

Abstract
The aggregating proteoglycans of the lectican family are important components of extracellular matrices. Aggrecan is the most well studied of these and is central to cartilage biomechanical properties and skeletal development. Key to its biological function is the fixed charge of the many glycosaminoglycan chains, that provide the basis for the viscoelastic properties necessary for load distribution over the articular surface. This review is focused on the globular domains of aggrecan and their role in anchoring the proteoglycans to other extracellular matrix components. The N-terminal G1 domain is vital in that it binds the proteoglycan to hyaluronan in ternary complex with link protein, retaining the proteoglycan in the tissue. The importance of the C-terminal G3 domain interactions has recently been emphasized by two different human hereditary disorders: autosomal recessive aggrecan-type spondyloepimetaphyseal dysplasia and autosomal dominant familial osteochondritis dissecans. In these two conditions, different missense mutations in the aggrecan C-type lectin repeat have been described. The resulting amino acid replacements affect the ligand interactions of the G3 domain, albeit with widely different phenotypic outcomes.
Keywords: extracellular matrix, cartilage, aggrecan, protein interactions, chondrodysplasia, osteochondritis dissecans
The chondroitin sulfate proteoglycans of the lectican family are vital components of extracellular matrices (ECMs) (Ruoslahti 1996). This family, also known as hyalectans, includes aggrecan, versican, neurocan, and brevican. Aggrecan, the best understood of the lecticans, is fundamental for cartilage function and skeletal development. This extremely highly charged proteoglycan was named for its ability to form large aggregates in the cartilage tissue. These proteoglycan complexes carry an enormous amount of fixed negative charge, attracting counter-ions and drawing water into the tissue. This is the biochemical basis for the viscoelastic properties of cartilage that allow load distribution in the skeletal joints. A key feature of osteoarthritis is the loss of aggrecan and thus tissue biomechanical function. Much attention is focused on aggrecan turnover and the mechanisms of proteolytic degradation in joint disease by matrix metalloproteinases and aggrecanases (see, e.g., Heinegård and Saxne 2011; Stanton et al. 2011; Troeberg and Nagase 2012).
Aggrecan is also of critical importance in skeletal development, as a key molecular component of the cartilage templates in the endochondral ossification process. Indeed, hereditary aggrecanopathies show that mutations that lead to impaired aggrecan function or diminished aggrecan levels result in severe or, in the case of complete absence of aggrecan, lethal chondrodysplasia. The mechanisms for this may involve both ECM expansion of the growth plate and chondroitin sulfate modulation of morphogen signaling (Cortes et al. 2009).
Furthermore, all lecticans, including aggrecan, are involved in regulating brain synaptic plasticity as components of perineuronal nets and in glial scar formation after brain injury (Dityatev et al. 2010; Kwok et al. 2011).
Given that the biological function of aggrecan depends on fixing charges in the cartilage ECM, tethering the proteoglycan is vital. The lectican proteoglycans are incorporated into the ECM through specific interactions with hyaluronan and other ECM components. These interactions are mediated through their globular domains. Recent findings of missense mutations in the aggrecan genes in patients with different skeletal disorders emphasize the importance of the globular domains of aggrecan. This review discusses the role of globular interaction domains for lectican function, with focus on aggrecan, and discusses some perspectives for the future.
Structure of Aggrecan
Pioneering studies in the 1950s to 1970s laid the foundation for proteoglycan biochemistry and the identity of aggrecan’s glycosaminoglycan chains. The aggrecan interaction with hyaluronan was discovered in around 1970 (outlined in Heinegård 2009). Electron microscopic studies in the 1980s revealed the domain organization of aggrecan, with an N-terminal G1 domain separated from a second globular domain (G2) by a short interglobular domain, an elongated domain carrying keratan sulfate and chondroitin sulfate chains, and a C-terminal globular G3 domain (Fig. 1) (Wiedemann et al. 1984; Paulsson et al. 1987). The structural protein units forming the globular domain were identified by the cloning of the aggrecan cDNA from different species in the 1980s, revealing homologies between the G1 and G2 domains with proteoglycan hyaluronan link protein (LP) and between the G3 domain and selectins (Doege et al. 1987, 1991; Hering et al. 1997; Krusius et al. 1987; Li et al. 1993; Watanabe H et al. 1994).
Figure 1.
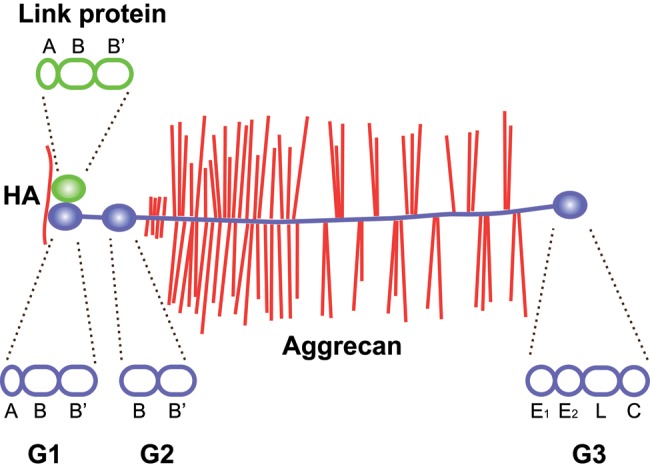
Domain structure of aggrecan and link protein. Aggrecan binds hyaluronan through its N-terminal G1 domain in ternary complex with link proteins. The G1 domain and link proteins are homologous, with an immunoglobulin-like repeat (A) followed by two proteoglycan tandem repeats (B and B′). In aggrecan, but not in other lecticans, the G1 is followed by a second globular domain (G2), separated from the G1 by an extended interglobular domain. The G2 domain consists of two proteoglycan tandem repeats (B and B′). The central, and largest, part of aggrecan is the glycosaminoglycan attachment region, an extended protein stretch carrying keratan sulfate and chondroitin sulfate chains. This is followed by the C-terminal G3 domain, which consists of an epidermal growth factor (EGF) repeat (E1), a calcium-binding EGF repeat (E2), a C-type lectin domain (L), and a complement regulatory protein repeat (C). Hyaluronan (HA) is shown in red, aggrecan in blue (core protein) with red glycosaminoglycan chains, link protein in green. The structural repeat composition of each globular domain of link protein and aggrecan is shown above and below the two proteins, respectively, as indicated by dashed lines.
Based on the amino acid sequence, the calculated molecular weights of the G1, G2, and G3 domains are approximately 37, 24, and 36 kDa, respectively. Together, this constitutes about 40% of the 250-kDa aggrecan core protein, but when taking the ~30 keratan sulfate and ~100 chondroitin sulfate chains on cartilage aggrecan into account, the globular domains only contribute a few percent of the 2.5 MDa (Hascall and Sajdera 1970). Indeed, the folded globular domains are spatially dwarfed by the glycosaminoglycan chains on the extended region of the aggrecan molecule, as shown by rotary shadowing electron microscopy (Wiedeman et al. 1984; Paulsson et al. 1987).
The N-terminal G1 Domain Anchors Aggrecan to Hyaluronan
Seminal work by Heinegård and Hascall in the 1970s showed that the cartilage proteoglycan aggregates were formed through interactions between hyaluronan, the aggrecan G1 domain, and cartilage link protein (Hascall and Heinegård 1974a, 1974b; Heinegård and Hascall 1974). Similar ternary complex formation was later shown for hyaluronan and other members of the lectican family proteoglycans and link proteins (Matsumoto et al. 2003; Rauch et al. 2004; Seyfried et al. 2005).
The aggrecan G1 domain is formed by an N-terminal immunoglobulin-like repeat (A subdomain) and two proteoglycan tandem repeats (PTR; B and B′ subdomains), as outlined in Figure 1. This is the same domain structure as in the LPs. Using mammalian expressed recombinant protein fragments, the hyaluronan interaction of aggrecan G1 was shown to be mediated by the B and B′ subdomains in concert, and the A subdomain appears to enhance the interaction (Watanabe H, Cheung, et al. 1997). In the case of aggrecan, the A subdomain mediates interaction with the LP A domain (Matsumoto et al. 2003). In contrast, in versican ternary complexes, both the LP and hyaluronan interactions occur through the PTR (Matsumoto et al. 2003; Shi et al. 2004).
The G2 Domain Is Unique to Aggrecan, Is Strongly Conserved, and Has No Known Function
The G2 domain is only found in aggrecan. It contains two PTRs similar to the G1 and LP PTRs but contains no Ig-like repeat. The G2 domain shows no interaction with hyaluronan or LP (Fosang and Hardingham 1989; Watanabe H, Cheung, et al. 1997). No other ECM interactions or functions have been described for this domain. The G2 domain may, however, be involved in regulating aggrecan progression through the secretory pathway (Kiani et al. 2001). Nevertheless, the G2 domain is conserved and is present in aggrecan from all species analyzed so far, suggesting it may have an important function in cartilage or the central nervous system.
The C-terminal G3 Domain Links the Proteoglycan Aggregates to the ECM
The presence of a C-terminal globular domain (G3) was deduced from sequence analysis of partial aggrecan cDNAs (Doege et al. 1986; Oldberg et al. 1987) and verified using rotary shadowing electron microscopy of aggrecan (Paulsson et al. 1987). The G3 domain consists of four structural motifs, two epidermal growth factor (EGF)–like repeats, followed by a C-type lectin domain (CLD) and a complement regulatory protein (CRP, also known as sushi) repeat (Doege et al. 1987; Zimmermann and Ruoslahti 1989; Rauch et al. 1992; Yamada et al. 1994).
Early work to identify functions for the lectican G3 domains focused on carbohydrate binding by the CLD. Indeed, calcium-dependent weak interactions of the aggrecan CLD with fucose and galactose were detected (Halberg et al. 1988; Saleque et al. 1993). Similarly, the versican CLD and G3 domain interacted with carbohydrates, including glycosaminoglycans (Aspberg et al. 1995; Ujita et al. 1994). In addition, sulfated glycolipids on the cell surface were found to interact with all four lectican CLDs (Miura et al. 1999).
Subsequently, ECM molecules such as tenascin-R (Aspberg et al. 1995, 1997), tenascin-C (Day et al. 2004; Rauch et al. 1997), fibulin-1 (Aspberg et al. 1999), fibulin-2 (Olin et al. 2001), and fibrillin-1 (Isogai et al. 2002) were identified as high-affinity ligands for the lectican CLDs, with KD values in the subnanomolar to nanomolar range (Fig. 2). As expected for a CLD, these interactions are calcium dependent. Using bacterially expressed recombinant domains of tenascin-R and C, the binding sites on both these proteins were mapped to fibronectin type III repeats 3 to 5 (Fig. 3). Surprisingly, this also showed that the tenascin interactions were carbohydrate independent (Aspberg et al. 1997; Rauch et al. 1997; Day et al. 2004). Indeed, the crystal structure of aggrecan CLD in complex with a tenascin-R fragment showed that a typical CLD carbohydrate interaction (i.e., via direct ligand coordination through the CLD binding site calcium ion) was sterically excluded in the complex (Lundell et al. 2004). The coordinated calcium ions are, however, critical for shaping the loops forming the CLD ligand-binding surface (Lundell et al. 2004). It remains unclear whether carbohydrates are involved in the lectican CLD interactions with fibulins and fibrillin. It should be noted that the CLD fold is a fairly common structural motif, and other examples of CLD proteins with protein ligands have been described (Lundell et al. 2004).
Figure 2.

Schematic of lectican C-type lectin domain (CLD) interactions with tenascin and fibulin ligand proteins. The different interactions between lectican CLDs, tenascins, and fibulins are shown as lines. Line thicknesses indicate relative binding strengths, and affinities (KD) are indicated. The lectican proteoglycans (blue core proteins with red glycosaminoglycan chains) are shown attached to hyaluronan (HA, red line at the bottom of the figure) through their respective G1 domains in complex with Link protein (green). The G3 domain CLD ligands are shown in black at the top of the figure.
Figure 3.

Lectican C-type lectin domain (CLD) binding sites on tenascins. Lectican CLD binding sites on tenascin-R and tenascin-C were mapped to fibronectin type III repeats 3 to 5 using panels of overlapping recombinant fragments. Affinities, determined by BIAcore surface plasmon resonance experiments, are in the low nanomolar range (KD:values for the aggrecan CLD are shown). The use of bacterially expressed tenascin proteins showed that the interactions were carbohydrate independent. The domain organization of the tenascins is shown with triangles for N-termini, spiral-filled circles for multimerization domains, diamonds for epidermal growth factor (EGF)–like repeats, ovals for fibronectin type III repeats, and hexagons for fibrinogen globules. Alternatively spliced fibronectin type III repeats are shadowed and shown with their insertion sites marked by dashed lines.
Interestingly, the CLD protein ligands are all dimeric or multimeric ECM proteins. This suggested a role in crosslinking or organizing the lectican-LP-hyaluronan aggregates (Fig. 4). Such interactions were indeed observed through molecular electron microscopy of aggrecan aggregates purified under native conditions and mixed with tenascins or fibulins (Olin et al. 2001; Lundell et al. 2004).
Figure 4.
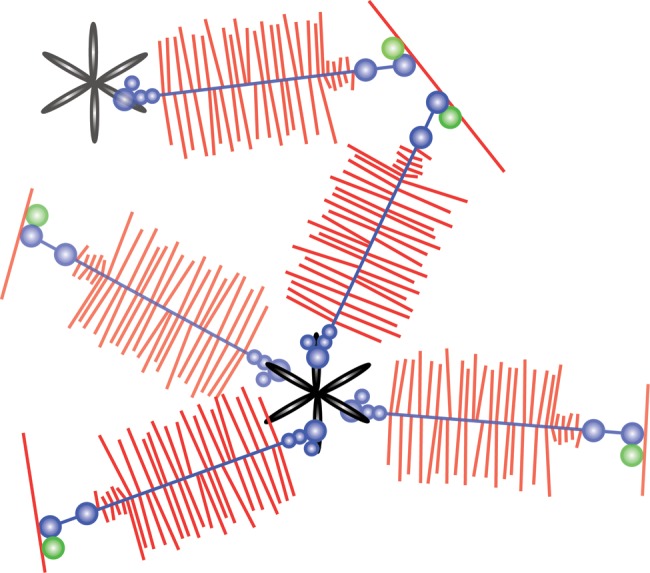
G3 domain-mediated organization of the aggrecan extracellular matrix. The model depicts how tenascin interaction through the lectican G3 domains may crosslink the proteoglycan aggregates and organize the extracellular matrix. Hyaluronan is shown in red (single red line forming the base), aggrecan in blue (core protein) with red glycosaminoglycan chains along the length of the core protein, link protein in green, and tenascin-C in black.
The CLD repeat may also be involved in the sorting and secretion of aggrecan, perhaps involving as yet unknown signals within the C-terminal regions of the CLD peptide (Domowicz et al. 2000).
The functions of the EGF and CRP repeats of the G3 domain are less well understood. In aggrecan, but apparently not in other lecticans, the G3 domain is subject to alternative splicing of the EGF repeats and the CRP repeat, whereas the CLD is constitutively present (Doege et al. 1991; Fulöp et al. 1993, 1996; Grover and Roughley 1993; Bonaventure et al. 1994). Interestingly, the EGF repeats of versican have been suggested to have growth factor activity (Zhang, Cao, Kiani, et al. 1998; Zhang, Cao, Yang, et al. 1998; Zhang et al. 1999, 2001).
Human Disorders Linked to Mutations in the Aggrecan G3 Domain
Chondrodysplasia due to mutations in genes for aggrecan and aggrecan synthesis has been reviewed in detail, but additional mutations have since been discovered (Schwartz and Domowicz 2002). Null and functional null mutations of the aggrecan gene have been described in humans, mice, cattle, and chick (see Table 1 for references). In homozygous animals, the resulting phenotype is lethal perinatal skeletal dysplasia, whereas animals heterozygous for the mutation show dwarfism to various degrees. In humans, heterozygosity for a single base pair insertion, causing a premature stop codon in the aggrecan gene, results in spondyloepiphyseal dysplasia (SED), Kimberley type (Gleghorn et al. 2005). Patients heterozygous for this mutation show short stature, stocky build, and early onset osteoarthritis.
Table 1.
Mutations Affecting Aggrecan Synthesis, Posttranslational Modification, or Interactions
| Type | Disorder or Mutation | Heterozygous Phenotype | Homozygous Phenotype | Reference |
|---|---|---|---|---|
| Loss of aggrecan (null) | Cartilage matrix deficiency mouse, Bc (cmdbc) | Mild dwarfism | Lethal chondrodysplasia | Krueger et al. 1999 |
| Loss of aggrecan (functional null) | Cartilage matrix deficiency mouse (cmd) | Mild dwarfism, shortened life span | Lethal chondrodysplasia | Watanabe H et al. 1994; Watanabe H, Nakata, et al. 1997 |
| Bulldog mutation, Dexter cattle (ACAN, two alleles) | Short limb dwarfism | Lethal chondrodysplasia | Cavanagh et al. 2007 | |
| SED, Kimberley type | SED, early OA | ? | Gleghorn et al. 2005 | |
| Nanomelia chick | Shortened legs | Lethal chondrodysplasia | Li et al. 1993 | |
| Impaired aggrecan glycosylation | Schneckenbecken syndrome (nucleotide-sugar transporter SLC35D1) | — | Lethal skeletal dysplasia | Hiraoka et al. 2007 |
| Chondroitin sulfate N-acetylgalactosaminyltransferase 1 knock-out mouse (CSGalNAcT-1) | — | Dwarfism | Sato et al. 2011; Watanabe Y et al. 2010 | |
| Impaired aggrecan sulfation | Diastrophic dysplasia (DTDST, SLC26A2) | — | Short stature, joint contractures, cleft palate | Hästbacka et al. 1994 |
| MED, recessive (DTDST, SLC26A2) | — | MED | Superti-Furga et al. 1999 | |
| Atelosteogenesis type II (DTDST, SLC26A2) | — | Lethal dysplasia | Hästbacka et al. 1996 | |
| Achondrogenesis type 1B (DTDST, SLC26A2) | — | Lethal dysplasia | Superti-Furga et al. 1996 | |
| SEMD Pakistani type (PAPS synthase 2) | — | SEMD | ul Haque et al. 1998 | |
| Brachymorph mouse (PAPS synthase 2) | — | Dome-shaped skull, shortened tail and limbs | Kurima et al. 1998; ul Haque et al. 1998 | |
| Brachyolmia, recessive (PAPS synthase 2 | — | Short trunk dysplasia | Miyake et al. 2012 | |
| SED Omani type (C6ST-1) | — | SED | Thiele et al. 2004 | |
| Chondrodysplasia, GPAPP type (IMPAD1) | — | Chondrodysplasia with joint dislocations | Vissers et al. 2011 | |
| Impaired G3 function | SEMD-ACAN | Possibly mild proportional short stature | SEMD | Tompson et al. 2009 |
| Familial OCD (ACAN) | OCD, short stature, OA | ? | Stattin et al. 2010 |
ACAN, aggrecan gene; C6ST, chondroitin-6-O-sulfotransferase; cmd, cartilage matrix deficiency; DTDST, diastrophic dysplasia sulfate transporter; GPAPP, Golgi-resident nucleotide phosphatase gene; IMPAD1, GPAPP gene; MED, multiple epiphyseal dysplasia; OA, osteoarthritis; OCD, osteochondritis dissecans; PAPS, 3′-phosphoadenosine 5′-phosphosulfate; SED, spondyloepiphyseal dysplasia; SEMD, spondyloepimetaphyseal dysplasia; SLC26A2, solute carrier family 26 (sulfate transporter), member 2; SLC35D1, solute carrier family 35 (UDP-glucuronic acid/UDP-N-acetylgalactosamine dual transporter), member D1. For recessively inherited conditions, the lack of phenotype in heterozygous individuals is indicated by a dash.
The molecular function of aggrecan is to produce a hydrodynamic swelling pressure in cartilage. This is achieved through the chondroitin sulfate (CS) chains linked to the central region of the aggrecan core protein. The many thousands of fixed negative charges on the CS chains of each aggrecan attract counter-ions and draw water into the extracellular matrix. Consequently, mutations in genes for proteins involved in CS synthesis and sulfation also result in different skeletal dysplasias of varying severity (Table 1).
No data are available on aggrecan G1 or LP mutation in humans. This likely reflects the fundamental importance of proteoglycan aggregate formation in skeletal development. Indeed, chondrodysplasia was observed in the cartilage LP gene knockout (Watanabe H and Yamada 1999), which was milder but similar to that in aggrecan functional null cmd mice (Watanabe H and Yamada 2002).
The functional importance of G3 domain interactions was recently emphasized by the identification of two different missense mutations in the aggrecan CLD. Surprisingly, the two mutations resulted in widely different phenotypes.
The first G3 mutation was identified in a family with autosomal recessive spondyloepimetaphyseal dysplasia (SEMD) (Tompson et al. 2009). The missense mutation results in an Asp to Asn replacement (D2267N), which affects one of the calcium coordinating residues of the CLD. It is unclear whether this amino acid replacement has any effect on CLD calcium binding, but the mutation results in a novel consensus sequence for N-linked glycosylation. Indeed, N-linked glycans were present on recombinant D2267N G3 domains expressed in mammalian cells (Tompson et al. 2009). Although not within the known ligand binding surface, the D2267 residue is situated in close proximity, and glycan substitution could present a steric hindrance to ligand interaction. This may well be the case, as surface plasmon resonance experiments on tenascin-C showed that the D2267N mutant protein reached a lower steady-state binding signal than the wild-type protein, although the binding strength was not determined. However, the shapes of the binding and dissociation curves are similar to the wild-type control curves, suggesting only minor differences in affinity. No data on aggrecan secretion or presence in patient cartilage are available, but the phenotypic similarity to other aggrecanopathies suggests that lowered aggrecan levels, glycosylation, or sulfation could contribute to the phenotype.
The second aggrecan G3 mutation was identified from a five-generation family with autosomal dominant familial osteochondritis dissecans (OCD) (Stattin et al. 2010). In OCD, cartilage and subchondral bone are dislodged from the joint surface. In familial OCD, this affects multiple joints and is accompanied by a disproportionate shortened stature and early onset osteoarthritis. The missense mutation leads to a Val to Met replacement in the aggrecan CLD. The mutated residue (V2303) is situated in the hydrophobic core of the CLD, right below the ligand binding surface. The V2303M replacement may disrupt the conformation of the binding surface, and loss of ligand interaction was verified biochemically using mammalian-expressed recombinant G3 fragments. Affinity measurements by surface plasmon resonance showed a complete loss of binding to fibulin-1 and -2 and an 8000-fold reduction in V2303M CLD affinity for tenascin-R. The presence of flanking EGF and CLD repeats appeared to stabilize the CLD fold, but affinities were still reduced. Due to the limited availability of patient material, no detailed analysis of aggrecan levels, glycosylation, or sulfation was performed. Nevertheless, proteoglycan extraction and purification from a familial OCD patient, followed by Ion Trap tandem mass spectrometry (MS/MS) analysis, confirmed the presence of the V2303M aggrecan in cartilage (see Fig. 5). This suggests that the loss of ECM ligand interactions in the V2303M aggrecan leads to a disturbed cartilage ECM assembly or organization and thus a less stable cartilage, predisposing the patient to OCD and early onset osteoarthritis.
Figure 5.
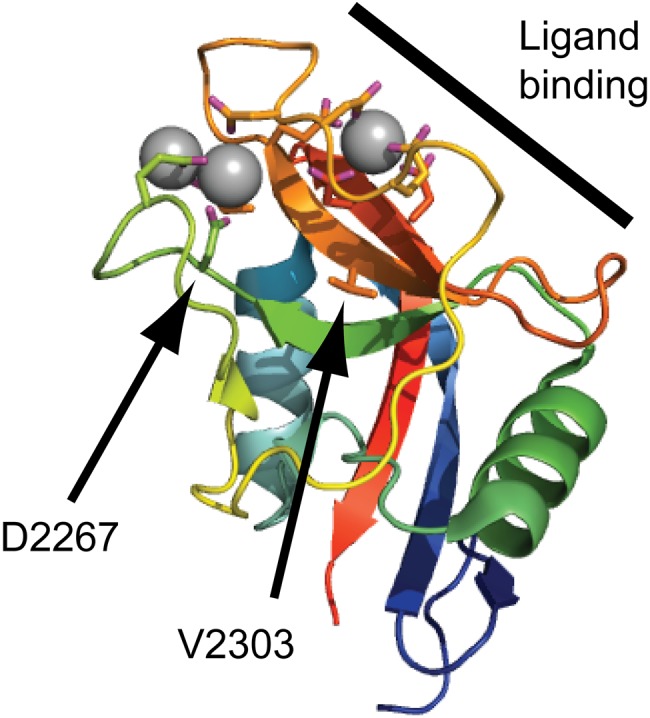
Disease-linked missense mutations in the aggrecan C-type lectin domain (CLD). The aggrecan C-type lectin domain structure determined by X-ray crystallography (Protein Data Bank ID: 1TDQ) is shown as a cartoon model. The coordinated calcium ions are shown as gray spheres. The side chains of amino acid residues coordinating the calcium ions or mutated in human disease are shown as sticks. The CLD binding surface for tenascin-R is marked by a thick line in the upper right corner of the cartoon. The amino acid residues mutated in spondyloepimetaphyseal dysplasia aggrecan type (D2267) and familial osteochondritis dissecans (V2303) are indicated by arrows.
It is unclear why the phenotypic outcome of these two missense mutations in the aggrecan CLD differs so widely. It is interesting to note that aggrecan-type SEMD is recessively inherited, whereas the few known individuals heterozygous for the D2267N mutation possibly display a mild proportionate shortened stature. In contrast, familial OCD is dominantly inherited, and no individuals homozygous for the V2303M mutations have been found. This raises the possibility that these two conditions reflect the two extremes along a scale of phenotypic severity, similar to that of mutations in the diastrophic dysplasia sulfate transporter (DTDST) gene, where different mutations give phenotypes ranging from mild recessive multiple epiphyseal dysplasia (MED) to lethal recessive chondrodysplasias (Rossi and Superti-Furga 2001). Alternatively, one or both of the aggrecan CLD mutations may affect as yet unknown molecular interactions or lead to gain of new functions.
Concluding Remarks
In the past decade, our understanding of ECM assembly as a whole has increased significantly. Aggrecan is no exception, with multiple interactions between the G3 domain CLD and other ECM molecules identified and their importance confirmed by the identification of missense mutations in patients with skeletal disorders.
The advent of novel sequencing technology will very likely lead to the identification of additional mutations in the aggrecan gene in patients and animals with different skeletal disorders. This will be helpful in answering some of the as yet unsolved questions concerning aggrecan function.
This includes why the clinical phenotypes of patients with the two identified aggrecan missense mutations are so divergent. Furthermore, as it is clear that anchoring aggrecan in the tissue is vital, it would be expected that the consequences of mutations impairing G1 or LP function result in severe phenotypes. Finally, the strong conservation of the G2 domain in aggrecan, but its absence in other members of the aggregating proteoglycan family (lecticans or hyalectans), suggests an important role for the G2 domain.
Another interesting question is why the aggrecan G3 is alternatively spliced when other lecticans are not. Given that the EGF repeats of the versican G3 domain appear to have growth factor activity, alternative splicing could reflect a regulatory mechanism.
This may also apply to the CRP repeat, which also is alternatively spliced in aggrecan. The complement system is emerging as a strong contributing factor in osteoarthritis development (Wang et al. 2011). Indeed, fragments released from cartilage ECM molecules can regulate complement activity (Happonen et al. 2009, 2010, 2012; Kalchishkova et al. 2011; Sjoberg et al. 2005, 2009). Fragments containing the G3 domain with its CRP repeat may thus be released into the synovial fluid. It will be interesting to determine whether the aggrecan G3 domain is involved in complement regulation and if this contributes to joint disease progression.
Acknowledgments
I would like to thank the patients, members of the laboratory and collaborators who have contributed to this line of research.
Footnotes
Declaration of Conflicting Interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Support from the Danish Research Council, the Lundbeckfonden, the Novo Nordisk Fonden and the Carlsbergfonden, the Gigtforeningen, and the Faculty of Science at the University of Copenhagen is gratefully acknowledged.
References
- Aspberg A, Adam S, Kostka G, Timpl R, Heinegård D. 1999. Fibulin-1 is a ligand for the C-type lectin domains of aggrecan and versican. J Biol Chem. 274:20444–20449 [DOI] [PubMed] [Google Scholar]
- Aspberg A, Binkert C, Ruoslahti E. 1995. The versican C-type lectin domain recognizes the adhesion protein tenascin-R. Proc Natl Acad Sci U S A. 92:10590–10594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aspberg A, Miura R, Bourdoulous S, Shimonaka M, Heinegård D, Schachner M, Ruoslahti E, Yamaguchi Y. 1997. The C-type lectin domains of lecticans, a family of aggregating chondroitin sulfate proteoglycans, bind tenascin-R by protein-protein interactions independent of carbohydrate moiety. Proc Natl Acad Sci U S A. 94:10116–10121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaventure J, Kadhom N, Cohen-Solal L, Ng KH, Bourguignon J, Lasselin C, Freisinger P. 1994. Reexpression of cartilage-specific genes by dedifferentiated human articular chondrocytes cultured in alginate beads. Exp Cell Res. 212:97–104 [DOI] [PubMed] [Google Scholar]
- Cavanagh JA, Tammen I, Windsor PA, Bateman JF, Savarirayan R, Nicholas FW, Raadsma HW. 2007. Bulldog dwarfism in Dexter cattle is caused by mutations in ACAN. Mamm Genome. 18:808–814 [DOI] [PubMed] [Google Scholar]
- Cortes M, Baria AT, Schwartz NB. 2009. Sulfation of chondroitin sulfate proteoglycans is necessary for proper Indian hedgehog signaling in the developing growth plate. Development. 136:1697–1706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day JM, Olin AI, Murdoch AD, Canfield A, Sasaki T, Timpl R, Hardingham TE, Aspberg A. 2004. Alternative splicing in the aggrecan G3 domain influences binding interactions with tenascin-C and other extracellular matrix proteins. J Biol Chem. 279:12511–12518 [DOI] [PubMed] [Google Scholar]
- Dityatev A, Schachner M, Sonderegger P. 2010. The dual role of the extracellular matrix in synaptic plasticity and homeostasis. Nat Rev Neurosci. 11:735–746 [DOI] [PubMed] [Google Scholar]
- Doege K, Fernandez P, Hassell JR, Sasaki M, Yamada Y. 1986. Partial cDNA sequence encoding a globular domain at the C terminus of the rat cartilage proteoglycan. J Biol Chem. 261:8108–8111 [PubMed] [Google Scholar]
- Doege K, Sasaki M, Horigan E, Hassell JR, Yamada Y. 1987. Complete primary structure of the rat cartilage proteoglycan core protein deduced from cDNA clones [published erratum appears in J Biol Chem. 1988;263(20):10040]. J Biol Chem. 262:17757–17767 [PubMed] [Google Scholar]
- Doege KJ, Sasaki M, Kimura T, Yamada Y. 1991. Complete coding sequence and deduced primary structure of the human cartilage large aggregating proteoglycan, aggrecan: human-specific repeats, and additional alternatively spliced forms. J Biol Chem. 266:894–902 [PubMed] [Google Scholar]
- Domowicz MS, Pirok EW, Novak TE, Schwartz NB. 2000. Role of the C-terminal G3 domain in sorting and secretion of aggrecan core protein and ubiquitin-mediated degradation of accumulated mutant precursors. J Biol Chem. 275:35098–35105 [DOI] [PubMed] [Google Scholar]
- Fosang AJ, Hardingham TE. 1989. Isolation of the N-terminal globular protein domains from cartilage proteoglycans: identification of G2 domain and its lack of interaction with hyaluronate and link protein. Biochem J. 261:801–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulöp C, Cs-Szabo G, Glant TT. 1996. Species-specific alternative splicing of the epidermal growth factor-like domain 1 of cartilage aggrecan. Biochem J. 319(Pt 3):935–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulöp C, Walcz E, Valyon M, Glant TT. 1993. Expression of alternatively spliced epidermal growth factor-like domains in aggrecans of different species: evidence for a novel module. J Biol Chem. 268:17377–17383 [PubMed] [Google Scholar]
- Gleghorn L, Ramesar R, Beighton P, Wallis G. 2005. A mutation in the variable repeat region of the aggrecan gene (AGC1) causes a form of spondyloepiphyseal dysplasia associated with severe, premature osteoarthritis. Am J Hum Genet. 77:484–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grover J, Roughley PJ. 1993. Versican gene expression in human articular cartilage and comparison of mRNA splicing variation with aggrecan. Biochem J. 291:361–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halberg DF, Proulx G, Doege K, Yamada Y, Drickamer K. 1988. A segment of the cartilage proteoglycan core protein has lectin-like activity. J Biol Chem. 263:9486–9490 [PubMed] [Google Scholar]
- Happonen KE, Furst CM, Saxne T, Heinegård D, Blom AM. 2012. PRELP protein inhibits the formation of the complement membrane attack complex. J Biol Chem. 287:8092–8100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Happonen KE, Saxne T, Aspberg A, Morgelin M, Heinegård D, Blom AM. 2010. Regulation of complement by cartilage oligomeric matrix protein allows for a novel molecular diagnostic principle in rheumatoid arthritis. Arthritis Rheum. 62:3574–3583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Happonen KE, Sjoberg AP, Morgelin M, Heinegård D, Blom AM. 2009. Complement inhibitor C4b-binding protein interacts directly with small glycoproteins of the extracellular matrix. J Immunol. 182:1518–1525 [DOI] [PubMed] [Google Scholar]
- Hascall VC, Heinegård D. 1974a. Aggregation of cartilage proteoglycans, I: the role of hyaluronic acid. J Biol Chem. 249:4232–4241 [PubMed] [Google Scholar]
- Hascall VC, Heinegård D. 1974b. Aggregation of cartilage proteoglycans, II: oligosaccharide competitors of the proteoglycan-hyaluronic acid interaction. J Biol Chem. 249:4242–4249 [PubMed] [Google Scholar]
- Hascall VC, Sajdera SW. 1970. Physical properties and polydispersity of proteoglycan from bovine nasal cartilage. J Biol Chem. 245:4920–4930 [PubMed] [Google Scholar]
- Hästbacka J, de la Chapelle A, Mahtani MM, Clines G, Reeve-Daly MP, Daly M, Hamilton BA, Kusumi K, Trivedi B, Weaver A, et al. 1994. The diastrophic dysplasia gene encodes a novel sulfate transporter: positional cloning by fine-structure linkage disequilibrium mapping. Cell. 78:1073–1087 [DOI] [PubMed] [Google Scholar]
- Hästbacka J, Superti-Furga A, Wilcox WR, Rimoin DL, Cohn DH, Lander ES. 1996. Atelosteogenesis type II is caused by mutations in the diastrophic dysplasia sulfate-transporter gene (DTDST): evidence for a phenotypic series involving three chondrodysplasias. Am J Hum Genet. 58:255–262 [PMC free article] [PubMed] [Google Scholar]
- Heinegård D. 2009. Proteoglycans and more—from molecules to biology. Int J Exp Pathol. 90:575–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinegård D, Hascall VC. 1974. Aggregation of cartilage proteoglycans, 3: characteristics of the proteins isolated from trypsin digests of aggregates. J Biol Chem. 249:4250–4256 [PubMed] [Google Scholar]
- Heinegård D, Saxne T. 2011. The role of the cartilage matrix in osteoarthritis. Nat Rev Rheumatol. 7:50–56 [DOI] [PubMed] [Google Scholar]
- Hering TM, Kollar J, Huynh TD. 1997. Complete coding sequence of bovine aggrecan: comparative structural analysis. Arch Biochem Biophys. 345:259–270 [DOI] [PubMed] [Google Scholar]
- Hiraoka S, Furuichi T, Nishimura G, Shibata S, Yanagishita M, Rimoin DL, Superti-Furga A, Nikkels PG, Ogawa M, Katsuyama K, et al. 2007. Nucleotide-sugar transporter SLC35D1 is critical to chondroitin sulfate synthesis in cartilage and skeletal development in mouse and human. Nat Med. 13:1363–1367 [DOI] [PubMed] [Google Scholar]
- Isogai Z, Aspberg A, Keene DR, Ono RN, Reinhardt DP, Sakai LY. 2002. Versican interacts with fibrillin-1 and links extracellular microfibrils to other connective tissue networks. J Biol Chem. 277:4565–4572 [DOI] [PubMed] [Google Scholar]
- Kalchishkova N, Furst CM, Heinegård D, Blom AM. 2011. NC4 domain of cartilage-specific collagen IX inhibits complement directly due to attenuation of membrane attack formation and indirectly through binding and enhancing activity of complement inhibitors C4B-binding protein and factor H. J Biol Chem. 286:27915–27926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiani C, Lee V, Cao L, Chen L, Wu Y, Zhang Y, Adams ME, Yang BB. 2001. Roles of aggrecan domains in biosynthesis, modification by glycosaminoglycans and product secretion. Biochem J. 354:199–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger RC, Jr, Kurima K, Schwartz NB. 1999. Completion of the mouse aggrecan gene structure and identification of the defect in the cmd-Bc mouse as a near complete deletion of the murine aggrecan gene. Mamm Genome. 10:1119–1125 [DOI] [PubMed] [Google Scholar]
- Krusius T, Gehlsen KR, Ruoslahti E. 1987. A fibroblast chondroitin sulfate proteoglycan core protein contains lectin-like and growth factor-like sequences. J Biol Chem. 262:13120–13125 [PubMed] [Google Scholar]
- Kurima K, Warman ML, Krishnan S, Domowicz M, Krueger RC, Jr, Deyrup A, Schwartz NB. 1998. A member of a family of sulfate-activating enzymes causes murine brachymorphism. Proc Natl Acad Sci U S A. 95:8681–8685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwok JC, Dick G, Wang D, Fawcett JW. 2011. Extracellular matrix and perineuronal nets in CNS repair. Dev Neurobiol. 71:1073–1089 [DOI] [PubMed] [Google Scholar]
- Li H, Schwartz NB, Vertel BM. 1993. cDNA cloning of chick cartilage chondroitin sulfate (aggrecan) core protein and identification of a stop codon in the aggrecan gene associated with the chondrodystrophy, nanomelia. J Biol Chem. 268:23504–23511 [PubMed] [Google Scholar]
- Lundell A, Olin AI, Morgelin M, Al-Karadaghi S, Aspberg A, Logan DT. 2004. Structural basis for interactions between tenascins and lectican C-type lectin domains: evidence for a crosslinking role for tenascins. Structure (Camb). 12:1495–1506 [DOI] [PubMed] [Google Scholar]
- Matsumoto K, Shionyu M, Go M, Shimizu K, Shinomura T, Kimata K, Watanabe H. 2003. Distinct interaction of versican/PG-M with hyaluronan and link protein. J Biol Chem. 278:41205–41212 [DOI] [PubMed] [Google Scholar]
- Miura R, Aspberg A, Ethell IM, Hagihara K, Schnaar RL, Ruoslahti E, Yamaguchi Y. 1999. The proteoglycan lectin domain binds sulfated cell surface glycolipids and promotes cell adhesion. J Biol Chem. 274:11431–11438 [DOI] [PubMed] [Google Scholar]
- Miyake N, Elcioglu NH, Iida A, Isguven P, Dai J, Murakami N, Takamura K, Cho TJ, Kim OH, Hasegawa T, et al. 2012. PAPSS2 mutations cause autosomal recessive brachyolmia. J Med Genet. 49:533–538 [DOI] [PubMed] [Google Scholar]
- Oldberg A, Antonsson P, Heinegård D. 1987. The partial amino acid sequence of bovine cartilage proteoglycan, deduced from a cDNA clone, contains numerous Ser-Gly sequences arranged in homologous repeats. Biochem J. 243:255–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olin AI, Morgelin M, Sasaki T, Timpl R, Heinegård D, Aspberg A. 2001. The proteoglycans aggrecan and versican form networks with fibulin-2 through their lectin domain binding. J Biol Chem. 276:1253–1261 [DOI] [PubMed] [Google Scholar]
- Paulsson M, Mörgelin M, Wiedemann H, Beardmore-Gray M, Dunham D, Hardingham T, Heinegård D, Timpl R, Engel J. 1987. Extended and globular protein domains in cartilage proteoglycans. Biochem J. 245:763–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch U, Clement A, Retzler C, Frohlich L, Fässler R, Göhring W, Faissner A. 1997. Mapping of a defined neurocan binding site to distinct domains of tenascin-C. J Biol Chem. 272:26905–26912 [DOI] [PubMed] [Google Scholar]
- Rauch U, Hirakawa S, Oohashi T, Kappler J, Roos G. 2004. Cartilage link protein interacts with neurocan, which shows hyaluronan binding characteristics different from CD44 and TSG-6. Matrix Biol. 22:629–639 [DOI] [PubMed] [Google Scholar]
- Rauch U, Karthikeyan L, Maurel P, Margolis RU, Margolis RK. 1992. Cloning and primary structure of neurocan, a developmentally regulated, aggregating chondroitin sulfate proteoglycan of brain. J Biol Chem. 267:19536–19547 [PubMed] [Google Scholar]
- Rossi A, Superti-Furga A. 2001. Mutations in the diastrophic dysplasia sulfate transporter (DTDST) gene (SLC26A2): 22 novel mutations, mutation review, associated skeletal phenotypes, and diagnostic relevance. Hum Mutat. 17:159–171 [DOI] [PubMed] [Google Scholar]
- Ruoslahti E. 1996. Brain extracellular matrix. Glycobiology. 6:489–492 [DOI] [PubMed] [Google Scholar]
- Saleque S, Ruiz N, Drickamer K. 1993. Expression and characterization of a carbohydrate-binding fragment of rat aggrecan. Glycobiology. 3:185–190 [DOI] [PubMed] [Google Scholar]
- Sato T, Kudo T, Ikehara Y, Ogawa H, Hirano T, Kiyohara K, Hagiwara K, Togayachi A, Ema M, Takahashi S, et al. 2011. Chondroitin sulfate N-acetylgalactosaminyltransferase 1 is necessary for normal endochondral ossification and aggrecan metabolism. J Biol Chem. 286:5803–5812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz NB, Domowicz M. 2002. Chondrodysplasias due to proteoglycan defects. Glycobiology. 12:57R–68R [DOI] [PubMed] [Google Scholar]
- Seyfried NT, McVey GF, Almond A, Mahoney DJ, Dudhia J, Day AJ. 2005. Expression and purification of functionally active hyaluronan-binding domains from human cartilage link protein, aggrecan and versican: formation of ternary complexes with defined hyaluronan oligosaccharides. J Biol Chem. 280:5435–5448 [DOI] [PubMed] [Google Scholar]
- Shi S, Grothe S, Zhang Y, O’Connor-McCourt MD, Poole AR, Roughley PJ, Mort JS. 2004. Link protein has greater affinity for versican than aggrecan. J Biol Chem. 279:12060–12066 [DOI] [PubMed] [Google Scholar]
- Sjoberg A, Onnerfjord P, Morgelin M, Heinegård D, Blom AM. 2005. The extracellular matrix and inflammation: fibromodulin activates the classical pathway of complement by directly binding C1q. J Biol Chem. 280:32301–32308 [DOI] [PubMed] [Google Scholar]
- Sjoberg AP, Manderson GA, Morgelin M, Day AJ, Heinegård D, Blom AM. 2009. Short leucine-rich glycoproteins of the extracellular matrix display diverse patterns of complement interaction and activation. Mol Immunol. 46:830–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton H, Melrose J, Little CB, Fosang AJ. 2011. Proteoglycan degradation by the ADAMTS family of proteinases. Biochim Biophys Acta. 1812:1616–1629 [DOI] [PubMed] [Google Scholar]
- Stattin EL, Wiklund F, Lindblom K, Onnerfjord P, Jonsson BA, Tegner Y, Sasaki T, Struglics A, Lohmander S, Dahl N, et al. 2010. A missense mutation in the aggrecan C-type lectin domain disrupts extracellular matrix interactions and causes dominant familial osteochondritis dissecans. Am J Hum Genet. 86:126–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Superti-Furga A, Hästbacka J, Wilcox WR, Cohn DH, van der Harten HJ, Rossi A, Blau N, Rimoin DL, Steinmann B, Lander ES, et al. 1996. Achondrogenesis type IB is caused by mutations in the diastrophic dysplasia sulphate transporter gene. Nat Genet. 12:100–102 [DOI] [PubMed] [Google Scholar]
- Superti-Furga A, Neumann L, Riebel T, Eich G, Steinmann B, Spranger J, Kunze J. 1999. Recessively inherited multiple epiphyseal dysplasia with normal stature, club foot, and double layered patella caused by a DTDST mutation. J Med Genet. 36:621–624 [PMC free article] [PubMed] [Google Scholar]
- Thiele H, Sakano M, Kitagawa H, Sugahara K, Rajab A, Hohne W, Ritter H, Leschik G, Nurnberg P, Mundlos S. 2004. Loss of chondroitin 6-O-sulfotransferase-1 function results in severe human chondrodysplasia with progressive spinal involvement. Proc Natl Acad Sci U S A. 101:10155–10160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompson SW, Merriman B, Funari VA, Fresquet M, Lachman RS, Rimoin DL, Nelson SF, Briggs MD, Cohn DH, Krakow D. 2009. A recessive skeletal dysplasia, SEMD aggrecan type, results from a missense mutation affecting the C-type lectin domain of aggrecan. Am J Hum Genet. 84:72–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troeberg L, Nagase H. 2012. Proteases involved in cartilage matrix degradation in osteoarthritis. Biochim Biophys Acta. 1824:133–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ujita M, Shinomura T, Ito K, Kitagawa Y, Kimata K. 1994. Expression and binding activity of the carboxyl-terminal portion of the core protein of PG-M, a large chondroitin sulfate proteoglycan. J Biol Chem. 269:27603–27609 [PubMed] [Google Scholar]
- ul Haque MF, King LM, Krakow D, Cantor RM, Rusiniak ME, Swank RT, Superti-Furga A, Haque S, Abbas H, Ahmad W, et al. 1998. Mutations in orthologous genes in human spondyloepimetaphyseal dysplasia and the brachymorphic mouse. Nat Genet. 20:157–162 [DOI] [PubMed] [Google Scholar]
- Vissers LE, Lausch E, Unger S, Campos-Xavier AB, Gilissen C, Rossi A, Del Rosario M, Venselaar H, Knoll U, Nampoothiri S, et al. 2011. Chondrodysplasia and abnormal joint development associated with mutations in IMPAD1, encoding the Golgi-resident nucleotide phosphatase, gPAPP. Am J Hum Genet. 88:608–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Rozelle AL, Lepus CM, Scanzello CR, Song JJ, Larsen DM, Crish JF, Bebek G, Ritter SY, Lindstrom TM, et al. 2011. Identification of a central role for complement in osteoarthritis. Nat Med. 17:1674–1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H, Cheung SC, Itano N, Kimata K, Yamada Y. 1997. Identification of hyaluronan-binding domains of aggrecan. J Biol Chem. 272:28057–28065 [DOI] [PubMed] [Google Scholar]
- Watanabe H, Kimata K, Line S, Strong D, Gao LY, Kozak CA, Yamada Y. 1994. Mouse cartilage matrix deficiency (cmd) caused by a 7 bp deletion in the aggrecan gene. Nat Genet. 7:154–157 [DOI] [PubMed] [Google Scholar]
- Watanabe H, Nakata K, Kimata K, Nakanishi I, Yamada Y. 1997. Dwarfism and age-associated spinal degeneration of heterozygote cmd mice defective in aggrecan. Proc Natl Acad Sci U S A. 94:6943–6947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H, Yamada Y. 1999. Mice lacking link protein develop dwarfism and craniofacial abnormalities. Nat Genet. 21:225–229 [DOI] [PubMed] [Google Scholar]
- Watanabe H, Yamada Y. 2002. Chondrodysplasia of gene knockout mice for aggrecan and link protein. Glycoconj J. 19:269–273 [DOI] [PubMed] [Google Scholar]
- Watanabe Y, Takeuchi K, Higa Onaga S, Sato M, Tsujita M, Abe M, Natsume R, Li M, Furuichi T, Saeki M, et al. 2010. Chondroitin sulfate N-acetylgalactosaminyltransferase-1 is required for normal cartilage development. Biochem J. 432:47–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedemann H, Paulsson M, Timpl R, Engel J, Heinegård D. 1984. Domain structure of cartilage proteoglycans revealed by rotary shadowing of intact and fragmented molecules. Biochem J. 224:331–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada H, Watanabe K, Shimonaka M, Yamaguchi Y. 1994. Molecular cloning of brevican, a novel brain proteoglycan of the aggrecan/versican family. J Biol Chem. 269:10119–10126 [PubMed] [Google Scholar]
- Zhang Y, Cao L, Kiani C, Yang BL, Hu W, Yang BB. 1999. Promotion of chondrocyte proliferation by versican mediated by G1 domain and EGF-like motifs. J Cell Biochem. 73:445–457 [PubMed] [Google Scholar]
- Zhang Y, Cao L, Kiani CG, Yang BL, Yang BB. 1998. The G3 domain of versican inhibits mesenchymal chondrogenesis via the epidermal growth factor-like motifs. J Biol Chem. 273:33054–33063 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Cao L, Yang BL, Yang BB. 1998. The G3 domain of versican enhances cell proliferation via epidermial growth factor-like motifs. J Biol Chem. 273:21342–21351 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Wu Y, Cao L, Lee V, Chen L, Lin Z, Kiani C, Adams ME, Yang BB. 2001. Versican modulates embryonic chondrocyte morphology via the epidermal growth factor-like motifs in G3. Exp Cell Res. 263:33–42 [DOI] [PubMed] [Google Scholar]
- Zimmermann DR, Ruoslahti E. 1989. Multiple domains of the large fibroblast proteoglycan, versican. Embo J. 8:2975–2981 [DOI] [PMC free article] [PubMed] [Google Scholar]