

Abstract
Diabetic nephropathy (DN) is a serious complication in diabetes. Major typical morphological changes are the result of changes in the extracellular matrix (ECM). Thus, basement membranes are thickened and the glomerular mesangial matrix and the tubulointerstitial space are expanded, due to increased amounts of ECM. One important ECM component, the proteoglycans (PGs), shows a more complex pattern of changes in DN. PGs in basement membranes are decreased but increased in the mesangium and the tubulointerstitial space. The amounts and structures of heparan sulfate chains are changed, and such changes affect levels of growth factors regulating cell proliferation and ECM synthesis, with cell attachment affecting endothelial cells and podocytes. Enzymes modulating heparan sulfate structures, such as heparanase and sulfatases, are implicated in DN. Other enzyme classes also modulate ECM proteins and PGs, such as matrix metalloproteinases (MMPs) and serine proteases, such as plasminogen activator, as well as their corresponding inhibitors. The levels of these enzymes and inhibitors are changed in plasma and in the kidneys in DN. Several growth factors, signaling pathways, and hyperglycemia per se affect ECM synthesis and turnover in DN. Whether ECM components can be used as markers for early kidney changes is an important research topic, whereas at present, the clinical use remains to be established.
Keywords: extracellular matrix, glycoconjugates, molecular pathology, MMP, fibrosis
A key element in diabetic nephropathy (DN) is changes in the extracellular matrix (ECM) of several of the components in the kidney. From a clinical perspective, the changes seen in the ECM are important both in diagnostics and for prognostic and therapeutic purposes. In the current review, we present some of the central clinical issues related to DN, as well as the most relevant changes to the ECM from a diagnostic point of view, and also discuss some of the changes observed in one of the important ECM components, the proteoglycans (PGs). Our aim is not to cover all relevant research in this rather wide field, ranging from clinical trials to studies on microRNA and other important regulators of kidney function, but to focus particularly on some key issues related to PG changes in DN.
Clinical Perspectives on DN
According to estimates from the International Diabetes Federation, the worldwide prevalence of diabetes is estimated to increase from 285 million persons in 2010 to 439 millions in 2030, a relative increase of 50% (Shaw et al. 2010). Among patients with type 1 diabetes, the incidence of DN has apparently decreased from 30-35% in the cohorts who developed diabetes 40 to 50 years ago to 10-15% in recent cohorts (Bojestig et al. 1994; Hovind et al. 2003). However, due to the increase in type 2 diabetes, the absolute prevalence of DN has increased over the past two decades. In 2009, DN was reported to be the cause of 44% of all cases of end-stage renal disease (ESRD) in the United States (www.usrds.org), with an incidence of 155 diabetic patients developing ESRD per million each year. This fact was earlier announced as a “medical catastrophe of world-wide dimensions” (Ritz et al. 1999). Nowadays, a slight decrease has been noted in the number of patients with type 2 diabetes who develop ESRD both in the United States (Burrows et al. 2010) and Europe (Zoccali et al. 2009). Although modern treatment of diabetes to some degree may have stabilized the occurrence of DN, the condition is still a leading cause in patients who require dialysis and transplantation in the Western world.
Definitions
Diabetic nephropathy is characterized by a progressive increase in the levels of albuminuria, hypertension, glomerulosclerosis, and an eventual reduction in glomerular filtration rate (GFR), leading to ESRD. Early DN is defined as persistent microalbuminuria measured on at least two different occasions as an albumin excretion rate of 20 to 200 µg/min or 30 to 300 mg/24 hr (Mogensen 1984; Mogensen and Christensen 1984). Today, most clinical centers would use spot urine measurements of the albumin-to-creatinine ratio to define microalbuminuria: 2.5 to 25 mg/mmol (Europe) or, alternatively, 30 to 300 mg/g (United States) (Mogensen et al. 1995). Overt DN is defined as albumin excretion beyond the microalbuminuric range, or urinary protein excretion >500 mg/24 hr. DN in type 1 diabetes usually takes more than 5 to 10 years to develop, whereas it may be present already when type 2 diabetes is diagnosed, due to several years of unrecognized hyperglycemia. Diagnostic criteria are summarized in Table 1.
Table 1.
Diagnosis of Diabetic Nephropathy
| Prerequisite |
| Type 1 diabetes >5 yearsa–c |
| Type 2 diabetesa–c |
| Probable diabetic nephropathy |
| Microalbuminuria and diabetic retinopathyb,c |
| Proteinuria and diabetic retinopathyb,c |
| Glomerular filtration rate <60 ml/min/1.73 m2 and diabetic retinopathyb,c |
| Definite diabetic nephropathy |
| Morphological changes in renal biopsyb,c |
DN has identical characteristics in type 1 and type 2 diabetes. On the other hand, a substantial amount of patients may develop kidney disease that does not pertain to diabetes; for example, hypertensive kidney changes (nephrosclerosis) or glomerulonephritis. Most of these patients will not have other signs of microvascular disease (e.g., retinopathy or peripheral neuropathy). However, it has become clear that some patients may develop biopsy-proven diabetic nephropathy even without a concomitant increase in the urinary albumin excretion rate. This has been reported in both type 1 (Lane et al. 1992) and type 2 diabetes (MacIsaac et al. 2004). Of clinical significance is the fact that renal function tends to decline at a faster rate in those with high-grade albuminuria (5%–6%/year) than in those with low-grade albuminuria (1%–2%/year) (Molitch et al. 2010).
Treatment and Mechanisms
Diabetic nephropathy develops partly because of changes in glucose metabolism with hyperglycemia and partly because of a genetic predisposition. Hyperglycemia is pivotal for the initiation of the pathological process, in that excess glucose intermediaries enter pathways such as the polyol pathway, lead to the activation of protein kinase C (PKC), and result in the formation of advanced glycation end products (AGEs). The primary injury is believed to take place in the glomerular tuft, disrupting the integrity of the basement membrane and subsequently expanding the mesangial volume, both leading to glomerular leakage of albumin and an eventual decline in renal function. However, it is evident that all components of the kidney are exposed to hyperglycemia, and consequently, changes in tubular and interstitial structures are also seen.
In fact, more than 40 years ago, Schainuck et al. (1970) reported that in different nephropathies, including DN, the fall in the glomerular filtration rate was more closely related to changes in the tubules and the interstitium than to changes in the glomeruli. This was later confirmed by Bohle et al. (1990) in that severe diabetic glomerulosclerosis alone, without significant tubular or interstitial changes, was not associated with increased plasma creatinine concentrations. It has been suggested that increased tubular trafficking of filtrated proteins may be responsible for tubular and interstitial changes taking place in different nephropathies (Nangaku 2004); alternatively, peritubular leakage of glomerular filtrate could be a mediating mechanism for tubulointerstitial inflammation and fibrosis (Kriz and LeHir 2005).
From a therapeutic point of view, this process is prevented by strict metabolic control, keeping the glucose levels as much as possible within the normal day- and nighttime ranges. Successful results for preventing DN with intensive insulin therapy were reported more than 15 years ago among type 1 (The Diabetes Control and Complications Trial Research Group 1993) and type 2 diabetic persons (UK Prospective Diabetes [UKPDS] Group 1998). However, when microalbuminuria develops, blood pressure is usually increased, and with overt nephropathy, hypertension is present in 80% of patients (Tarnow et al. 1994). The renal activity of the renin-angiotensin-system (RAS) is inappropriately high in DN. Therefore, either angiotensin-converting enzyme (ACE) inhibitors (Lewis et al. 1993) or angiotensin II receptor blockers (Lewis et al. 1993; Brenner et al. 2001) are the cornerstones of renoprotective therapy once increased albuminuria has developed.
Both hyperglycemia and increased RAS activity induce glomerular hypertension and hyperfiltration, leading to a mechanical stress on the glomerular structures. This works in concert with pathways activated by the hyperglycemic condition and induces increased transcription of transforming growth factor–β1 (TGF-β1) together with decreased action of matrix-degrading metalloproteinases (for a detailed review, see Kanwar et al. 2011). The net effect is an accumulation of the ECM. Increased fibroblast proliferation with subsequent fibrosis may also be mediated by activation of the serine/threonine kinase mammalian target of rapamycin (mTOR) through hyperglycemia (Lieberthal and Levine 2012). Inflammatory pathways are also activated, involving chemokines, adhesion molecules, transcription factors (nuclear factor κB), and inflammatory cytokines such as interleukin (IL)–1, IL-6, IL-18, and tumor necrosis factor–α (TNF-α) (Navarro-Gonzalez et al. 2011). Therefore, new approaches to the treatment of DN are currently under investigation (e.g., AGE inhibitors, inhibitors of fibrosis, and protein kinase C inhibitors). However, strict glycemic control and a reduction in the patient’s blood pressure, primarily by ACE inhibitors or angiotensin II receptor blockers, are currently the treatments of choice for DN.
The Pathology of DN
In DN, changes in the ECM are noted in the glomerular tuft, the tubulointerstitium, and vessels (Tervaert et al. 2010). The most prominent ones are presented in Table 2. In short, increased glomerular basement membrane thickness, both in the glomeruli and in the tubular system, is seen in both type 1 and type 2 diabetes. Furthermore, expansion of the ECM in the mesangium of the glomeruli is prominent. Interstitial fibrosis and tubular atrophy follow glomerular changes. Inflammation in the interstitium can also be observed, evident through the presence of T cells and macrophages. In more advanced stages of DN, glomerulosclerosis is evident, with a massive accumulation of ECM components in the mesangium at the expense of capillary volume. Moreover, hyaline arteriolosclerosis is often prominent.
Table 2.
Extracellular Matrix Changes in Diabetic Nephropathy
| Glomerular basement membranesa | Increase in collagen IV and VI |
| Increased glycation of collagen IV | |
| Increased crosslinking of collagen IV | |
| Increase in laminin and fibronectin | |
| Decrease in agrin, perlecan, and collagen XVIII | |
| Mesangial matrixb | Increase in collagen type I |
| Increase in decorin and biglycan | |
| Tubulointerstitial matrixc | Increase in collagen type I and III |
| Increase in decorin and biglycan | |
| Glycocalyxd | Decrease in content of glycocalyx proteins |
Several of these changes can be documented in biopsies obtained from kidneys of patients with DN, as illustrated in Figures 1 and 2. Thus, typical glomerular changes with mesangial expansion are seen in the light micrograph in Figure 1A. Furthermore, in Figure 1B, both mesangial expansion and global glomerulosclerosis are observed as well as tubulointerstitial and vascular lesions. Here interstitial fibrosis and tubular atrophy are evident. Focal interstitial inflammation is also seen, as well as severe arteriosclerosis in an arteriole.
Figure 1.
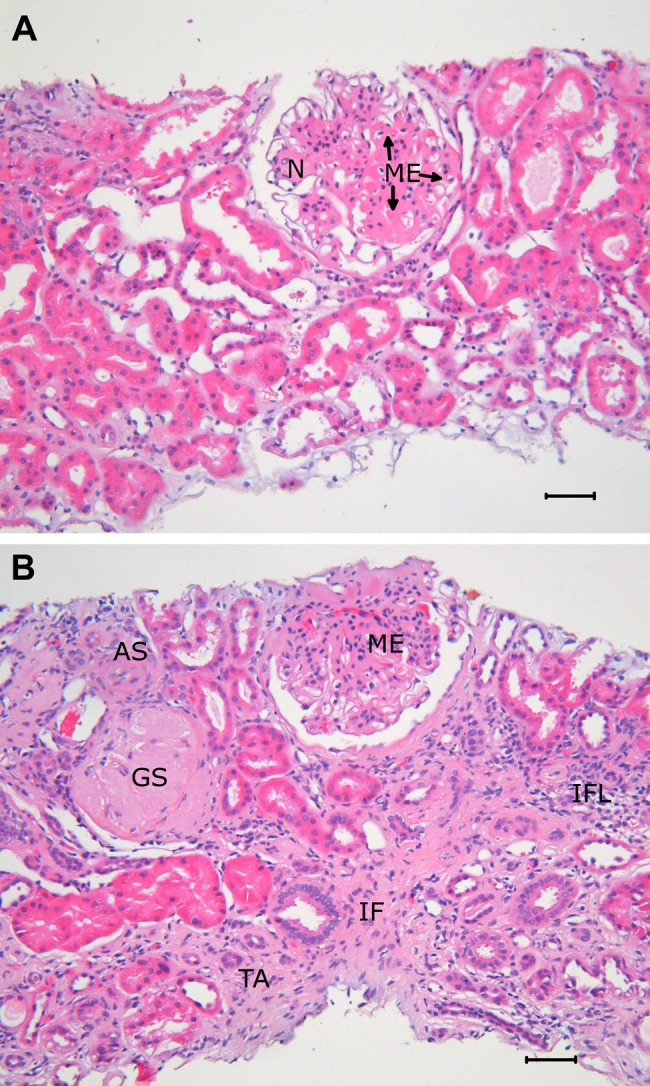
(A) Light micrograph of a renal biopsy in diabetes illustrating glomerular changes in an otherwise nearly normal kidney. The glomerular lesion is characterized by mesangial expansion (ME) with a partly nodular pattern (N). Paraffin-embedded tissue, hematoxylin and eosin staining, original magnification ×200. (B) Light micrograph of a renal biopsy in diabetes showing glomerular, tubulointerstitial, and vascular lesions. One glomerulus shows global sclerosis (GS), the other ME. There is tubular atrophy (TA), interstitial fibrosis (IF), and focal interstitial inflammation (IFL). In an arteriole, there is substantial arteriolosclerosis (AS). Paraffin-embedded tissue, hematoxylin and eosin staining, original magnification ×200.
Figure 2.
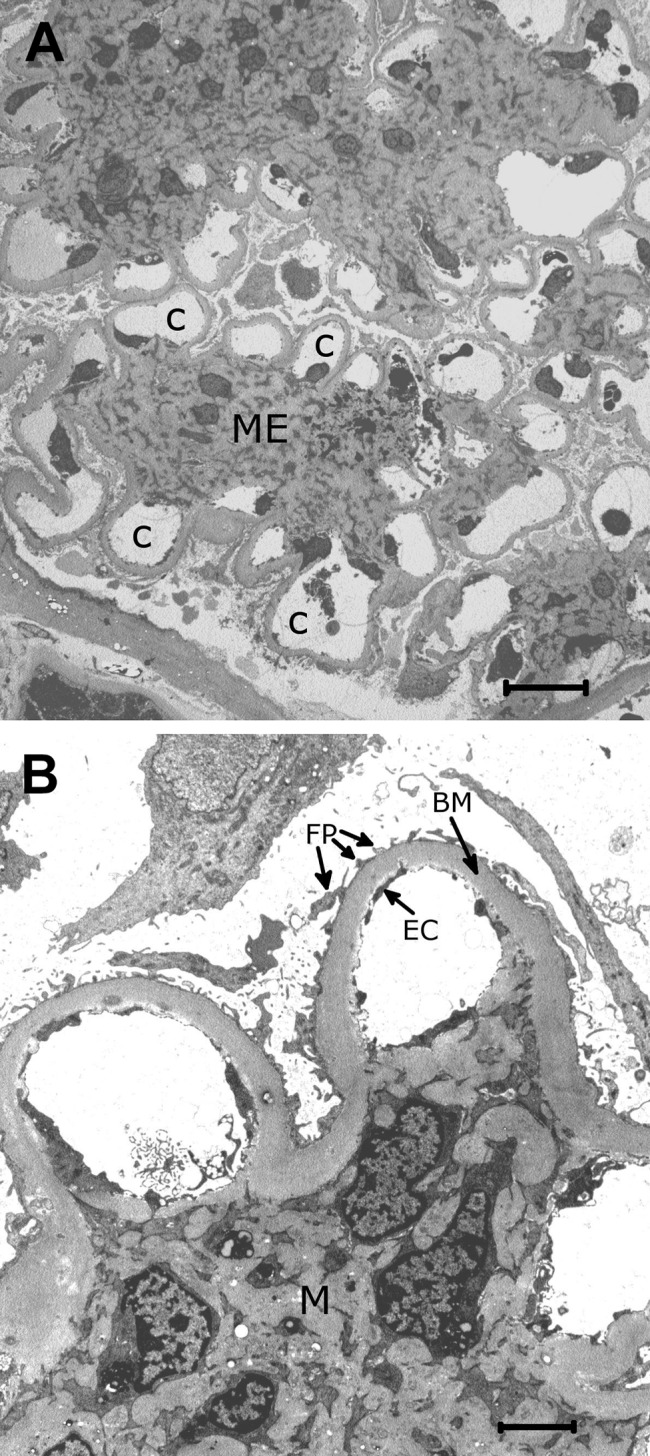
(A) Low-power electron micrograph of a segment of a glomerulus in diabetes. The mesangial expansion (ME) shows a partly nodular pattern. The mesangial expansion appears to take place at the expense of the size of the capillary lumens (C). Epoxy-embedded tissue, original magnification ×1700. (B) Higher power electron micrograph showing thickened basement membranes (BM) typical in diabetic nephropathy. The endothelial cells (EC) and foot processes of epithelial cells (FP) are normal, whereas the mesangium (M) is expanded. Epoxy-embedded tissue, original magnification ×8000.
By electron microscopy, more detailed analyses of such biopsies can be performed. The nodular pattern of mesangial expansion is clearly seen (Fig. 2A), as well as the thickened basement membrane with a homogeneous electron-dense structure typical for DN (Fig. 2B).
Proteoglycans and the Kidney
The major ECM components that are increased in DN are shown in Table 2. There is also an increase of PGs in the mesangium and the tubulointerstitium, whereas the PG content of the BMs is decreased. The increased amounts can be the result of increased synthesis of each individual component. However, ECMs are dynamic entities with different turnover rates in different tissues. Such turnover is dependent on a well-regulated balance between biosynthesis of each component and the degradation of these to ensure proper tissue dynamics and functions. Degradation of ECM components depends on two major classes of enzymes: the matrix metalloproteinases (MMPs) and serine proteases (Thrailkill et al. 2007). Furthermore, these enzymes are tightly regulated by tissue inhibitors of metalloproteinases (TIMPs), of which there are four types, TIMP1–4, and serine protease inhibitors. The latter group comprises a wide range of inhibitors, and many of these are detectable in the circulation. In relation to studies on ECM turnover, major focus has been placed on plasminogen activator inhibitor–1 (PAI-1). With hyperglycemia, there is increased generation of AGEs, and both plasma and ECM proteins are affected (Goh and Cooper 2008). Collagen IV in BM can be glycated, which leads to increased protease resistance, contributing to the thickening of glomeruli BM. In diabetic in rats treated with AGE inhibitors, AGE accumulation in glomeruli and tubules is reduced, as is microalbuminuria (Figarola et al. 2005). The use of such inhibitors in humans is still debated (Turgut and Bolton 2010).
With regard to PGs, the expression patterns differ in DN, depending on the type of PG and the part of the kidney where they are predominantly expressed. The major PGs in the BM are perlecan, collagen XVIII, and agrin, all of which show decreased concentration in DN. All these three PGs carry heparan sulfate (HS) chains (Raats et al. 2000). In the mesangium and the connective tissue of the tubulointerstitial space in DN, there is increased expression of the collagen-binding PGs decorin and biglycan, which both belong to the small leucine-rich repeat family of PGs, named SLRPs. Both SLRPs are substituted with chondroitin/dermatan sulfate (CS/DS) chains. There are also changes to the HSPGs in the mesangium (Wijnhoven et al. 2006).
Both BM and connective tissue PGs are expressed and secreted from endothelial cells, podocytes, mesangial cells, and tubular epithelial cells after completed biosynthesis. The regulation of PG biosynthesis in the kidneys is complex and depends on cell type, cellular signaling, and differences in the functions of BM PGs and the connective tissue PGs in the mesangium and tubulointerstitial space. Major focus has been on the HSPGs in the BM (Iozzo 2005) and, in particular, on their importance for glomerular filtration. A decrease in the concentration of such PGs and in the level and/or sulfation of their HS chains are regarded as important factors contributing to a decrease in filtration capacity. The highly negatively charged glycosaminoglycan (GAG) chains are a major contributor to the net negative charge in the BM, preventing negatively charged serum proteins, such as albumin, from passing from the bloodstream to the urinary space. A series of studies have addressed this issue (e.g., in experimental animal models and in studies of human kidney biopsies). Removal or inactivation of kidney HS has been demonstrated to induce proteinuria in several studies (van den Born et al. 1992; Raats et al. 2000). However, the generation of mice with a deleted version of podocyte-expressed agrin did not result in proteinuria (Harvey et al. 2007). Also, the use of the endoglucuronidase heparanase in experimental mouse systems, which leads to the generation of soluble HS oligosaccharides, did not lead to proteinuria, whereas treatment with neuraminidase, an enzyme that cleaves off terminal sialic acid residues on glycoproteins present on the cell surface and in the ECM, did indeed result in proteinuria (Wijnhoven, Lensen, Wismans, et al. 2007). Furthermore, in streptozotocin-induced diabetic mice, no decrease in the sulfation of liver heparan sulfate was found (Bishop et al. 2010). Also, using a panel of HS antibodies, no sulfation changes were detected in the glomeruli of diabetic rat renal tissue (van den Born et al. 2006).The issue of the importance of HS as a filtration barrier in the kidney is debated (Kanwar et al. 2007; Harvey and Miner 2008). In addition to the BM, the endothelial cell layer with the glycocalyx facing the bloodstream, other negatively charged ECM molecules than PGs, and the podocytes, with their associated slit diaphragms, all represent parts of a barrier system in the kidneys (Kanwar et al. 2007; Kanwar et al. 2011; Miner 2012). Early work suggested that low-dose heparin could reduce proteinuria in patients with type 1 diabetes (Myrup et al. 1995). Furthermore, the use of GAG-based drugs, such as sulodexide, has been shown to have antiproteinuric effects (Wijnhoven, Lensen, Rops, et al. 2007). Clinical trials will reveal if such an approach will have protective effects in DN (Lewis and Xu 2008). It is quite evident from several studies that the function of HS in the BM is not only a question of stabilizing a filtration barrier. Several other aspects of the functions of PGs carrying HS chains need to be considered when discussing their relevance in the development of microalbuminuria, DN, and finally diabetic ESRD. This also has to be taken into consideration when evaluating the role of the other major class of PGs, the SLRPs, in the development of DN.
HS Changes in DN
Much focus on HSPGs in the kidney has been on the filtration properties. In patients with diabetes, elevated levels of heparanase in the plasma have been demonstrated. Furthermore, staining of biopsies from patients with type 1 diabetes showed changes in the sulfation patterns of HS compared with controls (Wijnhoven et al. 2006), and another study supported such changes and also showed increased levels of heparanase in human kidney sections (Wijnhoven et al. 2008). This was also found in mice and rats with experimental DN (van den Hoven et al. 2006). These data suggest that heparanase may play a role in modifying HS structures in glomeruli, which might contribute to the development of DN. Studies on heparanase knockout mice have demonstrated that this enzyme is essential for the development of DN, as deletion of the gene protected mice from developing the lesions. Furthermore, in streptozotocin-induced diabetic mice, the heparanase inhibitor SST0001, which is modified heparin, decreased albumin secretion significantly and inhibited the increase in serum creatinine seen in the diabetic mice (Gil et al. 2012). Future studies will show if targeting heparanase to prevent the development of DN will be a useful treatment option.
Another family of enzymes that has attracted attention recently is the sulfatases. These enzymes also modify HS posttranslationally in a dynamic fashion, depending on stimuli. The 6-O-sulfatase has been the focus of several studies (Gorsi and Stringer 2007), some also related to diabetes. This enzyme removes sulfate groups from position 6 on the glucosamine entities in the repeating disaccharide units of HS. Mice with a deleted version of Sulf 1 and 2 developed proteinuria (Schumacher et al. 2011). Furthermore, both glomerular endothelial cells and podocytes were affected. Support for the importance of these sulfatases for the development of proteinuria is found in children with Wilms tumor. The transcription factor Wilms tumor gene 1 (WT1) regulates the expression level of the 6-O-sulfatase, and children with this tumor also have a kidney phenotype similar to the knockout mice (Schumacher et al. 2011). Finally, an important aspect addressed in the study was the interaction of HS with growth factors in kidney disease. Vascular endothelial growth factor–A (VEGF-A) and fibroblast growth factor–2 (FGF-2) signaling were affected by the lack of 6-O-sulfate groups in HS, highlighting novel functions of HS in the kidney other than those implicated in glomerular and tubular filtration and reabsorption.
HS interacts, both in the matrix and in cellular signaling, through collaboration with both growths factors, such as FGF-2, and their receptors (Esko and Selleck 2002; Manon-Jensen et al. 2010). Furthermore, cell adhesion is also dependent on cell surface HS (Xian et al. 2010). Cell surface HSPGs have been reviewed by others (Alexopoulou et al. 2007; Filmus et al. 2008; Manon-Jensen et al. 2010; Fico et al. 2011), but the implications of cell surface HS in kidney endothelial and epithelial cells have not been addressed to a large extent in DN. Endothelial dysfunction and loss of podocytes in DN may involve cell surface HSPGs in the syndecan and glypican families. A few studies have addressed syndecan-1 in serum as a possible marker for kidney changes in diabetes. Patients with type 1 diabetes and microalbuminuria showed higher levels of syndecan-1 in serum than patients without microalbuminuria and controls (Svennevig et al. 2006). Shedding of syndecan-1 may be regarded as a protective mechanism during heart surgery (Rehm et al. 2007; Svennevig et al. 2008), infections (Park et al. 2000), and possibly also in diabetes. Only a limited number of studies have addressed syndecan shedding in diabetes (Wang J-B et al. 2009).
Glycocalyx and DN
The possible increase in syndecan-1 in relation to kidney complications in diabetes may be the result of changes in the endothelial glycocalyx (Salmon and Satchell 2012), the protective coat on the apical membrane of endothelial cells. This coat contains cell surface PGs, associated GAGs, and serum components, and may be regarded as a specialized ECM and having a role in, for example, vascular permeability, adhesion of immune cells, ischemia-reperfusion injuries, and diabetic microalbuminuria (Salmon and Satchell 2012). The demonstrated shedding of syndecan-1 in patients with microalbuminuria (Svennevig et al. 2006) may be a marker for changes in endothelial glycocalyx and functions. Interestingly, the glycocalyx of the glomerular endothelial cells has been shown to be important for filtration in the kidneys. Removing this layer experimentally in rat kidneys resulted in proteinuria. Identification of proteins in this glycocalyx by mass spectrometry (MS) analysis showed that orosomucoid and lumican were two of the components (Friden et al. 2011). If there is a decrease in the glycocalyx in the glomerular capillaries, this may affect filtration properties and possibly contribute to increased leakage of serum proteins. Further research should provide us with new insight into the possible role of the glycocalyx in DN. Implications of HS changes for different processes are presented in Table 3.
Table 3.
Processes Affected by Changes in Kidney Heparan Sulfate
| Filtration in glomeruli and reabsorption in tubuli |
| Regulation of growth factor activities, such as transforming growth factor–β (TGF-β) and fibroblast growth factor–2 (FGF-2) |
| Proper assembly of basement membrane and interactions with other extracellular matrix components |
| Adhesion of endothelial and epithelial cells to basement membranes |
| Shedding of syndecans from endothelial cells |
| Protection of endothelial cells through the glycocalyx layer |
| Regulation of inflammation through changes in adhesion and regulation of inflammatory cells |
MMPs and DN
MMPs have been linked to kidney changes in DN, and several MMPs are increased in plasma and urine from patients with diabetes (Tan and Liu 2012). Increased levels of both MMP-2 and MMP-9 have been demonstrated in serum from patients with type 1 diabetes (Gharagozlian et al. 2009), and the level of urine MMP-9 was correlated with the degree of albuminuria (Tashiro et al. 2004; Thompson et al. 2011). The regulation of MMP activities is complex and depends on a balance with inhibitors in the TIMP family. Whether MMPs and TIMPs may serve as useful markers for DN is still a matter of debate (Tan and Liu 2012). However, these enzymes are potent modulators of ECM turnover and also of shedding of syndecans (Manon-Jensen et al. 2010), and further studies on their possible involvement in DN are warranted. In an attempt to study the relationship between MMPs in serum and ECM changes in diabetes, serum and kidney tissue from the db/db and db+ mice strains were compared. Higher gelatinase activities were detected in serum from the diabetic mice, but these were found to be serine proteases and not MMPs. In contrast, extracts from kidney tissue from db/db mice showed decreased MMP-9 activity compared with tissue from control db+ mice (Hadler-Olsen et al. 2011). This study suggests that local changes in ECM turnover are not necessarily translated into similar changes in the circulation. The possible connection between these two classes of proteases in relation to DN should be studied further in mouse models of diabetes and in clinical studies.
CS/DS Changes in DN
The SLRPs decorin and biglycan carry CS/DS chains. A major finding in DN is the increase in the mesangial matrix and the tubulointerstitial matrix concomitant with increased amounts of both these PGs (Schaefer et al. 2001). These increases are in parallel with an increase in the content of collagen I and III in the same locations (Stokes et al. 2000). Furthermore, biglycan was found to accumulate in glomeruli of diabetic mice fed a high cholesterol diet and to co-localize with apolipoprotein B, suggesting that renal biglycan contributes to diabetic nephropathy (Thompson et al. 2011). The transmembrane CSPG NG2 has also been shown to increase in the mesangium, Bowman’s capsule, and interstitial vessels in diabetic rats (Xiong et al. 2007). In diabetic nephropathy in mice, it has been shown that mesangial cells can transdifferentiate to cells expressing chondrocyte markers. Such changes could contribute to the development of mesangial sclerotic lesions (Kishi et al. 2011).
A limited number of studies have addressed changes in CS/DS structures in the diabetic kidney. One recent study demonstrated decreased CS/DS content in diabetic kidneys compared with controls and a decrease in the content of the highly sulfated CS-E (with sulfate groups in positions 4 and 6 on the galactosamine units) (Joladarashi et al. 2011). Such a decrease in CS sulfation may affect interactions with other ECM components and growth factors.
However, the presence of CS/DS in glomerular BM has also been addressed in the WBN/Kob rat model of chronic pancreatitis and late-onset diabetes. The BM of kidney sections was analyzed by electron microscopy after immunogold labeling with antibodies against HS and CS. A decrease in HS labeling in the BM was observed over time (2–19 months), whereas CS labeling increased from 2 to 10 months, after which it decreased at 19 months to the level found at 2 months (Karasawa et al. 1997). It has also been demonstrated that CSPG in BM is increased in diabetes, and it was noted that the CSPG increase was in those parts of the BM with increased thickness (McCarthy et al. 1994). Both these studies suggest that CS changes in the BM may be an early marker for kidney changes.
The major part of GAGs in the adult kidney is HS, but the content of CS can be up to 75% in the embryonic kidney (Steer et al. 2004). Connective tissue repair and inflammation often mimic embryonic events, and further studies on CS in DN and in other kidney conditions would therefore be of interest. The presence of CSPG in glomerular BM (Couchman et al. 1993) deserves more attention, particularly in light of the debated importance of HSPGs as a filtration barrier in the BM in DN (Kanwar et al. 2007; Harvey and Miner 2008).
The glycocalyx is an important barrier on the apical side of the endothelial cells. In vivo studies on mice and in perfused kidneys have demonstrated that CS/DS and hyaluronic acid are important components of this glycocalyx. Removing these components with specific enzymes increased the GFR (Jeansson and Haraldsson 2003), supporting the notion that the glycocalyx is a part of the charge barrier in glomerular filtration and that CS/DS and hyaluronic acid are also important for kidney filtration.
ECM Changes and Signaling
The most potent stimulator of ECM accumulation in the kidney is TGF-β (Yamamoto et al. 1993). It has been widely studied and reviewed, and earlier it was suggested to be a potential target for treatment (Border and Noble 1998). Studies with mouse models of diabetes using antibodies against TGF-β showed prevention of mesangial matrix accumulation (Ziyadeh et al. 2000). It remains to be demonstrated that this therapeutic approach is feasible in humans with DN. Another kidney pro-fibrotic factor is connective tissue growth factor (CTGF), also referred to as CCN-2 (Mason 2009). This factor has many of the same effects as TGF-β, and elevated levels of CTGF in plasma are correlated to ESRD in DN (Nguyen et al. 2008). The upregulation of these pro-fibrotic signaling molecules has been widely reviewed elsewhere (see, e.g., Kanwar et al. 2011).
Most of the kidney changes in DN are closely linked to hyperglycemia, which, if poorly controlled, will lead to a series of changes in cellular signaling, both systemically and locally in the kidney. Studies on renal biopsies in type 1 diabetes patients undergoing two different treatment regimes during 3 years showed that the group with HbA1c at 8.1 developed a much smaller increase in their BM (56 nm) compared with the group with an HbA1c value of 10.1 (140 nm) at the end of the study (Bangstad et al. 1994).
Hyperglycemia leads to the generation of AGEs (Busch et al. 2010), reactive oxygen species, increased flux through the hexosamine pathway, and activation of PKC (Brownlee 2005). It may also increase the secretion of renin from the kidney, leading to generation of angiotensin II and ultimately resulting in increased systemic blood pressure (Kanwar et al. 2011). All of these pathways can be modified through tight blood glucose regulation (Brownlee 2005). In addition, the role of inflammatory molecules in the development of DN is receiving increased attention, and several of the signaling molecules involved also regulate cell adhesion and ECM turnover (Navarro-Gonzalez et al. 2011). Activation of monocytes and an increase in apoptosis can also be the result of signaling pathways upregulated by hyperglycemia where intracellular accumulation of hyaluronan may play a part (Wang A and Hascall 2009). Whether ECM markers may be used for early detection of kidney complications is still a matter of debate. Several changes in levels of such markers have been shown to correlate to microalbuminuria. ECM markers are not in standard clinical use but are used in clinical trials and in experimental animal studies. Such studies are continuously throwing new light on the complexity of DN development and, it is hoped, will provide us with new tools in the future for early detection of kidney complications in patients with type 1 and type 2 diabetes.
Footnotes
Declaration of Conflicting Interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from The Throne Holst Foundation, The South Eastern Norway Regional Health Authority.
References
- Alexopoulou AN, Multhaupt HA, Couchman JR. 2007. Syndecans in wound healing, inflammation and vascular biology. Int J Biochem Cell Biol. 39:505–528 [DOI] [PubMed] [Google Scholar]
- American Diabetes Association 2010. Standards of medical care in diabetes—2010. Diabetes Care. 2010;33:S11–S61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bangstad HJ, Osterby R, Dahl-Jorgensen K, Berg KJ, Hartmann A, Hanssen KF. 1994. Improvement of blood glucose control in IDDM patients retards the progression of morphological changes in early diabetic nephropathy. Diabetologia. 37:483–490 [DOI] [PubMed] [Google Scholar]
- Bishop JR, Foley E, Lawrence R, Esko JD. 2010. Insulin-dependent diabetes mellitus in mice does not alter liver heparan sulfate. J Biol Chem. 285:14658–14662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohle A, Mackensen-Haen S, Von Gise H, Grund KE, Wehrmann M, Batz C, Bogenschutz O, Schmitt H, Nagy J, Muller C, et al. 1990. The consequences of tubulo-interstitial changes for renal function in glomerulopathies: A morphometric and cytological analysis. Pathol Res Pract. 186:135–144 [DOI] [PubMed] [Google Scholar]
- Bojestig M, Arnqvist HJ, Hermansson G, Karlberg BE, Ludvigsson J. 1994. Declining incidence of nephropathy in insulin-dependent diabetes mellitus. N Engl J Med. 330:15–18 [DOI] [PubMed] [Google Scholar]
- Border WA, Noble NA. 1998. Evidence that TGF-beta should be a therapeutic target in diabetic nephropathy. Kidney Int. 54:1390–1391 [DOI] [PubMed] [Google Scholar]
- Brenner BM, Cooper ME, De Zeeuw D, Keane WF, Mitch WE, Parving H-H, Remuzzi G, Snapinn SM, Zhang Z, Shahinfar S. 2001. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 345:861–869 [DOI] [PubMed] [Google Scholar]
- Brownlee M. 2005. The pathobiology of diabetic complications. Diabetes. 54:1615–1625 [DOI] [PubMed] [Google Scholar]
- Burrows NR, Li Y, Geiss LS. 2010. Incidence of treatment for end-stage renal disease among individuals with diabetes in the U.S. continues to decline. Diabetes Care. 33:73–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch M, Franke S, Rüster C, Wolf G. 2010. Advanced glycation end-products and the kidney. Eur J Clin Invest. 40:742–755 [DOI] [PubMed] [Google Scholar]
- Conde-Knape K. 2001. Heparan sulfate proteoglycans in experimental models of diabetes: a role for perlecan in diabetes complications. Diabetes Metab Res Rev. 17:412–421 [DOI] [PubMed] [Google Scholar]
- Couchman JR, Abrahamson DR, McCarthy KJ. 1993. Basement membrane proteoglycans and development. Kidney Int. 43:79–84 [DOI] [PubMed] [Google Scholar]
- The Diabetes Control and Complications Trial Research Group 1993. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. 1993. N Engl J Med. 329:977–986 [DOI] [PubMed] [Google Scholar]
- Esko JD, Selleck SB. 2002. Order out of chaos: assembly of ligand binding sites in heparan sulfate. Annu Rev Biochem. 71:435–471 [DOI] [PubMed] [Google Scholar]
- Fico A, Maina F, Dono R. 2011. Fine-tuning of cell signaling by glypicans. Cell Mol Life Sci. 68:923–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figarola JL, Scott S, Loera S, Xi B, Synold T, Rahbar S. 2005. Renoprotective and lipid-lowering effects of LR Compounds, novel advanced glycation end product inhibitors, in streptozotocin-induced diabetic rats. Ann N Y Acad Sci. 1043:767–776 [DOI] [PubMed] [Google Scholar]
- Filmus J, Capurro M, Rast J. 2008. Glypicans. Genome Biol. 9:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friden V, Oveland E, Tenstad O, Ebefors K, Nystrom J, Nilsson UA, Haraldsson B. 2011. The glomerular endothelial cell coat is essential for glomerular filtration. Kidney Int. 79:1322–1330 [DOI] [PubMed] [Google Scholar]
- Gharagozlian S, Svennevig K, Bangstad HJ, Winberg JO, Kolset SO. 2009. Matrix metalloproteinases in subjects with type 1 diabetes. BMC Clin Pathol. 9:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil N, Goldberg R, Neuman T, Garsen M, Zcharia E, Rubinstein AM, van Kuppevelt T, Meirovitz A, Pisano C, Li JP, et al. 2012. Heparanase is essential for the development of diabetic nephropathy in mice. Diabetes. 61:208–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh S-Y, Cooper ME. 2008. The role of advanced glycation end products in progression and complications of diabetes. J Clin Endocrinol Metab. 93:1143–1152 [DOI] [PubMed] [Google Scholar]
- Gorsi B, Stringer SE. 2007. Tinkering with heparan sulfate sulfation to steer development. Trends Cell Biol. 17:173–177 [DOI] [PubMed] [Google Scholar]
- Gross JL, De Azevedo MJ, Silveiro SP, Canani LH, Caramori ML, Zelmanovitz T. 2005. Diabetic nephropathy: diagnosis, prevention, and treatment. Diabetes Care. 28:164–176 [DOI] [PubMed] [Google Scholar]
- Hadler-Olsen E, Winberg JO, Reinholt FP, Larsen T, Uhlin-Hansen L, Jenssen T, Berg E, Kolset SO. 2011. Proteases in plasma and kidney of db/db mice as markers of diabetes-induced nephropathy. ISRN Endocrinol. 2011:832642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haraldsson B, Nystrom J. 2012. The glomerular endothelium: new insights on function and structure. Curr Opin Nephrol Hypertens. 21:258–263 [DOI] [PubMed] [Google Scholar]
- Harvey SJ, Jarad G, Cunningham J, Rops AL, van der Vlag J, Berden JH, Moeller MJ, Holzman LB, Burgess RW, Miner JH. 2007. Disruption of glomerular basement membrane charge through podocyte-specific mutation of agrin does not alter glomerular permselectivity. Am J Pathol. 171:139–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey SJ, Miner JH. 2008. Revisiting the glomerular charge barrier in the molecular era. Curr Opin Nephrol Hypertens. 17:393–398 [DOI] [PubMed] [Google Scholar]
- Hill RE, Williams PE. 2002. A quantitative analysis of perineurial cell basement membrane collagen IV, laminin and fibronectin in diabetic and non-diabetic human sural nerve. J Anat. 201:185–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmquist P, Torffvit O. 2009. Urinary transforming growth factor-beta(1), collagen IV and the effect of insulin in children at diagnosis of diabetes mellitus. Scand J Urol Nephrol. 43:142–147 [DOI] [PubMed] [Google Scholar]
- Hovind P, Tarnow L, Rossing K, Rossing P, Eising S, Larsen N, Binder C, Parving HH. 2003. Decreasing incidence of severe diabetic microangiopathy in type 1 diabetes. Diabetes Care. 26:1258–1264 [DOI] [PubMed] [Google Scholar]
- Iozzo RV. 2005. Basement membrane proteoglycans: from cellar to ceiling. Nat Rev Mol Cell Biol. 6:646–656 [DOI] [PubMed] [Google Scholar]
- Jeansson M, Haraldsson B. 2003. Glomerular size and charge selectivity in the mouse after exposure to glucosaminoglycan-degrading enzymes. J Am Soc Nephrol. 14:1756–1765 [DOI] [PubMed] [Google Scholar]
- Joladarashi D, Salimath PV, Chilkunda ND. 2011. Diabetes results in structural alteration of chondroitin sulfate/dermatan sulfate in the rat kidney: effects on the binding to extracellular matrix components. Glycobiology. 21:960–972 [DOI] [PubMed] [Google Scholar]
- Kanwar YS, Danesh FR, Chugh SS. 2007. Contribution of proteoglycans towards the integrated functions of renal glomerular capillaries: a historical perspective. Am J Pathol. 171:9–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanwar YS, Sun L, Xie P, Liu F-Y, Chen S. 2011. A glimpse of various pathogenetic mechanisms of diabetic nephropathy. Annu Rev Pathol. 6:395–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karasawa R, Nishi S, Suzuki Y, Imai N, Arakawa M. 1997. Early increase of chondroitin sulfate glycosaminoglycan in the glomerular basement membrane of rats with diabetic glomerulopathy. Nephron. 76:62–71 [DOI] [PubMed] [Google Scholar]
- KDOQI 2007. KDOQI clinical practice guidelines and clinical practice recommendations for diabetes and chronic kidney disease. Am J Kidney Dis. 49:S12–S154 [DOI] [PubMed] [Google Scholar]
- Kishi S, Abe H, Akiyama H, Tominaga T, Murakami T, Mima A, Nagai K, Kishi F, Matsuura M, Matsubara T, et al. 2011. SOX9 protein induces a chondrogenic phenotype of mesangial cells and contributes to advanced diabetic nephropathy. J Biol Chem. 286:32162–32169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriz W, Lehir M. 2005. Pathways to nephron loss starting from glomerular diseases: insights from animal models. Kidney Int. 67:404–419 [DOI] [PubMed] [Google Scholar]
- Lane PH, Steffes MW, Mauer SM. 1992. Glomerular structure in IDDM women with low glomerular filtration rate and normal urinary albumin excretion. Diabetes. 41:581–586 [DOI] [PubMed] [Google Scholar]
- Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. 1993. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. N Engl J Med. 330:1456–1462 [DOI] [PubMed] [Google Scholar]
- Lewis EJ, Xu X. 2008. Abnormal glomerular permeability characteristics in diabetic nephropathy. Diabetes Care. 31:S202–S207 [DOI] [PubMed] [Google Scholar]
- Lieberthal W, Levine JS. 2012. Mammalian target of rapamycin and the kidney part II: pathophysiology and therapeutic implications. Am J Physiol Renal Physiol. 303:F180–F191 [DOI] [PubMed] [Google Scholar]
- MacIsaac RJ, Tsalamandris C, Panagiotopoulos S, Smith TJ, McNeil KJ, Jerums G. 2004. Nonalbuminuric renal insufficiency in type 2 diabetes. Diabetes Care. 27:195–200 [DOI] [PubMed] [Google Scholar]
- Manon-Jensen T, Itoh Y, Couchman JR. 2010. Proteoglycans in health and disease: the multiple roles of syndecan shedding. FEBS J. 277:3876–3889 [DOI] [PubMed] [Google Scholar]
- Mason R. 2009. Connective tissue growth factor (CCN2), a pathogenic factor in diabetic nephropathy. What does it do? How does it do it? J Cell Commun Signal. 3:95–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy KJ, Abrahamson DR, Bynum KR, John PL, Couchman JR. 1994. Basement membrane-specific chondroitin sulfate proteoglycan is abnormally associated with the glomerular capillary basement membrane of diabetic rats. J Histochem Cytochem. 42:473–484 [DOI] [PubMed] [Google Scholar]
- Miner JH. 2012. The glomerular basement membrane. Exp Cell Res. 318:973–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogensen CE. 1984. Microalbuminuria predicts clinical proteinuria and early mortality in maturity-onset diabetes. N Engl J Med. 310:356–360 [DOI] [PubMed] [Google Scholar]
- Mogensen CE, Christensen CK. 1984. Predicting diabetic nephropathy in insulin-dependent patients. N Engl J Med. 311:89–93 [DOI] [PubMed] [Google Scholar]
- Mogensen CE, Keane WF, Bennett PH, Striker GE, Jerums G, Parving HH, Passa P, Steffes MW, Viberti GC. 1995. Prevention of diabetic renal disease with special reference to microalbuminuria. Lancet. 346:1080–1084 [DOI] [PubMed] [Google Scholar]
- Molitch ME, Steffes M, Sun W, Rutledge B, Cleary P, De Boer IH, Zinman B, Lachin J. 2010. Development and progression of renal insufficiency with and without albuminuria in adults with type 1 diabetes in the diabetes control and complications trial and the epidemiology of diabetes interventions and complications study. Diabetes Care. 33:1536–1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriya T, Groppoli TJ, Kim Y, Mauer M. 2001. Quantitative immunoelectron microscopy of type VI collagen in glomeruli in type I diabetic patients. Kidney Int. 59:317–323 [DOI] [PubMed] [Google Scholar]
- Myrup B, Hansen PM, Jensen T, Kofoed-Enevoldsen A, Feldt-Rasmussen B, Gram J, Kluft C, Jespersen J, Deckert T. 1995. Effect of low-dose heparin on urinary albumin excretion in insulin-dependent diabetes mellitus. Lancet. 345:421–422 [DOI] [PubMed] [Google Scholar]
- Nakajima T, Hasegawa G, Kamiuchi K, Fukui M, Yamasaki M, Tominaga M, Asano M, Hosoda H, Yoshikawa T, Nakamura N. 2006. Differential regulation of intracellular redox state by extracellular matrix proteins in glomerular mesangial cells: potential role in diabetic nephropathy. Redox Report. 11:223–230 [DOI] [PubMed] [Google Scholar]
- Nangaku M. 2004. Mechanisms of tubulointerstitial injury in the kidney: final common pathways to end-stage renal failure. Intern Med. 43:9–17 [DOI] [PubMed] [Google Scholar]
- Navarro-Gonzalez JF, Mora-Fernandez C, De Fuentes MM, Garcia-Perez J. 2011. Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nat Rev Nephrol. 7:327–340 [DOI] [PubMed] [Google Scholar]
- Nguyen TQ, Tarnow L, Jorsal A, Oliver N, Roestenberg P, Ito Y, Parving HH, Rossing P, van Nieuwenhoven FA, Goldschmeding R. 2008. Plasma connective tissue growth factor is an independent predictor of end-stage renal disease and mortality in type 1 diabetic nephropathy. Diabetes Care. 31:1177–1182 [DOI] [PubMed] [Google Scholar]
- Park PW, Pier GB, Preston MJ, Goldberger O, Fitzgerald ML, Bernfield M. 2000. Syndecan-1 shedding is enhanced by LasA, a secreted virulence factor of Pseudomonas aeruginosa. J Biol Chem. 275:3057–3064 [DOI] [PubMed] [Google Scholar]
- Raats CJ, van den Born J, Berden JH. 2000. Glomerular heparan sulfate alterations: mechanisms and relevance for proteinuria. Kidney Int. 57:385–400 [DOI] [PubMed] [Google Scholar]
- Rehm M, Bruegger D, Christ F, Conzen P, Thiel M, Jacob M, Chappell D, Stoeckelhuber M, Welsch U, Reichart B, et al. 2007. Shedding of the endothelial glycocalyx in patients undergoing major vascular surgery with global and regional ischemia. Circulation. 116:1896–1906 [DOI] [PubMed] [Google Scholar]
- Ritz E, Rychlík I, Locatelli F, Halimi S. 1999. End-stage renal failure in type 2 diabetes: a medical catastrophe of worldwide dimensions. Am J Kidney Dis. 34:795–808 [DOI] [PubMed] [Google Scholar]
- Salmon AHJ, Satchell SC. 2012. Endothelial glycocalyx dysfunction in disease: albuminuria and increased microvascular permeability. J Pathol. 226:562–574 [DOI] [PubMed] [Google Scholar]
- Schaefer L, Raslik I, Grone HJ, Schonherr E, Macakova K, Ugorcakova J, Budny S, Schaefer RM, Kresse H. 2001. Small proteoglycans in human diabetic nephropathy: discrepancy between glomerular expression and protein accumulation of decorin, biglycan, lumican, and fibromodulin. FASEB J. 15:559–561 [DOI] [PubMed] [Google Scholar]
- Schainuck LI, Striker GE, Cutler RE, Benditt EP. 1970. Structural-functional correlations in renal disease, II: the correlations. Hum Pathol. 1:631–641 [DOI] [PubMed] [Google Scholar]
- Schumacher VA, Schlötzer-Schrehardt U, Karumanchi SA, Shi X, Zaia J, Jeruschke S, Zhang D, Pavenstädt H, Drenckhan A, Amann K, et al. 2011. WT1-dependent sulfatase expression maintains the normal glomerular filtration barrier. J Am Soc Nephrol. 22:1286–1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw JE, Sicree RA, Zimmet PZ. 2010. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res Clin Pract. 87:4–14 [DOI] [PubMed] [Google Scholar]
- Shimizu F, Sano Y, Haruki H, Kanda T. 2011. Advanced glycation end-products induce basement membrane hypertrophy in endoneurial microvessels and disrupt the blood-nerve barrier by stimulating the release of TGF-beta and vascular endothelial growth factor (VEGF) by pericytes. Diabetologia. 54:1517–1526 [DOI] [PubMed] [Google Scholar]
- Steer DL, Shah MM, Bush KT, Stuart RO, Sampogna RV, Meyer TN, Schwesinger C, Bai X, Esko JD, Nigam SK. 2004. Regulation of ureteric bud branching morphogenesis by sulfated proteoglycans in the developing kidney. Dev Biol. 272:310–327 [DOI] [PubMed] [Google Scholar]
- Stokes MB, Holler S, Cui Y, Hudkins KL, Eitner F, Fogo A, Alpers CE. 2000. Expression of decorin, biglycan, and collagen type I in human renal fibrosing disease. Kidney Int. 57:487–498 [DOI] [PubMed] [Google Scholar]
- Svennevig K, Hoel TN, Thiara AS, Kolset SO, Castelheim A, Mollnes TE, Brosstad F, Fosse E, Svennevig JL. 2008. Syndecan-1 plasma levels during coronary artery bypass surgery with and without cardiopulmonary bypass. Perfusion. 23:165–171 [DOI] [PubMed] [Google Scholar]
- Svennevig K, Kolset SO, Bangstad HJ. 2006. Increased syndecan-1 in serum is related to early nephropathy in type 1 diabetes mellitus patients. Diabetologia. 49:2214–2216 [DOI] [PubMed] [Google Scholar]
- Tan RJ, Liu Y. 2012. Matrix metalloproteinases in kidney homeostasis and diseases. Am J Physiol Renal Physiol. 302:F1351–F1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarnow L, Rossing P, Gall M-A, Nielsen FS, Parving H-H. 1994. Prevalence of arterial hypertension in diabetic patients before and after the JNC-V. Diabetes Care. 17:1247–1251 [DOI] [PubMed] [Google Scholar]
- Tashiro K, Koyanagi I, Ohara I, Ito T, Saitoh A, Horikoshi S, Tomino Y. 2004. Levels of urinary matrix metalloproteinase-9 (MMP-9) and renal injuries in patients with type 2 diabetic nephropathy. J Clin Lab Anal. 18:206–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tervaert TWC, Mooyaart AL, Amann K, Cohen AH, Cook HT, Drachenberg CB, Ferrario F, Fogo AB, Haas M, De Heer E, et al. 2010. Pathologic classification of diabetic nephropathy. J Am Soc Nephrol. 21:556–563 [DOI] [PubMed] [Google Scholar]
- Thompson J, Wilson P, Brandewie K, Taneja D, Schaefer L, Mitchell B, Tannock LR. 2011. Renal accumulation of biglycan and lipid retention accelerates diabetic nephropathy. Am J Pathol. 179:1179–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thrailkill KM, Bunn RC, Moreau CS, Cockrell GE, Simpson PM, Coleman HN, Frindik JP, Kemp SF, Fowlkes JL. 2007. Matrix metalloproteinase-2 dysregulation in type 1 diabetes. Diabetes Care. 30:2321–2326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turgut F, Bolton WK. 2010. Potential new therapeutic agents for diabetic kidney disease. Am J Kidney Dis. 55:928–940 [DOI] [PubMed] [Google Scholar]
- UK Prospective Diabetes Study (UKPDS) Group 1998. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet. 352:854–865 [PubMed] [Google Scholar]
- van den Born J, Pisa B, Bakker MAH, Celie JWAM, Straatman C, Thomas S, Viberti GC, Kjellen L, Berden JHM. 2006. No change in glomerular heparan sulfate structure in early human and experimental diabetic nephropathy. J Biol Chem. 281:29606–29613 [DOI] [PubMed] [Google Scholar]
- van den Born J, van den Heuvel LP, Bakker MA, Veerkamp JH, Assmann KJ, Berden JH. 1992. A monoclonal antibody against GBM heparan sulfate induces an acute selective proteinuria in rats. Kidney Int. 41:115–123 [DOI] [PubMed] [Google Scholar]
- van den Hoven MJ, Rops AL, Bakker MA, Aten J, Rutjes N, Roestenberg P, Goldschmeding R, Zcharia E, Vlodavsky I, van der, Vlag J, et al. 2006. Increased expression of heparanase in overt diabetic nephropathy. Kidney Int. 70:2100–2108 [DOI] [PubMed] [Google Scholar]
- Wang A, Hascall VC. 2009. Hyperglycemia, intracellular hyaluronan synthesis, cyclin D3 and autophagy. Autophagy. 5:864–865 [DOI] [PubMed] [Google Scholar]
- Wang J-B, Guan J, Shen J, Zhou L, Zhang Y-J, Si Y-F, Yang L, Jian X-H, Sheng Y. 2009. Insulin increases shedding of syndecan-1 in the serum of patients with type 2 diabetes mellitus. Diabetes Res Clin Pract. 86:83–88 [DOI] [PubMed] [Google Scholar]
- Wijnhoven T, van den Hoven M, Ding H, van Kuppevelt T, van der Vlag J, Berden J, Prinz R, Lewis E, Schwartz M, Xu X. 2008. Heparanase induces a differential loss of heparan sulphate domains in overt diabetic nephropathy. Diabetologia. 51:372–382 [DOI] [PubMed] [Google Scholar]
- Wijnhoven TJ, Lensen JF, Rops AL, McCarthy KJ, van der Vlag J, Berden JH, van den Heuvel LP, van Kuppevelt TH. 2007. Anti-proteinuric effects of glycosaminoglycan-based drugs. Curr Opin Mol Ther. 9:364–377 [PubMed] [Google Scholar]
- Wijnhoven TJ, Lensen JF, Rops AL, van der Vlag J, Kolset SO, Bangstad HJ, Pfeffer P, van den Hoven MJ, Berden JH, van den Heuvel LP, et al. 2006. Aberrant heparan sulfate profile in the human diabetic kidney offers new clues for therapeutic glycomimetics. Am J Kidney Dis. 48:250–261 [DOI] [PubMed] [Google Scholar]
- Wijnhoven TJ, Lensen JF, Wismans RG, Lamrani M, Monnens LA, Wevers RA, Rops AL, van der Vlag J, Berden JH, van den Heuvel LP, et al. 2007. In vivo degradation of heparan sulfates in the glomerular basement membrane does not result in proteinuria. J Am Soc Nephrol. 18:823–832 [DOI] [PubMed] [Google Scholar]
- Xian X, Gopal S, Couchman J. 2010. Syndecans as receptors and organizers of the extracellular matrix. Cell Tissue Res. 339:31–46 [DOI] [PubMed] [Google Scholar]
- Xiong J, Wang Y, Zhu Z, Liu J, Wang Y, Zhang C, Hammes H-P, Lang F, Feng Y. 2007. NG2 proteoglycan increases mesangial cell proliferation and extracellular matrix production. Biochem Biophys Res Commun. 361:960–967 [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Nakamura T, Noble NA, Ruoslahti E, Border WA. 1993. Expression of transforming growth factor beta is elevated in human and experimental diabetic nephropathy. Proc Natl Acad Sci U S A. 90:1814–1818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yard BA, Kahlert S, Engelleiter R, Resch S, Waldherr R, Groffen AJ, van den Heuvel LPWJ, van der Born J, Berden JHM, Kröger S, et al. 2001. Decreased glomerular expression of agrin in diabetic nephropathy and podocytes, cultured in high glucose medium. Exp Nephrol. 9:214–222 [DOI] [PubMed] [Google Scholar]
- Ziyadeh FN, Hoffman BB, Han DC, Iglesias-De La, Cruz MC, Hong SW, Isono M, Chen S, McGowan TA, Sharma K. 2000. Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor–beta antibody in db/db diabetic mice. Proc Natl Acad Sci U S A. 97:8015–8020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoccali C, Kramer A, Jager K. 2009. The databases: renal replacement therapy since 1989—The European Renal Association and European Dialysis and Transplant Association (ERA-EDTA). Clin J Am Soc Nephrol. 4:S18–S22 [DOI] [PubMed] [Google Scholar]