

Abstract
TMPRSS2-Ets gene fusions were identified in prostate cancers where the promoter of transmembrane protease, serine 2 (TMPRSS2) fused with coding sequence of the erythroblastosis virus E26 (Ets) gene family members. TMPRSS2 is an androgen responsive transmembrane serine protease. Ets family members are oncogenic transcription factors that contain a highly conserved Ets DNA binding domain and an N-terminal regulatory domain.
Fusion of these gene results in androgen dependent transcription of Ets factor in prostate tumor cells. The ERG is the most common fusion partner with TMPRSS2 promoter in prostate cancer patients. The high prevalence of these gene fusions, in particular TMPRSS2-ERG, makes them attractive as potential diagnostic and prognostic indicators, as well as making them a potential target for tailored therapies.
This review focuses on the clinical and biological significance of TMPRSS2-ERG fusions and their role in PC development and progression.
Keywords: TMPRSS2-ERG, Prostate Cancer, Clinical, Biological significance
Introduction
Prostate cancer (PC) is the most common form of cancer found in American men and the second leading cause of cancer death. The American Cancer Society [1] estimates that in 2012 there were 241,740 newly diagnosed cases of PC, with an estimated 28,170 men dying as a result of the disease. This means that approximately 28.5% of cancers and 3.5% of cancer related deaths in men are due to PC. Because of research that has generated improved treatments and earlier diagnosis, the five-year survival rate has significantly increased in PC patients [1]. However, the key molecular mechanisms responsible for the initiation and progression of PC remain largely unknown.
Gene fusions were known to be prevalent in liquid tumors, such as BCR-ABL in chronic myelogenous leukemia, but had not been identified in solid epithelial tumors. The first PC fusion genes, involving the promoter of transmembrane protease, serine 2 (TMPRSS2) fused with coding sequence of the erythroblastosis virus E26 (Ets) gene family members, were identified in 2005 by Tomlins et al. [2]. TMPRSS2 is a prostate specific, androgen responsive, transmembrane serine protease. Ets family members are oncogenic transcription factors that contain a highly conserved Ets DNA binding region and an N-terminal regulatory domain. The ETS domain serves as a DNA binding recognition site, as well as a protein-protein interaction site commonly used for interactions with other transcription factors [3-5]. Therefore, the fusion of these genes leads to the production of Ets transcription factors under the control of the androgen sensitive promoter elements of TMPRSS2. This allows for a situation in which androgen-bound androgen receptor can bind these regions of TMPRSS2, resulting in the overexpression of Ets gene family members; these Ets members can then induce their target gene expression (Figure 1) [2]. The most common of these fusions is with ERG (Ets related gene), a member of the Ets family, resulting in the TMPRSS2-ERG fusion. The TMPRSS2-ERG fusion has been identified in approximately 50% of PC cases. TMPRSS2 has also been identified in fusions with Ets family members ETV1, ETV4, and ETV5 in PC. The prevalence of these gene fusions, in particular TMPRSS2-ERG, makes them attractive as potential diagnostic and prognostic indicators, as well as making them potential targets for tailored therapies.
Figure 1. Biology of TMPRSS2-ERG gene fusions in prostate cancer.
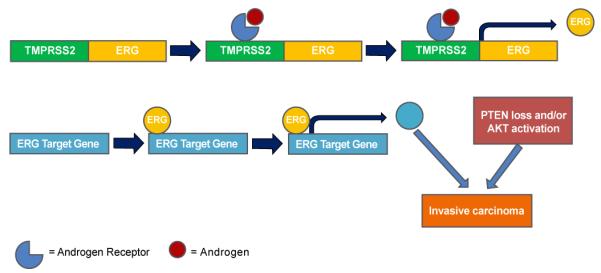
Association of bound androgen receptor with TMPRSS2 in fusion genes results in the upregulation of ERG transcription (or other Ets gene family members, i.e. ETV1, ETV4, and ETV5). This over production of ERG can then exert its effects by binding target gene promoter regions, which results in their activation or inhibition, and the generation of a neoplastic phenotype. Known direct target genes of ERG in TMPRSS2-ERG fusion positive tissues include MMP3, PLAU, LAMC2, KCNS3, PLA1A, C-MYC, GNMT, SARDH, CXCR4, ADAMTS1,TFF3, ERG, PLAT, MMP9, NDRG1, CUTL2, AR, KLK3 (PSA), KLK2, SLC43A1, FKBP5, EZH2, ZBTB16, HPGD, ZEB1, SPINT1, IL1R2, PSMA, and OPN. These and other up or down regulated genes in fusion positive cancers could facilitate PC progression. In addition, both PTEN loss and AKT overexpression facilitate the development of invasive carcinoma in TMPRSS2-ERG positive prostate tissues.
Due to its prevalence in PC, the primary focus of this review will be on the TMPRSS2-ERG fusion regarding its clinical significance, biological role in PC development, and progression. This review will therefore discuss the different forms of the TMPRSS2-ERG fusion found in PC patients, as well as the clinical associations found between fusion positive PCs and patient outcome and disease aggressiveness will be examined. Additionally, in vivo, in vitro, and gene expression studies will be evaluated to consider the biological role of the TMPRSS2-ERG fusion.
TMPRSS2-ERG Fusion Gene Express Alternatively Spliced Transcript Variants in Prostate Cancer
The first two fusion genes found in PC, TMPRSS2-ERG and TMPRSS2-ETV1, were discovered in 2005 by Tomlins et al. [2]. Shortly thereafter, TMPRSS2-ETV4 was identified [6]. Other fusions involving Ets family members found in PC include: HERV_K_22q11.23-ETV1, SLC45A3-ETV1, C15orf21-ETV1, HNRPA2B1-ETV1 [7], KLK2-ETV4, CANT1-ETV4 [8], TMPRSS2-ETV5, SLC45A3-ETV5 [9], SLC45A3-ERG, DDX5-ETV4, FLJ35294-ETV1 [10], and NDRG1-ERG [11] (Figure 2). These rearrangements and translocations can occur due to an intrachromosomal deletion, such as with TMPRSS2-ERG, as well as from an interchromosomal translocation, such as with TMPRSS2-ETV4. In the case of TMPRSS2-ERG, the rearrangement occurs either by a ~3 Mb interstitial deletion on a single copy of chromosome 21, or by a chromosomal translocation. The high prevalence of TMPRSS2-ERG fusions suggests that this region is a hot spot for chromosomal rearrangements in PC. Prostate cancer targeted exome sequence studies also confirm the expression of fusion genes in PC patient tissue [12]. The other 5′ fusion partners of Ets family members are only present in a small number of PC cases and tend to be located on different chromosomes.
Figure 2. Additional Ets fusions identified in prostate cancer.
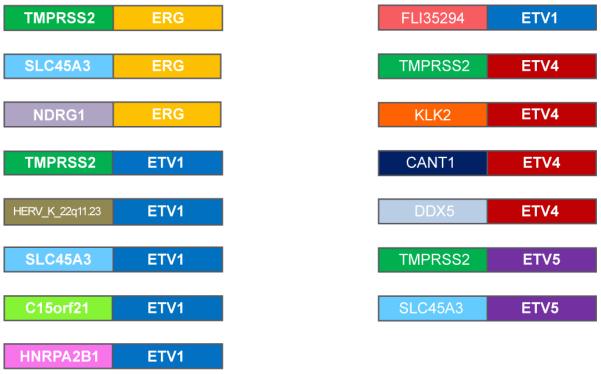
Several Ets factors have been identified in fusion genes in prostate cancer, including ERG, ETV1, ETV4, and ETV5. In addition to TMPRSS2, numerous other 5′ fusion partners of these Ets family members have been identified.
Wild-type ERG has been shown to exist as multiple different mRNA transcript variants due to alternative splicing. This alternative splicing of native ERG mRNA transcripts results in the expression of different isoforms of the ERG protein [3,13]. In accordance with this, many different splice variants of the TMPRSS2-ERG fusion have been identified. The differences between these variants can be found in the exons included in the transcript of that particular variant. These include T1-E4 [2], T1-E2, T4-E4, T4-E5, T5-E4 [14], T1-E5 [15], T1-E3 [16], T3-E4, T2-E2, T1-E3,5, T1-E2,3,4,6, T2-E4, T1-E6, T1-E3a4, T1-E3b4, T1-E3c4 [17], T1-E6,4 [18], T2-E5 [19] (T represents the last exon of TMPRSS2 in the fusion; E represents the first ERG exon included, as depicted in Figure 3). Among the TMPRSS2-ERG fusion transcript variants, T1-E4 is the most common. Additionally, a variably expressed 72-bp exon has been identified that can be expressed in several of the TMPRSS2-ERG isoforms [13].
Figure 3. TMPRSS2-ERG fusion variants.
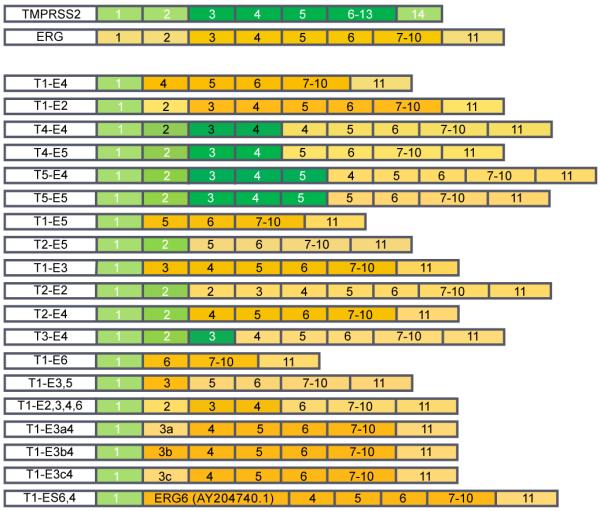
Numerous variations of the TMPRSS2-ERG fusion have been identified, with the most common being T1-E4. As shown, these variants differ in the exons from TMPRSS2 and ERG found in the fusion. T2-S6,4 is a variant in which the fusion is formed between exon 2 of TMPRSS2 and 95 nucleotides that were shown to be identical to a portion of ERG splice form 6, which is then followed by ERG exon 4. The native transcripts of TMPRSS2 and ERG are shown on top for reference, with TMPRSS2 in green and ERG in gold. Light shading represents untranslated regions; dark shading represents open reading frame. T indicates the last exon of TMPRSS2 in the fusion; E indicates the first ERG exon included.
The TMPRSS2-ERG fusion proteins are most commonly expressed as N-terminal truncated ERG proteins, due to the loss of the 5′ ERG native exons. The 5′ TMPRSS2 exons included in the fusion transcripts are generally non-coding and are usually not translated into protein product. However, there are several transcript variants that result in both TMPRSS2 and ERG exons being translated into protein. For example, fusion protein transcript variant T2-E4 is expressed as a true fusion protein product with part of TMPRSS2 exon 2 being expressed in the N-terminus of the fusion protein fused to N-terminal truncated ERG [16,17]. The DNA binding ETS domain and transactivation domain of the ERG protein are located in the C-terminus region of the protein [3]. Therefore, it is likely that, despite the N-terminal truncation, the biological functions of DNA binding and protein-protein interactions via the ETS domain remain intact in TMPRSS2-ERG fusion proteins. Due to this, it is possible that the different ERG fusion isoforms have similar biological functions as their wild-type ERG counterparts, and mediate PC progression due to overexpression. Whether novel biological functions of the ERG fusion isoforms lend to their oncogenic phenotype remains to be further elucidated.
The Role of TMPRSS2-ERG Fusion Genes in the Clinical Prognosis of Prostate Cancer Patients
Since the TMPRSS2-ERG fusion was identified in 2005, there have been conflicting reports regarding the effects of the fusion on PC development, progression, aggressiveness, and clinical outcome. Several groups found no association between TMPRSS2-ERG fusion positive cancers and stage, grade, Gleason score, PSA-indicated recurrence, progression, prognosis, and/or disease aggressiveness [20-28]. TMPRSS2-ERG fusion PCs have additionally been linked to favorable prognoses. One group found TMPRSS2-ERG fusion positive status to be associated with favorable outcomes for patients undergoing radical prostatectomy [29]. Another group found fusion positive tumors to be associated with better survival and lower Gleason scores [30]. Hermans et al. [31] found a novel TMPRSS2 transcript involved in fusion that starts at an alternative site, resulting in the inclusion of what they designated to be exon 0, which when included in the fusion is found to be associated with less aggressive behavior and therefore more favorable prognosis.
Fusion status has also been linked to poor outcomes and prognostic indicators. Fitzgerald et al. [28] found copy number increases involving the fusion gene are associated with decreased survival. A study by Gopalan et al. [32] also found copy number increase of fusions generated by deletion to be linked with aggressive disease. Another group identified a similar group of fusion positive tumors, identified as 2+Edel (signifying duplicated copies of fusions generated by deletion), to be associated with only a 25% survival rate after 8 years [33]. An increase in fusion copy number is also associated with poor prognosis [34]. As mentioned above, there are also reports that have indicated fusion formed via deletion, rather than translocation, to be associated with aggressive disease. One such report, by Mehra et al. [35] found that fusion positive metastatic sites were uniformly generated via deletion. Deletion generated TMPRSS2-ERG fusions have also been associated with higher tumor stage and lymph node involvement in PC patients [36].
TMPRSS2-ERG fusions have also been associated with biochemical or PSA indicated recurrence [37-39]. One study found 58.4% of fusion positive, versus 8.1% of fusion negative, patients to have recurrence within 5 years of treatment [38]. Another found that those with fusion positive tumors had a 5-year recurrence rate of 79.5% compared to 37.5% for fusion negative patients [40]. Barwick et al. [37] also found TMPRSS2-ERG fusion positive cancers to be statistically associated with biochemical recurrence. Rostad et al. [41] found TMPRSS2-ERG fusions to be associated with high levels of PSA, advanced stage, and high Gleason scores. Another group also found a link between TMPRSS2-ERG positive tumors and high Gleason scores [42]. In addition, cancers with TMPRSS2-ERG fusions have been associated with high grade tumors [43], and are more prone to metastasis [44]. Another study showed that there is a link between TMPRSS2-ERG fusion positive tumors and prostate specific death [45].
Different TMPRSS2-ERG transcript variants have also been linked to poor prognostic indicators. Wang et al. [13] found a variably expressed 72 base pair region, that, when present, was found to increase proliferation, invasiveness, and mortality. This group also found T2-E4 fusion to be associated with aggressive disease [13]. It has also been found that having more full length ERG than truncated ERG, lacking the Ets domain, is associated with poor differentiation, higher Gleason scores, and biochemical recurrence [46]. Additionally, T1-E2, T1-E3, and T2-E4 fusions all are associated with more aggressive disease, seminal vesicle invasion, and early PSA recurrence [16].
The tumor suppressor PTEN (phosphatase and tensin homolog) is a critical regulator of growth factors and inhibitor of PI3K. Loss of PTEN is frequently observed in prostate cancer, resulting in the deregulation of cell survival, growth, and proliferation. Previous studies have found that PTEN is lost or mutated in 30-80% of primary prostate cancer, and 50% of prostate cancer bone metastases. Concomitant PTEN loss and TMPRSS2-ERG fusion are associated with poor outcome. Carver et al. [47] found that PTEN loss combined with ERG rearrangement are statistically independent predictors of biochemical recurrence. Another study showed that cancers with no rearranged ERG and normal PTEN expression were statistically associated with good prognoses [48]. A study by Mosquera et al. [49] also shows the TMPRSS2-ERG fusion to be associated with certain morphological features. These features, including cribriform growth, blue tinged mucin, macronucleoli, and collagenous micronodules, are characteristic of PC.
It seems that the majority of findings indicate that the presence of TMPRSS2-ERG fusion gene expression in PC patients is associated with poor clinical prognosis. Evidence suggests that different subclasses of the ERG fusion transcripts may yield different clinical outcomes, such as 2+Edel, T2-E4, and +72bp transcripts being associated with worse clinical outcome. If the latter is true, then future prognostic studies may reconsider grouping all ERG fusion transcript variants into one prognostic parameter when analyzing PC cohort samples, as this may skew statistical results. A better understanding of the biological mechanisms employed by the ERG fusion proteins in PC progression may help to divide large cohorts into more effective subsets for prognostic analyses. With emerging evidence suggesting the involvement of the ERG fusion proteins in metastatic and advanced PC, it seems likely that the presence of the fusion proteins in PC contributes to a poor clinical prognosis. However, the fact that several large cohort studies have revealed opposing results regarding the prognosis value of TMPRSS2-ERG fusion gene expression in PC warrants further investigation into this matter.
Role of TMPRSS2-ERG gene fusions in prostate cancer progression
It has been known since 2005 that gene fusions are found in the majority of PCs [2]. The role that these fusions play in the development and progression of PC is much less understood. Since the discovery of the gene fusions, several groups have identified a collection of pathways and effects associated with the overexpression of ERG found in TMPRSS2-ERG fusion positive PCs. Recent studies have revealed several of the protein-protein interactions and target genes of ERG fusions, lending insight into ERG transcriptional regulation. Many of these studies have elucidated a key involvement of ERG fusions with the androgen receptor (AR) in hormone naïve, androgen sensitive prostate cancers. Despite castration resistant prostate cancer (CRPC) displaying partial independence from AR regulation, the expression of TMPRSS2-ERG fusion proteins in castration resistant prostate cancer (hormone refractory PC) has been shown to persist.
ERG overexpression in mouse models induces prostate cancer progression
Tomlins et al. [50] was one of the first groups to study the biological effects of TMPRSS2-ERG fusions in PC. To this end, transgenic mice expressing a truncated version of ERG (exon 2 thru the native stop codon) under the control of a probasin promoter were generated and identified as ARR2Pb-ERG mice. In these ARR2Pb-ERG mice, 3/8 developed murine prostatic intraepithelial neoplasia (mPIN) by 12-14 weeks of age. Klezovitch et al. [51] found that mice expressing high levels of ERG under probasin promoter control developed mPIN by five to six months of age, and mice expressing comparatively lower, but still higher than normal, levels of ERG developed mPIN in 10-12 months. In ERG overexpressing murine prostate, there was disruption of the basal cell layer when compared to benign glands, an indicator of early stages of PC development. These observations were further supported by tissue recombination studies with ERG overexpressing prostate epithelial cells implanted in renal capsule showing disruption of basal epithelial cells [52]. Subsequently, further studies showed that ERG overexpression in mouse prostate does not result in high grade PIN lesions but rather subtle changes in mice prostate epithelial cells [47,53]. These differences in observed phenotypes were attributed to the different genetic backgrounds of the mice used in these studies and different isoforms of ERG gene used in overexpression studies. Carver et al. [47] found that ERG levels in PTEN knockout mice to be significantly higher than in controls. This group additionally generated PTEN haploinsufficient mice with ERG expression driven by the probasin promoter. These mice developed HGPIN at approximately two months of age and multifocal adenocarcinoma by six months of age, whereas control PTEN heterozygous mice developed HGPIN at approximately eight months of age and did not develop adenocarcinoma [47]. Comparable results were found by King et al. [53] in a study in which TMPRSS2-ERG mice were crossed with PTEN haploinsufficient mice. All of these mice (8/8) showed PIN development by six months of age, compared with only 1/8 littermate controls. These studies demonstrate that ERG cooperates with PTEN loss leading to the development of adenocarcinoma. This observation was also supported by tissue recombinant studies, where ERG overexpression in combination with either PTEN knockdown or expression of constitutively activated Akt resulted in development of adenocarcinoma [52]. The studies considered so far have all examined ERG overexpression alone or in combination with other genetic alterations. Alternatively, Sun et al. [54] and Wang et al. [13] investigated the biological effects of ERG knockdown using TMPRSS2-ERG fusion positive VCaP cells in xenograft model systems. When ERG expression was knocked down using siRNA in VCaP cells in a xenograft model, only 2/9 of the ERG siRNA SCID mice developed tumors by day 42, compared to 5/5 SCID mice in the control group [54]. Wang et al. [13] similarly investigated ERG expression knockdown in a mouse orthotropic model using stably transfected shRNA (short hairpin RNA) VCaP cell lines. After four weeks the scrambled shRNA control animals had a luciferase signal and tumor weight 4-fold that of the ERG shRNA animals. Together, these xenograft studies show that ERG expression in VCaP cells promotes tumor growth.
The findings from these mouse studies indicate that TMPRSS2-ERG fusion is not, on its own, sufficient to induce the development of invasive carcinoma. However, these fusions can cause the formation of PIN lesions. These studies also indicate that PTEN loss and ERG overexpression cooperate in the formation of PIN and invasive carcinoma, indicating that PTEN loss could function as a “second hit” in TMPRSS2-ERG fusion positive PC. TMPRSS2-ERG fusions seem to further associate with the PI3K pathway through cooperation with active Akt, the combination resulting in the development of invasive carcinoma.
ERG overexpression in prostate cell culture models increases cell invasiveness
In addition to animal models, cell culture-based studies have also been used to elucidate the biological role of ERG. Knockdown of ERG by siRNA in VCaP cells significantly inhibits their invasiveness [13,50,54]. Blocking urokinase plasminogen activator (uPA) and plasminogen activator pathways, which have been associated with ERG overexpression, as well as siRNA knockdown of uPA, were also shown to significantly inhibit cellular invasiveness in RWPE, VCaP, and BPH-1 cells [50,51].
Conversely, ERG overexpression has also been found to have many effects in cell culture studies. ERG overexpression in RWPE and PrEC cells was found to significantly increase their invasion [50]. ERG overexpression was also shown to increase invasiveness in PNT1a cells [13]. Additionally, ERG overexpression in BPH-1 cells was shown to increase proliferation rates and invasiveness [51]. Proliferation rates were also shown to increase in ERG overexpressing PrEC cells [13]. The chemokine receptor CXCR4 increases cellular invasiveness in VCaP cells; gene expression of CXCR4 is directly upregulated in the presence of R1881 treatment via the ERG fusion protein binding to the CXCR4 promoter [55]. In an in vitro chemoinvasion assay, VCaP cells invaded through a matrigel chamber at a significantly higher rate in the presence of the CXCR4 ligand, CXCL12, as a chemoattractant. The chemoinvasion rate was enhanced further when VCaP cells were treated with synthetic androgen R1881, suggesting that AR activation and subsequent ERG fusion upregulation induced increased CXCR4 cell surface expression, leading to increased chemoinvasion [55]. This suggests another mechanism of enhanced cellular invasion mediated by the ERG fusion proteins. ERG overexpression in either BPH-1 or PNTla cells enhances cellular migration [13,47]. Interestingly, co-overexpression of multiple ERG transcript variants increases cell proliferation rates, suggesting a potential synergism between co-expressed ERG variants [13]. These cell culture studies indicate that ERG overexpression mediated by TMPRSS2-ERG fusion can result in increased invasiveness, cell proliferation, and cellular migration. These effects are due to the aberrant regulation of downstream target genes of ERG, which in turn are able to alter normal cellular activity.
ERG mediates transcriptional regulation at specific gene loci via direct protein-protein and DNA binding interactions
In vitro studies on ERG protein structure and interactions using co-immunoprecipitation (Co-IP) revealed that the ETS domain is involved in DNA binding and protein interactions. Studies showed that wild-type ERG proteins physically interacted through their ETS domain via heterodimerization with other ETS transcription factor family members such as Fli-1, ETS-2, Er81, and Pu-1 [3,5]. Different ERG isoforms are also involved in the formation of a ternary complex with AP1 family members Fos and Jun, forming an ERG/Fos/Jun complex. In the same study, ERG proteins were shown to physically interact via homodimerization and homo-iso-dimerization with the same isoform or with other ERG isoforms [3]. Another study verified that these wild-type ERG protein-protein interactions influenced the regulation and expression of the MMP1 and MMP3 gene promoter regions. ERG interacted with Fos and Jun in order to activate MMP1 promoter activity, and in contrast, ERG inhibited ETS-2-induced activation of MMP3 promoter activity [56]. As shown by Co-IP followed by mass spectrometry analysis or western blot analysis in VCaP cells and ERG fusion positive human prostate cancer tissues, ERG physically interacts with PARP1 and DNA-PKcs in a DNA independent manner and with Ku70 and Ku80 in a DNA dependent manner. ERG interacts with all four proteins through its C-terminal region, and ERG interacts with DNA-PKcs specifically through the ERG-ETS domain amino acid Y373. These protein interactions mediate DNA double stranded breaks and transcriptional regulation. In this same study, PARP1, DNA-PKcs, Ku70, and Ku80 were shown via chromatin immunoprecipitation assay to bind to ERG target genes, and PARP1 and DNA-PKcs were required for ERG transcriptional activation of the gene PLA1 [57]. These results suggest that ERG can regulate gene transcriptional activity in a protein interaction dependent manner, which is most likely coupled with DNA binding transcriptional regulation.
ERG is associated with the aberrant expression of many genes. Several of these genes have been identified through chromatin immunoprecipitation (ChIP) assays to be direct binding targets of ERG. Tomlins et al. [50] used such an assay to show that ERG can directly bind the proximal promoter of Urokinase Plasminogen Activator (PLAU) and MMP3. ChIP has also identified the recruitment of ERG to the C-MYC promoter upstream Ets element [54]. This study demonstrated that ERG was recruited to the PSA enhancer and to the prostein promoter upstream Ets element, showing direct interaction of ERG with PSA expression [54]. Additionally, ERG binds the GNMT promoter and the SARDH promoter [58]. A ChIP assay also found that ERG directly binds both CXCR4 and ADAMTS1 promoter regions, and the expression of these two genes is upregulated in the presence of ERG overexpression [47]. Additionally, ERG binds directly to the CXCR4 gene promoter region in response to R1881 treatment [55]. In a study by Tomlins et al. [50] ChIP identified LAMC2, KNCS3, and PLA1A as direct ERG targets. ERG was shown to be recruited to many gene regions including, PLAU, PLAT, MMP9, NDRG1, CUTL2, AR, KLK3 (PSA), KLK2, SLC45A1, FKBP5, EZH2, and ZBTB16 [59]. ERG also cooperates with AR during the recruitment of ERG to the TFF3 gene locus [60]. One study identified ERG as a modulator of prostaglandin signaling and showed that ERG directly bound to the core gene promoter region of the HPGD gene. In this study, ERG siRNA in VCaP cells resulted in an increase in HPGD expression, suggesting that ERG negatively regulates expression of the HPGD promoter [61]. ERG also promotes epithelial to mesenchymal transition, and ERG directly binds the ZEB1, SPINT1, and IL1R2 gene regions [62]. A study by Flajollet et al. [63] revealed that ERG directly bound to an ETS binding sequence in the promoter of the osteopontin gene (OPN), which resulted in an increase in OPN gene expression. In another study, ERG was shown to negatively regulate the expression of the prostate specific membrane antigen (PSMA) by direct binding to the PSMA gene locus [64]. One recent study revealed an important novel finding that wild-type ERG gene expression is under the control of the TMPRSS2-ERG fusion protein. This was verified by ChIP assay showing that ERG bound directly to the wild-type ERG locus in VCaP fusion positive prostate cancers, but not in LNCaP fusion negative cancers [65]. This finding is important as it reveals a feed-forward mechanism of ERG overexpression in TMPRSS2-ERG fusion positive PC cells, further suggesting an important role of ERG overexpression in the progression of PC. These ChIP assays demonstrate that many genes are direct targets of ERG, providing a potential explanation for their aberrant regulation.
ERG and AR co-mediate transcriptional regulation of the ERG and AR gene loci, as well as their target genes in prostate cancer
The role of AR in TMPRSS2-ERG fusion positive PC cells has also been evaluated using cell culture studies. In VCaP cells, ERG physically interacts with AR, as verified by Co-IP. In vitro studies revealed that the ERG-AR protein-protein interaction was mediated through the ERG-ETS domain in a DNA independent manner. This same study revealed that ERG transcriptional regulation often occurred concomitantly with AR. ChIP assays demonstrate that ERG and AR co-occupy many androgen regulated genes, such as KLK3 (PSA) and AR. In this study, ERG had an inhibitory effect on AR mRNA and protein expression, as well as an inhibitory effect on the mRNA expression of several AR regulated genes, some of which were co-occupied by ERG and AR. ERG mediates its inhibitory effects via direct DNA binding and transcriptional repression of AR target genes and by protein-protein interactions with AR [59]. Another study showed that ERG regulated expression of TFF3 (an androgen regulated gene) via direct binding to the TFF3 gene region in hormone-naïve PC and in CRPC [60]. Interestingly, ERG suppressed TFF3 expression in hormone naïve prostate cancer, and induced TFF3 expression in CRPC.
It has been shown that AR positively regulates ERG expression and possibly ERG target gene expression. As mentioned previously, a study by Cai et al. [55] showed that the synthetic androgen R1881 upregulates ERG and CXCR4 expression in fusion positive VCaP cells. In addition, R1881 activation of TMPRSS2-ERG fusions functionally activates CXCR4 expression in VCaP cells [55]. These data suggest that increased CXCR4 levels following R1881 treatment are indirectly attributed to AR activation via the R1881/AR induced upregulation of ERG. Consistent with these results, treatment of VCaP cells with R1881 increased TMPRSS2-ERG expression in several other independent studies [2,64,66,67]. However, TMPRSS2-ERG expression was not increased after R1881 treatment in AR negative, fusion positive NCI-H660 cells [66]. As mentioned previously, treatment of androgen sensitive VCaP cells with R1881 increased both TMPRSS2-ERG and wild-type ERG transcript expression. However, only the TMPRSS2-ERG transcript was directly increased by AR signaling, whereas wild-type ERG transcript was increased indirectly via the TMPRSS2-ERG fusion binding directly to the wild-type ERG locus [65]. Interestingly, AR signaling is implicated in a causal role in the formation of the TMPRSS2-ERG gene fusion rearrangements, further suggesting a strong involvement of AR with TMPRSS2-ERG gene fusions [68-71].
Expression of the TMPRSS2-ERG gene fusion persists in patients with Castration Resistant Prostate Cancer
TMPRSS2 gene expression is normally under the control of AR, so whether TMPRSS2-ERG fusions play a role in CRPC (hormone-refractory prostate cancer) has remained a controversial topic that is currently being investigated. A study by Hermans et al. [19] showed that although TMPRSS2-ERG gene fusions were present in four androgen independent prostate tumor xenografts, ERG fusion mRNA transcript was not detected in any of these fusion positive samples. Three of the four androgen independent prostate tumor xenografts also showed little to no expression of AR mRNA transcript, with the fourth showing only a low level of AR transcript expression. Another study showed that two AR negative, hormone-refractory xenografts LuCaP49 and LuCaP93 [72,73] were also TMPRSS2-ERG fusion positive, but did not express the ERG fusion transcript [29]. Thus, it seems that with the exception of the NCI-H660 cell line, AR negative PC cells harboring the TMPRSS2-ERG gene fusion generally do not express ERG fusion transcripts.
Despite these findings, several other studies have consistently shown that TMPRSS2-ERG fusion gene is present and expressed in AR positive CRPC patient tumors and xenogrfts [74]. Cai et al. [74] showed that TMPRSS2-ERG gene fusion mRNA expression was present in VCaP xenografts from castrated mice as well as in 11 of 29 (38%) of CRPC patient samples. These same fusion positive CRPC patient samples also showed increased AR mRNA expression levels relative to androgen dependent prostate cancer samples. This suggests that AR may continue to play a role in TMPRSS2-ERG gene expression even in a CRPC state. Additionally, ERG mRNA expression in VCaP mouse xenografts was decreased four days after mice underwent castration surgery. However, after approximately six weeks mice experienced tumor relapse, and ERG mRNA expression increased above levels seen prior to the castration surgery. These same xenograft mice showed progressively increasing AR mRNA and protein levels throughout the six week relapse period, consistent with the AR overexpression seen in the CRPC patient samples [74]. Relapse is a common occurrence among prostate cancer patients who undergo androgen deprivation therapies (ADT) in the clinic, so these findings seem to closely mimic the CRPC clinical setting. At the time of relapse, this same xenograft study found that there was an upregulation in the mRNA expression of several enzymes present in the DHT biosynthetic pathway [74]. Additionally, several studies have also shown that prostate cancer cells can synthesize androgens de novo [75,76]. Therefore, upregulation of DHT biosynthetic enzymes and de novo androgen synthesis by prostate cancer cells suggests a possible mechanism in which AR signaling and TMPRSS2-ERG expression may be reactivated in a CRPC state, possibly contributing to ADT resistance. These findings suggest a role for TMPRSS2-ERG in the progression of CRPC, although the mechanism of how CRPC cells bypass ADT and continue the AR induced expression of TMPRSS2-ERG remains to be fully characterized. It has been suggested that TMPRSS2-ERG expression may eventually become androgen independent in CRPC, but no data have yet confirmed this hypothesis; further studies will be needed in order to investigate this possibility [77].
Mehra et al. [35] found that TMPRSS2-ERG mRNA expression was present in 10 of 27 (37%) of hormone-refractory metastatic prostate cancer patient samples. Of these 10 patients, 100% of metastatic sites contained a TMPRSS2-ERG fusion formed through interstitial deletion (Edel). In accordance with these findings, all metastatic tumor sites in one individual harbored the same fusion subtype, and this same fusion subtype was also found in the primary tumor site of the prostate. This suggests a mechanism of clonal selection and clonal expansion of the TMPRSS2-ERG fusions in metastatic fusion-positive CRPC. Attard et al. [67] found similar results, with 9 of 15 (60%) CRPC tumor samples expressing the TMPRSS2-ERG transcript, and circulating tumor cells (CTCs) collected from 11 CRPC patients showing the same ERG gene status as prostate tumor tissues from the same individual.
Many studies have revealed the presence of TMPRSS2-ERG transcript in CRPC; Rickman et al. [60] revealed a unique biological role of the TMPRSS2-ERG fusions in CRPC. In this study, 14 of 19 (73%) TMPRSS2-ERG fusion positive CRPC samples were found to express the ERG fusion transcript. Importantly, this study found that ERG fusions induced expression of TFF3 in CRPC, but inhibited TFF3 expression in hormone-naïve PC samples. Additionally, ERG fusion regulation of TFF3 was dependent on AR signaling, and overexpression of TFF3 in fusion positive CRPC resulted in an increase in cellular invasiveness. This study revealed a novel functional role of TMPRSS2-ERG fusion proteins in the progression of CRPC, and that the function of ERG fusions can change depending on the selective pressures and progressive state of the disease.
Concluding Remarks
Overall, the studies discussed here support a model (Figure 1) in which circulating androgens in PC patients activate TMPRSS2-ERG fusions, with the resulting ERG protein regulating genes whose expression and function facilitate PC progression. As mentioned above, this can increase invasiveness, cellular motility, and disease aggressiveness. In addition, it has been shown to cause the upregulation of oncogenes, as well as the downregulation of prostate differentiation genes. This, as well as the other evidence outlined above, indicates the importance of TMPRSS2-ERG fusions in PC. Considering the prevalence of TMPRSS2-ERG fusion in PC, and the large number of men who develop PC each year, the further study of these fusions is vital to the understanding of PC biology. Once better elucidated, the associations between this fusion and prognostic indicators and disease aggressiveness could give clinicians a more informed way of classifying PC for prognostic and treatment purposes. The furthered understanding of ERG’s biological roles in PC will augment this and potentially provide novel target pathways for future therapies, potentially through inhibition of ERG itself. Continued research of gene fusions, in particular TMPRSS2-ERG, is needed to better understand their biological roles and relationship to PC development and progression.
Acknowledgement
This study was supported by U.S. Department of Defense, W81XWH-09-1-0250, NIH-NCI Grant R01CA151557 and Fund for Cancer Research foundation.
References
- 1.Cancer facts & figures 2012. The American Cancer Society; Atlanta, GA: 2012. [Google Scholar]
- 2.Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 3.Carrère S, Verger A, Flourens A, Stehelin D, Duterque-Coquillaud M. Erg proteins, transcription factors of the Ets family, form homo, heterodimers and ternary complexes via two distinct domains. Oncogene. 1998;16:3261–3268. doi: 10.1038/sj.onc.1201868. [DOI] [PubMed] [Google Scholar]
- 4.Verger A, Duterque-Coquillaud M. When Ets transcription factors meet their partners. Bioessays. 2002;24:362–370. doi: 10.1002/bies.10068. [DOI] [PubMed] [Google Scholar]
- 5.Basuyaux JP, Ferreira E, Stéhelin D, Butticè G. The Ets transcription factors interact with each other and with the c-Fos/c-Jun complex via distinct protein domains in a DNA-dependent and -independent manner. J Biol Chem. 1997;272:26188–26195. doi: 10.1074/jbc.272.42.26188. [DOI] [PubMed] [Google Scholar]
- 6.Tomlins SA, Mehra R, Rhodes DR, Smith LR, Roulston D, et al. TMPRSS2:ETV4 gene fusions define a third molecular subtype of prostate cancer. Cancer Res. 2006;66:3396–3400. doi: 10.1158/0008-5472.CAN-06-0168. [DOI] [PubMed] [Google Scholar]
- 7.Tomlins SA, Laxman B, Dhanasekaran SM, Helgeson BE, Cao X, et al. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature. 2007;448:595–599. doi: 10.1038/nature06024. [DOI] [PubMed] [Google Scholar]
- 8.Hermans KG, Bressers AA, van der Korput HA, Dits NF, Jenster G, et al. Two unique novel prostate-specific and androgen-regulated fusion partners of ETV4 in prostate cancer. Cancer Res. 2008;68:3094–3098. doi: 10.1158/0008-5472.CAN-08-0198. [DOI] [PubMed] [Google Scholar]
- 9.Helgeson BE, Tomlins SA, Shah N, Laxman B, Cao Q, et al. Characterization of TMPRSS2:ETV5 and SLC45A3:ETV5 gene fusions in prostate cancer. Cancer Res. 2008;68:73–80. doi: 10.1158/0008-5472.CAN-07-5352. [DOI] [PubMed] [Google Scholar]
- 10.Han B, Mehra R, Dhanasekaran SM, Yu J, Menon A, et al. A fluorescence in situ hybridization screen for E26 transformation-specific aberrations: identification of DDX5-ETV4 fusion protein in prostate cancer. Cancer Res. 2008;68:7629–7637. doi: 10.1158/0008-5472.CAN-08-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pflueger D, Rickman DS, Sboner A, Perner S, LaFargue CJ, et al. N-myc downstream regulated gene 1 (NDRG1) is fused to ERG in prostate cancer. Neoplasia. 2009;11:804–811. doi: 10.1593/neo.09572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lonigro RJ, Grasso CS, Robinson DR, Jing X, Wu YM, et al. Detection of somatic copy number alterations in cancer using targeted exome capture sequencing. Neoplasia. 2011;13:1019–1025. doi: 10.1593/neo.111252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang J, Cai Y, Yu W, Ren C, Spencer DM, et al. Pleiotropic Biological Activities of Alternatively Spliced TMPRSS2/ERG Fusion Gene Transcripts. Cancer Res. 2008;68:8516–8524. doi: 10.1158/0008-5472.CAN-08-1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soller MJ, Isaksson M, Elfving P, Soller W, Lundgren R, et al. Confirmation of the high frequency of the <I>TMPRSS2/ERG</I> fusion gene in prostate cancer. Genes Chromosomes Cancer. 2006;45:717–719. doi: 10.1002/gcc.20329. [DOI] [PubMed] [Google Scholar]
- 15.Yoshimoto M, Joshua AM, Chilton-Macneill S, Bayani J, Selvarajah S, et al. Three-color FISH analysis of TMPRSS2/ERG fusions in prostate cancer indicates that genomic microdeletion of chromosome 21 is associated with rearrangement. Neoplasia. 2006;8:465–469. doi: 10.1593/neo.06283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang J, Cai Y, Ren C, Ittmann M. Expression of Variant TMPRSS2/ERG Fusion Messenger RNAs Is Associated with Aggressive Prostate Cancer. Cancer Res. 2006;66:8347–8351. doi: 10.1158/0008-5472.CAN-06-1966. [DOI] [PubMed] [Google Scholar]
- 17.Clark J, Merson S, Jhavar S, Flohr P, Edwards SM, et al. Diversity of TMPRSS2-ERG fusion transcripts in the human prostate. Oncogene. 2007;26:2667–2673. doi: 10.1038/sj.onc.1210070. [DOI] [PubMed] [Google Scholar]
- 18.Iljin K, Wolf M, Edgren H, Gupta S, Kilpinen S, et al. TMPRSS2 Fusions with Oncogenic ETS Factors in Prostate Cancer Involve Unbalanced Genomic Rearrangements and Are Associated with HDAC1 and Epigenetic Reprogramming. Cancer Res. 2006;66:10242–10246. doi: 10.1158/0008-5472.CAN-06-1986. [DOI] [PubMed] [Google Scholar]
- 19.Hermans KG, van Marion R, van Dekken H, Jenster G, van Weerden WM, et al. TMPRSS2:ERG fusion by translocation or interstitial deletion is highly relevant in androgen-dependent prostate cancer, but is bypassed in late-stage androgen receptor-negative prostate cancer. Cancer Res. 2006;66:10658–10663. doi: 10.1158/0008-5472.CAN-06-1871. [DOI] [PubMed] [Google Scholar]
- 20.Esgueva R, Perner S, J LaFargue C, Scheble V, Stephan C, et al. Prevalence of TMPRSS2-ERG and SLC45A3-ERG gene fusions in a large prostatectomy cohort. Mod Pathol. 2010;23:539–546. doi: 10.1038/modpathol.2009.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mwamukonda K, Chen Y, Ravindranath L, Furusato B, Hu Y, et al. Quantitative expression of TMPRSS2 transcript in prostate tumor cells reflects TMPRSS2-ERG fusion status. Prostate Cancer Prostatic Dis. 2009;13:47–51. doi: 10.1038/pcan.2009.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mosquera JM, Mehra R, Regan MM, Perner S, Genega EM, et al. Prevalence of TMPRSS2-ERG Fusion Prostate Cancer among Men Undergoing Prostate Biopsy in the United States. Clin Cancer Res. 2009;15:4706–4711. doi: 10.1158/1078-0432.CCR-08-2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Darnel AD, Lafargue CJ, Vollmer RT, Corcos J, Bismar TA. TMPRSS2-ERG fusion is frequently observed in Gleason pattern 3 prostate cancer in a Canadian cohort. Cancer Biol Ther. 2009;8:125–130. doi: 10.4161/cbt.8.2.7134. [DOI] [PubMed] [Google Scholar]
- 24.Rouzier C, Haudebourg J, Carpentier X, Valério L, Amiel J, et al. Detection of the TMPRSS2-ETS fusion gene in prostate carcinomas: retrospective analysis of 55 formalin-fixed and paraffin-embedded samples with clinical data. Cancer Genet Cytogenet. 2008;183:21–27. doi: 10.1016/j.cancergencyto.2008.01.021. [DOI] [PubMed] [Google Scholar]
- 25.Furusato B, Gao CL, Ravindranath L, Chen Y, Cullen J, et al. Mapping of TMPRSS2-ERG fusions in the context of multi-focal prostate cancer. Mod Pathol. 2008;21:67–75. doi: 10.1038/modpathol.3800981. [DOI] [PubMed] [Google Scholar]
- 26.Tu JJ, Rohan S, Kao J, Kitabayashi N, Mathew S, et al. Gene fusions between TMPRSS2 and ETS family genes in prostate cancer: frequency and transcript variant analysis by RT-PCR and FISH on paraffin-embedded tissues. Mod Pathol. 2007;20:921–928. doi: 10.1038/modpathol.3800903. [DOI] [PubMed] [Google Scholar]
- 27.Albadine R, Latour M, Toubaji A, Haffner M, Isaacs WB, et al. TMPRSS2-ERG gene fusion status in minute (minimal) prostatic adenocarcinoma. Mod Pathol. 2009;22:1415–1422. doi: 10.1038/modpathol.2009.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.FitzGerald LM, Agalliu I, Johnson K, Miller MA, Kwon EM, et al. Association of TMPRSS2-ERG gene fusion with clinical characteristics and outcomes: results from a population-based study of prostate cancer. BMC Cancer. 2008;8:230. doi: 10.1186/1471-2407-8-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saramäki OR, Harjula AE, Martikainen PM, Vessella RL, Tammela TL, et al. TMPRSS2: ERG Fusion Identifies a Subgroup of Prostate Cancers with a Favorable Prognosis. Clin Cancer Res. 2008;14:3395–3400. doi: 10.1158/1078-0432.CCR-07-2051. [DOI] [PubMed] [Google Scholar]
- 30.Winnes M, Lissbrant E, Damber JE, Stenman G. Molecular genetic analyses of the TMPRSS2-ERG and TMPRSS2-ETV1 gene fusions in 50 cases of prostate cancer. Oncol Rep. 2007;17:1033–1036. [PubMed] [Google Scholar]
- 31.Hermans KG, Boormans JL, Gasi D, van Leenders GJ, Jenster G, et al. Overexpression of Prostate-Specific TMPRSS2(exon 0)-ERG Fusion Transcripts Corresponds with Favorable Prognosis of Prostate Cancer. Clin Cancer Res. 2009;15:6398–6403. doi: 10.1158/1078-0432.CCR-09-1176. [DOI] [PubMed] [Google Scholar]
- 32.Gopalan A, Leversha MA, Satagopan JM, Zhou Q, Al-Ahmadie HA, et al. TMPRSS2-ERG Gene Fusion Is Not Associated with Outcome in Patients Treated by Prostatectomy. Cancer Res. 2009;69:1400–1406. doi: 10.1158/0008-5472.CAN-08-2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Attard G, Clark J, Ambroisine L, Fisher G, Kovacs G, et al. Duplication of the fusion of TMPRSS2 to ERG sequences identifies fatal human prostate cancer. Oncogene. 2007;27:253–263. doi: 10.1038/sj.onc.1210640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yoshimoto M, Joshua AM, Cunha IW, Coudry RA, Fonseca FP, et al. Absence of TMPRSS2:ERG fusions and PTEN losses in prostate cancer is associated with a favorable outcome. Mod Pathol. 2008;21:1451–1460. doi: 10.1038/modpathol.2008.96. [DOI] [PubMed] [Google Scholar]
- 35.Mehra R, Tomlins SA, Yu J, Cao X, Wang L, et al. Characterization of TMPRSS2-ETS Gene Aberrations in Androgen-Independent Metastatic Prostate Cancer. Cancer Res. 2008;68:3584–3590. doi: 10.1158/0008-5472.CAN-07-6154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perner S, Demichelis F, Beroukhim R, Schmidt FH, Mosquera JM, et al. TMPRSS2:ERG Fusion-Associated deletions provide insight into the heterogeneity of prostate Cancer. Cancer Res. 2006;66:8337–8341. doi: 10.1158/0008-5472.CAN-06-1482. [DOI] [PubMed] [Google Scholar]
- 37.Barwick BG, Abramovitz M, Kodani M, Moreno CS, Nam R, et al. Prostate cancer genes associated with TMPRSS2-ERG gene fusion and prognostic of biochemical recurrence in multiple cohorts. Br J Cancer. 2010;102:570–576. doi: 10.1038/sj.bjc.6605519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nam RK, Sugar L, Yang W, Srivastava S, Klotz LH, et al. Expression of the TMPRSS2:ERG fusion gene predicts cancer recurrence after surgery for localised prostate cancer. Br J Cancer. 2007;97:1690–1695. doi: 10.1038/sj.bjc.6604054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bonaccorsi L, Nesi G, Nuti F, Paglierani M, Krausz C, et al. Persistence of expression of the TMPRSS2:ERG fusion gene after pre-surgery androgen ablation may be associated with early prostate specific antigen relapse of prostate cancer: preliminary results. J Endocrinol Invest. 2009;32:590–596. doi: 10.1007/BF03346514. [DOI] [PubMed] [Google Scholar]
- 40.Nam RK, Sugar L, Wang Z, Yang W, Kitching R, et al. Expression of TMPRSS2:ERG gene fusion in prostate cancer cells is an important prognostic factor for cancer progression. Cancer Biol Ther. 2007;6:40–45. doi: 10.4161/cbt.6.1.3489. [DOI] [PubMed] [Google Scholar]
- 41.Rostad K, Hellwinkel OJ, Haukaas SA, Halvorsen OJ, Øyan AM, et al. TMPRSS2:ERG fusion transcripts in urine from prostate cancer patients correlate with a less favorable prognosis. APMIS. 2009;117:575–582. doi: 10.1111/j.1600-0463.2009.02517.x. [DOI] [PubMed] [Google Scholar]
- 42.Hofer MD, Kuefer R, Maier C, Herkommer K, Perner S, et al. Genome-Wide Linkage Analysis of TMPRSS2-ERG Fusion in Familial Prostate Cancer. Cancer Res. 2009;69:640–646. doi: 10.1158/0008-5472.CAN-08-2008. [DOI] [PubMed] [Google Scholar]
- 43.Mehra R, Tomlins SA, Shen R, Nadeem O, Wang L, et al. Comprehensive assessment of TMPRSS2 and ETS family gene aberrations in clinically localized prostate cancer. Mod Pathol. 2007;20:538–544. doi: 10.1038/modpathol.3800769. [DOI] [PubMed] [Google Scholar]
- 44.Perner S, Svensson MA, Hossain RR, Day JR, Groskopf J, et al. ERG Rearrangement Metastasis Patterns in Locally Advanced Prostate Cancer. Urology. 2010;75:762–767. doi: 10.1016/j.urology.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Demichelis F, Fall K, Perner S, Andrén O, Schmidt F, et al. TMPRSS2:ERG gene fusion associated with lethal prostate cancer in a watchful waiting cohort. Oncogene. 2007;26:4596–4599. doi: 10.1038/sj.onc.1210237. [DOI] [PubMed] [Google Scholar]
- 46.Hu Y, Dobi A, Sreenath T, Cook C, Tadase AY, et al. Delineation of TMPRSS2-ERG Splice Variants in Prostate Cancer. Clin Cancer Res. 2008;14:4719–4725. doi: 10.1158/1078-0432.CCR-08-0531. [DOI] [PubMed] [Google Scholar]
- 47.Carver BS, Tran J, Gopalan A, Chen Z, Shaikh S, et al. Aberrant ERG expression cooperates with loss of PTEN to promote cancer progression in the prostate. Nat Genet. 2009;41:619–624. doi: 10.1038/ng.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reid AH, Attard G, Ambroisine L, Fisher G, Kovacs G, et al. Molecular characterisation of ERG, ETV1 and PTEN gene loci identifies patients at low and high risk of death from prostate cancer. Br J Cancer. 2010;102:678–684. doi: 10.1038/sj.bjc.6605554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mosquera JM, Perner S, Demichelis F, Kim R, Hofer MD, et al. Morphological features of <I>TMPRSS2-ERG</I> gene fusion prostate cancer. J Pathol. 2007;212:91–101. doi: 10.1002/path.2154. [DOI] [PubMed] [Google Scholar]
- 50.Tomlins SA, Laxman B, Varambally S, Cao X, Yu J, et al. Role of the TMPRSS2-ERG gene fusion in prostate cancer. Neoplasia. 2008;10:177–188. doi: 10.1593/neo.07822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Klezovitch O, Risk M, Coleman I, Lucas JM, Null M, et al. A causal role for ERG in neoplastic transformation of prostate epithelium. Proc Natl Acad Sci USA. 2008;105:2105–2110. doi: 10.1073/pnas.0711711105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zong Y, Xin L, Goldstein AS, Lawson DA, Teitell MA, et al. ETS family transcription factors collaborate with alternative signaling pathways to induce carcinoma from adult murine prostate cells. Proc Natl Acad Sci USA. 2009;106:12465–12470. doi: 10.1073/pnas.0905931106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.King JC, Xu J, Wongvipat J, Hieronymus H, Carver BS, et al. Cooperativity of TMPRSS2-ERG with PI3-kinase pathway activation in prostate oncogenesis. Nat Genet. 2009;41:524–526. doi: 10.1038/ng.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sun C, Dobi A, Mohamed A, Li H, Thangapazham RL, et al. TMPRSS2-ERG fusion, a common genomic alteration in prostate cancer activates C-MYC and abrogates prostate epithelial differentiation. Oncogene. 2008;27:5348–5353. doi: 10.1038/onc.2008.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cai J, Singareddy R, Kropinski A, Sheng S, Cher ML, et al. Androgens Induce Functional CXCR4 through ERG Factor Expression in TMPRSS2-ERG Fusion-Positive Prostate Cancer Cells. Transl Oncol. 2010;3:195–203. doi: 10.1593/tlo.09328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Butticè G, Duterque-Coquillaud M, Basuyaux JP, Carrère S, Kurkinen M, et al. Erg, an Ets-family member, differentially regulates human collagenase1 (MMP1) and stromelysin1 (MMP3) gene expression by physically interacting with the Fos/Jun complex. Oncogene. 1996;13:2297–2306. [PubMed] [Google Scholar]
- 57.Brenner JC, Ateeq B, Li Y, Yocum AK, Cao Q, et al. Mechanistic rationale for inhibition of poly(ADP-ribose) polymerase in ETS gene fusion-positive prostate cancer. Cancer Cell. 2011;19:664–678. doi: 10.1016/j.ccr.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sreekumar A, Poisson LM, Rajendiran TM, Khan AP, Cao Q, et al. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature. 2009;457:910–914. doi: 10.1038/nature07762. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 59.Yu J, Yu J, Mani RS, Cao Q, Brenner CJ, et al. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell. 2010;17:443–454. doi: 10.1016/j.ccr.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rickman DS, Chen YB, Banerjee S, Pan Y, Yu J, et al. ERG cooperates with androgen receptor in regulating trefoil factor 3 in prostate cancer disease progression. Neoplasia. 2010;12:1031–1040. doi: 10.1593/neo.10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mohamed AA, Tan SH, Sun C, Shaheduzzaman S, Hu Y, et al. ERG oncogene modulates prostaglandin signaling in prostate cancer cells. Cancer Biol Ther. 2011;11:410–417. doi: 10.4161/cbt.11.4.14180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Leshem O, Madar S, Kogan-Sakin I, Kamer I, Goldstein I, et al. TMPRSS2/ERG promotes epithelial to mesenchymal transition through the ZEB1/ZEB2 axis in a prostate cancer model. PLoS One. 2011;6:e21650. doi: 10.1371/journal.pone.0021650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Flajollet S, Tian TV, Flourens A, Tomavo N, Villers A, et al. Abnormal expression of the ERG transcription factor in prostate cancer cells activates osteopontin. Mol Cancer Res. 2011;9:914–924. doi: 10.1158/1541-7786.MCR-10-0537. [DOI] [PubMed] [Google Scholar]
- 64.Yin L, Rao P, Elson P, Wang J, Ittmann M, et al. Role of TMPRSS2-ERG gene fusion in negative regulation of PSMA expression. PLoS One. 2011;6:e21319. doi: 10.1371/journal.pone.0021319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mani RS, Iyer MK, Cao Q, Brenner JC, Wang L, et al. TMPRSS2-ERG-mediated feed-forward regulation of wild-type ERG in human prostate cancers. Cancer Res. 2011;71:5387–5392. doi: 10.1158/0008-5472.CAN-11-0876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mertz KD, Setlur SR, Dhanasekaran SM, Demichelis F, Perner S, et al. Molecular characterization of TMPRSS2-ERG gene fusion in the NCI-H660 prostate cancer cell line: a new perspective for an old model. Neoplasia. 2007;9:200–206. doi: 10.1593/neo.07103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Attard G, Swennenhuis JF, Olmos D, Reid AH, Vickers E, et al. Characterization of ERG, AR and PTEN gene status in circulating tumor cells from patients with castration-resistant prostate cancer. Cancer Res. 2009;69:2912–2918. doi: 10.1158/0008-5472.CAN-08-3667. [DOI] [PubMed] [Google Scholar]
- 68.Mani RS, Tomlins SA, Callahan K, Ghosh A, Nyati MK, et al. Induced chromosomal proximity and gene fusions in prostate cancer. Science. 2009;326:1230. doi: 10.1126/science.1178124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lin C, Yang L, Tanasa B, Hutt K, Ju BG, et al. Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell. 2009;139:1069–1083. doi: 10.1016/j.cell.2009.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bastus NC, Boyd LK, Mao X, Stankiewicz E, Kudahetti SC, et al. Androgen-induced TMPRSS2:ERG fusion in nonmalignant prostate epithelial cells. Cancer Res. 2010;70:9544–9548. doi: 10.1158/0008-5472.CAN-10-1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Haffner MC, De Marzo AM, Meeker AK, Nelson WG, Yegnasubramanian S. Transcription-induced DNA double strand breaks: both oncogenic force and potential therapeutic target? Clin Cancer Res. 2011;17:3858–3864. doi: 10.1158/1078-0432.CCR-10-2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Perner S, Demichelis F, Beroukhim R, Schmidt FH, Mosquera JM, et al. TMPRSS2:ERG fusion-associated deletions provide insight into the heterogeneity of prostate cancer. Cancer Res. 2006;66:8337–8341. doi: 10.1158/0008-5472.CAN-06-1482. [DOI] [PubMed] [Google Scholar]
- 73.Linja MJ, Savinainen KJ, Saramäki OR, Tammela TL, Vessella RL, et al. Amplification and overexpression of androgen receptor gene in hormone-refractory prostate cancer. Cancer Res. 2001;61:3550–3555. [PubMed] [Google Scholar]
- 74.Cai C, Wang H, Xu Y, Chen S, Balk SP. Reactivation of Androgen Receptor-Regulated TMPRSS2:ERG Gene Expression in Castration-Resistant Prostate Cancer. Cancer Res. 2009;69:6027–6032. doi: 10.1158/0008-5472.CAN-09-0395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lubik AA, Gunter JH, Hendy SC, Locke JA, Adomat HH, et al. Insulin increases de novo steroidogenesis in prostate cancer cells. Cancer Res. 2011;71:5754–5764. doi: 10.1158/0008-5472.CAN-10-2470. [DOI] [PubMed] [Google Scholar]
- 76.Locke JA, Guns ES, Lubik AA, Adomat HH, Hendy SC, et al. Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer Res. 2008;68:6407–6415. doi: 10.1158/0008-5472.CAN-07-5997. [DOI] [PubMed] [Google Scholar]
- 77.Baldi E, Bonaccorsi L, Nesi G, Serni S, Forti G, et al. TMPRSS2:ERG fusion gene and androgen-ablation therapy in prostate cancer. Arch Pathol Lab Med. 2010;134:964–965. doi: 10.5858/2009-0499-LE.1. [DOI] [PubMed] [Google Scholar]