

Background: The bifunctional AAC(6′)-Ie/APH(2″)-Ia enzyme was reported to phosphorylate all classes of aminoglycoside antibiotics using ATP.
Results: GTP, and not ATP, is the cosubstrate of the enzyme. 4,5-disubstituted and atypical aminoglycosides are not substrates.
Conclusion: The enzyme is a narrow spectrum GTP-dependent kinase that phosphorylates 4,6-disubstituted aminoglycosides exclusively.
Significance: Knowledge of enzyme activity is essential for developing novel antibiotics and conducting effective antimicrobial therapy.
Keywords: Antibiotic Resistance, Enzyme Catalysis, Enzyme Kinetics, GTPase, Kinetics, Aminoglycosides, Phosphotransferase
Abstract
The bifunctional aminoglycoside-modifying enzyme aminoglycoside acetyltransferase(6′)-Ie/aminoglycoside phosphotransferase(2″)-Ia, or AAC(6′)-Ie/APH(2″)-Ia, is the major source of aminoglycoside resistance in Gram-positive bacterial pathogens. In previous studies, using ATP as the cosubstrate, it was reported that the APH(2″)-Ia domain of this enzyme is unique among aminoglycoside phosphotransferases, having the ability to inactivate an unusually broad spectrum of aminoglycosides, including 4,6- and 4,5-disubstituted and atypical. We recently demonstrated that GTP, and not ATP, is the preferred cosubstrate of this enzyme. We now show, using competition assays between ATP and GTP, that GTP is the exclusive phosphate donor at intracellular nucleotide levels. In light of these findings, we reevaluated the substrate profile of the phosphotransferase domain of this clinically important enzyme. Steady-state kinetic characterization using the phosphate donor GTP demonstrates that AAC(6′)-Ie/APH(2″)-Ia phosphorylates 4,6-disubstituted aminoglycosides with high efficiency (kcat/Km = 105-107 m−1 s−1). Despite this proficiency, no resistance is conferred to some of these antibiotics by the enzyme in vivo. We now show that phosphorylation of 4,5-disubstituted and atypical aminoglycosides are negligible and thus these antibiotics are not substrates. Instead, these aminoglycosides tend to stimulate an intrinsic GTPase activity of the enzyme. Taken together, our data show that the bifunctional enzyme efficiently phosphorylates only 4,6-disubstituted antibiotics; however, phosphorylation does not necessarily result in bacterial resistance. Hence, the APH(2″)-Ia domain of the bifunctional AAC(6′)-Ie/APH(2″)-Ia enzyme is a bona fide GTP-dependent kinase with a narrow substrate profile, including only 4,6-disubstituted aminoglycosides.
Introduction
Aminoglycosides are broad spectrum antibiotics used to treat a wide variety of bacterial infections caused by both Gram-negative and Gram-positive pathogens (1). Based on their structure, they are broadly classified into three major classes: 4,6-disubstituted, 4,5-disubstituted, and atypical (Fig. 1). The most important mechanism of bacterial resistance to this class of antibiotics is enzymatic modification, which greatly diminishes the affinity of aminoglycosides for the bacterial 30 S ribosomal subunit, their target in vivo (2, 3). Three families of aminoglycoside-modifying enzymes are known: aminoglycoside O-nucleotidyltransferases, aminoglycoside N-acetyltransferases, and aminoglycoside O-phosphotransferases. Most of these enzymes are monofunctional; however, a few bifunctional enzymes, thought to result from the fusion of two genes, are known to exist (4).
FIGURE 1.
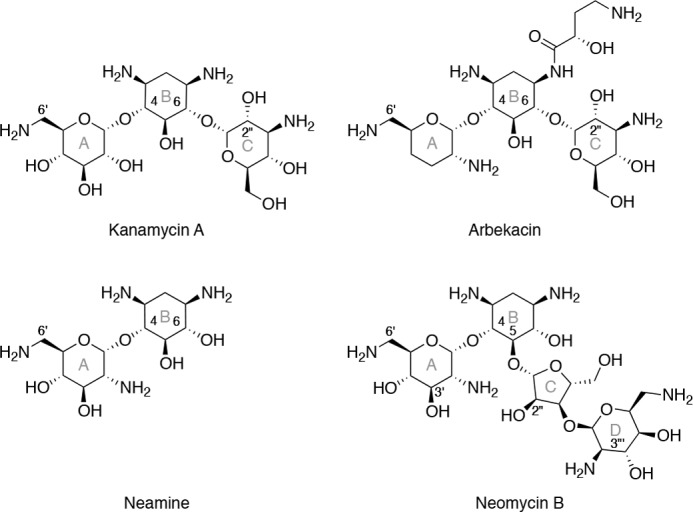
The structures of several aminoglycoside antibiotics are shown, representing the three structural classes: 4,6-disubstituted (kanamycin A and arbekacin), atypical (neamine), and 4,5-disubstituted (neomycin B). Numerals are employed to denote certain ring positions, whereas letters (in gray) are used to indicate the rings.
The bifunctional AAC(6′)-Ie/APH(2″)-Ia2 enzyme is the most clinically important aminoglycoside-modifying enzyme in Gram-positive bacteria, responsible for high-level resistance in both Enterococcus and Staphylococcus (3, 4). The gene for this unique enzyme encodes an N-terminal AAC(6′) domain and a C-terminal APH(2″) domain. Both domains can function independently (5) and have been reported to inactivate, through phosphorylation and/or acetylation, most aminoglycoside antibiotics (6, 7) (Fig. 2).
FIGURE 2.
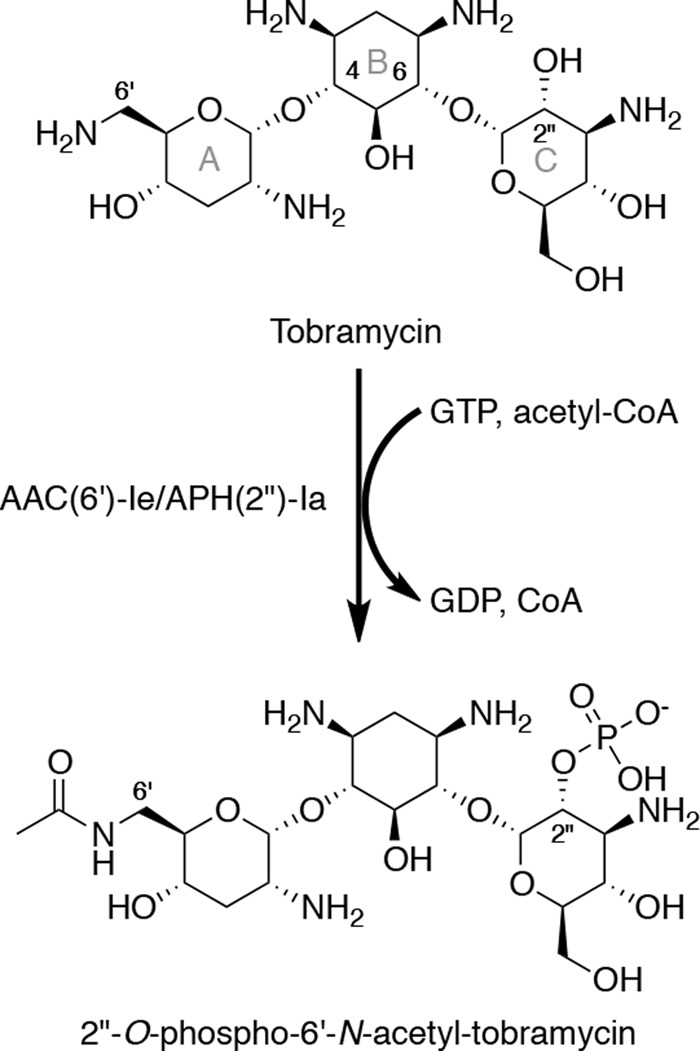
2″-O-phosphorylation and 6′-N-acetylation of tobramycin by AAC(6′)-Ie/APH(2″)-Ia.
Earlier enzymological studies of AAC(6′)-Ie/APH(2″)-Ia have led to the notion that APH(2″)-Ia is unique among aminoglycoside phosphotransferases due to its extremely broad substrate specificity (6). It was reported that both 4,6-disubstituted and 4,5-disubstituted aminoglycosides and neamine, an atypical aminoglycoside, are substrates for APH(2″)-Ia when ATP is used as the cosubstrate (6). This has led to the assumption that aminoglycoside antibiotics cannot be used for the treatment of infections caused by bacteria producing AAC(6′)-Ie/APH(2″)-Ia (1).
Since the discovery of AAC(6′)-Ie/APH(2″)-Ia, three additional APH(2″) enzymes (APH(2″)-IIa, -IIIa, and -IVa) have been identified. These members of the aminoglycoside 2″ kinase subfamily share only 28–32% amino acid sequence identity among themselves and the APH(2″) domain of the bifunctional enzyme (8). We and others have shown that APH(2″)-IIa and -IVa have comparable relative affinities for both ATP and GTP, whereas the two others preferentially bind GTP, an indication that GTP and not ATP is the physiological cosubstrate for these enzymes (9, 10). Contrary to what has been reported for the APH(2″)-Ia domain of the bifunctional enzyme, our kinetic characterization of APH(2″)-IIa, -IIIa, and -IVa demonstrated that 4,5-disubstituted antibiotics and the atypical aminoglycoside neamine are not substrates, but inhibitors of these enzymes (11–13). In light of the substantial differences in the reported substrate specificities of APH(2″)-Ia and the remaining members of the APH(2″) subfamily of aminoglycoside kinases and the discovery that GTP, and not ATP, is the preferred cosubstrate for APH(2″)-Ia, we sought to reevaluate the nucleotide and aminoglycoside substrate profiles of the clinically important AAC(6′)-Ie/APH(2″)-Ia enzyme.
EXPERIMENTAL PROCEDURES
Aminoglycoside Reagents
Amikacin was purchased from the United States Pharmacopeial Convention (Rockville, MD). Arbekacin, dibekacin, and kanamycin B were obtained from Meiji Seika Kaisha, Ltd. (Tokyo, Japan). Neamine was a gift from S. Mobashery (University of Notre Dame). Kanamycin A, gentamicin C, sisomicin, tobramycin, butirosin A and B, lividomycin A, paromomycin, and neomycin B were bought from Sigma. Netilmicin was obtained from Schering-Plough (Kenilworth, NJ).
MIC Determinations
The native gene for AAC(6′)-Ie/APH(2″)-Ia was custom-synthesized (GenScript) and cloned between the NdeI and HindIII restriction sites of pBluescript II KS (+) under the control of the promoter for aph(2″)-IIIa (pBluescript::AAC(6′)-Ie/APH(2″)-Ia) (10). The MICs of selected aminoglycoside antibiotics were determined by the broth microdilution method as recommended by the Clinical and Laboratory Standards Institute (14). AAC(6′)-Ie/APH(2″)-Ia was expressed in Escherichia coli JM83 using the above plasmid. E. coli JM83 without the plasmid was used as a control. The MICs were evaluated in Mueller-Hinton II broth (Difco) using a bacterial inoculum of 5 × 105 cfu/ml. All plates were incubated at 37 °C for 16–20 h before results were interpreted.
Cloning, Expression, and Purification of AAC(6′)-Ie/APH(2″)-Ia
The gene for AAC(6′)-Ie/APH(2″)-Ia, optimized for expression in E. coli, was custom-synthesized (GenScript) and cloned between the NdeI and HindIII restriction sites of pET22b(+). The recombinant plasmid (pET22::AAC(6′)-Ie/APH(2″)-Ia) was transformed into E. coli BL21 (DE3), and transformants were selected on LB agar plates supplemented with 100 μg/ml ampicillin. Selected cells were grown in LB medium (300 ml) supplemented with 100 μg/ml ampicillin at 37 °C until A600 = 0.7. Isopropyl-β-d-thiogalactoside was added to a final concentration of 0.6 mm to induce protein expression, and the cells were further grown at 22 °C for an additional 20 h. The bacteria were harvested by centrifugation at 4000 × g, 4 °C for 15 min, resuspended in 30 ml of 25 mm HEPES (pH 7.5) containing 1 mm EDTA and 0.2 mm DTT, and disrupted by sonication at 4 °C. The insoluble material was removed by centrifugation at 20,000 × g, 4 °C for 30 min, and nucleic acids were precipitated from the soluble fraction using 1.5% (w/v) streptomycin sulfate. The precipitate was removed by centrifugation, and the sample was dialyzed against 25 mm HEPES (pH 7.5) at 4 °C. After clarifying the soluble proteins by centrifugation at 20,000 × g, 4 °C for 30 min, the sample was loaded onto an Affi-Gel 15-gentamicin affinity column equilibrated with 25 mm HEPES (pH 7.5) (10). AAC(6′)-Ie/APH(2″)-Ia was eluted using a linear NaCl gradient (200–800 mm) in 25 mm HEPES (pH 7.5), and fractions positive for aminoglycoside phosphotransferase activity were analyzed by 12% SDS-PAGE. Fractions containing AAC(6′)-Ie/APH(2″)-Ia were pooled, concentrated, and dialyzed against 25 mm HEPES (pH 7.5). The dialyzed sample was loaded onto a DEAE anion exchange column equilibrated with 25 mm HEPES (pH 7.5), and AAC(6′)-Ie/APH(2″)-Ia was eluted in 25 mm HEPES (pH 7.5) using a linear NaCl gradient (0–800 mm). Purified AAC(6′)-Ie/APH(2″)-Ia (>95% pure as judged by SDS-PAGE) was recovered from fractions containing 400–450 mm NaCl. These fractions were pooled, concentrated, and dialyzed against 25 mm HEPES (pH 7.5) and stored in liquid N2. The protein concentration was evaluated using the Pierce® BCA protein assay kit (Thermo Scientific).
Coupled Phosphate Transfer Assay
Enzyme activity was continuously monitored at room temperature by coupling the production of GDP, from AAC(6′)-Ie/APH(2″)-Ia-catalyzed phosphate transfer, to pyruvate kinase and lactate dehydrogenase as described previously (10). Reaction mixtures containing 100 mm HEPES (pH 7.0), 10 mm MgCl2, 20 mm KCl, 2 mm phosphoenolpyruvate, 100 μm NADH, 20 units/ml pyruvate kinase, 25 units/ml lactate dehydrogenase, saturating GTP (100 μm), and varying concentrations of aminoglycoside(s) were initiated by the addition of enzyme (5–200 nm final) and monitored at 340 nm (Δϵ340 = −6200 cm−1 m−1) on a Cary 60 UV-visible spectrophotometer (Agilent). The steady-state velocities (v) were plotted versus the concentration of aminoglycoside and fit nonlinearly with the Michaelis-Menten equation using Prism 5 (GraphPad Software, Inc.) to determine the steady-state kinetic parameters kcat and Km.
The inhibitor dissociation constants for butirosin A and B, lividomycin A, and paromomycin were determined by the method of Dixon (15) using two fixed concentrations of amikacin (40 and 60 μm) and varying concentrations of the inhibitor. The inhibitor dissociation constants for neomycin B and neamine were evaluated using a single fixed concentration of substrate (amikacin, 40 μm, or sisomicin, 20 μm) and varying concentrations of the inhibitor and fitting the fractional activity (vi/vo) with Equation 1 using Prism 5 (16).
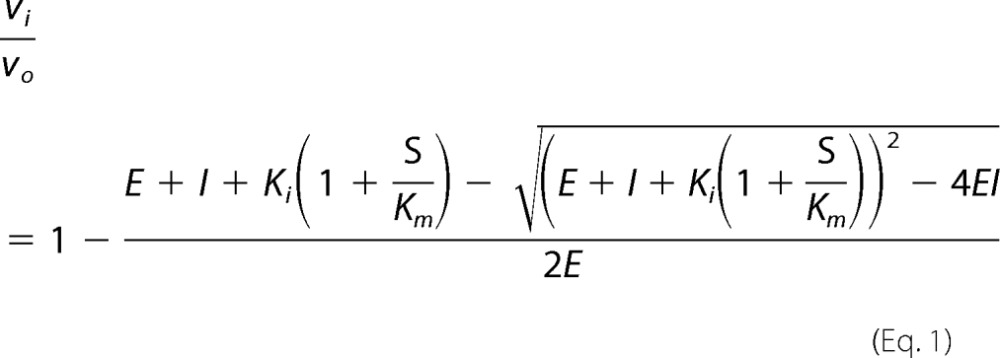 |
where vi is the steady-state velocity in the presence of inhibitor, vo is the steady-state velocity in the absence of inhibitor, E is the concentration of enzyme, I is the concentration of inhibitor, Ki is the inhibitor dissociation constant, S is the concentration of substrate, and Km is the Michaelis-Menten constant for the substrate.
Radiolabeled Phosphate Transfer Assay
Enzyme activity was monitored discontinuously at room temperature using a modified version of a previously described assay (17). Reaction mixtures containing 100 mm HEPES (pH 7.0), 10 mm MgCl2, and varying concentrations of GTP, ATP, and aminoglycoside were initiated by the addition of enzyme (0.25–3 μm final). The GTP stock contained 5–10% (v/v) [γ-32P]GTP (PerkinElmer Life Sciences). At various time points, aliquots were quenched in an equal volume of 0.5 n formic acid and spotted on a PEI-cellulose TLC plate (10 cm × 20 cm). The plates were developed in 0.3 m potassium Pi (pH 3.4) for 7 min followed by quantification of the radiolabeled substrate, [γ-32P]GTP, and products, [32P]Pi and 32P-labeled aminoglycoside, using a Storm 840 PhosphorImager and the ImageQuant 5.2 software (Amersham Biosciences). The concentration of substrate or product was calculated using Equations 2–4.
 |
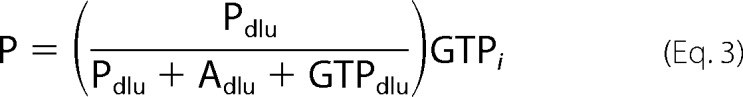 |
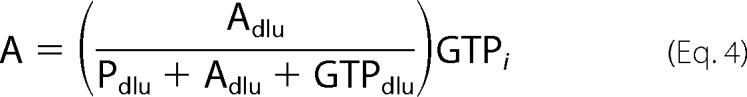 |
where GTP is the concentration of GTP, Pdlu is the amount of [32P]Pi in DLU, Adlu is the amount of 32P-labeled aminoglycoside in DLU, GTPdlu is the amount of [γ-32P]GTP in DLU, GTPi is the initial concentration of GTP, P is the concentration of Pi, and A is the concentration of phosphorylated aminoglycoside.
RESULTS AND DISCUSSION
Aminoglycoside Resistance Profile
Comparison of the resistance profiles conferred by various aminoglycoside-modifying enzymes is complicated by the fact that the genes for these enzymes are expressed in different strains, under different promoters, and from different locations (chromosomal or plasmid), which can all significantly affect their levels of expression. We earlier reported the aminoglycoside resistance profile of the monofunctional APH(2″)-Ia (10) but were unable to evaluate the contribution of this domain to the resistance conferred by the bifunctional AAC(6′)-Ie/APH(2″)-Ia using the available MIC data due to these issues (5, 18, 19). As such, we cloned the gene for AAC(6′)-Ie/APH(2″)-Ia into the plasmid pBluescript II KS (+) under the promoter for aph(2″)-IIIa we previously used for the monofunctional enzyme (10), transformed into the same host strain, E. coli JM83, and evaluated the MIC of several aminoglycosides (Table 1). With the exception of tobramycin, the bifunctional enzyme conferred a level of resistance equivalent (within 2-fold) to the monofunctional enzyme. Thus, in vivo it would appear that the level of aminoglycoside resistance observed with E. coli JM83 harboring the bifunctional enzyme is mostly due to the activity of the APH(2″)-Ia domain. This agrees with studies that could only detect phosphorylated kanamycin A in vivo using a strain expressing AAC(6′)-Ie/APH(2″)-Ia (7).
TABLE 1.
MIC data, steady-state kinetic parameters, and dissociation constants for AAC(6′)-Ie/APH(2″)-Ia
| Aminoglycoside | MICa | kcat | Km | kcat/Km | Kib |
|---|---|---|---|---|---|
| μg/ml | s−1 | μm | m−1 s−1 | μm | |
| 4,6-Disubstituted | |||||
| Kanamycin A | 512/4 | 0.57 ± 0.02 | <1 | >5.7 × 105 | |
| Kanamycin B | 64/1 | 0.12 ± 0.01 | <1 | >1.2 × 105 | |
| Tobramycin | 64/0.5 | 0.11 ± 0.01 | <1 | >1.1 × 105 | |
| Dibekacin | 32/0.5 | 0.15 ± 0.01 | 0.3 ± 0.2 | (5 ± 4) × 105 | |
| Arbekacin | 0.5/0.25 | 2.3 ± 0.1 | 0.8 ± 0.2 | (2.9 ± 0.7) × 106 | |
| Amikacin | 4/2 | 3.7 ± 0.1 | 33 ± 4 | (1.1 ± 0.1) × 105 | |
| Gentamicin C | 32/0.25 | 3.0 ± 0.1 | 0.6 ± 0.3 | (5 ± 2) × 106 | |
| Sisomicin | 16/0.25 | 8.1 ± 0.2 | 0.8 ± 0.2 | (1.0 ± 0.2) × 107 | |
| Netilmicin | 2/0.25 | 6.5 ± 0.1 | 0.41 ± 0.05 | (1.6 ± 0.2) × 107 | |
| 4,5-Disubstituted | |||||
| Neomycin B | 1/0.5 | 0.26 ± 0.02 | |||
| Paromomycin | 4/4 | 5.3 ± 0.3 | |||
| Lividomycin A | 8/4 | 11 ± 1 | |||
| Butirosin A and B | 140 ± 10 | ||||
| Atypical | |||||
| Neamine | 16/8 | 0.10 ± 0.01 | |||
a Ratio of the MICs with E. coli JM83 harboring pBluescript::AAC(6′)-Ie/APH(2″)-Ia to the E. coli JM83 host strain.
b Dissociation constant for the binary enzyme·GTP complex.
GTP as the Phosphate Donor
Steady-state kinetic studies of the bifunctional AAC(6′)-Ie/APH(2″)-Ia and the monofunctional APH(2″)-Ia enzymes have demonstrated a selectivity for GTP (Km = 3 and 3.5 μm, respectively) over ATP (Km = 3600 and 870 μm, respectively) as the phosphate donor (9, 10). Whether this preference manifests itself in vivo has yet to be explored. The concentrations of ATP and GTP in the bacterial cell have been measured in various microorganisms, with estimates ranging from 1 to 10 mm for ATP and from 0.5 to 5 mm for GTP (20–23). At these concentrations, GTP is saturating and ATP is at least at Km level for AAC(6′)-Ie/APH(2″)-Ia; thus, there is the potential for both to be used as substrates in vivo.
To test whether GTP, ATP, or both could serve as the phosphate donor(s) for AAC(6′)-Ie/APH(2″)-Ia in the presence of both NTPs, we devised an assay to determine the rate of phosphate transfer from GTP in the presence and absence of ATP under conditions where the relative ratios of the two nucleotides are as measured in vivo. Four ratios of ATP and GTP were evaluated; one was determined in Staphylococcus aureus (24), two were determined in E. coli (21, 22), and a condition using the lowest concentration of GTP (500 μm) and highest concentration of ATP (10 mm) reported (23). We followed phosphorylation of tobramycin by AAC(6′)-Ie/APH(2″)-Ia using [γ-32P]GTP as the radiolabeled phosphate donor, thus detecting only GTP-dependent phosphorylation, even in the presence of ATP. Using PEI-cellulose TLC plates, we were able to separate the [γ-32P]GTP and 32P-labeled tobramycin, and then the amount of tobramycin phosphorylated by GTP was quantified by phosphorimaging (Fig. 3).
FIGURE 3.
Phosphor image of GTP-dependent phosphorylation of tobramycin and water by AAC(6′)-Ie/APH(2″)-Ia after separation of the reaction mixture on a PEI-cellulose TLC plate.
If AAC(6′)-Ie/APH(2″)-Ia was using both ATP and GTP as the phosphate donor, we would expect to see a decrease in the rate of GTP-dependent phosphorylation when ATP was present in the reaction. Comparison of the rate of GTP-dependent phosphorylation of tobramycin in the absence and presence of ATP showed no differences when the three ratios of NTPs mimicking in vivo levels were used (Fig. 4, A–C). More importantly, even under conditions in which ATP was in 20-fold excess over GTP and 3-fold above the Km level, we still saw no difference in the rate of GTP-dependent phosphorylation of tobramycin (Fig. 4D). This argues that only GTP is used by AAC(6′)-Ie/APH(2″)-Ia as a source of phosphate.
FIGURE 4.

GTP-dependent phosphorylation of tobramycin by AAC(6′)-Ie/APH(2″)-Ia in the absence and presence of various concentrations of ATP. The relative concentrations of each nucleotide were 1.2 mm GTP and 3.6 mm ATP (24) (A), 1.8 mm GTP and 3.6 mm ATP (21) (B), 5 mm GTP and 10 mm ATP (22) (C), and 500 μm GTP and 10 mm ATP (D) (adapted from Refs. 22 and 23). In each panel are time courses for the formation of 2″-O-phosphotobramycin in the absence (blue circle) and presence (red circle) of ATP. Error bars in all panels indicate S.E.
Recent kinetic and x-ray crystallographic studies of three APH(2″) phosphotransferases, APH(2″)-IIa, -IIIa, and -IVa, revealed that these enzymes possess structural templates for the binding of both ATP and GTP (13, 25, 26). The availability of both structural templates accounts for the ability of APH(2″)-IIa and -IVa to use both ATP and GTP as cosubstrates. On the other hand, the structure of APH(2″)-IIIa, an enzyme that has a 400-fold higher relative affinity for GTP when compared with ATP (4 and 1600 μm, respectively), revealed that the ATP-binding template in this enzyme is obstructed by the presence of a bulky tyrosine residue, whereas the GTP-binding template is fully accessible (10). Although the structure of AAC(6′)-Ie/APH(2″)-Ia has not yet been solved, amino acid alignment of the phosphotransferase domain of the bifunctional enzyme and APH(2″)-IIIa shows that a tyrosine is also conserved in the NTP-binding site of APH(2″)-Ia, an indication that tyrosine also blocks the ATP-binding template of this enzyme and provides the structural basis for GTP specificity in vivo.
Phosphorylation of 4,6-Disubstituted Aminoglycosides
The earlier reported substrate profiles of AAC(6′)-Ie/APH(2″)-Ia were determined using ATP as the cosubstrate (6, 7). Our competition experiments highlight the fact that when both ATP and GTP are present at intracellular levels, only GTP is used as the phosphate donor. With this in mind, we evaluated the steady-state kinetic parameters for phosphorylation of 4,6-disubstituted aminoglycosides using GTP. Phosphotransferase activity of AAC(6′)-Ie/APH(2″)-Ia is known to follow a random sequential Bi Bi mechanism (9), meaning that under physiological conditions, in which the concentration of GTP is over 100-fold above the Km level, the enzyme will exist in a binary complex with GTP. As such, all kinetic parameters were evaluated under conditions in which GTP was saturating. As expected, the enzyme phosphorylated all 4,6-disubstituted aminoglycosides tested and did so with catalytic efficiencies ranging from 105 to 107 m−1 s−1 (Table 1).
An analysis of the turnover numbers for 4,6-disubstituted aminoglycosides reveals that garosamine-type aminoglycosides (gentamicin C, sisomicin, and netilmicin) have kcat values ranging from 2.9 to 8.1 s−1, whereas most glucosamine-type antibiotics (kanamycin, tobramycin, and dibekacin) have noticeably lower kcat values (0.11–0.57 s−1). Thus, the former are better substrates for phosphorylation by AAC(6′)-Ie/APH(2″)-Ia. Arbekacin and amikacin seem to be exceptions; their turnover numbers elevated to the level of garosamine-type antibiotics (kcat = 2.3 and 3.7 s−1, respectively). Structurally, dibekacin and arbekacin differ only in the N1 position, the latter having an additional 4-amino-2-hydroxybutyric acid moiety (Fig. 1). This addition results in a 15-fold increase in kcat for arbekacin, whereas the Km remains unchanged. This moiety is also present at the N1 position of amikacin. Amikacin, however, has a much higher Km (33 μm) than arbekacin (0.8 μm). These aminoglycosides differ only in the substituents on the A-ring (2′-NH2, 3′-H, and 4′-H in arbekacin versus 2′-, 3′-, and 4′-OH in amikacin), which presumably accounts for the differences in their relative affinity.
The Km values of 4,6-disubstituted aminoglycosides for the APH(2″)-Ia domain of the bifunctional enzyme, with the exception of amikacin, are less than 1 μm (Table 1). Thus, the relative affinity of all 4,6-disubstituted antibiotics determined in the presence of GTP is higher than that evaluated in the presence of ATP (6).
One would expect that AAC(6′)-Ie/APH(2″)-Ia would confer resistance to those aminoglycosides that it proficiently modifies. As anticipated, the high catalytic efficiency of the enzyme against most 4,6-disubstituted aminoglycosides translates into a 64–128-fold increase in the MICs of these antibiotics over background levels (Table 1); however, there are exceptions. Although the catalytic efficiencies of AAC(6′)-Ie/APH(2″)-Ia for amikacin and arbekacin are 105 and 106 m−1 s−1, respectively, the enzyme does not confer resistance to these antibiotics. Previous studies have demonstrated that arbekacin acetylated at the 3-, 2′-, or 6′-positions retains antibiotic activity against Gram-positive and Gram-negative bacteria, despite modification (27, 28). Our data suggest that the product of arbekacin phosphorylation may also retain antibacterial activity. This can be seen with netilmicin as well, which has a low MIC value despite the high catalytic efficiency (107 m−1 s−1) of the enzyme against this antibiotic. Poor correlation between MIC values and catalytic efficiency of the enzyme can also be found with APH(2″)-IIa, which does not confer resistance to amikacin or arbekacin despite kcat/Km values of 105-106 m−1 s−1 (12). These findings are encouraging as they indicate that some aminoglycoside antibiotics retain efficacy even upon modification by aminoglycoside resistance enzymes.
Phosphorylation of 4,5-Disubstituted and Atypical Aminoglycosides
An interesting feature reported for the AAC(6′)-Ie/APH(2″)-Ia enzyme is its ability to use ATP to phosphorylate 4,5-disubstituted aminoglycosides, most with catalytic efficiencies comparable with 4,6-disubstituted antibiotics (6). These antibiotics differ in the C-ring, 4,5-disubstituted aminoglycosides having a pentose sugar versus a hexose sugar in 4,6-disubstituted aminoglycosides. The three other members of the APH(2″) subfamily, APH(2″)-IIa, -IIa, and -IVa, have recently been characterized by our laboratory, and we have shown that 4,5-disubstituted aminoglycosides are not substrates, but inhibitors of these enzymes (11–13). As ATP is not the phosphate donor for AAC(6′)-Ie/APH(2″)-Ia, we wanted to investigate whether the enzyme would phosphorylate 4,5-disubstiuted aminoglycosides using GTP as the cosubstrate. Employing the coupled assay, we could detect a change in absorbance (due to the formation of GDP) in the presence of 4,5-disubstuted aminoglycosides and neamine; however, this signal did not correlate with the concentration of aminoglycoside in the reaction. The amount of GDP generated in the reaction was always in excess of the concentration of aminoglycoside used. If the aminoglycoside was phosphorylated at one specific position (i.e. the 2″-OH), we would expect to see GDP generated in an equimolar quantity to the aminoglycoside, as we observed with 4,6-disubstituted antibiotic substrates. This implies either that multiple sites on the 4,5-disubstituted aminoglycoside are being phosphorylated, as has been observed for neomycin B (6), or that there is an alternate phosphate acceptor in the reaction. The coupled assay does not provide any information as to where the phosphate from GTP was transferred as it simply measures the amount of GDP generated.
We hypothesized that in addition to the aminoglycoside, water could act as a secondary phosphate acceptor. This is not unprecedented for APH enzymes as APH(3′)-Ia and -IIa are known to possess significant ATPase activity (29). To test whether AAC(6′)-Ie/APH(2″)-Ia exhibited GTPase activity, we used [γ-32P]GTP to detect phosphate transfer to water in both the presence and the absence of various 4,5-disubstituted aminoglycosides and neamine (an atypical aminoglycoside lacking a C-ring possessing a 2″-OH) (Fig. 1) under conditions where both the aminoglycoside and the GTP were saturating. PEI-cellulose TLC plates were used to separate the [γ-32P]GTP, [γ-32P]Pi, and γ-32P-labeled aminoglycosides, and the amount of each was quantified by phosphorimaging. These experiments have demonstrated that AAC(6′)-Ie/APH(2″)-Ia has intrinsic GTPase activity (i.e. in the absence of aminoglycoside) (kobs = 0.012 s−1, Fig. 5A). AAC(6′)-Ie/APH(2″)-Ia, like most aminoglycoside-modifying enzymes, is expressed constitutively in vivo (19, 30). As such, its intrinsic GTPase activity results in the continuous depletion of GTP within the bacterial cell, regardless of the presence or absence of aminoglycoside antibiotic. An intrinsic ATPase activity was earlier reported for APH(3′)-Ia (kcat = 0.020 s−1) (29). Moreover, it was shown that continuous turnover of ATP by APH(3′)-Ia resulted in a metabolic burden on the bacteria such that there was selective pressure in the absence of aminoglycoside antibiotic resulting in the loss of the plasmid harboring the aph(3′)-Ia gene. We also detected GTPase activity in the presence of all 4,5-disubstituted aminoglycosides tested, as well as with the atypical aminoglycoside, neamine. In fact, neamine and the 4,5-disubstituted antibiotics neomycin B and paromomycin all enhanced the GTPase activity of the enzyme above the intrinsic level (kobs = 0.082, 0.040, and 0.032 s−1, respectively). This implies that the binding of these aminoglycosides causes a conformational change in the active site of the enzyme that facilitates hydrolysis of GTP. Moreover, neamine, which is not even a substrate for phosphorylation (Fig. 5A), has the highest propensity to stimulate the GTPase activity of the enzyme. Further structural studies of AAC(6′)-Ie/APH(2″)-Ia in complex with these 4,5-disubstituted and atypical aminoglycosides are warranted to elucidate how they alter the arrangement of active site residues.
FIGURE 5.
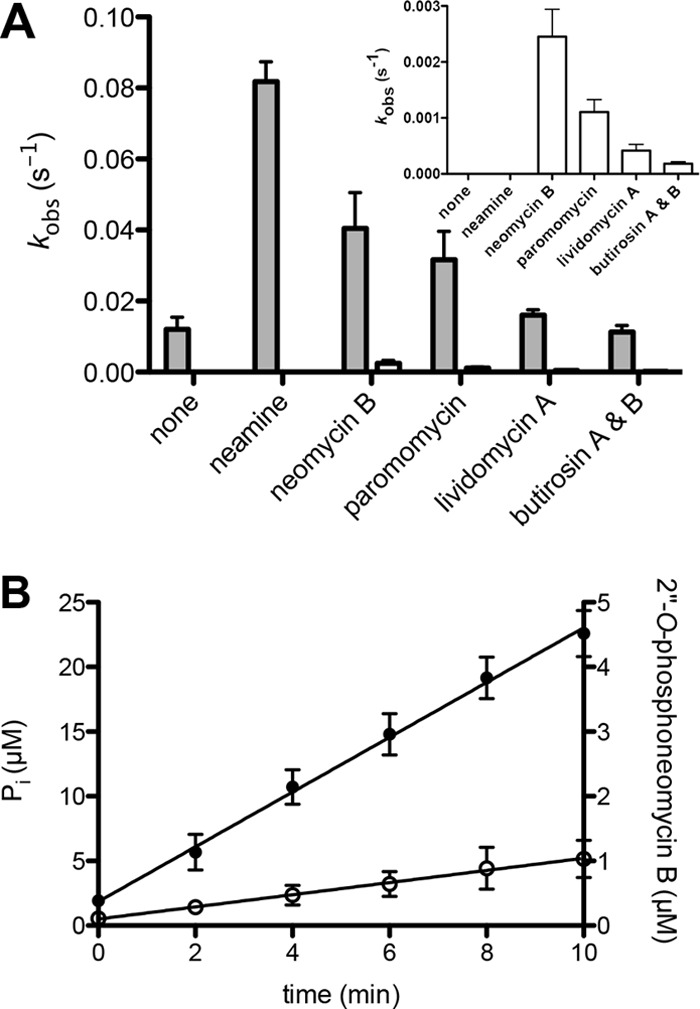
GTP-dependent phosphotransferase and GTPase activities of AAC(6′)-Ie/APH(2″)-Ia in the presence of atypical and 4,5-disubstituted aminoglycosides. A, the rate constants for GTP-dependent phosphate transfer by AAC(6′)-Ie/APH(2″)-Ia to water (gray) and aminoglycosides (white). The rate constants for phosphotransferase activity are also presented separately (inset) to show their magnitude. B, time courses for the GTPase (●) and phosphotransferase (○) activity of AAC(6′)-Ie/APH(2″)-Ia in the presence of neomycin B. Error bars in both panels indicate S.E.
Previous studies, using ATP as the phosphate donor, showed that AAC(6′)-Ie/APH(2″)-Ia has an unusual substrate profile, extending to 4,5-disubstituted aminoglycosides and the atypical aminoglycoside neamine (6). Using GTP, we were also able to detect some phosphorylation of 4,5-disubstituted antibiotics, but not neamine (Fig. 5A, inset). The phosphorylation of neomycin B was the fastest of all the 4,5-disubstituted aminoglycosides tested (kobs = 0.0025 s−1). This was still 55-fold slower than phosphorylation of tobramycin (kcat = 0.11 s−1), the slowest kcat we evaluated for a 4,6-disubstituted aminoglycoside. Furthermore, the rates of phosphate transfer to 4,5-disubstituted aminoglycosides were 16–61-fold slower than the rate of phosphate transfer to water (Fig. 5A). As a result, 94–100% of all observed enzymatic transfer of the γ-phosphate was to water and not to the aminoglycosides. Taken together, these results clearly demonstrate that neither neamine nor 4,5-disubstituted aminoglycosides are substrates of phosphorylation by the APH(2″)-Ia domain of the bifunctional enzyme. This is corroborated by the fact that the MICs for these aminoglycosides are at background levels (Table 1). This experiment also indicates that the majority of activity we observed using 4,5-disubstituted aminoglycosides in the coupled assay was not due to phosphotransferase activity, but rather the GTPase activity of the enzyme.
As atypical and 4,5-disubstituted aminoglycosides are, at most, extremely poor substrates for phosphorylation by AAC(6′)-Ie/APH(2″)-Ia, we evaluated their dissociation constants from the binary AAC(6′)-Ie/APH(2″)-Ia·GTP complex (Table 1). The range of affinities is quite broad, ranging from high micromolar (butirosin A and B) to mid nanomolar (neamine), similar to what has been measured for APH(2″)-IIa, -IIIa, and -IVa (11–13). The dissociation constants for APH(2″)-Ia are comparable with the relative affinities (Km values) of 4,6-disustituted substrates, implying that binding is not the reason for their poor phosphorylation, but likely the position of the 2″-OH.
Conclusion
The bifunctional enzyme AAC(6′)-Ie/APH(2″)-Ia is an important determinant of aminoglycoside resistance due to its widespread occurrence in nature (4). The current literature considers the APH(2″)-Ia domain of this enzyme unique due to its reported promiscuous nature with respect to both its NTP and its aminoglycoside substrate usage. The in vitro studies have shown it to utilize both ATP and GTP as the phosphate donor (9, 10). Our studies have demonstrated that GTP, and not ATP, is the exclusive cosubstrate for APH(2″)-Ia. Earlier in vitro studies have also demonstrated ATP-dependent phosphorylation of 4,5-disubstituted, 4,6-disubstituted, and atypical aminoglycosides (6). We have shown that in the presence of the GTP, most 4,6-disubstituted aminoglycosides are phosphorylated with high catalytic efficiency, whereas 4,5-disubstituted and atypical antibiotics are not substrates. We have also demonstrated that APH(2″)-Ia has a weak intrinsic GTPase activity, which may be stimulated by aminoglycoside antibiotics. Taken together, these studies have shown that APH(2″)-Ia is a bona fide GTP-dependent kinase with the narrow substrate profile characteristic of other members of the APH(2″) subfamily of aminoglycoside-modifying enzymes.
This work was supported, in whole or in part, by National Institutes of Health Grant RO1 AI057393 (to S. B. V.).
- AAC(6′)-Ie/APH(2″)-Ia
- aminoglycoside(6′) acetyltransferase Ie/aminoglycoside(2″) phosphotransferase Ia
- AAC
- aminoglycoside N-acetyltransferase
- APH
- aminoglycoside O-phosphotransferase
- DLU
- density light unit
- MIC
- minimal inhibitory concentration.
REFERENCES
- 1. Vakulenko S. B., Mobashery S. (2003) Versatility of aminoglycosides and prospects for their future. Clin. Microbiol. Rev. 16, 430–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Llano-Sotelo B., Azucena E. F., Jr., Kotra L. P., Mobashery S., Chow C. S. (2002) Aminoglycosides modified by resistance enzymes display diminished binding to the bacterial ribosomal aminoacyl-tRNA site. Chem. Biol. 9, 455–463 [DOI] [PubMed] [Google Scholar]
- 3. Culebras E., Martínez J. L. (1999) Aminoglycoside resistance mediated by the bifunctional enzyme 6′-N-aminoglycoside acetyltransferase-2"-O-aminoglycoside phosphotransferase. Front Biosci. 4, D1–D8 [DOI] [PubMed] [Google Scholar]
- 4. Zhang W., Fisher J. F., Mobashery S. (2009) The bifunctional enzymes of antibiotic resistance. Curr. Opin. Microbiol. 12, 505–511 [DOI] [PubMed] [Google Scholar]
- 5. Boehr D. D., Daigle D. M., Wright G. D. (2004) Domain-domain interactions in the aminoglycoside antibiotic resistance enzyme AAC(6′)-APH(2″). Biochemistry 43, 9846–9855 [DOI] [PubMed] [Google Scholar]
- 6. Daigle D. M., Hughes D. W., Wright G. D. (1999) Prodigious substrate specificity of AAC(6′)-APH(2″), an aminoglycoside antibiotic resistance determinant in enterococci and staphylococci. Chem. Biol. 6, 99–110 [DOI] [PubMed] [Google Scholar]
- 7. Azucena E., Grapsas I., Mobashery S. (1997) Properties of a bifunctional bacterial antibiotic resistance enzyme that catalyzes ATP-dependent 2"-phosphorylation and acetyl-CoA-dependent 6′-acetylation of aminoglycosides. J. Am. Chem. Soc. 119, 2317–2318 [Google Scholar]
- 8. Chow J. W. (2000) Aminoglycoside resistance in enterococci. Clin. Infect. Dis. 31, 586–589 [DOI] [PubMed] [Google Scholar]
- 9. Martel A., Masson M., Moreau N., Le Goffic F. (1983) Kinetic studies of aminoglycoside acetyltransferase and phosphotransferase from Staphylococcus aureus RPAL. Relationship between the two activities. Eur. J. Biochem. 133, 515–521 [DOI] [PubMed] [Google Scholar]
- 10. Toth M., Chow J. W., Mobashery S., Vakulenko S. B. (2009) Source of phosphate in the enzymic reaction as a point of distinction among aminoglycoside 2"-phosphotransferases. J. Biol. Chem. 284, 6690–6696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Badarau A., Shi Q., Chow J. W., Zajicek J., Mobashery S., Vakulenko S. (2008) Aminoglycoside 2"-phosphotransferase type IIIa from Enterococcus. J. Biol. Chem. 283, 7638–7647 [DOI] [PubMed] [Google Scholar]
- 12. Toth M., Zajicek J., Kim C., Chow J. W., Smith C., Mobashery S., Vakulenko S. (2007) Kinetic mechanism of enterococcal aminoglycoside phosphotransferase 2"-Ib. Biochemistry 46, 5570–5578 [DOI] [PubMed] [Google Scholar]
- 13. Toth M., Frase H., Antunes N. T., Smith C. A., Vakulenko S. B. (2010) Crystal structure and kinetic mechanism of aminoglycoside phosphotransferase-2"-IVa. Protein Sci. 19, 1565–1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Clinical and Laboratory Standards Institute (2009). Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically: Approved Standard, 8th Ed., Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 15. Dixon M. (1953) The determination of enzyme inhibitor constants. Biochem. J. 55, 170–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Copeland R. A. (2000) Enzymes: A Practical Introduction to Structure, Mechanism, and Data Analysis, 2nd Ed., pp. 310–313, John Wiley & Sons, Inc., New York [Google Scholar]
- 17. Gilbert S. P., Mackey A. T. (2000) Kinetics: a tool to study molecular motors. Methods 22, 337–354 [DOI] [PubMed] [Google Scholar]
- 18. Ubukata K., Yamashita N., Gotoh A., Konno M. (1984) Purification and characterization of aminoglycoside-modifying enzymes from Staphylococcus aureus and Staphylococcus epidermidis. Antimicrob. Agents Chemother. 25, 754–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rouch D. A., Byrne M. E., Kong Y. C., Skurray R. A. (1987) The aacA-aphD gentamicin and kanamycin resistance determinant of Tn4001 from Staphylococcus aureus: expression and nucleotide sequence analysis. J. Gen. Microbiol. 133, 3039–3052 [DOI] [PubMed] [Google Scholar]
- 20. Bochner B. R., Ames B. N. (1982) Complete analysis of cellular nucleotides by two-dimensional thin layer chromatography. J. Biol. Chem. 257, 9759–9769 [PubMed] [Google Scholar]
- 21. Buckstein M. H., He J., Rubin H. (2008) Characterization of nucleotide pools as a function of physiological state in Escherichia coli. J. Bacteriol. 190, 718–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bennett B. D., Kimball E. H., Gao M., Osterhout R., Van Dien S. J., Rabinowitz J. D. (2009) Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat. Chem. Biol. 5, 593–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. De Mey M., Taymaz-Nikerel H., Baart G., Waegeman H., Maertens J., Heijnen J. J., van Gulik W. M. (2010) Catching prompt metabolite dynamics in Escherichia coli with the BioScope at oxygen rich conditions. Metab. Eng. 12, 477–487 [DOI] [PubMed] [Google Scholar]
- 24. Liebeke M., Meyer H., Donat S., Ohlsen K., Lalk M. (2010) A metabolomic view of Staphylococcus aureus and its Ser/Thr kinase and phosphatase deletion mutants: involvement in cell wall biosynthesis. Chem. Biol. 17, 820–830 [DOI] [PubMed] [Google Scholar]
- 25. Young P. G., Walanj R., Lakshmi V., Byrnes L. J., Metcalf P., Baker E. N., Vakulenko S. B., Smith C. A. (2009) The crystal structures of substrate and nucleotide complexes of Enterococcus faecium aminoglycoside-2"-phosphotransferase-IIa [APH(2″)-IIa] provide insights into substrate selectivity in the APH(2″) subfamily. J. Bacteriol. 191, 4133–4143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Smith C. A., Toth M., Frase H., Byrnes L. J., Vakulenko S. B. (2012) Aminoglycoside 2"-phosphotransferase IIIa (APH(2″)-IIIa) prefers GTP over ATP: structural templates for nucleotide recognition in the bacterial aminoglycoside-2" kinases. J. Biol. Chem. 287, 12893–12903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hotta K., Zhu C. B., Ogata T., Sunada A., Ishikawa J., Mizuno S., Ikeda Y., Kondo S. (1996) Enzymatic 2′-N-acetylation of arbekacin and antibiotic activity of its product. J. Antibiot. 49, 458–464 [DOI] [PubMed] [Google Scholar]
- 28. Hotta K., Sunada A., Ikeda Y., Kondo S. (2000) Double stage activity in aminoglycoside antibiotics. J. Antibiot. 53, 1168–1174 [DOI] [PubMed] [Google Scholar]
- 29. Kim C., Cha J. Y., Yan H., Vakulenko S. B., Mobashery S. (2006) Hydrolysis of ATP by aminoglycoside 3′-phosphotransferases: an unexpected cost to bacteria for harboring an antibiotic resistance enzyme. J. Biol. Chem. 281, 6964–6969 [DOI] [PubMed] [Google Scholar]
- 30. Matsuo H., Kobayashi M., Kumagai T., Kuwabara M., Sugiyama M. (2003) Molecular mechanism for the enhancement of arbekacin resistance in a methicillin-resistant Staphylococcus aureus. FEBS Lett. 546, 401–406 [DOI] [PubMed] [Google Scholar]