

Background: Tau aggregation is a multistep process. The identity of Tau species compromising cell viability remains largely unknown.
Results: Analysis of Tau aggregation dynamic identifies oligomeric Tau aggregates as toxic species that impair viability.
Conclusion: Membrane leakage induced by oligomeric Tau is a mechanism for toxicity.
Significance: Tau belongs to the class of amyloidogenic proteins that share a common toxicity-mediating mechanism.
Keywords: Alzheimer Disease, Chromatography, Electron Microscopy (EM), Phospholipid Vesicle, Protein Aggregation, Tau, SH-SY5Y Cells, beta-Sheet Structure, Cytotoxicity, Vesicle Leakage
Abstract
The microtubule-associated protein Tau is mainly expressed in neurons, where it binds and stabilizes microtubules. In Alzheimer disease and other tauopathies, Tau protein has a reduced affinity toward microtubules. As a consequence, Tau protein detaches from microtubules and eventually aggregates into β-sheet-containing filaments. The fibrillization of monomeric Tau to filaments is a multistep process that involves the formation of various aggregates, including spherical and protofibrillar oligomers. Previous concepts, primarily developed for Aβ and α-synuclein, propose these oligomeric intermediates as the primary cytotoxic species mediating their deleterious effects through membrane permeabilization. In the present study, we thus analyzed whether this concept can also be applied to Tau protein. To this end, viability and membrane integrity were assessed on SH-SY5Y neuroblastoma cells and artificial phospholipid vesicles, treated with Tau monomers, Tau aggregation intermediates, or Tau fibrils. Our findings suggest that oligomeric Tau aggregation intermediates are the most toxic compounds of Tau fibrillogenesis, which effectively decrease cell viability and increase phospholipid vesicle leakage. Our data integrate Tau protein into the class of amyloidogenic proteins and enforce the hypothesis of a common toxicity-mediating mechanism for amyloidogenic proteins.
Introduction
Alzheimer disease (AD)2 is a neurodegenerative disorder, histopathologically characterized by the formation of extracellular neuritic plaques and intracellular neurofibrillary tangles. Both lesions are made up by fibrillar aggregates of specific proteins. Whereas plaques contain the Aβ-peptide, derived by proteolytic cleavage from the amyloid precursor protein, tangles are composed of the microtubule-associated protein Tau (1). Although plaques and tangles are typical hallmarks of the disease, their precise role in the pathomechanism of the disease and how their formation is linked to neurodegeneration are not entirely clear.
Under normal conditions, Tau protein is predominantly localized in axons, where it binds to microtubules and promotes microtubule assembly (2–5). In AD and some other neurodegenerative disorders, such as frontotemporal dementia and parkinsonism linked to chromosome 17, Tau protein becomes hyperphosphorylated and detaches from microtubules. This hyperphosphorylated Tau protein, which shows reduced affinity toward microtubules, eventually sequesters into neurofibrillary tangles (6–7). The distribution of neurofibrillary tangles correlates well with the loss of neurons and cognitive dysfunction in AD (8, 9). The conversion from native soluble Tau to aggregated Tau seems to involve conformational changes from a random coil to a β-sheet structure and the assembly of β-sheet-containing Tau monomers into filaments (10–12). The increase of β-sheet structures in the Tau molecule and the formation of cross-β structures during Tau protein aggregation classify Tau protein as an amyloidogenic protein (13, 14).
Amyloidogenic proteins that can lose their native functional conformation and convert into β-sheet-containing deposits are a characteristic feature of a group of diseases, commonly referred to as amyloidosis (15, 16). Although a direct causal relationship has not unequivocally been proven, a number of indirect lines of evidence indicate a link between protein aggregation and degeneration in these disorders: (i) protein deposits and cell death are often found colocalized (17–19); (ii) in familial forms of amyloidosis, mutations frequently occur in the gene encoding for the amyloidogenic protein (20–23); (iii) these mutations increase aggregation propensity of recombinant proteins (24–27); and (iv) overexpression of amyloidogenic proteins in animal models can lead to a disease-associated phenotype (28–30).
The pathogenic cascade potentially linking amyloidosis to cell degeneration appears to be a multistep process. It might involve the formation of various aggregation intermediates, such as spherical oligomers, containing 15–40 monomers as well as chainlike protofibrils composed of several spherical oligomers. Protofibrils further convert into amyloid fibrils but in addition might form circular, porelike species, which could puncture the cell membrane (31–34). Thus, prefibrillar aggregation intermediates rather than the fibrillar aggregation end product more likely represent the cytotoxic species (35–38). Accordingly, it has been demonstrated that oligomeric forms of Aβ, α-synuclein, and IAPP are toxic to cells (39–41), findings that have provided the basis for a concept referred to as the “toxic oligomer hypothesis” (42, 43).
Still, for Tau protein, cytotoxicity of intermediates, generated in the process of fibrillogenesis, has not been investigated systematically. Therefore, in the present study, we analyzed whether a defined Tau species generated in the process of fibrillar aggregation can be identified, accounting for the deleterious consequences of Tau fibrillogenesis. In addition, we addressed the question of whether the cell membrane, which is generally considered to be a primary target of amyloidogenic compounds (44–46), might be involved in the cascade of pathogenic events.
To this end, viability and membrane integrity were assessed on SH-SY5Y neuroblastoma cells and artificial phospholipid vesicles, treated with Tau monomers, Tau aggregation intermediates, or Tau fibrils. Our data suggest that Tau aggregation intermediates are the most toxic compounds of Tau fibrillogenesis, which effectively decrease cell viability and increase phospholipid vesicle leakage. The present findings integrate Tau protein into the class of amyloidogenic proteins and enforce the hypothesis of a common toxicity-mediating mechanism for amyloidogenic proteins.
EXPERIMENTAL PROCEDURES
Expression and Purification of Recombinant Tau Protein
The prk172-ht40 plasmid, encoding the longest human Tau isoform (Tau; 441 amino acids, including exons 2, 3, and 10) was provided courtesy of M. Goedert (Cambridge, UK) (1). The integrity of the plasmid was confirmed by automated sequencing. Tau purification was performed using a modified procedure described by Yoshida and Goedert (47). Briefly, prk172-ht40 transformed E. coli were extracted with extraction buffer (50 mm Tris, pH 7.4, 5 mm EDTA, 0.1 mm DTT, 1% Triton X-100, 1 mm PMSF). Supernatant was cleared using a diethylaminoethyl cellulose column and afterward loaded onto a phosphocellulose column. Tau was eluted stepwise in 5-ml fractions with NaCl (0.1–0.5 m). The purest fractions containing Tau were pooled, and Tau was precipitated by ammonium sulfate with 60% saturation. Tau precipitate was solubilized in gel filtration buffer (50 mm MES, pH 6.5, 50 mm NaCl, 0.1 mm EDTA, and 1 mm PMSF) with 0.5% β-mercaptoethanol, heat-denatured, cleared, and loaded onto a HiPrep Sephacryl S300 HR gel filtration column. Tau fractions were pooled, concentrated, and dialyzed against 5 mm MOPS, pH 7.0, 50 mm NaCl, 0.1 mm EDTA, and 0.1 mm PMSF).
Tau Filament Assembly
Aggregation of 400 μl of recombinant Tau protein (100 μm) was carried out with the aid of heparin (∼14 kDa; BDH) in a final concentration of 0.4 mg/ml as described by Haase et al. (48). Aggregation was performed for 0, 6, 24, 48, 72, or 144 h.
Detection of Tau Filament Assembly
Aggregation of recombinant Tau protein was monitored by SDS-PAGE, 4,4′-dianilino-1,1′-binaphthyl-5,5′-disulfonic acid (bis-ANS), and thioflavin T (ThT) fluorescence spectrometry and electron microscopy. A 2-μl aliquot of each aggregation sample was separated on 10% SDS-PAGE and stained with Coomassie Brilliant Blue R250 followed by a densitometric analysis. Quantitative aggregation analysis via bis-ANS fluorescence spectrometry (excitation 360 nm, emission 485 nm) and ThT fluorescence spectrometry (excitation 430 nm, emission 485 nm) was carried out in a volume of 100 μl containing 27 mm MOPS (pH 7.4), 20 μm bis-ANS, or 5 μm ThT and 5% (v/v) aggregation sample in a black 96-well plate (HTS 7000 Bio Assay Reader, PerkinElmer Life Sciences). For electron microscopy, aggregation samples were diluted 1:5 in aqua dest (B. Braun Melsungen AG) and placed on carbon-coated 200-mesh gold grids with a diameter of 3.05 mm (S160A, Plano) for 2 min. Grids were washed with aqua dest for 2 min and stained with 2% (w/v) uranyl acetate in aqua dest for a further 2 min. Grids were analyzed in a LEO 912 OMEGA electron microscope (Zeiss), and microphotographs were taken. Diluted samples from FPLC purification were concentrated by centrifugation for 2 h at 200,000 × g before transfer of 1.5 μl on Formvar-coated copper grids for ultrastructural analysis. After uranyl acetate staining, samples were analyzed with a Zeiss EM900 transmission electron microscope (Zeiss NTS, Oberkochen, Germany).
Characterization of Tau Aggregation Intermediates by Size Exclusion Chromatography (SEC)
400 μl of aggregated Tau protein was centrifuged at 1000 × g for 10 min and then loaded onto a HiPrep 16/60 Sephacryl S-500 HR and eluted with gel filtration buffer (50 mm MOPS, pH 7.0, 50 mm NaCl, 0.1 mm EDTA at 0.5 ml/min). Elution of Tau protein was monitored at 280 nm, and 2-ml fractions were collected and stored at 4 °C. Each fraction was analyzed for protein content using Fluram (Fluka) and for β-sheet content using thioflavin S and bis-ANS. Fluram is a sensitive fluorimetric reagent for detecting primary amines. Using a black microtiter plate, we combined 80 μl of a FPLC fraction with 20 μl of 100 mm phosphate buffer, pH 8.0, and 50 μl of 1 mm Fluram in acetone. After 5 min of incubation, we measured fluorescence using 355-nm excitation and 480-nm emission filters. Protein content was calculated using BSA as a dilution curve because content of the primary amines in both proteins is similar (arginine content 3.2% versus 3.9% and lysine content 10% versus 10.1% in 441-amino acid (2 N-terminal inserts 4 microtubule-binding repeats (2N4R)) Tau protein and BSA, respectively). β-Sheet content was measured with ThT and bis-ANS as described above using 80 μl of each FPLC fraction.
Dynamic Light Scattering
Dynamic light scattering measurements of FPLC fractions were performed with a Zetasizer Nano ZS (Malvern, Herrenberg, Germany) containing a 5-milliwatt helium-neon 633-nm laser at 173° measurement angle. Sample were thermally equilibrated at 25 °C for 2 min. Particle size was obtained as an average of two measurements with 25 runs each and expressed as volume distribution.
Detection of Tau Oligomers via Sandwich ELISA
A 96-microwell plate with MaxiSorb surface and flat bottom cavities (Nunc) was coated overnight with 5 μg/ml AffiniPure goat anti-rabbit IgG (H+L) (Jackson ImmunoResearch Laboratories) in coating buffer (50 mm Tris, pH 8.0, 1 mm EDTA). Further washing and incubation steps were carried out using Tris-buffered saline containing 0.1% (v/v) Tween 20 (TBS-T). Coated plate was washed and incubated for 1 h with 2 μg/ml rabbit (polyclonal) anti-oligomer antibody (A11) (Invitrogen). The plate was washed, blocked for 2 h with 2% (w/v) skimmed milk powder, and afterward incubated overnight with Tau aggregation samples diluted 1:1000 in aqua dest. After washing, the plate was incubated for 1 h with 0.25 μg/ml detection antibody anti-TAU1 monoclonal antibody (mouse IgG2a) (Chemicon), followed by a 1-h incubation with ImmunoPure antibody goat anti-mouse IgG+IgM (H+L), horseradish peroxidase-labeled (Pierce) and diluted 1:3000. Detection was performed with 100 μl/well chromogen solution (2.4 mm o-phenylenediamine, 0.03% H2O2 in 0.1 m acetate buffer, pH 5.0). The reaction was stopped after 15 min with 6 m HCl. Absorption was analyzed at 492 nm (HT3 microtiter plate reader, Anthos Mikrosysteme).
Cell Culture, 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), and Lactate Dehydrogenase (LDH) Assay
For the incubation of SH-SY5Y cells with aggregated Tau protein, cells were maintained in a 96-well plate in DMEM/Ham's F-12 medium supplemented with 15% (v/v) fetal bovine serum (FBS), 2 mm l-glutamine, non-essential amino acids (Biochrom AG), 50 μg/ml gentamicin (PAA Laboratories), and 15 mm HEPES, pH 7.4, in a 5% CO2 environment at 37 °C for 24 h. Afterward, cells were maintained for 24 h in 185 μl of the described medium containing only 2% (v/v) FBS. Then 15 μl/well aggregated Tau protein were added to the cells for 48 h. MTT (Sigma) was added in triplicate to cells to a final concentration of 1 mg/ml, and cells were incubated for 2 h. After incubation, cell culture medium with MTT was removed, 200 μl of DMSO and 25 μl of Soerensen's glycine buffer (0.1 m glycine, pH 10.5, 0.1 m NaCl) per well were added, and absorbance values were measured at 550 nm (HT3 microtiter plate reader). Absorbance values of SH-SY5Y cells cultures treated with Tau protein were normalized to heparin-only-treated SH-SY5Y cell cultures at each aggregation time point. The LDH assay was performed according to the manufacturer's recommendations (Roche Applied Science). Absorbance values were measured at 492 nm (HT3 microtiter plate reader). The obtained absorbance values were normalized as mentioned for the MTT assay.
Cellular Tau Protein Uptake
400 μl of 48-h preaggregated Tau was mixed with 1 mg of Alexa488-SDP ester (Invitrogen) after adjusting pH to 7.6 and allowed to react for 1 h. The reaction was terminated with hydroxylamine, and labeled Tau monomer and oligomer fractions were purified by gel filtration using Sephacryl S-500 HR matrix. Equal quantities of Alexa488-labeled Tau monomer or oligomer were applied to SH-SY5Y cells for 24 h. Cellular localization of labeled Tau was analyzed by optical sectioning with a Zeiss Apotome fluorescence microscope after washing cells with 0.1 m acetate buffer, pH 5.0.
Vesicle Leakage Assay with Preaggregated Tau
The vesicle leakage assay was performed as described by Zschörnig et al. (49). Large unilamellar vesicles filled with a 20 mm NaCl solution containing a 12.5 mm concentration of the fluorescence dye 8-aminonaphthalene-1,3,6-trisulfonic acid (ANTS) and a 45 mm concentration of the quencher p-xylene-bis-pyridinium bromide (DPX) were used. For the measurement, 30 μl of the large unilamellar vesicle suspension were transferred to 2.5 ml of 10 mm HEPES (pH 7.4) and 150 μl of Tau protein preaggregated for 0, 24, or 144 h was added. Dialysis buffer containing all ingredients needed for Tau protein aggregation but without Tau protein incubated for 24 h served as a control. ANTS fluorescence was measured every 15 s over a time of 220 min with an excitation wavelength of 360 nm and an emission wavelength of 530 nm. After 220 min, 100% leakage was determined by adding 0.1% (v/v) Triton X-100.
RESULTS
Characterization of Tau Aggregation Intermediates
The innate ability of recombinant Tau to aggregate in vitro into high molecular weight adducts is low. Thus, aggregation of recombinant human 441 amino acid Tau isoform (Tau) was triggered using the polyanionic aggregation inducer heparin (50). Tau aggregation was allowed to proceed for up to 144 h at 37 °C. Aggregation of Tau over time in the presence and absence of heparin was monitored by SDS-PAGE, SEC on Sephacryl S-500, ThT and bis-ANS fluorescence spectroscopy, electron microscopy, and ELISA with oligomer-specific antibodies.
On SDS-PAGE, monomeric Tau runs as a single band at around 63 kDa. After a 24-h incubation of Tau with heparin, additional bands occurred in the range of 130–200 kDa and increased with further incubation time (Fig. 1A). Most likely, they represent SDS-stable aggregation products. Alzheimer-like Tau filaments arising during Tau aggregation do not enter the stacking gel and therefore cannot be quantified via SDS-PAGE. It is noteworthy that monomeric Tau decreases with increasing incubation time due to the formation of aggregated, high molecular weight Tau species. In contrast, Tau incubated without heparin exhibits a low aggregation tendency. Besides visualization of Tau aggregation via SDS-PAGE and protein staining, it could also be confirmed that Tau remains stable during incubation time and is not degraded. The densitometric analysis of shifted Tau (smear and bands) above the monomer band, representing high molecular weight Tau species, affirms the increase of Tau aggregation in the presence of heparin with increasing incubation time. Aggregated Tau above the monomer band occurs only marginally with proceeding incubation time in the absence of heparin (Fig. 1C).
FIGURE 1.

Monitoring Tau aggregation. Tau was aggregated for different periods of time (0, 6, 24, 48, 72, and 144 h) with (A) and without (B) heparin. Tau aggregation was visualized by separating aggregation samples by 10% SDS-PAGE, followed by protein staining with Coomassie Brilliant Blue R250. A, increasing aggregation time leads to an increase of high molecular weight Tau aggregates above the Tau monomer band at 63 kDa. B, incubation of Tau without heparin does not lead to formation of high molecular weight products. Please note shifted Tau bands in the range of 65–72 kDa in A and B, which probably represent conformationally changed monomeric Tau. C, densitometric analysis also reveals an increase of high molecular weight Tau aggregates with increasing incubation time if Tau fibrillogenesis is induced by heparin. Tau aggregation with and without heparin was also measured by ThT (D) and bis-ANS fluorescence spectrometry (E). Both fluorescence dyes indicate Tau protein aggregation in the presence of heparin, whereas Tau protein aggregation does not occur in the absence of heparin. Data are expressed as mean values ± S.D. (error bars).
Tau aggregation was also monitored using the fluorescence dyes ThT and bis-ANS, detecting β-sheet content and exposed hydrophobic patches, respectively (Fig. 1, D and E). Both ThT and bis-ANS fluorescence intensity gradually increased with time, when Tau was incubated with heparin. ThT fluorescence and bis-ANS fluorescence intensity reached a steady-state level at 48 h (Fig. 1D) and between 72 and 144 h (Fig. 1E), respectively. Fluorescence intensity of both dyes did not increase with time for Tau incubated without heparin (Fig. 1, D and E).
To obtain more precise information on the size distribution and kinetics in the formation of Tau aggregation intermediates, aggregates were sampled after different periods of time (24, 48, 72, and 120 h) and subjected to SEC on Sephacryl S-500. Each chromatographic fraction was characterized with respect to protein content, β-sheet content, and exposed hydrophobic patches by fluorescence spectroscopy (Fig. 2). The hydrodynamic radius of peak fractions was determined by dynamic light scattering, which allowed us to discriminate between Tau monomers (hydrodynamic radius (RH) = 4.7 nm), oligomers (RH = 14 nm), and higher aggregates (RH = 92 nm) (Fig. 3). Integrating the protein peaks obtained by SEC allowed us to quantify the relative amount of each of these aggregation forms (Table 1). Oligomeric Tau showed its highest amount after 48 h of aggregation and decreased again thereafter. 72 h of aggregation led to the formation of Tau fibrils eluting in the void volume of the SEC column. At the same time point, monomeric Tau had almost completely disappeared. However, Tau monomers reappeared again after 120 h, although with a higher bis-ANS signal than unaggregated Tau, which most likely is due to an altered secondary structure. A prominent finding of Tau aggregate size exclusion chromatography is the presence of a single, defined oligomeric Tau species with a uniform hydrodynamic radius of 14 nm, which can be calculated to consist of 40 Tau protein molecules.
FIGURE 2.
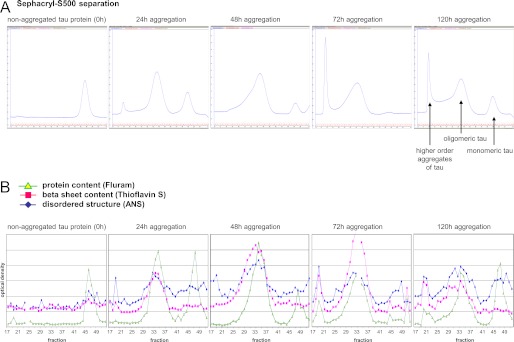
Characterization of Tau aggregation intermediates. Tau samples aggregated for different periods of time were separated by SEC on Sephacryl-S500, and fractions were analyzed for protein content (Fluram), β-sheet content (thioflavin S), and disordered structure (bis-ANS). Tau aggregation for 48 h leads to the highest relative amount of oligomeric Tau. After 72-h aggregation, fibrillar Tau appears eluting in the void volume of the SEC column, whereas monomeric Tau disappears. After 120 h, disaggregation of Tau adducts leads to a reappearance of Tau monomers, which confer a higher bis-ANS signal than unaggregated Tau protein, probably due to altered secondary structure.
FIGURE 3.

RH of peak fractions was determined by dynamic light scattering, which allowed identification of the peak for monomeric Tau (4.7 nm), oligomeric Tau (14 nm), and higher molecular aggregates (mean value, 92 nm). Monomeric and oligomeric Tau peaks each consist of molecules of similar molecular size, whereas the high molecular weight peak represents a more heterogeneous mixture of different sized aggregates.
TABLE 1.
Determination of relative protein content of eluted protein fractions after size exclusion chromatography
| Aggregation time | Relative amount of molecular Tau species |
||
|---|---|---|---|
| Monomer | Oligomer | Filament | |
| h | % | % | % |
| 0 | 100 | ||
| 24 | 28.2 | 66.5 | 5.3 |
| 48 | 9.0 | 87.5 | 3.5 |
| 72 | 1.8 | 56.3 | 41.9 |
| 120 | 25.4 | 46.7 | 27.9 |
For the qualitative analysis of Tau aggregates, electron micrographs were taken at different time points (Fig. 4). Small spherical structures, most likely representing Tau oligomers, were visible after 6 h of Tau aggregation in the presence of heparin. Short filaments with straight appearance were clearly visible after 48 h of Tau aggregation. With proceeding aggregation time up to 144 h, an elongation of the filaments could be observed. Similar results were obtained using gel filtration-purified aggregation products, which, however, had to be centrifuged before coating grids (supplemental Fig. S5). In these electron micrographs, oligomeric Tau appears also as spherical structures but less complex and with a smaller diameter of 12 nm. This value is very close to the hydrodynamic radius obtained by light scattering.
FIGURE 4.
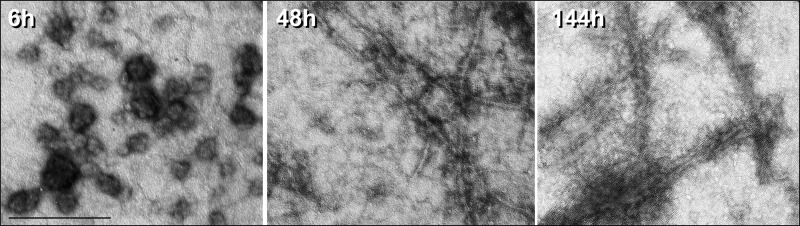
Visualization of Tau aggregation intermediates by electron microscopy. For qualitative analysis, Tau aggregation samples were applied to carbon-coated gold grids, stained with 2% uranyl acetate, and analyzed using a LEO 912 OMEGA transmission electron microscope. After 6 h of aggregation (left), spherical oligomeric aggregates predominate, which after longer aggregation time progressively convert to fibrillar aggregates (48 and 144 h) (middle and right). Magnification was ×50,000; scale bar, 242 nm.
To validate the kinetics of oligomer formation with an independent method, we quantified the relative amounts of Tau oligomers after different periods of aggregation by a sandwich ELISA (Fig. 5). The oligomer-specific antibody described by Kayed et al. (46) was used for capturing Tau oligomers. Preincubated Tau samples were allowed to aggregate in the presence or absence of heparin for up to 144 h and were subsequently diluted 1:1000 to stop aggregation. Tau oligomer content in the presence of heparin increased after 6 h and reached its maximum at 48 h. It decreased thereafter and remained stable at an intermediate level. This decrease of Tau oligomer content at an aggregation time of 72 and 144 h might be due to the progressive formation of fibrils, which depletes Tau oligomers. The fact that Tau oligomers after 72 h decrease toward a level that is well above the initial level at 0 h might indicate an equilibrium reaction maintained by the still ongoing conversion from monomers to oligomers and its further incorporation into fibrils.
FIGURE 5.

Quantification of Tau oligomers by ELISA. The oligomer content in Tau protein samples aggregated for different periods of time (0, 6, 24, 48, 72, and 144 h) with or without heparin was quantified by a sandwich ELISA. Tau oligomers were captured with the anti-oligomer antibody A11 and detected with anti-TAU1 antibody. In the presence of heparin, the Tau oligomer content rapidly increased up to 48 h of aggregation and decreased thereafter toward an intermediate level. Formation of Tau oligomers in the absence of heparin remains marginal. Error bars, S.D.
Tau Oligomers Decrease SH-SY5Y Cell Viability
SH-SY5Y cells were incubated for 48 h with Tau samples preaggregated for different periods of time. The stability of Tau protein during incubation of SH-SY5Y cell culture has been confirmed (supplemental Fig. S1). The neurotoxicity of preaggregated Tau was determined by the MTT assay, measuring mitochondrial dehydrogenase activity of treated cells (Fig. 6A). Controls such as Tau protein only and heparin only were included for each time point to account for the trophic effect of heparin (supplemental Fig. S2). SH-SY5Y cells incubated with non-aggregated Tau (0 h) exhibit the highest MTT turnover. With increasing preaggregation time of Tau, MTT turnover by SH-SY5Y cells decreased and reached its minimum for a Tau preaggregation time of 24 h. With further increasing preaggregation time of Tau, MTT turnover increased again slightly without reaching the initial level at 0 h. This time curve of decreased cell viability matches the formation of oligomeric Tau (Table 1), which shows its highest amount after 48 h of aggregation, in contrast to monomeric Tau (0 h) or Tau filaments (144 h). We compared the size distribution of 6-h preaggregated Tau protein after a 48-h incubation of SH-SY5Y cells with non-incubated Tau protein and can confirm that Tau aggregation does not progress in cell culture medium (supplemental Fig. S3).
FIGURE 6.

Effect of aggregated Tau protein on SH-SY5Y cell viability. Tau aggregation triggered by heparin was allowed to proceed for different periods of time up to 144 h. SH-SY5Y cells were incubated for 48 h with the different preaggregated Tau samples or preincubated with heparin only as a control. Cell viability was assessed with MTT and LDH assays. Data were normalized to heparin-only-treated SH-SY5Y cell cultures at each preaggregation time point. A, MTT assay. Tau protein preaggregated for 6 h or more decreases cell viability significantly compared with non-aggregated Tau protein (0 h). B, LDH assay. Tau protein preaggregated for 24 h or more increases LDH leakage significantly compared with non-aggregated Tau protein (0 h). MTT and LDH assays were performed in triplicate. Data are mean values ± S.D. (error bars) (S.D. includes error propagation in consequence of normalization); differences are significant at p < 0.05 (*) and p < 0.01 (**) (Student's t test).
The membrane integrity of SH-SY5Y cells treated with preaggregated Tau was assayed by LDH leakage (Fig. 6B). There was only a low LDH leakage for cells incubated with Tau preaggregated for 0 and 6 h, which increased with increasing preaggregation time. It reached its maximum for a Tau preaggregation time of 48 h and decreased again thereafter slightly (Fig. 6B). This overall pattern of LDH leakage matches the pattern of cell viability obtained by the MTT assay (see Fig. 6A). Thus, the LDH assay and MTT assay, both indicators of cell viability, show a highly significant correlation with content of oligomeric Tau (Fig. 7). To analyze the mode of action, we investigated whether oligomeric Tau gains access to the cell membrane. Alexa488-labeled Tau monomer and oligomer were applied to SH-SY5Y cells for 24 h. Fluorescence microscopy revealed more intracellular residence of oligomeric Tau protein compared with monomeric Tau protein (supplemental Fig. S4). Monomeric Tau protein is concentrated in the perinuclear region, where endocytic vesicles are located. We find oligomeric Tau protein more evenly distributed with some association with the cell membrane.
FIGURE 7.

High Tau oligomer content correlates with decreased cell viability. Oligomer content of Tau samples preaggregated for different periods of time and membrane integrity of treated SH-SY5Y measured by LDH release cells are highly correlated (r = 0.956, T = 5.61, p < 0.01).
Tau Oligomers Disturb Membrane Integrity of Artificial Phospholipid Vesicles
To further specify the effects of Tau aggregates on membrane integrity, we analyzed its influence on leakage of artificial phospholipid vesicles (Fig. 8). Phospholipid vesicles were filled with the fluorescence dye ANTS and the fluorescence quencher DPX. Upon vesicle leakage by pore formation, ANTS and DPX became separated, and the ANTS fluorescence yield increases. Phospholipid vesicles were treated with Tau preaggregated for 0, 24, or 144 h, and the increase of fluorescence dye intensity was monitored over a time of 220 min. In Fig. 8, the leakage of phospholipid vesicles is plotted as a function of time after applying preaggregated Tau. The vesicle leakage caused by preaggregated Tau appears to be a slow process. After 50–60 min of application of Tau preaggregated for 0, 24, and 144 h, a difference in vesicle leakage becomes visible. Tau preaggregated for 24 h causes the most dramatic vesicle leakage, which after 220 min reaches almost 100%. Tau preaggregated for 144 h and non-aggregated Tau (0 h) lead to a vesicle leakage of about 70 and 20%, respectively. This probably indicates that oligomeric Tau, which represents the major constituent of the Tau sample aggregated for 24 h, evokes the greatest disturbance of membrane integrity.
FIGURE 8.

Leakage of phospholipid vesicles treated with preaggregated Tau protein. Large unilamellar vesicles filled with the fluorescence dye ANTS and the quencher DPX were treated for 220 min with Tau protein preaggregated for 0, 24, or 144 h. During the time of treatment, the fluorescence yield of ANTS was measured. ANTS fluorescence increases upon separation from DPX, which is caused by vesicle leakage. Tau aggregated for 24 h induces the greatest vesicle leakage compared with long-aggregated and non-aggregated Tau.
DISCUSSION
The cytotoxic effects of amyloidogenic proteins have been explained by the concept that oligomers formed during the process of amyloidogenesis lead to a disturbance of membrane integrity by a non-channel mechanism or by forming pores into the membrane (39, 51).
The aim of the present study was to analyze whether this concept can also be applied to the Tau protein, representing an integral part of Alzheimer pathology. We thus analyzed whether, during Tau aggregation, a toxic species is generated, which can attack cellular membranes. To this end, we incubated the longest isoform of recombinant human Tau in the presence of heparin for different periods of time and analyzed the amount of oligomeric Tau that had formed after this time. Subsequently, we used these probes and tested their effects on cell viability and phospholipid vesicle leakage. As a result, we obtained a clear cut relationship between the content of oligomeric Tau contained in the probe and its deleterious effect on cell viability and phospholipid vesicle leakage.
Tau Aggregation and Oligomer Detection
For our experiments, we preaggregated Tau for different time periods in the presence of heparin. Subsequently, we characterized aggregation intermediates obtained after defined aggregation periods by (i) SDS-PAGE, (ii) bis-ANS and ThT fluorescence spectrometry; (iii) electron microscopy, (iv) quantification of Tau oligomers with an ELISA using an anti-oligomer antibody, and (v) size exclusion chromatography, followed by an analysis of protein content, β-sheet content, and exposed hydrophobic patches of each fraction. The hydrodynamic radius of peak fractions was measured by dynamic light scattering, which allowed the allocation of protein peaks to monomeric Tau, oligomeric Tau, and higher molecular Tau aggregates.
The biochemical mechanisms that lead to aggregation of Tau into paired helical filaments remain not fully understood. It has been shown that sulfated glycosaminoglycans like heparin or heparan sulfate can trigger Tau aggregation in vitro and that heparan sulfate is coexistent with hyperphosphorylated Tau in pretangle neurons (50). We thus used heparin as an effective trigger to accelerate Tau aggregation in vitro. Our SDS-PAGE data indicate that in the presence of heparin, Tau first undergoes conformational changes and then forms high molecular size aggregates in a time-dependent manner.
Bis-ANS and ThT fluorescence spectrometry enabled us to detect the partial structural transition from a random coil to a more hydrophobic cross-β structure in our preaggregated Tau samples. The hydrophobic fluorescent probe bis-ANS binds to solvent-accessible hydrophobic protein structures, resulting in an increase of bis-ANS fluorescence intensity (52). ThT binds to β-sheet structures, whereupon ThT fluorescence intensity increases (53, 54). Up to 48 h of Tau preaggregation, we observed a strong increase of fluorescence intensity with increasing preaggregation time for both dyes. From 48 to 144 h of Tau preaggregation, we found a slight increase of fluorescence intensity with increasing preaggregation time for both dyes. This result might indicate an equilibrium of Tau fibrillogenesis that is reached after about 48 h of preaggregation. Still, our SDS-PAGE data indicate a progression of fibrillogenesis up to 144 h. Here we observed a continuous depletion of monomeric Tau and an increase of high molecular smear up to 144 h of preaggregation. These findings might suggest that fluorescence dyes have a higher affinity to oligomers and small fibrils, which are more easily accessible than the long and compact fibrils. To quantify the content of oligomers in preaggregated Tau samples, we used two independent methods, which yielded consistent results (i.e. SEC combined with bis-ANS and ThT fluorescence spectrometry and dynamic light scattering of peak fractions and ELISA with an anti-oligomer antibody, A11).
The A11 antibody is known to recognize oligomers of amyloidogenic proteins independent of the amino acid sequence (46). With a sandwich ELISA, using the A11 antibody for capturing oligomers, we detected the highest oligomer content in the Tau samples aggregated for 24 and 48 h. Tau samples preaggregated for more than 48 h showed a reduced oligomer content, probably due to increasing filament formation, which consumes Tau oligomers. Tau protein samples preaggregated for less than 24 h exhibited the lowest oligomer content, indicating that a certain period of time needs to elapse before oligomers are formed to a detectable amount. These results are in agreement with the findings of Maeda et al. (55), who obtained the highest level of granular Tau oligomers consisting of 40 ± 3 Tau molecules after 21 h. The peak formation of oligomeric Tau after 24–48 h of aggregation time was confirmed through dynamic light scattering analysis of protein fractions separated by SEC. The oligomeric Tau species possesses an RH of 14 nm, which amounts to 40 monomers (55). Formation of Tau oligomers continuously increased until this time point, when oligomers accounted for about two-thirds of total Tau, and slightly decreased thereafter.
Influence of Preaggregated Tau on Cell Viability and Artificial Phospholipid Vesicles
Effects of Tau aggregation intermediates on cell viability and membrane integrity were analyzed on SH-SY5Y neuroblastoma cells and artificial phospholipid vesicles. SH-SY5Y cells were treated for 48 h with Tau preaggregated for up to 144 h. To characterize the effects on cell viability, we performed two assays. We measured MTT reduction and LDH release. MTT turnover of SH-SY5Y cells treated with Tau that were preaggregated for at least 6 h was significantly reduced compared with SH-SY5Y cells treated with non-aggregated Tau. Of note, effects were most pronounced for cells treated with probes that had been preaggregated for 24 or 48 h (i.e. which contained the highest amount of Tau oligomers). Similar results were obtained with the LDH assay, where the oligomer content of probes highly significantly correlated with its deleterious effects on membranes, leading to LDH release. Permeabilizing effects of Tau oligomers on membranes were corroborated further in the vesicle permeabilization assay, where we measured the leakage of the fluorescence dye ANTS out of phospholipid vesicles. In agreement with results from the LDH assay, effects were most pronounced for preaggregated Tau with the highest content of oligomers.
The present results suggest that Tau oligomers are the cytotoxic species of Tau fibrillogenesis that lead to an impairment of cell viability. Furthermore, our results identify the cellular membrane as a primary target for Tau oligomers.
These findings support previous concepts that amyloidogenic proteins are produced as soluble proteins that can progressively convert into lower molecular weight oligomers that represent the primary toxic species that contribute to disease progression (43, 51, 56). Amyloid-related diseases might thus be characterized by a common mechanism of aggregation, cytotoxicity and propagation of pathology, which critically involves permeabilization of membranes (57, 58). Several models have been proposed on how oligomers might lead to a disruption of cellular membranes, including transmembrane oligomeric pore structures reminiscent of those of core-forming toxins (57, 58), nonspecific binding of amyloid oligomers to the membrane surface (51, 56), and detergent-like membrane dissolution by amyloid fibrils growing on the membrane surface (59). Oligomers of amyloidogenic proteins, for example, are suspected to form circular porelike structures, which could puncture the cell membrane (31–34). Since the original finding by Arispe et al. (39) that Aβ exhibits ion channel activity in lipid bilayers, channel activity has been described for other amyloidogenic proteins, such as IAPP (60), α-synuclein (61–63), polyglutamine (64), and prion-derived peptides (65).
If one assumes that Tau oligomers might form a discrete pore into the cell membrane that would allow for the permeation of LDH (121 kDa; estimated radius = 4.6 nm), an inner diameter of at least 4.6 nm would be required. Lashuel et al. (34) showed that aggregation of 20–26 monomers of β-synuclein can form a pore with an inner diameter of 2.0–2.5 nm. Because granular Tau oligomers are proposed to consist of ∼40 monomers (55), formation of an inner pore with diameter of around 4.6 nm is not unlikely.
The effects of Tau aggregates on cell viability and vesicle leakage in the present experiments were assessed after external application of Tau samples. This external application was chosen because (i) recombinant overexpression of Tau in SH-SY5Y cells would implicate a time lag in expression, which could be avoided with an external treatment; (ii) the aggregation state of Tau can be determined precisely prior to external application; (iii) external application more likely attacks the membrane, thereby generating effects that can be related to Tau and its aggregation state; and (iv) effects are independent of posttranslational modifications, which might modulate intracellular Tau protein. Testing potential cytotoxic effects of Tau as an intracellular protein by external application might be counterintuitive. Still, recent results on dynamic spreading of Tau pathology in mouse models of Alzheimer disease (66, 67) indicate a transcellular propagation of Tau aggregates, which might require an inside-out and outside-in passage through the cell membrane. Because of phospholipid asymmetry in the plasma membrane, it remains unclear whether these two passages rely on the same mechanism. Accordingly, cell culture models have shown that extracellular Tau aggregates can be internalized, transmitting Tau misfolding from the outside to the inside of the cell (68). The present experimental paradigm of external application of oligomeric Tau aggregates, thus, very likely, might play a pathophysiological role in progression of AD. Current models on spreading of Tau pathology suggest a prion-like mechanism, where misfolded Tau might act as seed propagating further aggregation of Tau. Transmembrane spreading of Tau oligomers might well be involved in this initiation of widespread formation of fibrillar Tau pathology in AD. In support of this, formation of the MC1 epitope, as one of the earliest pathological alterations of Tau in AD (69), has been identified as occurring during oligomerization of Tau (70).
In summary, our results suggest Tau aggregation intermediates to be the toxic species of Tau fibrillogenesis. Tau aggregation intermediates disturb the integrity of biological membranes, probably by a pore-forming mechanism.
Acknowledgments
We thank M. Goedert for providing plasmid prk172ht40. We also thank B. Würz for providing help with the Zetasizer measurements.
This work was supported, in part, by Deutsche Forschungsgemeinschaft Grant HO2368-4/1 and by Alzheimer Forschung Initiative Project 06825.

This article contains supplemental Figs. S1–S5.
- AD
- Alzheimer disease
- bis-ANS
- 4,4′-dianilino-1,1′-binaphthyl-5,5′-disulfonic acid
- ANTS
- 8-aminonaphthalene-1,3,6-trisulfonic acid
- DPX
- p-xylene-bis-pyridinium bromide
- LDH
- lactate dehydrogenase
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- ThT
- thioflavin T
- SEC
- size exclusion chromatography
- RH
- hydrodynamic radius.
REFERENCES
- 1. Goedert M., Spillantini M. G., Jakes R., Rutherford D., Crowther R. A. (1989) Multiple isoforms of human microtubule-associated protein Tau. Sequences and localization in neurofibrillary tangles of Alzheimer's disease. Neuron 3, 519–526 [DOI] [PubMed] [Google Scholar]
- 2. Weingarten M. D., Lockwood A. H., Hwo S. Y., Kirschner M. W. (1975) A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. U.S.A. 72, 1858–1862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cleveland D. W., Hwo S. Y., Kirschner M. W. (1977) Physical and chemical properties of purified Tau factor and the role of Tau in microtubule assembly. J. Mol. Biol. 116, 227–247 [DOI] [PubMed] [Google Scholar]
- 4. Binder L. I., Frankfurter A., Rebhun L. I. (1985) The distribution of Tau in the mammalian central nervous system. J. Cell Biol. 101, 1371–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brandt R., Lee G. (1993) Functional organization of microtubule-associated protein Tau. Identification of regions which affect microtubule growth, nucleation, and bundle formation in vitro. J. Biol. Chem. 268, 3414–3419 [PubMed] [Google Scholar]
- 6. Bramblett G. T., Goedert M., Jakes R., Merrick S. E., Trojanowski J. Q., Lee V. M. (1993) Abnormal Tau phosphorylation at Ser396 in Alzheimer's disease recapitulates development and contributes to reduced microtubule binding. Neuron 10, 1089–1099 [DOI] [PubMed] [Google Scholar]
- 7. Buée L., Bussière T., Buée-Scherrer V., Delacourte A., Hof P. R. (2000) Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Brain Res. Rev. 33, 95–130 [DOI] [PubMed] [Google Scholar]
- 8. Braak H., Braak E. (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259 [DOI] [PubMed] [Google Scholar]
- 9. Holzer M., Holzapfel H. P., Zedlick D., Brückner M. K., Arendt T. (1994) Abnormally phosphorylated Tau protein in Alzheimer's disease. Heterogeneity of individual regional distribution and relationship to clinical severity. Neuroscience 63, 499–516 [DOI] [PubMed] [Google Scholar]
- 10. Bancher C., Brunner C., Lassmann H., Budka H., Jellinger K., Wiche G., Seitelberger F., Grundke-Iqbal I., Iqbal K., Wisniewski H. M. (1989) Accumulation of abnormally phosphorylated Tau precedes the formation of neurofibrillary tangles in Alzheimer's disease. Brain Res. 477, 90–99 [DOI] [PubMed] [Google Scholar]
- 11. von Bergen M., Friedhoff P., Biernat J., Heberle J., Mandelkow E. M., Mandelkow E. (2000) Assembly of Tau protein into Alzheimer paired helical filaments depends on a local sequence motif (306VQIVYK311) forming β structure. Proc. Natl. Acad. Sci. U.S.A. 97, 5129–5134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. von Bergen M., Barghorn S., Biernat J., Mandelkow E. M., Mandelkow E. (2005) Tau aggregation is driven by a transition from random coil to β sheet structure. Biochim. Biophys. Acta 1739, 158–166 [DOI] [PubMed] [Google Scholar]
- 13. Berriman J., Serpell L. C., Oberg K. A., Fink A. L., Goedert M., Crowther R. A. (2003) Tau filaments from human brain and from in vitro assembly of recombinant protein show cross-β structure. Proc. Natl. Acad. Sci. U.S.A. 100, 9034–9038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Andronesi O. C., von Bergen M., Biernat J., Seidel K., Griesinger C., Mandelkow E., Baldus M. (2008) Characterization of Alzheimer's-like paired helical filaments from the core domain of Tau protein using solid-state NMR spectroscopy. J. Am. Chem. Soc. 130, 5922–5928 [DOI] [PubMed] [Google Scholar]
- 15. Ross C. A., Poirier M. A. (2004) Protein aggregation and neurodegenerative disease. Nat. Med. 10, S10–S17 [DOI] [PubMed] [Google Scholar]
- 16. Chiti F., Dobson C. M. (2006) Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75, 333–366 [DOI] [PubMed] [Google Scholar]
- 17. Alzheimer A. (1907) Über eine eigenartige Erkrankung der Hirnrinde. Allg. Z. Psychatr. 64, 146–148 [Google Scholar]
- 18. Price D. L., Cork L. C., Struble R. G., Kitt C. A., Walker L. C., Powers R. E., Whitehouse P. J., Griffin J. W. (1987) Dysfunction and death of neurons in human degenerative neurological diseases and in animal models. Ciba Found. Symp. 126, 30–48 [DOI] [PubMed] [Google Scholar]
- 19. Ihara Y. (1988) Massive somatodendritic sprouting of cortical neurons in Alzheimer's disease. Brain Res. 459, 138–144 [DOI] [PubMed] [Google Scholar]
- 20. Chartier-Harlin M. C., Crawford F., Houlden H., Warren A., Hughes D., Fidani L., Goate A., Rossor M., Roques P., Hardy J. (1991) Early-onset Alzheimer's disease caused by mutations at codon 717 of the β-amyloid precursor protein gene. Nature 353, 844–846 [DOI] [PubMed] [Google Scholar]
- 21. Polymeropoulos M. H., Lavedan C., Leroy E., Ide S. E., Dehejia A., Dutra A., Pike B., Root H., Rubenstein J., Boyer R., Stenroos E. S., Chandrasekharappa S., Athanassiadou A., Papapetropoulos T., Johnson W. G., Lazzarini A. M., Duvoisin R. C., Di Iorio G., Golbe L. I., Nussbaum R. L. (1997) Mutation in the α-synuclein gene identified in families with Parkinson's disease. Science 276, 2045–2047 [DOI] [PubMed] [Google Scholar]
- 22. Hutton M., Lendon C. L., Rizzu P., Baker M., Froelich S., Houlden H., Pickering-Brown S., Chakraverty S., Isaacs A., Grover A., Hackett J., Adamson J., Lincoln S., Dickson D., Davies P., Petersen R. C., Stevens M., de Graaff E., Wauters E., van Baren J., Hillebrand M., Joosse M., Kwon J. M., Nowotny P., Che L. K., Norton J., Morris J. C., Reed L. A., Trojanowski J., Basun H., Lannfelt L., Neystat M., Fahn S., Dark F., Tannenberg T., Dodd P. R., Hayward N., Kwok J. B., Schofield P. R., Andreadis A., Snowden J., Craufurd D., Neary D., Owen F., Oostra B. A., Hardy J., Goate A., van Swieten J., Mann D., Lynch T., Heutink P. (1998) Association of missense and 5′-splice-site mutations in Tau with the inherited dementia FTDP-17. Nature 393, 702–705 [DOI] [PubMed] [Google Scholar]
- 23. Krüger R., Kuhn W., Müller T., Woitalla D., Graeber M., Kösel S., Przuntek H., Epplen J. T., Schöls L., Riess O. (1998) Ala30Pro mutation in the gene encoding α-synuclein in Parkinson's disease. Nat. Genet. 18, 106–108 [DOI] [PubMed] [Google Scholar]
- 24. Conway K. A., Harper J. D., Lansbury P. T. (1998) Accelerated in vitro fibril formation by a mutant α-synuclein linked to early-onset Parkinson disease. Nat. Med. 4, 1318–1320 [DOI] [PubMed] [Google Scholar]
- 25. Goedert M., Jakes R., Crowther R. A. (1999) Effects of frontotemporal dementia FTDP-17 mutations on heparin-induced assembly of Tau filaments. FEBS Lett. 450, 306–311 [DOI] [PubMed] [Google Scholar]
- 26. Barghorn S., Zheng-Fischhöfer Q., Ackmann M., Biernat J., von Bergen M., Mandelkow E. M., Mandelkow E. (2000) Structure, microtubule interactions, and paired helical filament aggregation by Tau mutants of frontotemporal dementias. Biochemistry 39, 11714–11721 [DOI] [PubMed] [Google Scholar]
- 27. Johansson A. S., Berglind-Dehlin F., Karlsson G., Edwards K., Gellerfors P., Lannfelt L. (2006) Physiochemical characterization of the Alzheimer's disease-related peptides A β 1–42Arctic and A β 1–42wt. FEBS J. 273, 2618–2630 [DOI] [PubMed] [Google Scholar]
- 28. Chen G., Chen K. S., Knox J., Inglis J., Bernard A., Martin S. J., Justice A., McConlogue L., Games D., Freedman S. B., Morris R. G. (2000) A learning deficit related to age and β-amyloid plaques in a mouse model of Alzheimer's disease. Nature 408, 975–979 [DOI] [PubMed] [Google Scholar]
- 29. Wong P. C., Cai H., Borchelt D. R., Price D. L. (2002) Genetically engineered mouse models of neurodegenerative diseases. Nat. Neurosci. 5, 633–639 [DOI] [PubMed] [Google Scholar]
- 30. Santacruz K., Lewis J., Spires T., Paulson J., Kotilinek L., Ingelsson M., Guimaraes A., DeTure M., Ramsden M., McGowan E., Forster C., Yue M., Orne J., Janus C., Mariash A., Kuskowski M., Hyman B., Hutton M., Ashe K. H. (2005) Tau suppression in a neurodegenerative mouse model improves memory function. Science 309, 476–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Conway K. A., Lee S. J., Rochet J. C., Ding T. T., Williamson R. E., Lansbury P. T., Jr. (2000) Acceleration of oligomerization, not fibrillization, is a shared property of both α-synuclein mutations linked to early-onset Parkinson's disease. Implications for pathogenesis and therapy. Proc. Natl. Acad. Sci. U.S.A. 97, 571–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ding T. T., Lee S. J., Rochet J. C., Lansbury P. T., Jr. (2002) Annular α-synuclein protofibrils are produced when spherical protofibrils are incubated in solution or bound to brain-derived membranes. Biochemistry 41, 10209–10217 [DOI] [PubMed] [Google Scholar]
- 33. Lashuel H. A., Hartley D. M., Petre B. M., Walz T., Lansbury P. T., Jr. (2002) Neurodegenerative disease. Amyloid pores from pathogenic mutations. Nature 418, 291. [DOI] [PubMed] [Google Scholar]
- 34. Lashuel H. A., Petre B. M., Wall J., Simon M., Nowak R. J., Walz T., Lansbury P. T., Jr. (2002) α-Synuclein, especially the Parkinson's disease-associated mutants, forms pore-like annular and tubular protofibrils. J. Mol. Biol. 322, 1089–1102 [DOI] [PubMed] [Google Scholar]
- 35. Klein W. L., Krafft G. A., Finch C. E. (2001) Targeting small Aβ oligomers. The solution to an Alzheimer's disease conundrum? Trends Neurosci. 24, 219–224 [DOI] [PubMed] [Google Scholar]
- 36. Kirkitadze M. D., Bitan G., Teplow D. B. (2002) Paradigm shifts in Alzheimer's disease and other neurodegenerative disorders. The emerging role of oligomeric assemblies. J. Neurosci. Res. 69, 567–577 [DOI] [PubMed] [Google Scholar]
- 37. Bucciantini M., Giannoni E., Chiti F., Baroni F., Formigli L., Zurdo J., Taddei N., Ramponi G., Dobson C. M., Stefani M. (2002) Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 416, 507–511 [DOI] [PubMed] [Google Scholar]
- 38. Caughey B., Lansbury P. T. (2003) Protofibrils, pores, fibrils, and neurodegeneration. Separating the responsible protein aggregates from the innocent bystanders. Annu. Rev. Neurosci. 26, 267–298 [DOI] [PubMed] [Google Scholar]
- 39. Arispe N., Pollard H. B., Rojas E. (1994) β-Amyloid Ca2+-channel hypothesis for neuronal death in Alzheimer disease. Mol. Cell Biochem. 140, 119–125 [DOI] [PubMed] [Google Scholar]
- 40. Kostka M., Högen T., Danzer K. M., Levin J., Habeck M., Wirth A., Wagner R., Glabe C. G., Finger S., Heinzelmann U., Garidel P., Duan W., Ross C. A., Kretzschmar H., Giese A. (2008) Single particle characterization of iron-induced pore-forming α-synuclein oligomers. J. Biol. Chem. 283, 10992–11003 [DOI] [PubMed] [Google Scholar]
- 41. Butterfield S. M., Lashuel H. A. (2010) Amyloidogenic protein-membrane interactions. Mechanistic insight from model systems. Angew. Chem. Int. Ed. Engl. 49, 5628–5654 [DOI] [PubMed] [Google Scholar]
- 42. Pountney D. L., Voelcker N. H., Gai W. P. (2005) Annular α-synuclein oligomers are potentially toxic agents in α-synucleinopathy. Hypothesis Neurotox. Res. 7, 59–67 [DOI] [PubMed] [Google Scholar]
- 43. Haataja L., Gurlo T., Huang C. J., Butler P. C. (2008) Islet amyloid in type 2 diabetes, and the toxic oligomer hypothesis. Endocr. Rev. 29, 303–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Goedert M. (1999) Filamentous nerve cell inclusions in neurodegenerative diseases. Tauopathies and α-synucleinopathies. Philos. Trans. R. Soc. Lond. B Biol. Sci. 354, 1101–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bodles A. M., Guthrie D. J., Harriott P., Campbell P., Irvine G. B. (2000) Toxicity of non-Aβ component of Alzheimer's disease amyloid, and N-terminal fragments thereof, correlates to formation of β-sheet structure and fibrils. Eur. J. Biochem. 267, 2186–2194 [DOI] [PubMed] [Google Scholar]
- 46. Kayed R., Head E., Thompson J. L., McIntire T. M., Milton S. C., Cotman C. W., Glabe C. G. (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300, 486–489 [DOI] [PubMed] [Google Scholar]
- 47. Yoshida H., Goedert M. (2002) Molecular cloning and functional characterization of chicken brain Tau. Isoforms with up to five tandem repeats. Biochemistry 41, 15203–15211 [DOI] [PubMed] [Google Scholar]
- 48. Haase C., Stieler J. T., Arendt T., Holzer M. (2004) Pseudophosphorylation of Tau protein alters its ability for self-aggregation. J. Neurochem. 88, 1509–1520 [DOI] [PubMed] [Google Scholar]
- 49. Zschörnig O., Paasche G., Thieme C., Korb N., Arnold K. (2005) Modulation of lysozyme charge influences interaction with phospholipid vesicles. Colloids Surf. B Biointerfaces 42, 69–78 [DOI] [PubMed] [Google Scholar]
- 50. Goedert M., Jakes R., Spillantini M. G., Hasegawa M., Smith M. J., Crowther R. A. (1996) Assembly of microtubule-associated protein Tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature 383, 550–553 [DOI] [PubMed] [Google Scholar]
- 51. Kayed R., Sokolov Y., Edmonds B., McIntire T. M., Milton S. C., Hall J. E., Glabe C. G. (2004) Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J. Biol. Chem. 279, 46363–46366 [DOI] [PubMed] [Google Scholar]
- 52. Semisotnov G. V., Rodionova N. A., Razgulyaev O. I., Uversky V. N., Gripas' A. F., Gilmanshin R. I. (1991) Study of the “molten globule” intermediate state in protein folding by a hydrophobic fluorescent probe. Biopolymers 31, 119–128 [DOI] [PubMed] [Google Scholar]
- 53. LeVine H., 3rd (1993) Thioflavine T interaction with synthetic Alzheimer's disease β-amyloid peptides. Detection of amyloid aggregation in solution. Protein Sci. 2, 404–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Krebs M. R., Bromley E. H., Donald A. M. (2005) The binding of thioflavin-T to amyloid fibrils. Localisation and implications. J. Struct. Biol. 149, 30–37 [DOI] [PubMed] [Google Scholar]
- 55. Maeda S., Sahara N., Saito Y., Murayama M., Yoshiike Y., Kim H., Miyasaka T., Murayama S., Ikai A., Takashima A. (2007) Granular Tau oligomers as intermediates of Tau filaments. Biochemistry 46, 3856–3861 [DOI] [PubMed] [Google Scholar]
- 56. Sokolov Y., Kozak J. A., Kayed R., Chanturiya A., Glabe C., Hall J. E. (2006) Soluble amyloid oligomers increase bilayer conductance by altering dielectric structure. J. Gen. Physiol. 128, 637–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Quist A., Doudevski I., Lin H., Azimova R., Ng D., Frangione B., Kagan B., Ghiso J., Lal R. (2005) Amyloid ion channels. A common structural link for protein-misfolding disease. Proc. Natl. Acad. Sci. U.S.A. 102, 10427–10432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lashuel H. A., Lansbury P. T., Jr. (2006) Are amyloid diseases caused by protein aggregates that mimic bacterial pore-forming toxins? Q. Rev. Biophys. 39, 167–201 [DOI] [PubMed] [Google Scholar]
- 59. Engel M. F., Khemtémourian L., Kleijer C. C., Meeldijk H. J., Jacobs J., Verkleij A. J., de Kruijff B., Killian J. A., Höppener J. W. (2008) Membrane damage by human islet amyloid polypeptide through fibril growth at the membrane. Proc. Natl. Acad. Sci. U.S.A. 105, 6033–6038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mirzabekov T. A., Lin M. C., Kagan B. L. (1996) Pore formation by the cytotoxic islet amyloid peptide amylin. J. Biol. Chem. 271, 1988–1992 [DOI] [PubMed] [Google Scholar]
- 61. Volles M. J., Lee S. J., Rochet J. C., Shtilerman M. D., Ding T. T., Kessler J. C., Lansbury P. T., Jr. (2001) Vesicle permeabilization by protofibrillar α-synuclein. Implications for the pathogenesis and treatment of Parkinson's disease. Biochemistry 40, 7812–7819 [DOI] [PubMed] [Google Scholar]
- 62. Volles M. J., Lansbury P. T., Jr. (2002) Vesicle permeabilization by protofibrillar α-synuclein is sensitive to Parkinson's disease-linked mutations and occurs by a pore-like mechanism. Biochemistry 41, 4595–4602 [DOI] [PubMed] [Google Scholar]
- 63. Kim H. Y., Cho M. K., Kumar A., Maier E., Siebenhaar C., Becker S., Fernandez C. O., Lashuel H. A., Benz R., Lange A., Zweckstetter M. (2009) Structural properties of pore-forming oligomers of α-synuclein. J. Am. Chem. Soc. 131, 17482–17489 [DOI] [PubMed] [Google Scholar]
- 64. Monoi H., Futaki S., Kugimiya S., Minakata H., Yoshihara K. (2000) Poly-L-glutamine forms cation channels. Relevance to the pathogenesis of the polyglutamine diseases. Biophys. J. 78, 2892–2899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kourie J. I., Farrelly P. V., Henry C. L. (2001) Channel activity of deamidated isoforms of prion protein fragment 106–126 in planar lipid bilayers. J. Neurosci. Res. 66, 214–220 [DOI] [PubMed] [Google Scholar]
- 66. Clavaguera F., Bolmont T., Crowther R. A., Abramowski D., Frank S., Probst A., Fraser G., Stalder A. K., Beibel M., Staufenbiel M., Jucker M., Goedert M., Tolnay M. (2009) Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 11, 909–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. de Calignon A., Polydoro M., Suárez-Calvet M., William C., Adamowicz D. H., Kopeikina K. J., Pitstick R., Sahara N., Ashe K. H., Carlson G. A., Spires-Jones T. L., Hyman B. T. (2012) Propagation of Tau pathology in a model of early Alzheimer's disease. Neuron 73, 685–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Frost B., Jacks R. L., Diamond M. I. (2009) Propagation of Tau misfolding from the outside to the inside of a cell. J. Biol. Chem. 284, 12845–12852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Weaver C. L., Espinoza M., Kress Y., Davies P. (2000) Conformational change as one of the earliest alterations of Tau in Alzheimer's disease. Neurobiol. Aging 21, 719–727 [DOI] [PubMed] [Google Scholar]
- 70. Bibow S., Mukrasch M. D., Chinnathambi S., Biernat J., Griesinger C., Mandelkow E., Zweckstetter M. (2011) The dynamic structure of filamentous Tau. Angew. Chem. Int. Ed. Engl. 50, 11520–11524 [DOI] [PubMed] [Google Scholar]