon. This experimental design enabled us to normalize the amount of IIIc inclusion to the levels of total EGFP transcript. On average, there was a fivefold increase in exon IIIc inclusion upon induction of EMT by TGF-β (P < 0.01). Taken together, these in vitro findings provide evidence that FSRs can be useful tools for the study of epithelial plasticity.
We also attempted to confirm whether the splicing switch in the context of the FSR reflected that of endogenous FGFR2 transcripts; however, we observed a nearly complete loss of FGFR2 mRNAs by RT-PCR in cells exposed to TGF-β, which precluded us from monitoring the endogenous isoforms being produced by NMuMGs incubated in the presence of vehicle or TGF-β (data not shown). Horiguchi et al. (2011) also observed the same trend in NMuMGs treated with TGF-β, and subsequent experiments by this group suggested that a combination of ESRPs and unspecified transcription factors was responsible for the loss of FGFR2 transcripts (Horiguchi et al. 2011).
Alternative splicing reporters mark epithelial cells in vivo
The pGIIIcI2 and pGIIIcIΔΔ constructs were inserted by homologous recombination into the ROSA26 locus of A129S4JaeSor embryonic stem (ES) cells. The ROSA26 locus was selected for its robust and uniform spatio-temporal gene expression in mice (Strathdee et al. 2006). ES cells were injected into C57BL/6J blastocysts, and newborns were screened by PCR. Positive agouti pups were crossed to C57BL/6J mice, and agouti pups were screened for germline transmission of the pGIIIcI2 and pGIIIcIΔΔ reporters by PCR. Reporter mice were crossed to ROSA26pRint/pRint mice to give ROSA26pRint/pGIIIcI2 or ROSA26pRint/pGIIIcIΔΔ.
Pups between 3 and 5 d old were imaged for DsRed and EGFP fluorescence. As shown in Figure 4, we observed strong expression of DsRed throughout the entire skin of the pRint/pRint, pRint/pGIIIcIΔΔ, and pRint/pGIIIcI2 animals and expression of EGFP almost exclusively in the pRint/pGIIIcI2 animals. Interestingly, we noted rare EGFP-expressing cells on one pRint/pGIIIcIΔΔ animal during the course of the study. Yet, the fact that pRint/pGIIIcIΔΔ pups express high levels of DsRed but not EGFP strongly suggested that deletion of IAS2 and ISAR was sufficient to abrogate exon IIIc skipping in vivo and provided proof-of-principle that splicing reporters could be used to investigate the importance of intronic sequence elements to specific alternative splicing programs in cells and tissues in vivo.
FIGURE 4.
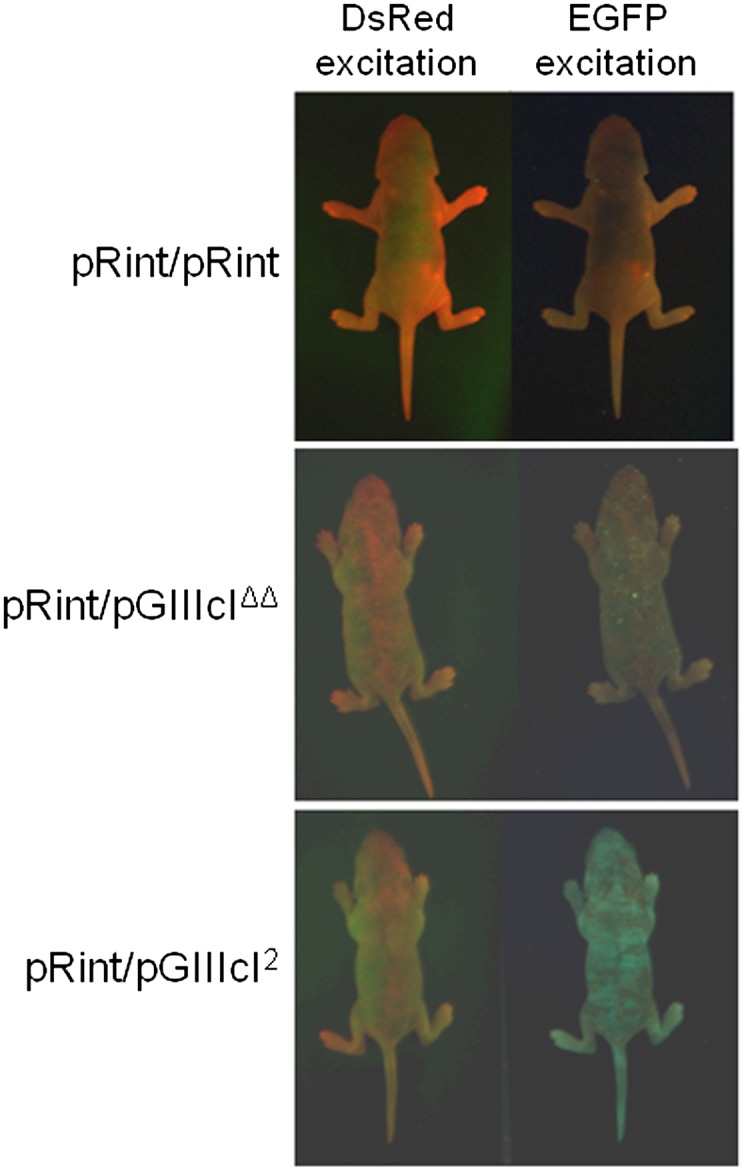
Whole body imaging of alternative splicing reporter mice. Three-day-old pups were imaged with a Pan-A-See-Ya imaging system. The pRint/pRint and pRint/pGIIIcIΔΔ mice express DsRed, but no EGFP, while the pRint/pGIIIcI2 animals display strong expression of DsRed and EGFP across the entire skin. Interestingly, both fluorescent proteins appear to exhibit a mottled pattern of expression, which does not appear to correlate with any specific physiological tissue or structure.
Intriguingly, a variegated, striped pattern of expression of both fluorescence signals was visualized on the skins of the pRint/pGIIIcI2 animals, which differed in position and intensity between individual mice. Although it is possible that the striping observed in EGFP is due to promoter shutdown, we noted that the pattern of EGFP expression did not correlate with that of DsRed, which is also driven by the CMV promoter at the ROSA26 locus, suggesting that shutdown of transcription was not the sole factor responsible for the varied signal. Although we could not pinpoint the causative factor(s) responsible for this pattern, we were able to use the allelic pRint control to interrogate splicing of DsRed-expressing cells.
We also sought to validate the utility of FSRs in tracing epithelial and mesenchymal cell types. To do this, we prepared primary keratinocyte and fibroblast cultures from skins of pRint/pGIIIcIΔΔ and pRint/pGIIIcI2 animals. Western blots from keratinocyte and fibroblast lysates revealed higher expression of E-cadherin, a fundamental marker of epithelial cells, in keratinocytes and higher N-cadherin and fibronectin in fibroblast primary cultures (Fig. 5A). Visualization of endogenous FGFR2 mRNAs by RT-PCR and isoform-specific restriction digestion revealed that the keratinocyte cultures are composed of cells that express the IIIb isoform, while the fibroblast cultures are predominantly made up of cells that express the IIIc isoform (Fig. 5B). The small percentage of exon IIIb expression is most likely due to the presence of a small number of contaminating keratinocytes that we observed in the fibroblast cultures. Next, we attempted to ensure that the splicing pattern of the pGIIIcI2 reporter could accurately predict that of the endogenous FGFR2 transcript. To do this, we performed RT-PCR to ensure that the splicing of the GIIIcI2 transcripts matched the expression of endogenous FGFR2 in keratinocyte and fibroblast cultures (Fig. 5C). Not surprisingly, the keratinocyte cultures efficiently skipped exon IIIc in the context of the pGIIIcI2 reporter, while dermal fibroblasts displayed markedly increased exon IIIc inclusion (Fig. 5C). The low level of keratinocytes in the fibroblast cultures likely explains the presence of skipped product in the RT-PCR from GIIIcI2 transcripts (Fig. 5B). Importantly, our results indicate that the splicing pattern of GIIIcI2 mRNAs was able to correctly predict the splicing of endogenous FGFR2 transcripts. Most importantly, despite the presence of high levels of DsRed in pRint/pGIIIcIΔΔ tails, epidermal and dermal skin layers, and primary cultures, none of these tissues or cell types expressed EGFP. The primary cultures from pRint/pGIIIcIΔΔ pups further verify the importance of IAS2 and ISAR in the proper regulation of exon IIIc (Fig. 5D, top panels). Conversely, tails, epidermal layers, and primary keratincoytes from pRint/pGIIIcI2 pups expressed high levels of DsRed and EGFP, while dermal layers and dermal-derived fibroblasts showed no EGFP expression, despite the clear presence of DsRed (Fig. 5D, bottom panels). Taken together, our results reveal that the pGIIIcI2 reporter can accurately predict the expression of epithelial and mesenchymal biomarkers in keratinocyte and fibroblast cultures, respectively.
FIGURE 5.

Validation of fluorescence reporters in vivo. (A) Western blots from primary cultures further verify enrichment of epithelial and mesenchymal markers and splicing factors in keratinocytes and fibroblasts, respectively. (K) Keratinocytes; (F) fibroblasts. (B) Endogenous FGFR2 transcripts from keratinocyte and fibroblast cultures contain restriction enzyme sites specific to the IIIb and IIIc isoforms, respectively. The fibroblast culture contains a minority of IIIb transcripts, which is likely due to a small amount of keratinocyte contamination observed in the fibroblast cultures. (L) 1 Kb Plus Ladder (Invitrogen); (M) mock-digested RT-PCR amplicon; (A) IIIb-specific restriction digestion with AvaI; (E) IIIc-specific restriction digestion with EcoRV. Positions and sizes of AvaI and EcoRV cleavage products are indicated with arrows to the left and right of the gel, respectively. (C) RT-PCR revealed that reporter transcripts from pRint/pGIIIcI2 keratinocyte primary cultures (K) predominantly skipped IIIc (bottom band), while transcripts from fibroblast primary cell lines (F) contained IIIc within the reporter (top band). The cartoon depiction to the right indicates the RT-PCR products of the EGFP transcript (gray boxes) with and without the IIIc exon (white box). (D) Despite robust expression of DsRed, almost no EGFP was observed in whole tails, epidermis, dermis, epidermal-derived keratinocytes, or dermal-derived fibroblasts from pRint/pGIIIcIΔΔ mice. Conversely, tails, epidermis, and primary keratinocytes displayed strong EGFP and DsRed expression, while dermis and dermal-derived primary fibroblasts nearly exclusively produced DsRed and not EGFP. Two contaminating keratinocytes in the pRint/pGIIIcI2 fibroblast culture are visible in the DsRed channel (white arrow).
To understand which splicing factors may be regulating exon IIIc splicing in mouse skin, we monitored expression levels of splicing factors known to regulate exon IIIc skipping or inclusion by Western blotting. ESRP1 and RBFOX2 are splicing factors known to induce exon IIIc skipping (Shapiro et al. 2011). Conversely, SRSF1 is a repressor of exon IIIc skipping (Mauger et al. 2008). Interestingly, ESRP1 expression was higher in keratinocytes, while no differences in RBFOX2 levels were observed (Fig. 5A). SRSF1 protein levels were slightly higher in fibroblast cultures (Fig. 5A), although the levels were very similar between the two cell types in one out of three replicate cultures. A loading control (β-actin) is shown for each different blot performed. These results suggest that, of the known exon IIIc regulatory factors, ESRP1 may be an important factor involved in the regulation of exon IIIc in keratinocytes and further demonstrates the utility of the reporters in predicting and segregating distinct cellular phenotypes.
Alternative splicing reporters reveal that FGFR2 isoform switching is not required for keratinocyte migration during wound healing
To determine whether the FSRs could be used to investigate phenotypic transitions in the transgenic animals, we applied the reporters to an ex vivo wound healing system. During cutaneous wound healing, keratinocytes are thought to undergo an EMT-like event whereby the cells lose cell-to-cell contacts and become motile to close the wound (Nakamura and Tokura 2011). Based on this, we postulated that migrating keratinocytes may undergo FGFR2 isoform switching during wound healing. To test this hypothesis, we utilized a previously described explant model of wound re-epithelialization (Wawersik et al. 2001; Mazzalupo et al. 2002) to test for EMT during keratinocyte migration. Full skin biopsy punches from GIIIcI2/Rint mice were cultured for 5 d in 24-well tissue culture plates. We did not observe cells on the perimeter of the explants earlier than ∼24–36 h after initial culture, but by 36 h, small numbers of cells were observed migrating from the epidermal surface of the skin (Fig. 6A). Surprisingly, we noted that many of these cells expressed EGFP, which suggested that migrating keratinocytes do not require FGFR2 isoform switching during wound closure (Fig. 6A) and may not have to undergo a full EMT in order to migrate. The EGFP-expressing keratinocytes were composed of cell types that expressed DsRed (Fig. 6A, white arrow) and those that did not express DsRed (Fig. 6A, arrowheads). Unfortunately, numerous migrating cells did not express either reporter particularly between days 3–5, after which fluorescence signal from both reporters began to appear, often simultaneously. As a result of this concomitant lack of fluorescence signal from both reporters, it cannot be ruled out that some keratinocytes, or subsets of keratinocytes, undergo isoform switching during wound closure; however, these cells cannot be interrogated for exon IIIc skipping because of the lack of signal from the pRint control reporter. Yet, despite this limitation, it is clear that FGFR2 isoform switching is not required for keratinocyte migration during cutaneous wound repair and calls into question the extent to which keratinocytes undergo EMT.
FIGURE 6.

Application of the fluorescence-based reporter mice to studies of EMT ex vivo. (A) Skin explants from pRint/pGIIIcI2 reporter mice were imaged daily from days 2–5. Keratinocytes expressing EGFP alone (white arrowheads) or in combination with DsRed (white arrow) were observed migrating from the explants, suggesting that FGFR2 isoform switching from IIIb to IIIc is not required for migration during wound healing. (B) Keratinocytes and fibroblasts from the explants were stained with E-cadherin and F-actin. Migrating keratinocytes show reductions in E-cadherin at the leading edge of the explants (dotted white line) and stress fiber formation throughout all keratinocytes. (C) To independently validate that isoform switching is not required during cutaneous wound healing, explants were treated with FGF-2 (IIIc-specific) or FGF-7 (IIIb-specific) either alone or in the presence of mitomycin C. The wound edges are indicated by dotted white lines. (D) Explants and cells were stained with Hoechst, and cells were quantified with ImageJ. Each bar represents the average of three explants. Error bars delineate the standard deviation of the mean. Where applicable, statistically significant relationships are indicated by brackets, and P-values are listed above the brackets. (*) P < 0.05; (**) P < 0.01.
Time-lapse microscopy was used to verify that isoform switching was not required for keratinocyte motility (Supplemental File 1). Multiple, EGFP-expressing cells were detected both moving from the explants and dividing. As witnessed in the daily images (Fig. 6A), the time-lapse also revealed several cells that began expressing DsRed (data not shown) and EGFP only after migrating away from the explants (Supplemental File 1), which we interpret as reactivation of a silenced CMV promoter rather than isoform switching because of the parallel behavior of EGFP and DsRed signals in these cells.
To gain further insight into the extent to which migrating keratinocytes undergo EMT, explants were stained with E-cadherin. We observed specific E-cadherin staining among the keratinocytes, with reductions in E-cadherin signal near the leading edge of the keratinocyte migration (Fig. 6B; the white dotted line indicates the leading edge of movement). Importantly, fibroblasts growing out from the explants were negative for E-cadherin (Fig. 6B). Keratinocytes and fibroblasts emanating from the explants were also stained with anti-phalloidin to detect F-actin. Although the intensity and morphology of F-actin staining were similar among all keratinocytes, including those near the explants and those at the leading edge, we noted a substantial difference in the patterns of F-actin localization between keratinocytes and fibroblasts (Fig. 6B, second set of panels). While the fibroblasts showed distinct localization of F-actin near focal adhesions, keratinocytes contained more evenly distributed stress fibers throughout the cell.
To independently test whether migrating keratinocytes undergo a transition from FGFR2-IIIb to -IIIc in an ex vivo model of wound healing, explants were treated with FGF-2 (IIIc specific) or FGF-7 (IIIb specific) either alone or in the presence of mitomycin C. Incubation with mitomycin C inhibits cell proliferation. Subtracting the number of cells after mitomycin C exposure from the untreated cells gave an estimate of the number of cells emanating from the explants. Subtracting the migrating cells from the control provided the number of proliferating cells that contribute to wound healing. As expected, mitomycin C dramatically reduced the number of cells originating from the explants, leaving only ∼25% (504/2043 cells) of migrating cells contributing to wound closure in our system. Based on our imaging data, we hypothesized that exposure to a IIIb-specific ligand would increase the number of keratinocytes, while a IIIc-specific ligand would have no effect. Interestingly, neither FGF-7 nor FGF-2 had a significant effect on the motile fraction of keratinocytes, and it is possible that migrating cells require a gradient chemotactic signal from available stroma at the site of the wound in vivo. In our assays, FGFs were placed throughout the media rather than from a specific site. Yet, given this potential difference between our system and in vivo conditions, FGF-7 treatment significantly increased the total number of proliferating cells, whereas FGF-2 did not significantly alter the proliferative capacity of the keratinocytes. Taken together, these results suggest that keratinocytes do not undergo FGFR2 isoform switching during wound healing, which may indicate a partial EMT, or lack of EMT, during wound closure. The ex vivo wound healing assays further demonstrate the utility of alternative splicing reporters, which allow interrogation of phenotypic transitions at the single-cell level and provide new insights into biological phenomena.
DISCUSSION
Alternative splicing imparts upon the mammalian genome the ability to amplify protein diversity from a restricted number of protein-coding genes. These splicing programs have been shown to be particularly important during EMT, and several investigators have identified splicing factors that regulate alternative exon usage during EMT (Baraniak et al. 2006; Warzecha et al. 2009; Valacca et al. 2010). Similarly, Shapiro et al. (2011) used an in vitro cell culture model of EMT in combination with RNA-seq to develop an alternative splicing signature of EMT. This investigation found that most EMT-based alternative splicing decisions were driven by specific groups of splicing factors, including the RBFOXs and ESRPs. Importantly, knockdown of RBFOX2 or overexpression of ESRP1 in mesenchymal cells was sufficient to partially restore an epithelial phenotype (Shapiro et al. 2011). In our analyses of keratinocyte and fibroblast primary cultures from Rint/GIIIcI2 reporter mice, we observed epithelial-specific expression of ESRP1, with no consistent changes in RBFOX2. Thus, our data confirm the previous findings of Warzecha et al. (2009) and Shapiro et al. (2011) that ESRP1 may be the predominant splicing factor in the regulation of epithelial-specific splicing and suggest that ESRP1 is involved in controlling cellular phenotypes in mouse skin. Perhaps more importantly, our findings also underscore the utility of FSRs in further understanding the fundamental sequence elements and protein factors that drive cellular phenotypes.
One of the drawbacks to the reporter system described in the present study is the variation observed in expression of both the pGIIIcI2 and pRint fluorescence-based constructs (Fig. 4). The fact that pRint animals also display variable expression of DsRed suggests that transcription of this open reading frame may be differentially silenced within a single cell type. Along these lines, others have observed down-regulation of the CMV promoter in vivo (Brooks et al. 2004; Stevanato et al. 2009); however, these effects are often observed during nontargeted insertions of transgenes. To mitigate any potential silencing of the CMV promoter, we chose to insert the fluorescence reporters into the ROSA26 locus, which is not susceptible to transcriptional shutdown due to chromatin remodeling (Chen et al. 2011). Despite these attempts, it appears that variability remains in expression from the CMV promoter, and future efforts to develop transgenic reporter mice should focus on identification of alternative promoters, such as the chicken β-actin promoter and CMV immediate early enhancer (CAGG), which has been shown to dramatically enhance expression over the CMV promoter in mouse embryonic stem cells at the ROSA26 locus (Chen et al. 2011).
To circumvent CMV-driven expression differences within and between tissues, we adopted several strategies, including the use of time-lapse microscopy, to track single migrating keratinocytes (Supplemental File 1) and the development and employment of methods that were independent of the FSRs, such as treatment with FGFs, to validate the conclusions made with the reporters ex vivo (Fig. 6B,C). Using these approaches and others, we were able to show that FSRs provide a unique tool to study phenotypic transitions in vitro, in vivo, and ex vivo. In particular, the reporter mice provided evidence that FGFR2 isoform switching is not required for keratinocyte migration and proliferation during wound healing (Fig. 6). Interestingly, however, keratinocytes have been shown to undergo an EMT-like event during wound re-epithelialization, and FGFR2 has been implicated in this process (Werner et al. 1994; Pilcher et al. 1997). Until now, however, it was not clear whether the EMT-like programs initiated during wound healing required a switch in FGFR2 isoforms.
It is, perhaps, not surprising that organisms utilize only a partial EMT during the normal physiological process of wound repair. Isoform switching during wound closure could desensitize keratinocytes to dermal cues and create an autocrine loop in which keratinocyte-derived FGFs drive proliferative signals via the IIIc receptor, an outcome that may lead to hyperplasia. Indeed, this is the scenario that appears to take place during EMT in a subset of carcinomas. For example, FGFR2 isoforms switch from IIIb to IIIc during EMT in rat bladder cancer cell lines (Savagner et al. 1994). Moreover, other investigations found that re-expression of IIIb in IIIc-specific rat prostate tumors substantially reduced tumor growth and led to restoration of epithelial cell differentiation and up-regulation of keratins (Feng et al. 1997). Additionally, FGFR2-IIIb was found to be down-regulated in poorly differentiated prostate tumors (Turner and Grose 2010). Without the appropriate paracrine dermal cues, epithelial cells can become hyperproliferative, leading to the initiating phases of carcinoma. The fact that FGFR2 isoform switching does not appear to be required for keratinocyte migration highlights the inherent differences between EMT-like events in various contexts, such as during wound healing and carcinoma progression.
By utilizing the knowledge garnered from multiple biochemical studies on FGFR2 alternative splicing, we were able to design alternative splicing constructs capable of accurately predicting cell types in vitro, in vivo, and ex vivo. We employed these FSRs in assays to reveal novel biological insights related to epithelial plasticity and phenotypic transitions. Future studies should be focused on improved technology to observe multiple exon choices within the same biologically relevant pathway.
MATERIALS AND METHODS
Generation of GIIIc splicing reporters
To create the pGIIIcI2 and pGIIIcIΔΔ in vivo splicing reporters, the FGFR2 IIIc exon and flanking introns from the pRIIIcI2 and pRIIIcΔ,Δ reporters (Oltean 2006) were subcloned into the EGFP reading frame at position 146 of the pEGFP-N1 vector. PGIIIcI2 and pGIIIcIΔΔ were inserted into the intermediate pBigT vector to add a neomycin-resistance gene, and the reporters were then transferred to the pROSA26PA plasmid via PacI and AscI restriction sites to add homologous recombination sites. The pBigT and pROSA26PA vectors were kind gifts of Dr. Frank Costantini (Columbia University, New York). The reporters pGIIIcI2 and pGIIIcIΔΔ were verified by sequencing.
Testing the FSRs in vitro
The pGIIIcI2 and pGIIIcIΔΔ splicing constructs were linearized with KpnI to interrupt the diphtheria toxin gene. The vectors were purified with the QIAGEN PCR purification kit and stably transfected into rat epithelial DT and mesenchymal AT3 cells using Lipofectamine 2000 following the manufacturer's recommendations. Cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin and streptomycin (Pen/Strep). Cells were passaged in the presence of 500 μg/mL G418 (Invitrogen) for 2 wk to select for integration of the reporters. Untransfected cells were gated out of the population by fluorescence activated cell sorting (FACS) at the Duke University Flow Cytometry Shared Resource. Images were captured using an Olympus IX 71 epifluorescence microscope with a DP70 digital camera and analyzed with CellSens software (Olympus).
Induction of EMT in stably transfected NMuMG cells
The pGIIIcI2 reporter was stably transfected into NMuMG cells following the methods outlined above. Untransfected cells were gated away by FACS as described above. A total of 1 × 105 NMuMG cells containing the pGIIIcI2 construct were seeded into six-well plates and cultured in 2.0 mL of DMEM (supplemented with 10% FBS and 1% Pen/Strep) in the presence of 2 ng/mL vehicle (4 mM HCl and 2% BSA) or recombinant human TGF-β (R&D Systems). Media was refreshed every other day, and cells were split upon reaching confluence. Cells were collected after 7 d for analysis.
NMuMG-pGIIIcI2 cells were also seeded into poly-D-lysine coated 35-mm glass bottom dishes (MatTek Corporation) for immunofluorescence imaging. After 7 d, the cells were fixed in 4% paraformaldehyde for 15 min and permeabilized with PBS containing 0.2% Triton X-100 for 15 min. Cells were stained with anti-E-cadherin using the Zenon Alexa Fluor 555-nm primary antibody labeling kit (Invitrogen) with a total of 1 μg of anti-E-cadherin (BD Biosciences). F-actin was stained with rhodamine-conjugated phalloidin (Invitrogen; 1:40 dilution in PBS) for 15 min. All nuclei were stained with Hoechst (Sigma-Aldrich; 1:2000 dilution in PBS) for 15 min. Fluorescence images were captured with 600× magnification under oil immersion using the microscope and camera described above.
For FACS analysis, cells were trypsinized and analyzed for EGFP intensity at the Duke University Flow Cytometry Shared Resource. Histograms were generated using WinMDI 2.9 software. Subsequent to FACS analysis, RNA was extracted with the RNeasy Mini kit (QIAGEN) following the manufacturer's recommendations. A total of 500 ng of RNA was used in each reverse transcription reaction containing 200 ng of random hexamer primers, 1× IMPROMII reverse transcriptase buffer, 10 μM dNTPs, 3.75 mM MgCl2, 0.1 μL of RNasin, and 1 μL of IMPROMII reverse transcriptase in a total volume of 20 μL. Quantitative, real-time PCRs were performed using ABI SYBR Green (Invitrogen) in 50-μL reaction volumes containing 1× Power SYBR Green PCR Master Mix and 0.3 μM each primer in a StepOnePlus real-time PCR thermocycler and analyzed using StepOne software (Applied Biosciences). Data were normalized to EGFP and represented as average fold change of exon IIIc-including transcripts compared with vehicle-treated wells. Error bars represent the standard deviation among three biological replicates. The primers used to detect exon IIIc inclusion were 5′-CCATCCTGGTCGAGCTGGACG-3′; and 5′-GTGTTAACACCGGCGGCCTCCATAC-3′. The primers used to detect EGFP were 5′-GAAGCGCGATCACATGGT-3′; and 5′-CCATGCCGAGAGTGATCC-3′.
Creation of transgenic mice
Both pGIIIcI2 and pGIIIcIΔΔ were linearized with KpnI to interrupt the diphtheria toxin reading frame and purified with the QIAGEN PCR purification kit. The reporter constructs were transfected into AK7 embryonic stem (ES) cells from 129S4 SvJaeSor mice, ES cell clones were screened for integration by PCR, and two positive clones were injected into C57/BL6J blastocysts by the Duke University Transgenic Mouse Facility. One clone produced viable chimeras, and tails from the pups were subsequently screened for germline integration by PCR and fluorescence microscopic imaging. Positive adults were crossed to 129S4 SvJaeSorpRint/pRint animals, which harbor the pRint reporter. The pRint construct is composed of the DsRed open reading frame interrupted by a short adenoviral intron (Fig. 1). The intron within pRint contains a consensus branch point and polypyrimidine tract as well as consensus 5′ and 3′ splice sites. Homozygous 129S4 SvJaeSorpRint/pRint and heterozygous 129S4 SvJaeSorpRint/pGIIIcI2 and 129S4 SvJaeSorpRint/pGIIIcIΔΔ mice were screened as delineated above. Whole body images of 3-d-old pups were captured using a Lightools Pan-A-See-Ya imaging station and Image Pro Plus (version 4.0) software.
RNA isolation and RT-PCR
Total RNA was extracted with TRIzol (Invitrogen) following the manufacturer's protocol with one exception: Following centrifugation of the TRIzol at 12,000g for 15 min, the aqueous phase was removed, 800 μL of chloroform was added, and the mixture was shaken vigorously for 15 sec and centrifuged for 1 min at 12,000g at 4°C. The aqueous phase was removed, and the TRIzol protocol was completed following the manufacturer's protocol. The RNA fraction from the TRIzol extraction was treated with Turbo DNase (Ambion) for 15 min at 37°C following the protocol recommended by the manufacturer. A total of 1 μg of DNase-treated RNA was used in each reverse transcription reaction using IMPROMII reverse transcriptase in a total volume of 20 μL as described above. PCRs were carried out in 50-μL volumes containing 4 μL of the reverse transcription reaction, 1× PFU Ultra AD buffer, 0.2 mM dNTPs, 0.2 μM each primer, and 2.5 units of PFU Ultra AD polymerase. The forward and reverse primers used to amplify EGFP with and without the IIIc exon were 5′-CGGCGACGTAAACGGCCACAAGTTC-3′ and 5′-GAAGTCGTGCTGCTTCATGTGGTCGG-3′, respectively. RT-PCR products were quantified using ImageJ. The average percent IIIc exon inclusion from three replicate RT-PCRs was calculated by dividing the total signal for skipped and included products by the intensity of the included product. Average percent inclusion and standard deviations were graphed in Excel. The forward and reverse primers used to monitor expression of endogenous mouse FGFR2 in primary cultures were 5′-GCAAGGTTTACAGCGATGCCCAGCC-3′ and 5′-GACTGTCACCACCATGCAGGGGATTA-3′, respectively. PCRs were electrophoretically separated in 1.5% agarose gels, stained with ethidium bromide, and visualized on a GelDoc imaging station. To detect the endogenous FGFR2 in DT and AT3 cells, PCRs were prepared as above, but with the addition of 0.2 μCi of [α-32P]CTP. PCR products were restricted with either AvaI (NEB) or EcoRV (NEB) and separated on 5.5% TBE acrylamide gels at 120 V. Gels were dried and visualized by autoradiography using Kodak Biomax MR film. The forward and reverse primers used to detect endogenous rat FGFR2 were 5′-CCCGGGTCTAGATTTATAGTGATGCC-3′ and 5′-CCCGGG GAATTCACCACCATGCAGG-3′, respectively.
Protein isolation and Western blotting
Total protein was isolated in radio-immunoprecipitation assay (RIPA) buffer (20 mM Tris-HCl at pH 7.5, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, and 1 μg/mL leupeptin) containing protease inhibitor cocktail (Roche). Proteins were separated in NuPAGE Novex 4%–12% Bis-Tris gels (Invitrogen), transferred to nitrocellulose, blocked for 1 h at room temperature in Licor Odyssey blocking buffer and incubated overnight in primary antibody at the appropriate dilution. Membranes were washed three times for 5 min each in PBS containing 0.05% Tween 20, incubated for 1 h at room temperature in secondary antibody diluted in Odyssey blocking buffer, washed three times as above, and visualized using the Licor Odyssey Imaging Station. Western blots using anti-ESRP1 were performed in the same way, except Tween 20 was removed from all buffers. The primary antibodies and dilutions used were mouse anti-E-cadherin (1:1000; BD Biosciences), mouse anti-N-cadherin (1:1000; BD Biosciences), rabbit anti-fibronectin (1:1000; Santa Cruz), mouse anti-ESRP1 (1:100; Abnova), rabbit anti-FOX2 (1:1000; Sigma-Aldrich), mouse anti-SRSF1 (1:1000; Invitrogen), and mouse anti-β-actin (1:3000; Santa Cruz).
Ex vivo wound healing assays
Full skin explants from 1- to 3-d-old mice were prepared and cultured following a previously detailed method (Mazzalupo et al. 2002). Briefly, pups were decapitated and soaked in 70% ethanol. In a tissue culture hood, limbs were removed, and skins were dissected and placed epidermis side down in six-well tissue culture plates (Corning). Skins were scanned for GFP and RFP using an ImageXpress Velos scanning cytometer and visualized with ImageXpress Velos software (Molecular Devices). Dual EGFP and DsRed positive sections of skin were removed with a 3-mm biopsy punch and placed dermis side down in 24-well tissue culture plates. Explants were allowed to adhere to the tissue culture plates for 5–10 min, after which 200 μL of explant medium was added. Explant medium was prepared as previously described (Mazzalupo et al. 2002). Explants were incubated overnight at 37°C with 5% CO2. The next day, explants were submerged by adding 800 μL of media to each well. Images were captured each day, and media was replaced every 2–3 d. To allow for proper focusing of keratinocytes, imaging was begun on day 2, and time-lapse images were taken every hour for 3 d (73 h in total) on a Zeiss AxioVert 200 M microscope equipped with an Orca ER camera. Time-lapse settings were controlled by Metamorph software (version 7.7.5). Images were compiled, and movies were created using ImageJ software. To treat with mitomycin C, explants were allowed to attach and grow for ∼16 h before addition of mitomycin C. After 2 h of incubation in the presence of mitomycin C or media alone, explants were washed with explant media and incubated with media or media containing either FGF-2 (10 nM) or FGF-7 (10 nM). Images were captured after 5 d at 40× total magnification, and keratinocytes surrounding each explant were quantified using ImageJ. Immunofluorescence staining with anti-E-cadherin and Alexafluor 647 conjugated anti-phalloidin was performed with the antibodies and conditions described above for NMuMG cells. An anti-mouse Alexafluor 647 conjugated secondary antibody (1:2000) was used to detect E-cadherin. Images were captured with 200× magnification as described.
Statistical analysis
A two-tailed t-test was used to compare the amount of IIIc inclusion in transcripts from NMuMG cells treated with vehicle or TGF-β. Differences in cell numbers among the explants were analyzed using a one-way ANOVA with Tukey's post-hoc analysis. JMP9 (SAS Institute Inc.) was used to perform all statistical tests.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health (NIH) grant R01 5R01-CA127727 to M.A.G.-B. J.A.S. acknowledges American Cancer Society Post-doctoral Fellowship #PF-11-036-01-DDC. D.S. acknowledges NIH T32 Training Grant #5T32CA009111. We kindly thank Dr. Andrew Armstrong for discussions and thoughtful suggestions and Dr. Shelton Bradrick for critical reading of the manuscript. We also acknowledge James Pearson and the Duke University RNAi Core Facility, Cheryl Bock and the Duke University Transgenic Mouse Facility, the Duke University Flow Cytometry Shared Resource, and the Duke University Light Microscopy Core Facility.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.035097.112.
REFERENCES
- Baraniak AP, Lasda EL, Wagner EJ, Garcia-Blanco MA 2003. A stem structure in fibroblast growth factor receptor 2 transcripts mediates cell-type-specific splicing by approximating intronic control elements. Mol Cell Biol 23: 9327–9337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baraniak AP, Chen JR, Garcia-Blanco MA 2006. Fox-2 mediates epithelial cell-specific fibroblast growth factor receptor 2 exon choice. Mol Cell Biol 26: 1209–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonano VI, Oltean S, Brazas RM, Garcia-Blanco MA 2006. Imaging the alternative silencing of FGFR2 exon IIIb in vivo. RNA 12: 2073–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonano VI, Oltean S, Garcia-Blanco MA 2007. A protocol for imaging alternative splicing regulation in vivo using fluorescence reporters in transgenic mice. Nat Protoc 2: 2166–2181 [DOI] [PubMed] [Google Scholar]
- Brooks AR, Harkins RN, Wang P, Qian HS, Liu P, Rubanyi GM 2004. Transcriptional silencing is associated with extensive methylation of the CMV promoter following adenoviral gene delivery to muscle. J Gene Med 6: 395–404 [DOI] [PubMed] [Google Scholar]
- Chen CM, Krohn J, Bhattacharya S, Davies B 2011. A comparison of exogenous promoter activity at the ROSA26 locus using a PhiC31 integrase mediated cassette exchange approach in mouse ES cells. PLoS ONE 6: e23376 doi: 10.1371/journal.pone.0023376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- David CJ, Manley JL 2010. Alternative pre-mRNA splicing regulation in cancer: Pathways and programs unhinged. Genes Dev 24: 2343–2364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell KR, Williams LT 1992. A novel form of fibroblast growth factor receptor 2. Alternative splicing of the third immunoglobulin-like domain confers ligand binding specificity. J Biol Chem 267: 21225–21229 [PubMed] [Google Scholar]
- Feng S, Wang F, Matsubara A, Kan M, McKeehan WL 1997. Fibroblast growth factor receptor 2 limits and receptor 1 accelerates tumorigenicity of prostate epithelial cells. Cancer Res 57: 5369–5378 [PubMed] [Google Scholar]
- Gal A, Sjoblom T, Fedorova L, Imreh S, Beug H, Moustakas A 2008. Sustained TGF β exposure suppresses Smad and non-Smad signalling in mammary epithelial cells, leading to EMT and inhibition of growth arrest and apoptosis. Oncogene 27: 1218–1230 [DOI] [PubMed] [Google Scholar]
- Horiguchi K, Sakamoto K, Koinuma D, Semba K, Inoue A, Inoue S, Fujii H, Yamaguchi A, Miyazawa K, Miyazono K, et al. 2011. TGF-β drives epithelial-mesenchymal transition through δEF1-mediated downregulation of ESRP. Oncogene 31: 3190–3201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp PR, Ellis PD, Smith CW 2005. Visualization of alternative splicing in vivo. Methods 37: 360–367 [DOI] [PubMed] [Google Scholar]
- Kuroyanagi H, Ohno G, Sakane H, Maruoka H, Hagiwara M 2010. Visualization and genetic analysis of alternative splicing regulation in vivo using fluorescence reporters in transgenic Caenorhabditis elegans. Nat Protoc 5: 1495–1517 [DOI] [PubMed] [Google Scholar]
- Marek L, Ware KE, Fritzsche A, Hercule P, Helton WR, Smith JE, McDermott LA, Coldren CD, Nemenoff RA, Merrick DT, et al. 2009. Fibroblast growth factor (FGF) and FGF receptor-mediated autocrine signaling in non-small-cell lung cancer cells. Mol Pharmacol 75: 196–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauger DM, Lin C, Garcia-Blanco MA 2008. hnRNP H and hnRNP F Complex with Fox2 to silence fibroblast growth factor receptor 2 exon IIIc. Mol Cell Biol 28: 5403–5419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzalupo S, Wawersik MJ, Coulombe PA 2002. An ex vivo assay to assess the potential of skin keratinocytes for wound epithelialization. J Invest Dermatol 118: 866–870 [DOI] [PubMed] [Google Scholar]
- Mehta AK, Majumdar SS, Alam P, Gulati N, Brahmachari V 2009. Epigenetic regulation of cytomegalovirus major immediate-early promoter activity in transgenic mice. Gene 428: 20–24 [DOI] [PubMed] [Google Scholar]
- Muh SJ 2002. A non-sequence-specific double-stranded RNA structural element regulates splicing of two mutually exclusive exons of fibroblast growth factor receptor 2 (FGFR2). J Biol Chem 277: 50143–50154 [DOI] [PubMed] [Google Scholar]
- Nakamura M, Tokura Y 2011. Epithelial–mesenchymal transition in the skin. J Dermatol Sci 61: 7–13 [DOI] [PubMed] [Google Scholar]
- Oltean S 2006. Alternative inclusion of fibroblast growth factor receptor 2 exon IIIc in Dunning prostate tumors reveals unexpected epithelial mesenchymal plasticity. Proc Natl Acad Sci 103: 14116–14121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oltean S, Febbo PG, Garcia-Blanco MA 2008. Dunning rat prostate adenocarcinomas and alternative splicing reporters: Powerful tools to study epithelial plasticity in prostate tumors in vivo. Clin Exp Metastasis 25: 611–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilcher BK, Gaither-Ganim J, Parks WC, Welgus HG 1997. Cell type-specific inhibition of keratinocyte collagenase-1 expression by basic fibroblast growth factor and keratinocyte growth factor. A common receptor pathway. J Biol Chem 272: 18147–18154 [DOI] [PubMed] [Google Scholar]
- Savagner P, Valles AM, Jouanneau J, Yamada KM, Thiery JP 1994. Alternative splicing in fibroblast growth factor receptor 2 is associated with induced epithelial–mesenchymal transition in rat bladder carcinoma cells. Mol Biol Cell 5: 851–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro IM, Cheng AW, Flytzanis NC, Balsamo M, Condeelis JS, Oktay MH, Burge CB, Gertler FB 2011. An EMT-driven alternative splicing program occurs in human breast cancer and modulates cellular phenotype. PLoS Genet 7: e1002218 doi: 10.1371/journal.pgen.1002218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevanato L, Corteling RL, Stroemer P, Hope A, Heward J, Miljan EA, Sinden JD 2009. c-MycERTAM transgene silencing in a genetically modified human neural stem cell line implanted into MCAo rodent brain. BMC Neurosci 10: 86 doi: 10.1186/1471-2202-10-86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strathdee D, Ibbotson H, Grant SG 2006. Expression of transgenes targeted to the Gt(ROSA)26Sor locus is orientation dependent. PLoS ONE 1: e4 doi: 10.1371/journal.pone.0000004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi A, Hosokawa M, Nojima T, Hagiwara M 2010. Splicing reporter mice revealed the evolutionally conserved switching mechanism of tissue-specific alternative exon selection. PLoS ONE 5: e10946 doi: 10.1371/journal.pone.0010946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner N, Grose R 2010. Fibroblast growth factor signalling: From development to cancer. Nat Rev Cancer 10: 116–129 [DOI] [PubMed] [Google Scholar]
- Valacca C, Bonomi S, Buratti E, Pedrotti S, Baralle FE, Sette C, Ghigna C, Biamonti G 2010. Sam68 regulates EMT through alternative splicing-activated nonsense-mediated mRNA decay of the SF2/ASF proto-oncogene. J Cell Biol 191: 87–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhusha VV, Kuznetsova IM, Stepanenko OV, Zaraisky AG, Shavlovsky MM, Turoverov KK, Uversky VN 2003. High stability of Discosoma DsRed as compared to Aequorea EGFP. Biochemistry 42: 7879–7884 [DOI] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB 2008. Alternative isoform regulation in human tissue transcriptomes. Nature 456: 470–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warzecha CC, Sato TK, Nabet B, Hogenesch JB, Carstens RP 2009. ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing. Mol Cell 33: 591–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wawersik MJ, Mazzalupo S, Nguyen D, Coulombe PA 2001. Increased levels of keratin 16 alter epithelialization potential of mouse skin keratinocytes in vivo and ex vivo. Mol Biol Cell 12: 3439–3450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner S, Smola H, Liao X, Longaker MT, Krieg T, Hofschneider PH, Williams LT 1994. The function of KGF in morphogenesis of epithelium and reepithelialization of wounds. Science 266: 819–822 [DOI] [PubMed] [Google Scholar]
- Yang J, Weinberg RA 2008. Epithelial–mesenchymal transition: At the crossroads of development and tumor metastasis. Dev Cell 14: 818–829 [DOI] [PubMed] [Google Scholar]