

Background: Human cystathionine β-synthase (CBS) catalyzes the condensation of homocysteine with serine or cysteine to give cystathionine and H2O or H2S.
Results: Difference stopped-flow spectroscopy was employed to characterize reaction intermediates.
Conclusion: An aminoacrylate intermediate common to both condensation reactions was detected.
Significance: This is the first pre-steady-state kinetic analysis of H2S production by human CBS.
Keywords: Heme, Homocysteine, Hydrogen Sulfide, Pre-steady-state Kinetics, Pyridoxal Phosphate
Abstract
Human cystathionine β-synthase (CBS), a novel heme-containing pyridoxal 5′-phosphate enzyme, catalyzes the condensation of homocysteine and serine or cysteine to produce cystathionine and H2O or H2S, respectively. The presence of heme in CBS has limited spectrophotometric characterization of reaction intermediates by masking the absorption of the pyridoxal 5′-phosphate cofactor. In this study, we employed difference stopped-flow spectroscopy to characterize reaction intermediates formed under catalytic turnover conditions. The reactions of l-serine and l-cysteine with CBS resulted in the formation of a common aminoacrylate intermediate (kobs = 0.96 ± 0.02 and 0.38 ± 0.01 mm−1 s−1, respectively, at 24 °C) with concomitant loss of H2O and H2S and without detectable accumulation of the external aldimine or other intermediates. Homocysteine reacted with the aminoacrylate intermediate with kobs = 40.6 ± 3.8 s−1 and re-formed the internal aldimine. In the reverse direction, CBS reacted with cystathionine, forming the aminoacrylate intermediate with kobs = 0.38 ± 0.01 mm−1 s−1. This study provides the first insights into the pre-steady-state kinetic mechanism of human CBS and indicates that the reaction is likely to be limited by a conformational change leading to product release.
Introduction
Cystathionine β-synthase (CBS)2 serves two major functions in the mammalian sulfur network. First, it plays a pivotal role in removing excess sulfur and maintaining low homocysteine levels by catalyzing its conversion to cystathionine (Fig. 1, Reaction 1) (1). Genetic defects in CBS, which catalyzes the first step in the transsulfuration pathway, are the single most common cause of homocystinuria, characterized by aggressive occlusive arterial disease and other severe pathologies (2). Second, CBS is one of two significant contributors to the biogenesis of H2S (3), a gaseous signaling molecule that has cardioprotective and other physiological effects (4). The role of CBS in H2S production is believed to be particularly important in brain, where H2S functions as a neuromodulator (5). The other major source of H2S, particularly in the periphery, is γ-cystathionase, the second enzyme in the transsulfuration pathway (6). A third enzyme, mercaptopyruvate sulfurtransferase, which is involved in cysteine catabolism, generates protein-bound persulfide, which could release H2S upon reduction (7, 8). The contribution of the sulfurtransferase at physiological concentrations of the substrate (mercaptopyruvate) remains to be evaluated (3).
FIGURE 1.

Reactions catalyzed by CBS. Reaction 1 represents the “canonical” reaction in the transsulfuration pathway, whereas Reactions 2–4 generate H2S.
CBS generates H2S via multiple reactions (Fig. 1, Reactions 2–4) (9). Each of these represents a β-replacement reaction, and under maximum velocity (Vmax) conditions, the turnover number for the condensation of cysteine and homocysteine (i.e. Reaction 2) is ∼20- and 40-fold faster than for Reactions 3 and 4, respectively. In fact, the canonical β-replacement reaction of serine by homocysteine (Reaction 1) proceeds ∼4-fold slower than Reaction 2 under Vmax conditions. At physiologically relevant substrate concentrations, the condensation of 2 mol of cysteine is predicted to be the least efficient route for H2S generation, accounting for 1.6% of the H2S produced, whereas the condensation of homocysteine and cysteine is by far the most efficient, accounting for 96% of the H2S (9).
Human CBS is unique in being a pyridoxal 5′-phosphate (PLP)-dependent enzyme that is also a heme protein. The heme cofactor resides ∼20 Å away from the active site (10, 11) and plays a regulatory role (12), switching off enzyme activity in response to CO (13–15) and NO (16). The heme in CBS is 6-coordinate in the ferric and ferrous states, and the redox potential for the ferrous/ferric couple for the full-length enzyme is estimated to be approximately ∼350 ± 4 mV (17). Kinetic coupling of reduction of the heme, which has a fairly low redox potential, to carbonylation allows formation of the ferrous CO species with physiological reductants (13). Changes in the heme ligation state are communicated to the active site and lead to a shift in the tautomeric equilibrium of the Schiff base of PLP from the predominantly active keto enamine to the inactive enol imine (14). The presence of the heme with a high extinction coefficient obscures the UV-visible spectrum of the PLP cofactor and renders challenging the direct observation of tautomeric states and reaction intermediates during the catalytic cycle. Indeed, the effect of ferrous CO on the PLP cofactor could be observed by fluorescence and resonance Raman spectroscopies rather than absorption spectroscopy (14).
CBS belongs to the fold II family of PLP enzymes, which includes well studied members, e.g. O-acetylserine sulfhydrylase (18), threonine deaminase (19), and tryptophan synthase (20). The PLP is tethered in the active site of human CBS via a Schiff base linkage with Lys-119 (Fig. 2). Binding of serine or cysteine leads to formation of the external aldimine in a multistep reaction involving a gem-diamine intermediate. Deprotonation of the α-proton followed by β-elimination of H2O from the external aldimine of serine (or H2S from the external aldimine of cysteine) yields the aminoacrylate intermediate. In the second half-reaction, the aminoacrylate intermediate undergoes nucleophilic attack by the thiolate of homocysteine to yield the external aldimine of the product, cystathionine. Alternatively, addition of cysteine or water yields lanthionine or serine, respectively, bound as external aldimines. Release of cystathionine, lanthionine, or serine by a second transchiffization reaction restores the resting internal aldimine state of the enzyme.
FIGURE 2.
Proposed catalytic cycle of CBS. The resting enzyme exists as the internal aldimine with Lys-119 in human CBS involved in Schiff base linkage. Addition of serine (or cysteine) gives a gem-diamine intermediate, which collapses to the external aldimine of serine (or cysteine). Elimination of water (or H2S) leads to the aminoacrylate intermediate to which homocysteine (cysteine or water) adds to form the external aldimine of the product cystathionine (lanthionine or serine). The latter is released via the transient formation of a gem-diamine intermediate in a transchiffization reaction involving the active site lysine. Elimination of cystathionine from the active site completes the catalytic cycle and regenerates the resting enzyme. The numbers in parentheses represent the absorption maxima of the intermediates.
Spectroscopic characterization of a hemeless variant of human CBS lacking the N-terminal heme-binding domain has been reported (21). This variant retains 40% of the activity of wild-type human CBS, and its characterization was limited by its instability and proteolytic susceptibility, leading to poor yields (21). Insights from studies on the hemeless variant of human CBS were also limited, as deletion of the heme domain unexpectedly resulted in the enzyme being inhibited rather than activated by S-adenosylmethionine. CBS contains a C-terminal regulatory domain to which S-adenosylmethionine binds, and the juxtaposition of the regulatory, catalytic, and heme domains was recently revealed in the structure of full-length Drosophila CBS (dCBS) (22). In contrast to human and fly CBS, the absence of heme in yeast CBS (yCBS) has permitted ready characterization of PLP-based intermediates. Detailed rapid reaction kinetics of the yCBS-catalyzed canonical and H2S-generating reactions have been reported (23–25).
In this study, we used difference electronic absorption spectroscopy to monitor the reaction kinetics of full-length human CBS under pre-steady-state conditions. We recently established the utility of this approach by spectroscopic detection of the aminoacrylate intermediate in dCBS, which was also captured in the crystal structure (22). The reactions catalyzed in the presence of serine or cysteine provide, for the first time, kinetic insights into the canonical transsulfuration reaction and the H2S-producing reactions catalyzed by human CBS.
EXPERIMENTAL PROCEDURES
Materials
l-Cysteine, dl-homocysteine, l-cystathionine, l-serine, and PLP were purchased from Sigma. [14C]Serine (164 mCi/mmol) was purchased from PerkinElmer Life Sciences. GST-Sepharose 4B was purchased from GE Healthcare. All other chemicals were purchased from Fisher.
Expression and Purification of CBS
Unless specified otherwise, CBS refers to the recombinant human enzyme used in this study. Wild-type CBS was expressed and purified as described previously (26). Briefly, the cell pellet obtained from a 6× 1 liter culture was resuspended in 300 ml of 50 mm Tris buffer (pH 8.0) containing a single protease inhibitor mixture tablet (Roche Applied Science) and 100 mg of lysozyme and stirred for 1 h at 4 °C. The cells were lysed by sonication, and the supernatant was obtained by centrifugation at 12,000 × g for 25 min. The supernatant was loaded onto a glutathione-Sepharose column pre-equilibrated with 1× PBS. The column was washed with 1× PBS, and the GST-fused CBS was eluted with 50 mm Tris (pH 8.0) containing 10 mm glutathione. This step was followed by thrombin digestion at 4 °C to remove the GST tag. The protein was loaded onto a Q-Sepharose column equilibrated with 50 mm Tris buffer (pH 8.0), and the column was then washed with the same buffer. The protein was eluted with a 500-ml gradient ranging from 0 to 500 mm NaCl in 50 mm Tris-HCl (pH 8.0). The fractions of interest were concentrated and stored at −80 °C.
Steady-state Enzyme Assay
Enzyme activity was measured as described previously (26) in the presence of varying concentrations of homocysteine and 30 mm [14C]serine (164 mCi/mmol) in 100 mm HEPES (pH 7.4) at 24 °C. The turnover number for the enzyme obtained under these conditions was used as a reference point for the observed reaction rates measured under pre-steady-state conditions.
Stopped-flow Spectroscopy
Pre-steady-state experiments were performed using an Applied Photophysics stopped-flow spectrophotometer (SX.MV18) in the photodiode array mode as described (22). In brief, a solution of CBS (60 μm calculated per 63-kDa monomer) in 100 mm HEPES (pH 7.4) was rapidly mixed with various concentrations of substrates (cysteine, serine, or cystathionine) in the same buffer. The time-dependent formation of reaction intermediates was followed by difference spectroscopy ((enzyme + substrate) − enzyme). The temperature was maintained at 24 °C using a circulating water bath. To ensure that changes in the heme spectrum did not occur during enzyme-monitored turnover, the spectrum of CBS (20 μm) was recorded following mixing with serine (60 mm) in a stopped-flow spectrophotometer. To verify that the kinetic analysis of difference spectra did not introduce artifacts, we also monitored the kinetics of formation of reaction intermediates at a single wavelength (465 or 406 nm) following mixing of 60 μm CBS with varying concentrations (2, 20, and 60 mm) of serine. When the reaction of preformed aminoacrylate with homocysteine was monitored, 60 μm enzyme was premixed with 25 mm serine in 100 mm HEPES (pH 7.4), and the resulting aminoacrylate intermediate was rapidly mixed with various concentrations of homocysteine as described previously (24).
For experiments in which cystathionine was employed, a stock solution was made in 100 mm HEPES (pH 7.4), followed by the gradual addition of 5 m NaOH until the solution was clear. Further dilutions of cystathionine were made in 100 mm HEPES (pH 7.4). Data from the stopped-flow experiments were fitted using SigmaPlot. The observed rate constants (kobs) for the reactions with serine, cysteine, homocysteine, and cystathionine were determined from fits using a single-exponential equation. The kobs values obtained at various concentrations of substrates were fitted to a linear equation to obtain the apparent second-order rate constants.
The observed rate constant for the disappearance of the aminoacrylate intermediate in the presence of homocysteine (Hyc) showed a hyperbolic dependence on substrate concentration, and the data were fitted using Equation 1, in which K21 = k2/k1.
RESULTS
Reaction of CBS with Serine
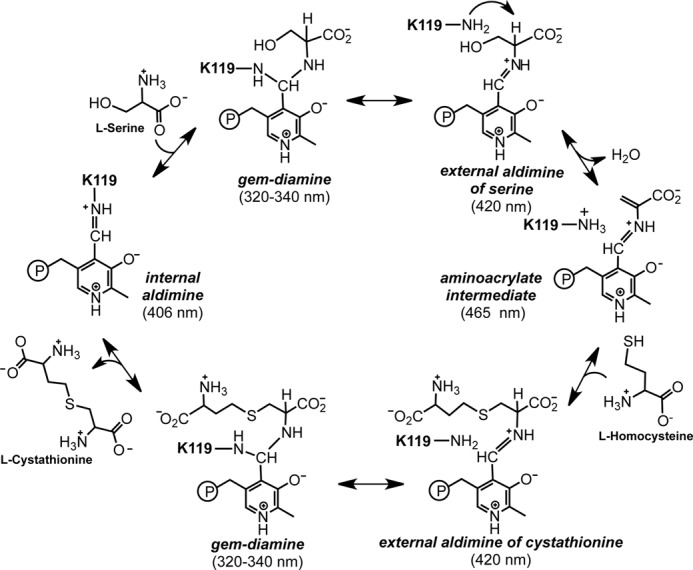
Rapid scanning stopped-flow spectra recorded after mixing CBS with serine (Fig. 3A) showed that although the absorption maxima associated with ferric heme (Soret peak at 428 nm and the α/β-bands centered at 550 nm) did not change, there was an increase in absorption at 465 nm (assigned to a PLP intermediate). To better characterize the PLP-dependent absorption changes elicited by serine, difference spectra were analyzed (Fig. 3B). A disappearance in the absorbance at 406 nm (assigned to the internal aldimine) and the appearance of absorbance at 465 nm (assigned to the aminoacrylate intermediate) were observed. The difference spectra exhibit two apparent isosbestic points at 357 and 422 nm, respectively, indicating that the external aldimine did not accumulate at measurable concentrations. For comparison, addition of serine led to the formation of a broad absorption peak between 460 and 510 nm with the hemeless variant of CBS (21) and at 465 nm with yCBS (23, 24). Rates for the disappearance of the internal aldimine (Fig. 3C) and the appearance of the aminoacrylate species (Fig. 3D) showed a linear dependence on the concentration of serine (1–55 mm). The absence of observable intermediates between the internal aldimine and the aminoacrylate species accounts for the linear dependence of kobs on serine concentration. From the analysis in Fig. 3 (C and D), bimolecular rate constants of comparable value were obtained: 1.08 ± 0.04 mm−1 s−1 for the disappearance of the internal aldimine and 0.96 ± 0.02 mm−1 s−1 for the formation of the aminoacrylate intermediate. To ensure that the kinetic analysis of difference spectrum data did not skew the estimates, the experiments were repeated under single-wavelength monitoring conditions at 406 and 465 nm, respectively, at three concentrations of serine (Fig. 3E). The bimolecular rate constant obtained from this analysis was identical (1.14 ± 0.13 mm−1 s−1) within experimental error to that estimated from multiple-wavelength monitoring.
FIGURE 3.

Spectroscopic and kinetic monitoring of the aminoacrylate intermediate formed in the presence of serine. A and B, spectra were recorded following mixing of 20 μm (before mixing) CBS with 60 mm serine (A), whereas difference spectra ((CBS + serine) − CBS) were acquired every 2 s following rapid mixing of CBS (60 μm in active sites, before mixing) with 50 mm serine in 100 mm HEPES (pH 7.4) at 24 °C (B). The internal aldimine has an absorption maximum at ∼406 nm, and upon addition of serine, it converts to the aminoacrylate intermediate with absorption maxima at 465 and 320 nm. C, dependence of the observed rates for disappearance of the internal aldimine on the concentration of serine. Inset, dependence of the absorbance change at 406 nm at 0.3 s after mixing on serine concentration. D, dependence of the observed rates for formation of the aminoacrylate on the concentration of serine obtained from the difference spectra. Inset, representative kinetic traces at 465 nm obtained in the presence of 1–55 mm serine (from bottom to top) are shown. E, dependence of the absorbance change at 465 nm at 0.1 s after mixing on serine concentration. F, the kinetics of aminoacrylate formation were recorded at single wavelengths (406 and 465 nm) at the indicated concentrations of serine. Inset, dependence of kobs at 465 nm on serine concentration.
From the ratio of the slope (1.05 ± 0.02 mm−1 s−1) to intercept (2.15 ± 0.82 s−1), the apparent Kd for serine was estimated to be 2.04 ± 0.72 mm (Fig. 3C). We also employed the dependence of the change in amplitude at 0.3 and 0.1 s at either 406 nm (Fig. 3C, inset) or 465 nm (Fig. 3E), respectively, to estimate the Kd for serine. This analysis yielded values of 2.3 ± 0.5 and 2.5 ± 0.6 mm, respectively. We note, however, that interpretation of the dependence of the observed rate constants on the concentration of serine is complex because kobs does not simply correspond to the binding of serine. Formation of the aminoacrylate from the internal aldimine involves multiple steps (e.g. the gem-diamine, external aldimine, and carbanion intermediates) that are spectroscopically and kinetically unresolved.
Addition of serine to CBS also gave rise to a second absorption peak centered at 320 nm, which we assigned as the enol imine tautomer of the aminoacrylate species (Fig. 3B). The rate of increase in absorbance at 465 and 320 nm was identical (not shown), consistent with our assignment that the internal aldimine converts to the tautomers of the aminoacrylate intermediate without detectable formation of the gem-diamine or the external aldimine intermediates. Formation of the aminoacrylate intermediate with absorption features at ∼320 and 460 nm has also been reported for yCBS (25) and dCBS (22).
Reaction of CBS with Cysteine
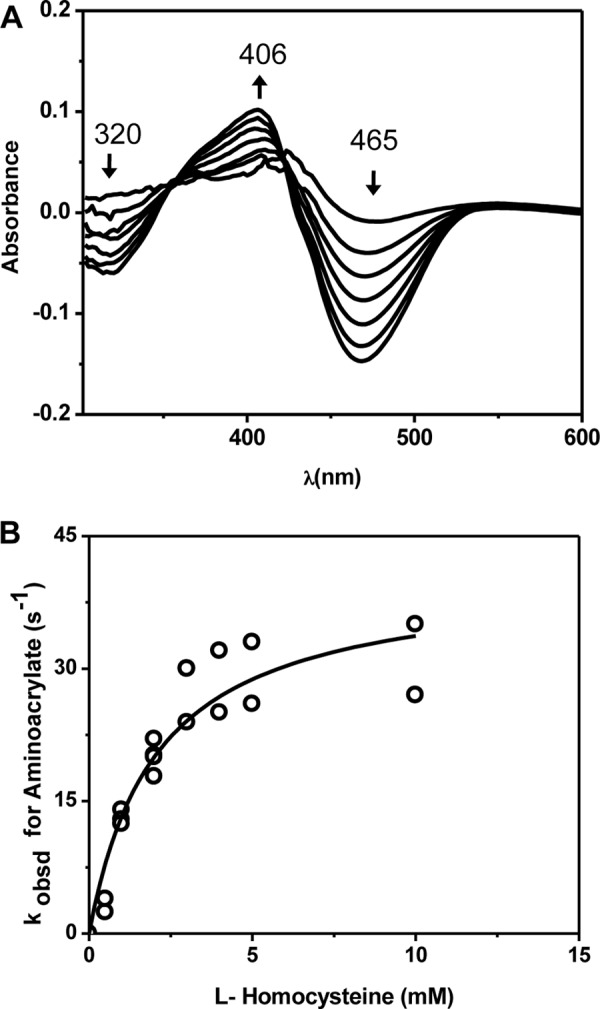
Addition of cysteine resulted in the appearance of difference absorption spectra (Fig. 4A) that were comparable with those seen in the presence of serine (Fig. 3B). The spectral changes were consistent with the conversion of the internal aldimine (406 nm) to the aminoacrylate intermediate (465 and 320 nm) with apparent isosbestic points at 357 and 422 nm. The observed rates for the disappearance of the internal aldimine and the formation of the aminoacrylate intermediate showed linear dependences on the concentration of cysteine (1–50 mm) (Fig. 4, B and C). The 320 nm peak formed at the same rate as the 465 nm peak (data not shown) but was of lower intensity. Data analysis at 406 and 465 nm yielded bimolecular rate constants that were of similar magnitude: 0.40 ± 0.03 mm−1 s−1 for the disappearance of the internal aldimine and 0.38 ± 0.01 mm−1 s−1 for the formation of the aminoacrylate, respectively. These rate constants were ∼2-fold lower than the corresponding biomolecular rate constants obtained with serine (Fig. 3). We note, however, that the reaction kinetics of cysteine with CBS are complicated by the simultaneous occurrence of two reactions, i.e. the β-replacement of cysteine with H2O and the β-replacement of cysteine with a second mole of cysteine (Fig. 1, Reactions 3 and 4). Hence, a simple comparison with the bimolecular rate constants obtained with serine and cysteine is not useful.
FIGURE 4.
Spectroscopic and kinetic monitoring of the aminoacrylate intermediate formed in the presence of cysteine. A, difference spectra ((CBS + cysteine) − CBS) obtained following rapid mixing of CBS with cysteine in 100 mm HEPES (pH 7.4) at 24 °C. The spectra were acquired every 2 s following mixing of 60 μm CBS with 50 mm cysteine. The internal aldimine has an absorption maximum at ∼406 nm, and upon addition of serine, it rapidly converts to the aminoacrylate intermediate with an absorption maximum at 465 nm. B and C, dependence of the apparent rates for the disappearance of the internal aldimine (B) and the formation of the aminoacrylate intermediate (C) on the concentration of cysteine. Insets, dependence of the absorbance changes at 406 and 465 nm, respectively, on cysteine concentration at 0.3 s after mixing.
From the ratio of the slope (0.4 ± 0.03 mm−1 s−1) to intercept (2.95 ± 0.58 s−1), the apparent Kd for cysteine was estimated to be 7.4 ± 1.5 mm (Fig. 3C). The dependence of the change in amplitude at 0.3 s at either 406 nm (Fig. 4B, inset) or 465 nm (Fig. 4C, inset) on cysteine concentration yielded Kd estimates of 7.4 ± 1.4 and 6.1 ± 1.8 mm, respectively. These values are similar to the Km1 value of 6.8 ± 1.7 mm for cysteine binding to the PLP site in CBS and distinct from the Km2 value of 27.3 ± 3.7 mm for cysteine binding to the second site in CBS (9).
Reaction of the Aminoacrylate Intermediate with Homocysteine
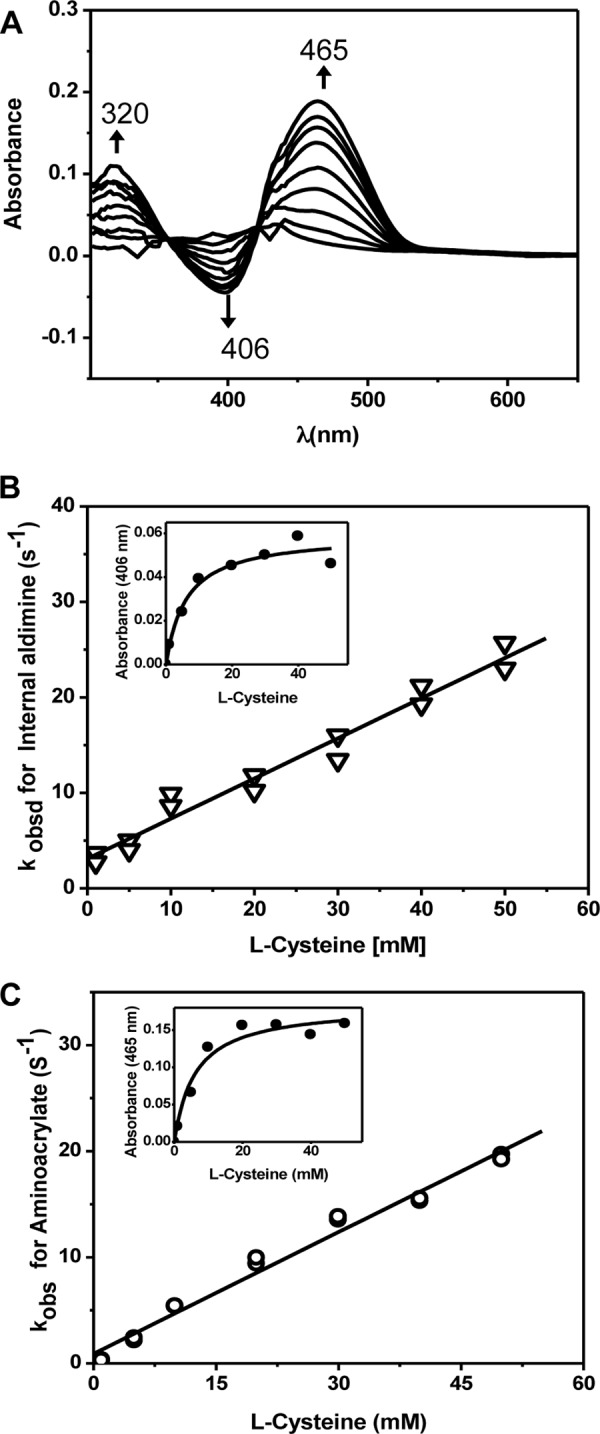
In the second half-reaction catalyzed by CBS, the aminoacrylate intermediate (generated by elimination of H2O from serine or H2S from cysteine) reacts with homocysteine to form cystathionine (Fig. 2). To determine the kinetics of cystathionine formation, the aminoacrylate intermediate was preformed by mixing 60 μm CBS and 25 mm serine, which were then rapidly mixed with varying concentrations of homocysteine. The difference spectra obtained following mixing of the aminoacrylate intermediate with 30 mm l-homocysteine show the disappearance of the 320 and 465 nm-absorbing aminoacrylate intermediate and the appearance of a 406 nm-absorbing aldimine species (Fig. 5A). These spectral changes occur with two apparent isosbestic points at 357 and 422 nm. The rate of disappearance of the aminoacrylate intermediate exhibits a hyperbolic dependence on homocysteine concentration and yields a maximum rate of 40.6 ± 3.8 s−1 for formation of the aldimine and a Km(app) for homocysteine of 2.1 ± 0.5 mm. The Km for homocysteine obtained in the steady-state assay using conditions identical to those used in the stopped-flow experiments is 2.8 ± 0.6 mm.
FIGURE 5.
Spectroscopic and kinetic monitoring of the conversion of the aminoacrylate intermediate in the presence of homocysteine. A, difference spectra ((CBS-aminoacrylate + homocysteine) − CBS-aminoacrylate) obtained following rapid mixing of preformed CBS-aminoacrylate with homocysteine in 100 mm HEPES (pH 7.4) at 24 °C. To obtain the spectra, 60 μm CBS was premixed with 25 mm serine and then mixed with 30 mm homocysteine The spectra were recorded every 2 s. Addition of homocysteine resulted in the disappearance of the aminoacrylate intermediate (320 and 465 nm) and the re-formation of the internal aldimine with an absorption maximum at ∼406 nm. B, dependence of the apparent rates for disappearance of the aminoacrylate intermediate on the concentration of homocysteine.
Reaction of CBS with l-Cystathionine
When CBS was rapidly mixed with l-cystathionine, the disappearance of the absorbance feature centered at 406 nm and the appearance of the 320 and 465 nm-absorbing aminoacrylate species were observed (Fig. 6A). Formation of the aminoacrylate intermediate from cystathionine indicates that the reaction is partially reversible. The observed rate of formation of the aminoacrylate intermediate was linearly dependent on the concentration of cystathionine (1–10 mm) (Fig. 6B). From these data, the apparent bimolecular rate constant for formation of the aminoacrylate intermediate from cystathionine was 0.38 ± 0.01 mm−1 s−1, and the Kd(app) for cystathionine was estimated to be 2.5 ± 0.7 mm from the ratio of the slope (0.38 ± 0.03 mm−1 s−1) to intercept (0.94 ± 0.26 s−1) (Fig. 6B). A Kd for cystathionine of 2.1 ± 0.7 mm was also determined from the dependence of the amplitude change at 465 nm at 0.5 s on the concentration of cystathionine (Fig. 6B, inset).
FIGURE 6.
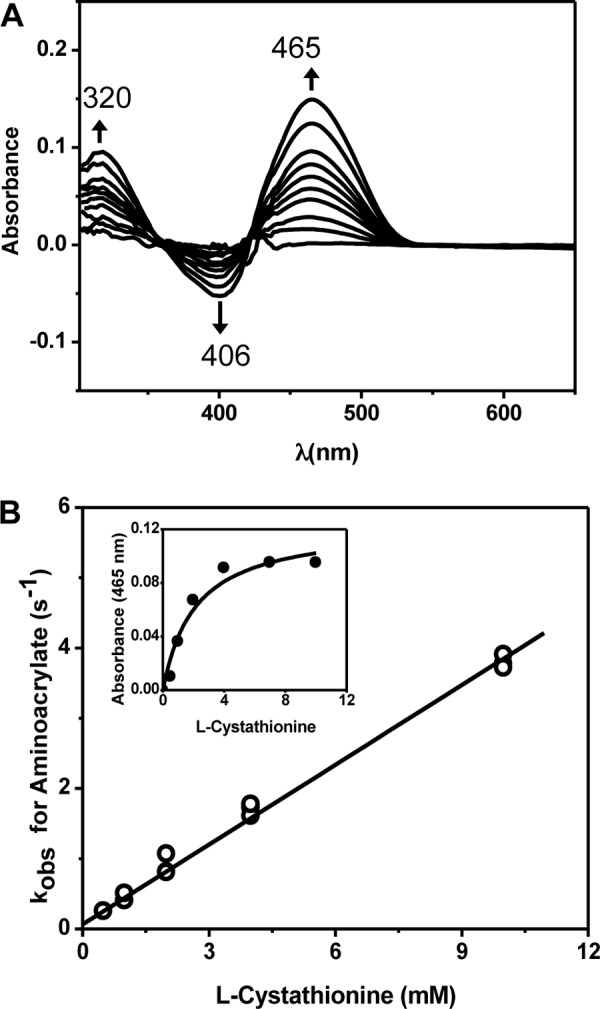
Spectroscopic and kinetic monitoring of the aminoacrylate intermediate formed in the presence of cystathionine. A, difference spectra ((CBS + cystathionine) − CBS) obtained following rapid mixing of CBS with cystathionine in 100 mm HEPES (pH 7.4) at 24 °C. The experiment was initiated by mixing 60 μm CBS with 20 mm l-cystathionine, and the spectra were recorded every 2 s. The internal aldimine has an absorption maximum at ∼406 nm, and upon addition of serine, it converts to the aminoacrylate intermediate with an absorption maximum at 320 or 465 nm. B, dependence of the apparent rate of formation of the aminoacrylate on the concentration of cystathionine. Inset, dependence of the absorbance changes at 465 nm on cystathionine concentration at 0.5 s after mixing.
DISCUSSION
The critical role of CBS in mammalian sulfur metabolism is exemplified by the pleiotropic clinical consequences of its impairment, inherited as an autosomal recessive disorder (27). Studies on the effects of CBS deficiency have been focused on the homocysteine management aspect of the reaction because homocystinuria is a hallmark of this condition. However, CBS also plays a role in H2S biogenesis, particularly in brain. Studying the detailed kinetic mechanism of human CBS is therefore of significant interest but has been limited by the presence of the heme cofactor, whose absorption spectrum masks that of the PLP cofactor. In contrast, yCBS, devoid of the heme cofactor, has been the subject of detailed kinetic analyses (23–25). However, significant differences exist in the catalytic and regulatory properties of yeast and human CBS (28), warranting direct characterization of the human enzyme.
Our study provides the first insights into the reaction catalyzed by full-length human CBS. The aminoacrylate is the only distinct intermediate detected in either direction by rapid scanning stopped-flow spectroscopy. The lack of detectable accumulation of the preceding external aldimine intermediate suggests that removal of the α-proton, needed to initiate formation of the aminoacrylate intermediate, might not be rate-limiting. This is consistent with the lack of a primary kinetic isotope effect on aminoacrylate formation in yCBS (25). In contrast, a primary kinetic isotope effect on formation of an aminoacrylate intermediate is seen in both O-acetylserine sulfhydrylase and tryptophan synthase, which are fold II PLP enzymes that catalyze β-replacement reactions (20, 29, 30).
In general, the rates of formation and disappearance of the aminoacrylate intermediate are slower for human versus yeast CBS (Table 1). This is consistent with the ∼5-fold lower kcat for Reaction 1 between serine and homocysteine and the ∼2.8-fold lower kcat for Reaction 2 between cysteine and homocysteine for the human versus yeast enzymes (23). Other differences in the pre-steady-state kinetics are also evident between yeast and human CBS. Although formation of the aminoacrylate intermediate by human CBS exhibited a linear dependence on the concentrations of serine (Fig. 3C), yCBS exhibited saturation kinetics with evidence for transient accumulation of an external aldimine, whose formation was linearly dependent on serine concentration (23, 24). In the presence of cysteine, a single linear dependence of the rate of aminoacrylate formation was observed with human CBS, whereas yCBS showed two phases, interpreted as representing Reactions 3 and 4, respectively (23).
TABLE 1.
Comparison of parameters obtained from pre-steady-state analyses of yeast and human CBS
| Parameter | Human CBS | yCBSa |
|---|---|---|
| Kd(app) for Serb | 2.04 ± 0.72 mm | 14.4 ± 1.7 mm |
| kobs (AA from Ser) | 0.96 ± 0.02 mm−1 s−1 | 38.0 ± 3.0 mm−1 s−1 |
| Kd(app) for Cysb | 7.4 ± 1.5 mm | ND |
| kobs (AA from Cys) | 0.38 ± 0.01 mm−1 s−1 | 1.61 ± 0.3 mm−1 s−1,c 4.1. ± 0.2 mm−1 s−1d |
| Kd(app) for cystathionineb | 2.5 ± 0.7 mm | 1.6 ± 0.3 mm |
| kobs (AA from cystathionine) | 0.38 ± 0.01 mm−1 s−1 | 4.1 ± 0.2 mm−1 s−1 |
| Km(app) for Hcy,e Kd(app) for Hcy,f | 2.1 ± 0.5 mm | 0.37 ± 0.02 mm |
| kobs (aldimine from AA)g | ND | 183 ± 4 mm−1 s−1 |
| kmax (aldimine from AA) | 40.6 ± 3.8 s−1 | ND |
a The values for yCBS are taken from Ref. 23.
b The Kd(app) values listed for human CBS were derived from the dependence of kobs on the respective substrate concentrations as discussed under “Results.”
c The values represent the rates for Reaction 3 in Fig. 1.
d The values represent the rates for Reaction 4 in Fig. 1.
e This parameter was determined for human CBS. Hyc, homocysteine.
f This parameter was determined for yCBS.
g AA, aminoacrylate; ND, determined.
Crystal structures of full-length dCBS have revealed details of how the carbanion and aminoacrylate intermediates interact with and are stabilized by neighboring residues (Fig. 7A) (22). The 1.7 Å structure of the carbanion intermediate generated upon removal of the Cα proton shows that the ϵ-amino group of the active site lysine (corresponding to Lys-119 in the human sequence) is positioned within 2.1 Å of the Cα of serine and 3.0 Å from the C4 of PLP, stabilizing the negative charge (Fig. 7A). On the opposite side of the lysine, the hydroxyl group of serine (Ser-147 in the human sequence) contacts the Cα and might play a role in activating the leaving group (OH in serine and SH in cysteine). The structure supports a direct role of the active site lysine (Lys-119 in the human sequence) as a general base for Cα proton abstraction and for carbanion stabilization. Mutation of Lys-119 to alanine results in a 1000-fold diminution in enzyme activity compared with wild-type CBS, which is alleviated by ∼2-fold by provision of an exogenous base, e.g. ethylamine, consistent with the additional role of Lys-119 as a general base during the catalytic cycle (21). The interactions between the carbanion intermediate and active site residues are consistent with a greater role for the Schiff base in providing ylide-type electrostatic stabilization versus delocalization of the negative charge through the extended conjugated ring system of the PLP to generate a quinonoid intermediate, which is not observed under pre-steady-state conditions with either human or yeast CBS (23–25). Furthermore, the presence of a serine residue (Ser-349 in the human sequence) proximal to N1 in the PLP ring (versus an aspartate, for example) is expected not to stabilize a quinonoid intermediate.
FIGURE 7.

Structures of reaction intermediates in CBS. The structures of the carbanion (A) and aminoacrylate (B) intermediates in dCBS show few key interactions. The numbering refers to the dCBS sequence, whereas the numbers in parentheses refer to the human CBS sequence. The figures were generated from Protein Data Bank code 3PC4 (A) and 3PC3 (B).
Although the carbanion intermediate was fortuitously trapped in the structure of dCBS, it is a transient and reactive intermediate that is expected to rapidly undergo β-elimination to yield the aminoacrylate intermediate. In the 1.55 Å structure of the aminoacrylate intermediate, the ϵ-amino group of the active site lysine has swung away from the Cα and interacts instead with the phosphate oxygens of PLP (Fig. 7B). The Cβ of the aminoacrylate intermediate is positioned for nucleophilic addition (22), which is expected to occur from the same face from which the leaving group departed based on stereochemical analyses reported by Borcsok and Abeles (31).
The kcat for the reaction of CBS (with serine and homocysteine) determined in the steady-state assay using the same buffer, pH, and temperature conditions as in the stopped-flow experiments is 0.60 ± 0.05 s−1. At 30 mm serine, the concentration used in the steady-state assay, the rate of aminoacrylate intermediate formation is estimated to be ∼32 s−1. Hence, steps between binding of serine and aminoacrylate formation are not rate-determining in the first half-reaction. The kobs for formation of the aminoacrylate from cysteine is ∼2.5-fold lower than that from serine. Because H2S is in fact a better leaving group than H2O at physiological pH, the higher kobs for the reaction with serine suggests that H2O is released faster than H2S from the active site. Similarly, the chemical steps between the aminoacrylate intermediate and re-formation of the internal aldimine are not rate-determining (kmax = 40.6 ± 3.8 s−1). Hence, a step following re-formation of the internal aldimine, e.g. product release or a conformational change, probably limits the reaction rate. A conformational change induced by substrate binding/product release is a common feature of PLP enzymes. Such a conformational change in CBS is evident upon comparing the crystal structures of the apo- and substrate-bound forms of dCBS (22). Binding of substrate triggers the closed conformation and repositioning of the active site serine in the proximity of the cofactor.
This work was supported, in whole or in part, by National Institutes of Health Grant HL58984.
- CBS
- cystathionine β-synthase
- PLP
- pyridoxal 5′-phosphate
- dCBS
- Drosophila CBS
- yCBS
- yeast CBS.
REFERENCES
- 1. Zou C.-G., Banerjee R. (2005) Homocysteine and redox signaling. Antioxid. Redox Signal. 7, 547–559 [DOI] [PubMed] [Google Scholar]
- 2. Kraus J. P., Janosík M., Kozich V., Mandell R., Shih V., Sperandeo M. P., Sebastio G., de Franchis R., Andria G., Kluijtmans L. A., Blom H., Boers G. H., Gordon R. B., Kamoun P., Tsai M. Y., Kruger W. D., Koch H. G., Ohura T., Gaustadnes M. (1999) Cystathionine β-synthase mutations in homocystinuria. Hum. Mutat. 13, 362–375 [DOI] [PubMed] [Google Scholar]
- 3. Singh S., Banerjee R. (2011) PLP-dependent H2S biogenesis. Biochim. Biophys. Acta 1814, 1518–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kabil O., Banerjee R. (2010) The redox biochemistry of hydrogen sulfide. J. Biol. Chem. 285, 21903–21907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Abe K., Kimura H. (1996) The possible role of hydrogen sulfide as an endogenous neuromodulator. J. Neurosci. 16, 1066–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kabil O., Vitvitsky V., Xie P., Banerjee R. (2011) The quantitative significance of the transsulfuration enzymes for H2S production in murine tissues. Antioxid. Redox Signal. 15, 363–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nagahara N., Okazaki T., Nishino T. (1995) Cytosolic mercaptopyruvate sulfurtransferase is evolutionarily related to mitochondrial rhodanese. Striking similarity in active site amino acid sequence and the increase in the mercaptopyruvate sulfurtransferase activity of rhodanese by site-directed mutagenesis. J. Biol. Chem. 270, 16230–16235 [DOI] [PubMed] [Google Scholar]
- 8. Shibuya N., Tanaka M., Yoshida M., Ogasawara Y., Togawa T., Ishii K., Kimura H. (2009) 3-Mercaptopyruvate sulfurtransferase produces hydrogen sulfide and bound sulfane sulfur in the brain. Antioxid. Redox Signal. 11, 703–714 [DOI] [PubMed] [Google Scholar]
- 9. Singh S., Padovani D., Leslie R. A., Chiku T., Banerjee R. (2009) Relative contributions of cystathionine β-synthase and γ-cystathionase to H2S biogenesis via alternative transsulfuration reactions. J. Biol. Chem. 284, 22457–22466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Meier M., Janosik M., Kery V., Kraus J. P., Burkhard P. (2001) Structure of human cystathionine β-synthase: a unique pyridoxal 5′-phosphate-dependent heme protein. EMBO J. 20, 3910–3916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Taoka S., Lepore B. W., Kabil O., Ojha S., Ringe D., Banerjee R. (2002) Human cystathionine β-synthase is a heme sensor protein. Evidence that the redox sensor is heme and not the vicinal cysteines in the CXXC motif seen in the crystal structure of the truncated enzyme. Biochemistry 41, 10454–10461 [DOI] [PubMed] [Google Scholar]
- 12. Singh S., Madzelan P., Banerjee R. (2007) Properties of an unusual heme cofactor in PLP-dependent cystathionine β-synthase. Nat. Prod. Rep. 24, 631–639 [DOI] [PubMed] [Google Scholar]
- 13. Kabil O., Weeks C. L., Carballal S., Gherasim C., Alvarez B., Spiro T. G., Banerjee R. (2011) Reversible heme-dependent regulation of human cystathionine β-synthase by a flavoprotein oxidoreductase. Biochemistry 50, 8261–8263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Weeks C. L., Singh S., Madzelan P., Banerjee R., Spiro T. G. (2009) Heme regulation of human cystathionine β-synthase activity: insights from fluorescence and Raman spectroscopy. J. Am. Chem. Soc. 131, 12809–12816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Taoka S., West M., Banerjee R. (1999) Characterization of the heme and pyridoxal phosphate cofactors of human cystathionine β-synthase reveals nonequivalent active sites. Biochemistry 38, 2738–2744 [DOI] [PubMed] [Google Scholar]
- 16. Taoka S., Banerjee R. (2001) Characterization of NO binding to human cystathionine β-synthase: possible implications of the effects of CO and NO binding to the human enzyme. J. Inorg. Biochem. 87, 245–251 [DOI] [PubMed] [Google Scholar]
- 17. Singh S., Madzelan P., Stasser J., Weeks C. L., Becker D., Spiro T. G., Penner-Hahn J., Banerjee R. (2009) Modulation of the heme electronic structure and cystathionine β-synthase activity by second coordination sphere ligands: the role of heme ligand switching in redox regulation. J. Inorg. Biochem. 103, 689–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rabeh W. M., Cook P. F. (2004) Structure and mechanism of O-acetylserine sulfhydrylase. J. Biol. Chem. 279, 26803–26806 [DOI] [PubMed] [Google Scholar]
- 19. Gallagher D. T., Gilliland G. L., Xiao G., Zondlo J., Fisher K. E., Chinchilla D., Eisenstein E. (1998) Structure and control of pyridoxal phosphate-dependent allosteric threonine deaminase. Structure 6, 465–475 [DOI] [PubMed] [Google Scholar]
- 20. Drewe W. F., Jr., Dunn M. F. (1985) Detection and identification of intermediates in the reaction of l-serine with Escherichia coli tryptophan synthase via rapid-scanning ultraviolet-visible spectroscopy. Biochemistry 24, 3977–3987 [DOI] [PubMed] [Google Scholar]
- 21. Evande R., Ojha S., Banerjee R. (2004) Visualization of PLP-bound intermediates in hemeless variants of human cystathionine β-synthase: evidence that lysine 119 is a general base. Arch. Biochem. Biophys. 427, 188–196 [DOI] [PubMed] [Google Scholar]
- 22. Koutmos M., Kabil O., Smith J. L., Banerjee R. (2010) Structural basis for substrate activation and regulation by cystathionine β-synthase (CBS) domains in cystathionine β-synthase. Proc. Natl. Acad. Sci. U.S.A. 107, 20958–20963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Singh S., Ballou D. P., Banerjee R. (2011) Pre-steady-state kinetic analysis of enzyme-monitored turnover during cystathionine β-synthase-catalyzed H2S generation. Biochemistry 50, 419–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Taoka S., Banerjee R. (2002) Stopped-flow kinetic analysis of the reaction catalyzed by the full-length yeast cystathionine β-synthase. J. Biol. Chem. 277, 22421–22425 [DOI] [PubMed] [Google Scholar]
- 25. Jhee K. H., Niks D., McPhie P., Dunn M. F., Miles E. W. (2001) The reaction of yeast cystathionine β-synthase is rate-limited by the conversion of aminoacrylate to cystathionine. Biochemistry 40, 10873–10880 [DOI] [PubMed] [Google Scholar]
- 26. Taoka S., Ohja S., Shan X., Kruger W. D., Banerjee R. (1998) Evidence for heme-mediated redox regulation of human cystathionine β-synthase activity. J. Biol. Chem. 273, 25179–25184 [DOI] [PubMed] [Google Scholar]
- 27. Mudd S. H., Levy H. L., Kraus J. P. (2001) in The Online Metabolic and Molecular Bases of Inherited Disease (Scriver C. R., Beaudet A. L., Sly W. S., Valle D., Vogelstein B., Kinzler K. W., Childs B., eds) pp. 2007–2056, McGraw-Hill, New York [Google Scholar]
- 28. Jhee K. H., McPhie P., Miles E. W. (2000) Domain architecture of the heme-independent yeast cystathionine β-synthase provides insights into mechanisms of catalysis and regulation. Biochemistry 39, 10548–10556 [DOI] [PubMed] [Google Scholar]
- 29. Woehl E. U., Tai C. H., Dunn M. F., Cook P. F. (1996) Formation of the α-aminoacrylate immediate limits the overall reaction catalyzed by O-acetylserine sulfhydrylase. Biochemistry 35, 4776–4783 [DOI] [PubMed] [Google Scholar]
- 30. Hwang C. C., Woehl E. U., Minter D. E., Dunn M. F., Cook P. F. (1996) Kinetic isotope effects as a probe of the β-elimination reaction catalyzed by O-acetylserine sulfhydrylase. Biochemistry 35, 6358–6365 [DOI] [PubMed] [Google Scholar]
- 31. Borcsok E., Abeles R. H. (1982) Mechanism of action of cystathionine synthase. Arch. Biochem. Biophys. 213, 695–707 [DOI] [PubMed] [Google Scholar]