

Background: Bispecific antibodies (bisAbs) enable novel therapeutic strategies by concomitantly binding two unique antigens.
Results: IgG-like bisAbs can be produced in mammalian cells through the use of cleavable leucine zippers and GGS tethers.
Conclusion: The LUZ-Y platform enables mammalian cell production of bisAbs.
Significance: bisAbs structurally similar to full-length hIgG should enable novel therapies with minimal risk of adverse immune response.
Keywords: Antibody Engineering, Protein Engineering, Protein Processing, Protein Self-assembly, Protein-Protein Interactions, Bispecific Antibodies, Leucine Zippers
Abstract
The ability of bispecific antibodies to simultaneously bind two unique antigens has great clinical potential. However, most approaches utilized to generate bispecific antibodies yield antibody-like structures that diverge significantly from the structure of archetype human IgG, and those that do approach structural similarity to native antibodies are often challenging to engineer and manufacture. Here, we present a novel platform for the mammalian cell production of bispecific antibodies that differ from their parental mAbs by only a single point mutation per heavy chain. Central to this platform is the addition of a leucine zipper to the C terminus of the CH3 domain of the antibody that is sufficient to drive the heterodimeric assembly of antibody heavy chains and can be readily removed post-purification. Using this approach, we developed various antibody constructs including one-armed Abs, bispecific antibodies that utilize a common light chain, and bispecific antibodies that pair light chains to their cognate heavy chains via peptide tethers. We have applied this technology to various antibody pairings and will demonstrate the engineering, purification, and biological activity of these antibodies herein.
Introduction
Since the first description for the production of a bispecific antibody >25 years ago (1, 2), a cornucopia of approaches to produce bispecific antibodies have been developed, including the generation of quadromas, chemical cross-linking of antibody fragments, and genetic engineering (for a review, see Refs. 3–5). Although some of these approaches yield antibody-like proteins that are structurally similar to native human IgG, most result in proteins that have a significantly different structure. The approaches that yield bispecific antibodies that most resemble full-length human IgG utilize modifications in the Fc region that favor heterodimerization of two distinct heavy chains. Such examples include knobs-into-holes (6), strand-exchange engineered domain (SEED) (7), and electrostatic steering technologies (8). Although these approaches enable effective heterodimerization of two distinct heavy chains, appropriate pairing of cognate light and heavy chains remains a problem. Utilization of a common light chain (LC)2 can solve this issue (9); however, the development of a common light chain for a given bispecific is technically challenging and not always possible. Recently, Schaefer et al. (10) described a method in which the various domains of the Fab are switched within one of the bispecific arms. This method switches either the entire heavy chain (HC) and LC Fab domains or individual variable or constant domains between a HC and its cognate LC, resulting in a LC that will only pair with the modified HC. When combined with a knobs-into-holes modified Fc to allow HC heterodimerization, this approach yielded bispecific antibodies that can be expressed in mammalian cells and exhibit characteristics similar to native IgG, although not strictly identical in structure. The approach that yields a human bispecific antibody that is most structurally similar to native human IgG is the two-in-one antibody, which is developed using phage display technology to engineer complementarity-determining regions (CDRs) that specifically react with two different antigens (11). Although this antibody exhibits all of the characteristics of native human IgG, generation of a dual-specific antibody using this approach can be technically challenging.
In an effort to develop a robust procedure for the production of bispecific antibodies that enables heterodimerization of two distinct HCs with appropriate LC/HC pairing, we have engineered a bispecific antibody platform that can be expressed in mammalian cells and contains only a single non-native amino acid per HC in its mature form. The heterodimeric assembly of the HCs is achieved through the addition of a leucine zipper to the C terminus of the CH3 domains that is readily removed by limited proteolysis post-purification. Leucine zippers have been previously used to generate bispecific F(ab′)2 (12) and scFvs (13). For these approaches, the zippers remained an integral part of the final product, and an in vitro redox step was necessary for correct assembly of the bispecific molecules. Our method allows for the in situ assembly of full-length bispecific antibodies without the potentially immunogenic zippers present in the final product.
To ensure appropriate pairing of HCs and LCs, we engineered in furin cleavable linkers between cognate HCs and LCs that are removed in situ during expression in mammalian cells. Using this approach, we produced bispecific antibodies from mammalian cells that differed from their parental mAbs by only a single point mutation per HC. Because we used a leucine zipper to induce heterodimerization of two different HCs, we refer to these antibodies as LUZ-Ys.
EXPERIMENTAL PROCEDURES
Construction of LUZ-Y HC Backbone Vector
Sense and antisense oligonucleotides were designed and synthesized to encode for either the ACID.p1 (Ap1) (GGSAQLEKELQALEKENAQLEWELQALEKELAQGAT) or BASE.p1 (Bp1) (GGSAQLKKKLQALKKKNAQLKWKLQALKKKLAQGAT) peptide with 5′ AscI and 3′ XbaI overhangs. The oligonucleotides were annealed, phosphorylated, and ligated into a digested and dephosphorylated pRK.sm (Genentech) plasmid. The CH1 through CH3 domain of a hIgG1 was prepared via PCR to include a 5′ MCS (ClaI-BamHI-KpnI-ApaI) and a 3′ AscI site and cloned into the previously prepared pRK-Ap1 or pRK-Bp1 vector via ClaI and AscI. Finally, the lysine residue at position HC222 (EU numbering as in Kabat) was mutated into an alanine residue using Agilent's QuikChange II XL site-directed mutagenesis kit (catalog no. 200522).
Construction of LUZ-Y Antibodies
For common LC and one-armed LUZ-Y, the VH domain of the desired antibody was prepared using PCR to include 5′ ClaI and 3′ ApaI restriction sites. The PCR fragments were digested and cloned into a similarly prepared LUZ-Y backbone vector. No changes had to be made to the LC constructs already available for these antibodies.
For the tethered LUZ-Ys, the VH domain (minus the signal sequence) of the desired antibody was first prepared using PCR wherein the 5′ primer contained the 3′ half of a GGS tether and terminated in a 5′ BamHI site and the 3′ primer terminated in a 3′ ApaI site. The fragments were digested and cloned into a similarly prepared LUZ-Y backbone vector. The cognate LC of the desired antibody was then prepared using PCR, wherein the 5′ primer terminated in a 5′ ClaI site and the 3′ primer contained the 5′ portion of the GGS tether and terminated in a 3′ BamHI. The LC fragment was joined to its cognate HC (now in the LUZ-Y backbone) by cloning the fragment in front of the VH via ClaI and BamHI. The completed tether sequence linking the CL to the VH was GGGSGGSGGSGGSGGSGGSGGSGGSG. Similar methods were used for the construction of the Furin cleavable tethers, differing only in that the final tether sequence was RKRKRRGGGSGGSGGSGGSGGSGGSGRSRKRR.
Expression and Purification of LUZ-Y Antibodies
LUZ-Y antibodies were expressed in CHO cells using standard transient transfection techniques. Antibodies were purified from cell-conditioned media using MabSelect SuRe resin (GE Healthcare). Medium was loaded onto a MabSelect SuRe column at 450 cm/hr at 4 °C and successively washed with two column volumes of PBS, 10 column volumes of PBS + 0.1% Triton X-114, 10 column volumes of 400 mm potassium phosphate + 5 mm EDTA, 0.2% Tween 20, and again with 20 column volumes of PBS. The column was eluted with 100 mm acetic acid, pH 2.7, directly into fractions containing 1 m arginine and 1 m Tris, pH 8.0, to achieve a final buffer concentration of 200 mm arginine and 200 mm Tris, pH 8.0. The leucine zippers were enzymatically removed from the antibodies by incubation with lysyl endopeptidase (Wako Chemicals, Inc., Richmond, VA) at 37 °C for 1.5 h at an enzyme to antibody ratio 1:500 (w/w) Lys-C. To remove the liberated zippers, the Lys-C processed antibody was loaded onto a MabSelect SuRe column, which was washed to base line with PBS and eluted with acetic acid as described above. The processed antibody was further purified by size exclusion using a Sephacryl S200 column (GE Healthcare) run in PBS, 150 mm NaCl, 100 mm arginine, pH 8.0, and peak fractions were collected and dialyzed into PBS. Yields varied anywhere from 8 to 60 mg/liter with an average yield of ∼10 mg/liter in the transient setting. To resolve homodimer (parental) antibodies from the heterodimeric LUZ-Y antibody, purified antibodies containing each species were chromatographed on an analytical 1.5/5 mono S column (GE Healthcare) in 20 mm sodium acetate, pH 5.0. The antibodies were eluted with a 62-column volume gradient of 0.25 m to 0.65 m NaCl in 20 mm sodium acetate, pH 5.0.
Carboxypeptidase Digestion
Enzymatic digestion with carboxypeptidase B was carried out using porcine CpB (Roche Applied Science, Indianapolis, IN) at a ratio of 1:5 (w/w) for 2.5 h at 37 °C, after dialyzing the sample against PBS to remove arginine. After digestion, the sample was subsequently run on a MabSelect SuRe column, which was washed to base line with PBS and eluted with acetic acid as described previously. The final product was dialyzed into PBS with 0.2 m arginine.
Fab Preparation
Fab fragments of the antibodies were prepared by incubating the antibody with lysyl endopeptidase (Wako Chemicals, Inc.) at 37 °C for 1 h at an enzyme to antibody ratio of 1:1000. To separate the Fab from the liberated Fc, the digested antibody was loaded onto a protein L-agarose column (1 ml of resin/2 mg of protein; Thermo Scientific), washed to base line with PBS, and eluted with 100 mm acetic acid, pH 2.7, directly into fractions containing 1 m Tris, pH 8.0, to achieve a final buffer concentration 200 mm Tris, pH 8.0.
Mass Spectroscopy
Intact molecular weight of antibodies both non-reduced and reduced (+0.1 m DTT) were determined using an Agilent 6210 TOF LC/MS (mass accuracy of 2 ppm, Agilent Technologies, Santa Clara, CA). All antibodies were chromatographed on a Varian PRLP-S, 1000 A microbore column (50–2.1 mm, Polymer Laboratories) and eluted with a linear gradient from 5–90% B (solvent A, 0.05% TFA; solvent B, 0.05% TFA in acetonitrile) into a dual-nebulizer electrospray ion source. Data were collected by the Agilent MassHunter Work station data acquisition software and deconvoluted with Agilent MassHunter Qualitative Analysis Software. Prior to LC/MS analysis all antibodies (50 ug) were treated with 10,000 units/ml PNGase F (New England Biolabs) overnight at 37 °C to remove N-linked carbohydrates. The discrepancies in mass assignment when comparing the theoretical m/z (supplemental Fig. 2) to the experimental m/z values can be attributed to incomplete reduction of intrachain disulfide bonds. All masses are deconvoluted using the maxEnt algorithm and fall within ± 1 Da average mass accuracy. All examples of LUZ-Y contain a +162/163-Da adduct that is seen in the non-reduced and sometimes in the reduced mass spectrometry analysis (Fig. 2, a, d, g, j, and k). These additional masses are the result of a C-mannosylation of a tryptophan residue that cannot be removed (14).
FIGURE 2.

Mass spectrometry and SDS-PAGE analysis of LUZ-Ys. a–c, common LC LUZ-Y with leucine zipper attached: non-reduced (a) and reduced yielding two HCs (b) and the common LC (c). d–f, common light chain LUZ-Y with leucine zipper removed non-reduced (d) and reduced (e and f). h and i, one-arm LUZ-Y with leucine zipper removed non-reduced (g) and reduced to yield HC (h), LC, and truncated Fc (i). j and k, tethered LUZ-Y with leucine zipper removed non-reduced (j) and reduced yielding two LC-HC-tethered antibody halves (k). A comparison of the experimentally determined masses to the theoretical masses is shown in supplemental Table 1.
Binding Assays
Biolayer interferometry was performed on a ForteBio Octet system. Binding kinetics were measured using anti-human IgG Fc biosensors (ForteBio, Menlo Park, CA) loaded in 250 μl of HBS-P containing 25 μg/ml of each Ab. To measure successive binding of two different antigens to one bispecific, the anti-human IgG Fc biosensors were incubated with 250 μl of LUZ-Y at 25 μg/ml for 15 min and subsequently rinsed for 30 s in PBS to remove any unbound sample. All associations to the second and successive third binding sample were carried out for 3–5 min, with a 30-s rinse in PBS performed between associations. No dissociation of the antibody loaded to the anti-human IgG Fc probe or of bound protein samples was observed.
Equilibrium cell-binding assays were performed in triplicate using antibodies, and Fabs were iodinated by the lactoperoxidase method (Biotrend, Cologne, Germany). Radiolabeled antibodies (0.05 nm to 0.5 nm) were incubated with 75,000 to 200,000 NR6-HER2, NR6-EGFR or HCA-7 (co-expressing HER2 and EGFR) cells in the presence of varying concentrations of unlabeled protein for 4 h at 4 °C. Cells were subsequently harvested using Millipore filter plates and washed three times with PBS. Dried filters were counted for 1 and 10 min using a Perkin Elmer Wizard 1470 Automatic Gamma Counter, and binding data were analyzed using NEWLIGAND 1.05 Software (Genentech).
Cell Lines
NR6 cells were kindly provided by Mark Pegram (UCLA, Los Angeles, CA) (15) and maintained in RPMI 1640 (Genentech, Inc.) supplemented with 10% heat-inactivated calf serum (HyClone catalog no. SH30073.03), 10 mm l-glutamine (Genentech, Inc.), penicillin, and streptomycin (Invitrogen catalog no. 15140-122). The stably expressing NR6-EGFR and NR6-HER2 cell lines were generated as described previously (16). HCA-7 cells were maintained in Dulbecco's modified Eagle' s medium (Genentech, Inc.) supplemented with 10% heat-inactivated fetal bovine serum (HyClone catalog no. SH30071.03). l-Glutamine (Genentech, Inc.), penicillin, and streptomycin (Invitrogen catalog no. 15140-122).
Histamine Release Assay
A rat basophil leukemia cell line generated to express human FcϵRIα and human FcγRIIb was cultured for 72 h at 37 °C with 1 μg/ml NP-specific human IgE (JW8.5.13) in complete growth media (minimum essential medium with Earle's salts, Invitrogen catalog no. 11090, 1 mm glutamine (Genentech, Inc.), 1 mm sodium pyruvate (Invitrogen catalog no. 11360-070), 0.1 mm nonessential amino acids (Invitrogen catalog no. 11140-050), 1.5g/liter sodium bicarbonate (Invitrogen catalog no. 25080-094), 15% fetal bovine serum (Hyclone catalog no. SH30071.03)). Cells were trypsinized and plated onto a 96-well, flat bottom tissue culture plate at 3.5 × 105 cells/ml in 200 μl of complete growth media containing 1 μg/ml NP-specific human IgE and allowed to adhere for 2 h. Next, the cells were washed three times with fresh media to remove unbound NP-specific human IgE. Cells were treated with 0–10 μg/ml of bispecific antibody and incubated for 1 h at 37 °C prior to activation with antigen. Cells were activated by incubation with 0.1 μg/ml NP-conjugated ovalbumin (Biosearch Technologies, Inc. catalog no. N-5051-10) for 45 min at 37 °C. Following incubation, the histamine level in the cell supernatants (cell culture medium) was measured by ELISA using a histamine ELISA kit (KMI Diagnostics, Minneapolis, MN). Background histamine levels were obtained from cells treated with NP-specific human IgE alone with no activation.
Cell Proliferation Assay
FaDu cells (ATCC, Manassas, VA) were maintained in RPMI medium (Genentech, Inc.) supplemented with 10% fetal bovine serum (HyClone catalog no. SH30073.03). 5000 cells/well were plated in 96-well plates. The next day cells were treated with indicated concentrations (200 nm to 0.004 nm) of LUZ-Y antibodies or control antibodies in 1% serum-containing medium, each condition was run in duplicate. After 3 days, 25 μl of AlamarBlue (Invitrogen) was added to the wells, and fluorescence was read using a 96-well fluorometer with excitation at 530 nm and emission of 590 nm. The results are expressed in relative fluorescence units.
RESULTS
LUZ-Y Design and Assembly
The leucine zipper structure that we utilized to induce heterodimerization of half-antibodies is based on the Acid.p1 (Ap1) and Base.p1 (Bp1) peptides described by O'Shea et al. (17). These peptides were designed to fold as parallel helical dimers with a preference for the heterodimeric state. We found that fusing the Ap1 peptide to the C terminus of the Fc domain of one of the half-antibodies and the Bp1 peptide to the other via a short GGS linker was sufficient to induce heterodimerization. Using this approach, we developed a one-armed antibody format (Fig. 1a), and two bispecific formats that utilize either a common light chain (Fig. 1b) or tethered light chains (Fig. 1, c and d). The one-armed LUZ-Y contains a truncated IgG1 Fc domain paired with a full-length HC and a single LC (Fig. 1a). The common LC LUZ-Y consists of two distinct full-length HCs and a common light chain. Correct assembly of HC/LC pairs is guaranteed in these formats because only one possible orientation exists. For formats containing two distinct LCs (Fig. 1, c and d), we utilized non-cleavable or cleavable GGS tethers to assure correct assembly of cognate LC-HC pairings. The non-cleavable tether (Fig. 1c) connects the C terminus of the LC to the N terminus of its cognate HC via a 26-amino acid GGS tether (G(GGS)8G), whereas the cleavable tether (Fig. 1d) contains furin protease recognition sequences (RXRXRR) at both ends of the GGS tether. Removal of the leucine zipper from all formats is achieved by taking advantage of the native C-terminal lysine of the CH3 domain of human IgG1 and performing proteolytic digestion using the bacterial endoproteinase Lys-C post purification. Because Lys-C also cleaves at Lys-222 (EU numbering as in Kabat) in the hinge region of IgG1, we mutated the Lys-222 residue to an alanine to prevent Fab release during protease treatment. This mutation is proximal to the region of IgG1 that is known to interact with Fc receptors (18); we therefore analyzed the binding to hFcγRI, hFcγRIIa (His-131 and Arg-131), hFcγRIIb, and hFcγRIIIa (Phe-158 and Val-158) and saw no difference in binding between parental Abs and their K222A variants (data not shown).
FIGURE 1.
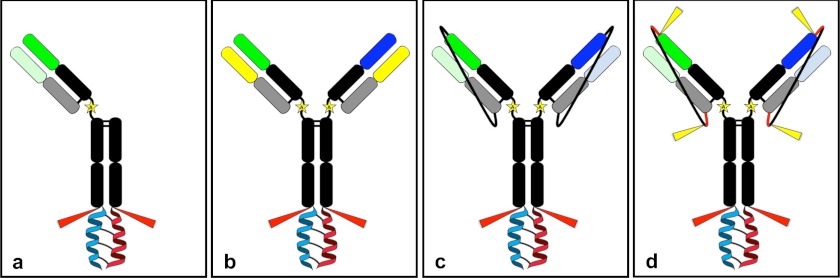
Schematic diagrams of the four LUZ-Y formats. a, the one-armed LUZ-Y; b, the common LC LUZ-Y; c, the tethered LUZ-Y; d, the tethered LUZ-Y with Furin cleavable tethers. K222A mutations are depicted in each figure as a star in the hinge region, Lys-C cleavage sites are indicated with red arrows and Furin recognition sites are indicated as a red tether segment with yellow arrows at a cleavage site.
Production and Characterization of LUZ-Ys
All LUZ-Ys were produced in CHO cells transiently transfected with the appropriate plasmids. Balancing the individual HC expression is imperative for the production of a homogeneous population of heterodimers. Although the leucine zippers direct heterodimeric Fc assembly, they do not prevent homodimerization and discordant expression of HC results in the formation of homodimers. To show this, we transfected cells with varying ratios of plasmid DNA encoding the two HCs for the anti-hFcϵRIα/hFcγRIIb common LC LUZ-Y (supplemental Fig. 2 and supplemental Table 2). This experiment demonstrates the impact of discordant expression on the balance between homodimer and heterodimer (LUZ-Y) production. Increasing the amount of DNA transfected for one HC results in either an increased production of that homodimer or the LUZ-Y, depending on the ratio transfected. Although balancing expression is best determined experimentally and varies with different constructs and conditions, the general ratio of plasmids transfected is as follows: common LC LUZ-Y HC1:HC2:CL, 1:1:2; one-armed LUZ-Y HC1:truncFc:LC, 1:0.6:0.6; tethered LUZ-Y tHC-LC1:tHC-LC2, 1:1; and furin-cleavable tethered LUZ-Y FtHC-LC1:FtHC-LC2, 1:1. Yields for the LUZ-Ys varied from 8–60 mg/liter. Because endogenous furin levels were insufficient to allow for complete tether cleavage, we co-transfected full-length human furin cDNA with the cleavable tether constructs.
Each LUZ-Y format was assembled using pre-existing recombinant mAb cDNAs as a template. The one-armed LUZ-Ys were constructed from either an anti-EGFR antibody (19) or an anti-HER2 (20) antibody. The common light chain LUZ-Y was derived from an anti-hFcϵRIα/hFcγRIIb bispecific that has been described previously (21), whereas the tethered LUZ-Ys were derived from anti-EGFR, anti-HER2, and anti-HER3 antibodies (19, 20, 22). Following expression in CHO cells and purification as described under “Experimental Procedures,” the various purified LUZ-Ys were characterized by SDS-PAGE and mass spectrometry analysis. Fig. 2, a–f, shows the results for the purified common LC anti-hFcϵRIα/hFcγRIIb LUZ-Y. The intact mass of the bispecific antibody is 153602.1 Da, whereas post Lys-C cleavage yields an intact antibody with a mass of 144,913.1 Da (Fig. 2, a and d). This reduction in mass corresponds to the complete removal of the leucine zipper, which has a mass of 8689 Da. This is further supported by MS analysis of the antibody following reduction (Fig. 2, b and e). MS analysis shows the expected mass reduction of each HC by either 4349.8 Da (for the Ap1 peptide) or 4342.3 Da (for the Bp1 peptide), whereas the LC remains unchanged (Fig. 2, c and f), demonstrating that zipper removal by Lys-C is complete without any off-target cleavage of the antibody. In addition, no peaks were observed at the predicted masses for the parent homodimers (145,159.6 Da for anti-hFcϵRIα and 144,666.6 Da for anti-hFcγRIIb post Lys-C digestion), confirming that the antibody is heterodimeric and therefore bispecific.
Lys-C cleavage of the one-armed anti-EGFR LUZ-Y yielded an intact antibody with the expected mass of 97,529.4 Da (Fig. 2, g–i). We did not observe the Fc dimer (50,240.4 Da) or full-length homodimer (144,814.8 Da) by SDS-PAGE or MS analysis (supplemental Fig. 1 and Fig. 2, h and i) and the appropriate HC (49,087.5 Da), LC (23,324.8 Da), and truncated Fc (25,124.6 Da) masses are observed upon reduction.
Digestion of the intact non-cleavable tethered LUZ-Y, anti-EGFR/HER2, by Lys-C yielded an intact antibody with a single mass peak that matches the heterodimer mass (148,478.1 Da), demonstrating that the antibody is exclusively heterodimeric and neither of the two potential homodimers (148,221.9 Da or 148,718.7 Da) were present. Upon reduction, the MS spectra of each tethered LC-HC arm show that their masses are also correct (anti-EGFR, 74,117.2 Da; anti-HER2, 74,365.8 Da, Fig. 2k).
We constructed anti-EGFR/HER3 LUZ-Y containing furin cleavable LC/HC linkers to allow for in situ removal of the linkers. Purification and Lys-C cleavage of the furin-cleavable tethered LUZ-Y reveals that in situ removal of the linker is complete as judged by SDS-PAGE and MS (Fig. 3, a–c). Analysis by SDS-PAGE shows that the anti-EGFR/HER3 LUZ-Y runs at the predicted mass corresponding to full-length intact antibody, and when resolved under reducing conditions, both the HC and LC are resolved (Fig. 3a), and there is no evidence of a tethered LC-HC peptide, which would have a mass of ∼75 kDa. However, the determined mass of the full-length antibody reveals multiple species (data not shown) and MS analysis under reducing conditions shows that this is due to remaining furin recognition site residues at the C terminus of both light chains (Fig. 3a). This is expected as furin cleaves at the C terminus of the sequence RXRXRR (23), and although this would liberate the linker exactly at the HC N terminus, the recognition site would remain attached to the C terminus of the LC. We also observe that this residual furin recognition sequence is partially degraded as MS analysis shows that the C termini of the two LCs are ragged with 2, 3, or 4 residues of the furin site remaining (Fig. 3a). The partial cleavage of the furin recognition site is likely due to endogenous carboxypeptidases in the cell culture fluid. To remove these remaining basic residues, we treated the antibody with carboxypeptidase B, which yielded a fully processed antibody with the expected theoretical mass of 144,533.1 Da and under reducing conditions HCs and LCs of the correct masses (Fig. 3b). The C-terminal lysine of each HC is also removed by the carboxypeptidase B treatment. Although this lysine is from the parental sequence, its removal from recombinant antibodies produced from mammalian cells is commonly observed and has no known functional consequences (24). Thus, this approach yields a bispecific antibody that is free of zippers and linkers and contains only a single mutation (K222A) when compared with the sequence of the parental antibodies. The overall approach is schematically represented in Fig. 3C and highlights the key proteolytic processing events.
FIGURE 3.

Anti-HER2/EGFR LUZ-Y with furin cleavable tethers. a, SDS-PAGE and MS analysis of anti-HER2/EGFR LUZ-Y LCs (containing residual furin site residues at the C terminus) and HCs following purification and Lys-C processing to remove the leucine zippers. Three major species of each LC are present after initial purification steps of the LUZ-Y Ab; LC + RK (i and ii), LC + RKR (iii and iv), and LC + RKRK (v and vi). b, SDS-PAGE and MS analysis of the anti-HER2/EGFR LUZ-Y following carboxypeptidase B treatment to remove the residual furin site from the C terminus of the light chains. Inset shows mass spectra for the non-reduced intact antibody post carboxypeptidase B (CpB) treatment. c, schematic representation of the overall process. *, additional mass is the LC+RKRK plus a sodium ion.
LUZ-Ys Exhibit Appropriate Binding Activity
To determine whether the LUZ-Y antibodies maintained their ability to specifically bind their cognate antigens, we analyzed their binding activity using biolayer interferometry and cell-based binding assays using radiolabeled antibodies. Analysis of the one armed anti-HER2 and the one-armed anti-EGFR by biolayer interferometry show these antibodies only bind to their respective antigens and do not cross-react (Fig. 4a).
FIGURE 4.

Biolayer interferometry analysis of the antigen binding activity of LUZ-Y. a, trace 1: one-armed anti-EGFR LUZ-Y loaded biosensor sequentially exposed to HER2 ECD (no binding) then to the EGFR ECD. Trace 2: one-armed anti-HER2 LUZ-Y loaded biosensor exposed first to EGFR ECD (no binding) followed with binding to HER2 ECD. b, common light anti-hFcϵRIα/hFcγRIIb LUZ-Y loaded biosensor probe sequentially exposed to hFcϵRIα ECD then to hFcγRIIb ECD. c, anti-EGFR/HER2 tethered LUZ-Y loaded biosensor, sequentially exposed to first to EGFR ECD then to HER2 ECD (1) or first to HER2 ECD then to the EGFR ECD (2). d, fully processed LUZ-Y (anti-EGFR/HER3) loaded biosensor exposed to EGFR ECD followed with exposure to HER3 ECD.
The common-LC bispecific LUZ-Y directed against hFcϵRIα and hFcγRIIb exhibited the desired binding activity to both of its antigens (Fig. 4b). In this experiment, the antibody is bound to the biosensor and is successively exposed to the soluble extracellular domain (ECD) of hFcϵRIα and then to the hFcγRIIb ECD. The binding response curve shows successive binding of both receptors without a corresponding dissociation of the first receptor upon exposure to the second, indicating that the antibody can simultaneously bind both antigens in a non-competitive association. We further observed that a non-cleavable tethered bispecific LUZ-Y directed against EGFR and HER2 also bound its antigens simultaneously (Fig. 4c). The LCs of this LUZ-Y are covalently linked to their cognate HCs by a 26-amino acid GGS tether (G(GGS)8G). Binding this antibody to a biosensor and exposing it to the extracellular domains of EGFR and HER2 in succession shows that both antigens bind successively and simultaneously, regardless of the order in which they are added. These results indicate that the antibody binds its antigens concomitantly and that the tether does not interfere with binding. Finally, the binding activity of a Furin cleavable tethered LUZ-Y against EGFR and HER3 is shown in Fig. 4d. As described above, this antibody has been fully processed with both the linkers and zippers removed through proteolytic digestion yielding a near native antibody (Fig. 3, a–c). Similar to the tethered LUZ-Y, the fully processed anti-EGFR/HER3 LUZ-Y simultaneously binds both its target antigens (Fig. 4d). The ability of the LUZ-Y formatted bispecific antibodies to simultaneously and discriminately bind two antigens may be important in cases where the specific cross-linking of two antigens is desired, such as neutralizing an activating receptor by bringing it together with an inhibitory receptor. This characteristic also allows LUZ-Ys to be used in settings where homodimeric cross-linking of an antigen could lead to an undesirable pathology, such as the inappropriate activation of an immune-activating receptor or growth factor receptor.
In addition to the biolayer interferometry studies, we also carried out direct radioligand cell-binding assays to compare the affinities of the LUZ-Y formatted antibodies with their parental antibodies. Cell-based affinity measurements were obtained for the tethered LUZ-Y anti-EGFR/HER2, the one-armed LUZ-Ys (anti-EGFR and anti-HER2), and the parental bivalent anti-EGFR and anti-HER2 mAbs. Fab fragments were also generated from each of the parental antibodies to compare with the one-armed LUZ-Ys. We analyzed binding to NR6 cells stably expressing either human EGFR or HER2, or to the colorectal cell line HCA-7 that co-expresses both EGFR and HER2 (25); the data are summarized in Table 1. Binding studies using the NR6-EGFR cell line show that the anti-EGFR Fab, one-armed anti-EGFR LUZ-Y, and the tethered anti-EGFR/HER2 LUZ-Y all bound with comparable affinities (∼1–3 nm) with the one-armed LUZ-Y demonstrating the highest affinity. As expected, the parental anti-EGFR antibody bound with a higher affinity of 0.56 nm, which is likely due to the bivalency of this antibody.
TABLE 1.
Equilibrium cell binding assays
Radiolabeled antibodies in the presence of varying concentrations of unlabeled antibodies were incubated with NR6 cells expressing either EGFR or HER2 or with HCA-7 cells that co-express both EGFR and HER2. Affinity determinations are an average of three assays shown with the S.E.
| Cell line/antibody | Kd |
|---|---|
| nm | |
| NR6 expressing EGFR | |
| WT anti-EGFR | 0.56 ± 0.19 |
| Anti-EGFR Fab | 2.07 ± 0.24 |
| One-armed LUZ-Y anti-EGFR | 1.15 ± 0.07 |
| Tethered LUZ-Y anti-EGFR/HER2 | 2.64 ± 0.30 |
| NR6 expressing HER2 | |
| wt anti-HER2 | 0.90 ± 0.13 |
| Anti-HER2 Fab | 2.76 ± 0.17 |
| one-armed LUZ-Y anti-HER2 | 1.72 ± 0.11 |
| Tethered LUZ-Y anti-EGFR/HER2 | 5.11 ± 0.20 |
| HCA-7 co-expressing | |
| Tethered LUZ-Y anti-EGFR/HER2 | 0.99 ± 0.13 |
| WT anti-EGFR | 0.40 ± 0.08 |
| WT anti-HER2 | 0.19 ± 0.02 |
| One-armed LUZ-Y anti-EGFR | 1.56 ± 0.09 |
| One-armed LUZ-Y anti-HER2 | 2.18 ± 0.04 |
The affinities of the anti-HER2 Fab, one-armed anti-HER2 LUZ-Y, and the tethered anti-EGFR/HER2 LUZ-Y to the NR6 HER2 expressing cells were 2.76 nm, 1.72 nm, and 5.11 nm respectively, whereas the affinity of the parent bivalent anti-HER2 antibody was 0.90 nm. Although there appears to be a roughly 2-fold lower affinity of the LUZ-Y than the Fab or one-armed anti-HER2, this difference is within range of the error for this assay.
The affinity of the anti-HER2/EGFR LUZ-Y binding to HCA-7 cells that co-express both receptors was 0.99 nm, which is ∼1.5 to two times higher affinity than the affinity of the corresponding one-armed antibodies on HCA-7 cells and could be due to an avidity effect conferred by the presence of both receptors on HCA-7 cells. However, both parental antibodies exhibited a 2- (anti-EGFR) to 5-fold (anti-HER2) higher affinity binding to HCA-7 cells than the LUZ-Y on the HCA-7 cells (Table 1).
Evaluation of Biological Activity of LUZ-Y
We evaluated the biological activity of the common light chain anti-hFcϵRIα/hFcγRIIb LUZ-Y using a histamine release assay. Previously, a knobs-into-holes version of this antibody was shown to limit histamine release in this assay by cross-linking the inhibitory receptor hFcγRIIb to the activating receptor hFcϵRIα, thus preventing its activation by IgE (21). Similarly, the anti-hFcϵRIα/hFcγRIIb CL LUZ-Y attenuated histamine release in a dose-dependent manner (Fig. 5a).
FIGURE 5.

Biological activity of LUZ-Y. a, effect of anti-hFcϵRIα/hFcγRIIb-bispecific antibodies on histamine release from IgE-activated rat basophil leukemia cells expressing hFcϵRIα and hFcγRIIb. Common light chain LUZ-Y (blue), knobs-into-holes antibody (red), no treatment (purple). Duplicate samples of each antibody were assayed, and data are shown for each of the duplicates. b, effect of LUZ-Y antibodies on the proliferation of FaDu cells. FaDu cells were incubated, in duplicate, with the indicated concentrations of the parental anti-EGFR and HER3 antibodies separately and in combination, or with the anti-EGFR/HER3 LUZ-Y in 1% serum containing media. Cell proliferation was analyzed after three days by AlamarBlue staining.
To evaluate the biological activity of anti-EGFR/HER3 furin cleavable tethered LUZ-Y, we performed cell proliferation assays using FaDu cells, whose cell growth is dependent on EGFR and HER3 activation (19, 20). As shown in Fig. 5b, the inhibitory activity of the fully processed anti-EGFR/HER3 LUZ-Y is comparable with the activity of the combination of the two parental antibodies, neither of which has a significant inhibitory effect as a single agent. In the experiment shown the IC50 for the LUZ-Y was 0.7 nm, whereas the observed IC50 for the combined parental antibodies was 0.35 nm.
DISCUSSION
Numerous formats have been reported for producing bispecific antibodies from either bacterial or mammalian cells (3, 4, 5). With the exception of the previously noted bispecific antibody formats, most of these formats yield antibodies that have significant structural differences when compared with normal IgG. Although the two-in-one Ab is structurally similar to the native IgG, development of this type of bispecific antibody is challenging. The goal of this study was to develop a format that allowed the rapid production of bispecific antibodies from mammalian cells that would have minimal structural differences to human IgG. In this report, we demonstrate the use of leucine zippers and peptide tethers to direct heterodimeric assembly of distinct HCs and appropriate pairing of cognate LCs and HCs and utilize a proteolytic processing scheme to generate a hIgG-like bispecific antibody. Following purification and proteolytic processing, LUZ-Ys differ from their parental mAbs by only a single Lys to Ala mutation at residue 222 in the hinge region of the HC. We demonstrate that these antibodies are able to bind their antigens specifically, with high affinity and, in the case of the bispecific formats, discriminately and non-competitively.
Although the LUZ-Y platform developed here enabled production of bispecific antibodies from transiently transfected cells, production of stable cell lines to allow manufacturing scale production of LUZ-Ys will require the isolation of a stable cell line that expresses roughly equimolar amounts of the two HCs and maintains this expression stoichiometry over time. Also unknown is the scalability of the enzymatic steps employed to completely remove the tether. Solving both of these issues will be required to allow production of LUZ-Ys at the manufacturing scale.
Acknowledgments
We thank Jean-Michel Vernes and Yu-Ju (Gloria) Meng for performing Fc receptor binding ELISAs and J. Michael Elliott for help with mass spectroscopy analysis.
The authors disclose potential conflicts of interest as all authors are or have been employed by Genentech, which develops and markets drugs for profit.

This article contains supplemental Tables 1 and 2 and Figs. 1 and 2.
- LC
- light chain
- HC
- heavy chain
- EGFR
- EGF receptor
- ECD
- extracellular domain
- NP
- Nitrophenyl.
REFERENCES
- 1. Staerz U. D., Kanagawa O., Bevan M. J. (1985) Hybrid antibodies can target sites for attack by T cells. Nature 314, 628–631 [DOI] [PubMed] [Google Scholar]
- 2. Staerz U. D., Bevan M. J. (1986) Hybrid hybridoma producing a bispecific monoclonal antibody that can focus effector T-cell activity. Proc. Natl. Acad. Sci. U.S.A. 83, 1453–1457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kontermann R. E. (2010) Alternative antibody formats. Curr. Opin. Mol. Ther 12, 176–183 [PubMed] [Google Scholar]
- 4. Fischer N., Léger O. (2007) Bispecific antibodies: molecules that enable novel therapeutic strategies. Pathobiology 74, 3–14 [DOI] [PubMed] [Google Scholar]
- 5. Hollander N. (2009) Bispecific antibodies for cancer therapy. Immunotherapy 1, 211–222 [DOI] [PubMed] [Google Scholar]
- 6. Ridgway J. B., Presta L. G., Carter P. (1996) ‘Knobs-into-holes’ engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng. 9, 617–621 [DOI] [PubMed] [Google Scholar]
- 7. Davis J. H., Aperlo C., Li Y., Kurosawa E., Lan Y., Lo K-M., Houston J. S. (2010) SEEDbodies: fusion proteins based on strand-exchange engineered domain (SEED) CH3 heterodimers in an Fc analogue platform for asymmetric binders or immunofusions and bispecific antibodies. Protein Engineering, Design and Selection 23, 195–202 [DOI] [PubMed] [Google Scholar]
- 8. Gunasekaran K., Pentony M., Shen M., Garrett L., Forte C., Woodward A., Ng S. B., Born T., Retter M., Manchulenko K., Sweet H., Foltz I. N., Wittekind M., Yan W. (2010) Enhancing antibody Fc heterodimer formation through electrostatic steering effects. J. Biol. Chem. 285, 19637–19646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Merchant A. M., Zhu Z., Yuan J. Q., Goddard A., Adams C. W., Presta L. G., Carter P. (1998) An efficient route to human bispecific IgG. Nat. Biotechnol. 16, 677–681 [DOI] [PubMed] [Google Scholar]
- 10. Schaefer W., Regula J. T., Bähner M., Schanzer J., Croasdale R., Dürr H., Gassner C., Georges G., Kettenberger H., Imhof-Jung S., Schwaiger M., Stubenrauch K. G., Sustmann C., Thomas M., Scheuer W., Klein C. (2011) Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies. Proc. Natl. Acad. Sci. U.S.A. 108, 11187–11192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bostrom J., Yu S. F., Kan D., Appleton B. A., Lee C. V., Billeci K., Man W., Peale F., Ross S., Wiesmann C., Fuh G. (2009) Variants of the antibody herceptin that interact with HER2 and VEGF at the antigen binding site. Science 323, 1610–1614 [DOI] [PubMed] [Google Scholar]
- 12. Kostelny S. A., Cole M. S., Tso J. Y. (1992) Formation of a bispecific antibody by the use of leucine zippers. J. Immunol. 148, 1547–1553 [PubMed] [Google Scholar]
- 13. de Kruif J., Logtenberg T. (1996) Leucine zipper dimerized bivalent and bispecific scFv antibodies from a semi-synthetic antibody phage display library. J. Biol. Chem. 271, 7630–7634 [DOI] [PubMed] [Google Scholar]
- 14. Li J. S., Cui L., Rock D. L., Li J. (2005) Novel glycosidic linkage in Aedes aegypti chorion peroxidase. J. Biol. Chem. 280, 38513–38521 [DOI] [PubMed] [Google Scholar]
- 15. Pruss R. M., Herschman H. R., Klement V. (1978) 3T3 variants lacking receptors for epidermal growth factor are susceptible to transformation by Kirsten sarcoma virus. Nature 274, 272–274 [DOI] [PubMed] [Google Scholar]
- 16. Schaefer G., Shao L., Totpal K., Akita R. W. (2007) Erlotinib directly inhibits HER2 kinase activation and downstream signaling events in intact cells lacking epidermal growth factor receptor expression. Cancer Res. 67, 1228–1238 [DOI] [PubMed] [Google Scholar]
- 17. O'Shea E. K., Lumb K. J., Kim P. S. (1993) Peptide 'Velcro': design of a heterodimeric coiled coil. Curr. Biol. 3, 658–667 [DOI] [PubMed] [Google Scholar]
- 18. Wines B. D., Powell M. S., Parren P. W., Barnes N., Hogarth P. M. (2000) The IgG Fc contains distinct Fc receptor (FcR) binding sites: The leukocyte receptors FcγRI and FcγRIIa bind to a region in the Fc distinct from that recognized by neonatal FcR and protein A. J. Immunol. 164, 5313–5318 [DOI] [PubMed] [Google Scholar]
- 19. Schaefer G., Haber L., Crocker L. M., Shia S., Shao L., Dowbenko D., Totpal K., Wong A., Lee C. V., Stawicki S., Clark R., Fields C., Lewis Phillips G. D., Prell R. A., Danilenko D. M., Franke Y., Stephan J. P., Hwang J., Wu Y., Bostrom J., Sliwkowski M. X., Fuh G., Eigenbrot C. (2011) A two-in-one antibody against HER3 and EGFR has superior inhibitory activity compared with monospecific antibodies. Cancer Cell 20, 472–486 [DOI] [PubMed] [Google Scholar]
- 20. Hudziak R. M., Lewis G. D., Winget M., Fendly B. M., Shepard H. M., Ullrich A. (1989) p185HER2 monoclonal antibody has antiproliferative effects in vitro and sensitizes human breast tumor cells to tumor necrosis factor. Mol. Cell. Biol. 9, 1165–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jackman J., Chen Y., Huang A., Moffat B., Scheer J. M., Leong S. R., Lee W. P., Zhang J., Sharma N., Lu Y., Iyer S., Shields R. L., Chiang N., Bauer M. C., Wadley D., Roose-Girma M., Vandlen R., Yansura D. G., Wu Y., Wu L. C. (2010) Development of a two-part strategy to identify a therapeutic human bispecific antibody that inhibits IgE receptor signaling. J. Biol. Chem. 285, 20850–20859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wilson T. R., Lee D. Y., Berry L., Shames D. S., Settleman J. (2011) Neuregulin-1-mediated autocrine signaling underlies sensitivity to HER2 kinase inhibitors in a subset of human cancers. Cancer Cell 20, 158–172 [DOI] [PubMed] [Google Scholar]
- 23. Krysan D. J., Rockwell N. C., Fuller R. S. (1999) Quantitative characterization of furin specificity. J. Biol. Chem. 274, 23229–23234 [DOI] [PubMed] [Google Scholar]
- 24. Harris R. J. (1995) Processing of C-terminal lysine and arginine residues of proteins isolated from mammalian cell culture. J. Chromatogr. A 705, 129–134 [DOI] [PubMed] [Google Scholar]
- 25. Kuwada S. K., Scaife C. L., Kuang J., Li X., Wong R. F., Florell S. R., Coffey R. J., Jr., Gray P. D. (2004) Effects of trastuzumab on epidermal growth factor receptor-dependent and -independent human colon cancer cells. Int. J. Cancer 109, 291–301 [DOI] [PubMed] [Google Scholar]