

Background: Condensin SMC proteins are frequently overexpressed in WNT-activated hyperplastic cells.
Results: The SMC2 promoter is a novel target on the β-catenin·TCF4 transcription complex.
Conclusion: β-Catenin·TCF4 may drive production of condensin in hyperplastic cells. SMC2 is required to ensure cellular mitosis and fast proliferation.
Significance: Down-regulation of SMC2 expression can repress cell proliferation in WNT-activated cells and represents a new therapeutic target in cancer treatment.
Keywords: Beta-Catenin, Chromosomes/Non-histone Chromosomal Proteins, T-cell Factor (TCF), Transcription Regulation, WNT Signaling, SMC Protein, WNT Signaling, Cell Division, Condensin
Abstract
Human SMC2 is part of the condensin complex, which is responsible for tightly packaging replicated genomic DNA prior to segregation into daughter cells. Engagement of the WNT signaling pathway is known to have a mitogenic effect on cells, but relatively little is known about WNT interaction with mitotic structural organizer proteins. In this work, we described the novel transcriptional regulation of SMC2 protein by direct binding of the β-catenin·TCF4 transcription factor to the SMC2 promoter. Furthermore, we identified the precise region in the SMC2 promoter that is required for β-catenin-mediated promoter activation. Finally, we explored the functional significance of down-regulating SMC2 protein in vivo. Treatment of WNT-activated intestinal tumor cells with SMC2 siRNA significantly reduced cell proliferation in nude mice, compared with untreated controls (p = 0.02). Therefore, we propose that WNT signaling can directly activate SMC2 transcription as a key player in the mitotic cell division machinery. Furthermore, SMC2 represents a new target for oncological therapeutic intervention.
Introduction
SMC (structural maintenance of chromosomes) proteins are a family of DNA-binding ATPases that are essential for maintenance of chromosomal integrity during cell division (1). Eukaryotes express at least six SMC proteins (SMC1–6), which form three heterodimers (SMC1/3, SMC2/4, and SMC5/6 (2)). SMC5/6 is part of a complex involved in DNA repair and checkpoint responses. The SMC1/3 heterodimer associates with two regulatory non-SMC proteins, SCC1 and SCC3, and collectively, this complex is known as cohesin. Cohesin holds sister chromatids together until they are physically segregated during anaphase (3). The SMC2/4 heterodimer associates with three non-SMC proteins to form a five-member complex known as condensin. Lower eukaryotes have a single condensin complex, but metazoans have two. In humans, both condensin I and condensin II contain the core SMC2/4 subunits, but have different regulatory non-SMC subunits. As the name suggests, condensin has DNA supercoiling activity, which is essential for packaging of chromatin prior to cell division. Condensin has also been shown to be necessary for resolution of sister chromatids during anaphase (4, 5). Condensin supercoiling activity is spatially and temporally regulated by mitotic kinases (6–10), which ensure DNA condensation only occurs at appropriate stages of the cell cycle.
Mutations in condensin subunits are likely to drive chromosomal destabilization and are found in some cancer genomes (11, 12). Furthermore, activated WNT signaling in colorectal tumors are considered to cause chromosomal instability. Upon investigation of normal human intestine and colorectal tumor samples, we noted that high SMC2 protein expression appeared to coincide with nuclear β-catenin localization in dividing cells. Therefore, we decided to investigate whether WNT signaling and β-catenin might transcriptionally regulate condensin.
Secreted WNT ligands are essential morphogens that control patterning and cell division during embryogenesis (13). WNT signals are principally transduced by two classes of cell surface receptors; Frizzled (Fz) proteins and low density lipoprotein receptor-related proteins 5 and 6 (LRP5/6). In canonical, β-catenin-dependent signaling, phosphorylation of LRP6 leads to release of cyctoplasmic β-catenin from the prodegradatory Axin complex (which includes glycogen synthase 3 and adenomatous polyposis coli (APC)3 protein). Free β-catenin translocates to the nucleus, where it acts as a transcriptional coactivator of target genes in combination with TCF/LEF transcription factors (14, 15).
WNT signaling is well known to promote cell cycle progression by up-regulating proliferation-stimulating target genes e.g. cyclin D and c-myc. However, it has become apparent that the cell cycle and WNT signaling are intrinsically linked (16). In a seminal study, WNT-β-catenin signaling (and β-catenin protein levels) were noted to oscillate during the cell cycle, peaking at the G2/M transition (17). Since that initial observation, many of the components of the WNT pathway have been shown to play an integral role during cell division. In addition to their function as activators of WNT target gene transcription, APC protein and β-catenin are important constituents of the centrosome complex (18–20). β-Catenin is also essential for centrosomal separation at the onset of spindle formation (21). Moreover, glycogen synthase 3 binds to and regulates microtubules, thereby contributing to mitotic spindle alignment (22).
For WNT-stimulated cells to undergo mitotic division, the genome must be faithfully replicated and packaged up prior to cytokinesis. By definition, this is a complex and highly regulated process, and failure to control each stage can lead to aneuploidy, chromosomal instability, and/or cell death. Chromosomal architecture during cell division is maintained in part by SMC proteins, and in this study, we provide evidence that canonical WNT signaling is directly driving SMC2 expression and that depleting tumor cells of SMC2 effectively drives a tumor xenograft model into mitotic catastrophe. Therefore, modulating cellular levels of condensin subunits may provide a novel chemotherapeutic tool for controlling the rate of cell division and/or critically destabilizing chromosomal organization.
EXPERIMENTAL PROCEDURES
Human Cancer Cell Lines and Cell Culture
Colorectal cancer (CRC) cell lines were purchased from the American Type Culture Collection (ATCC). Ls174T/dnTCF4 and Ls174T/pTER-β-catenin cells were kindly provided by Prof H. Clevers (Hubrecht Institute, Utrecht, The Netherlands). Cell lines were cultured in DMEM or RPMI 1640 (Ls174T variants) medium supplemented with 10% fetal bovine serum, 100 units/ml of penicillin, and 100 μg/ml of streptomycin at 37 °C under 5% CO2. To induce dnTCF4 or siRNA-BCAT, Ls174T cells were treated with 5 μg/ml doxycycline. Doubling time calculations were performed as described by Bex et al. (23).
Colorectal Tissue Samples
Tumor and counterpart normal samples were provided and analyzed by the Surgery and Pathology Departments of the Vall d'Hebron Universitary Hospital (Barcelona, Spain) respectively. Patients gave written consent before their inclusion in the analysis, and the study was approved by the Hospital Ethics Committee.
DNA Reagents
pTOPFLASH and pFOPFLASH plasmids were generously provided by Prof H. Clevers (24). VP16-TFC4 and pBCAT expression vectors were kindly supplied by Antonio García de Herreros (IMIM-Hospital del Mar, Barcelona, Spain). SMC2 promoter regions were amplified by PCR using the pairs of primers listed in supplemental Table 1. The products were directionally cloned in pGL3-basic vector (Promega) using KpnI and BglII restriction sites. Substitution mutants affecting the TCF4-binding sites on SMC2 promoter regions were generated with mutagenic oligonucleotides in supplemental Table 1 using QuikChange II XL site-directed mutagenesis kit (Stratagene). All constructs were confirmed by DNA sequencing under Big DyeTM cycling conditions on an Applied Biosystems 3730xl DNA Analyzer (Macrogen, Inc.).
RNA Extraction and Real-time PCR
Total RNA was extracted with Trizol® (Invitrogen) and further treated with DNase I amplification grade (Invitrogen) and retrotranscribed using a High Capacity cDNA reverse transcription kit (Applied Biosystems). Real time PCR reactions were performed in triplicate on an ABI PRISM 7500 real-time system (Applied Biosystems), using TaqMan gene expression assays (Applied Biosystems, catalog no. Hs00374522_m1, Hs00197593_m1, Hs00254617_m1, Hs00214861_m1, and Hs00379340_m1) according to the manufacturer's instructions. Data were normalized to 18 S rRNA (catalog no. 4333761F) expression but also confirmed with other endogenous controls: peptidylprolyl isomerase A (cyclophilin A) (catalog no. 4333763T) or β-actin (catalog no. 4333762T). The relative mRNA levels were calculated using the comparative Ct method (2−ΔΔCt) as described by Arango et al. (25).
Protein Extraction and Western Blotting (WB)
Cell pellets and tissue homogenates were lysed in radioimmune precipitation assay buffer (50 mm Tris-HCl at pH 8.0, 150 mm NaCl, 1 mm DTT, 1 mm sodium orthovanadate, 0.5% deoxycholate, 1% Triton X-100, 0.1% SDS) containing complete protease inhibitor mixture (Roche Diagnostics). Proteins in the crude lysates were quantified using the BCA protein assay (Pierce Biotechnology), and 50 μg of whole-cell lysates were separated by SDS-PAGE and transferred onto nitrocellulose filters. Blots were probed using antibodies against SMC2 (ab10412, Abcam; and 07-710, Upstate-Millipore, dilution factor of 1:1000), SMC4 (ab17958, Abcam, dilution factor of 1:1000), TCF4 (05-511, Upstate-Millipore, dilution factor, 1:500), NCAPH (HPA003008, Sigma Aldrich, dilution factor, 1:2000), β-catenin (610154, BD Transduction Laboratories, dilution factor, 1:1000) or c-Myc (monoclonal 9E10, sc-40, Santa Cruz Biotechnology, 1:100). Proteins were detected using corresponding HRP-conjugated secondary antibodies, anti-mouse (P0447, Dako), or anti-rabbit (P0217, Dako). Actin was used as loading control (CP01, Calbiochem, 1:5000). The intensity of the bands on the blots was quantified using the GeneTools Program (SynGene).
Immunohistochemistry
Paraffin-embedded tissues were provided by the archive tumor bank of the Department of Pathology of the Vall d'Hebron Universitary Hospital. Epitope retrival was heat induced in citrate buffer, pH 6.0. Immunohistochemistries were performed using EnVision + Dual Link System-HRP, DAB+ (Dako) according to the manufacturer's instructions, using the SMC2 antibody (ab10412, Abcam, 1:200), NCAPH antibody (HPA003008, Sigma Aldrich, dilution factor, 1:50), and β-catenin (610154, BD Transduction Laboratories, dilution factor, 1:X). Samples were additionally counterstained with hematoxilin. Anti-SMC2 antibody (ab10412) specificity was confirmed by immunocytochemistry of wt versus SMC2-depleted DLD-1 human colorectal cancer cells (supplemental Fig. 1).
Chromatin Immunoprecipitation (ChIP)
Cells were grown to 80% confluency in 15-cm dishes. Proteins and nucleic acids were cross-linked with formaldehyde (1%) for 10 min at 4 °C. Cross-linking was quenched by adding 125 mm glycine for 5 min. Following two washes with cold PBS containing protease inhibitors, cells were collected and resuspended in SDS lysis buffer (50 mm Tris-HCl, pH 8, 10 mm EDTA, 1% SDS). Lysates were sonicated 12× for 10 s (60-s interval on ice between pulses) at 8 Å on a Soniprep 150 (MSE, Ltd., Kent, U.K.). Chromatin samples were diluted with chromatin immunoprecipitation buffer (20 mm Tris-HCl, pH 8, 2 mm EDTA, 150 mm NaCl, 1% Triton X-100) supplemented with protease inhibitors. Samples were precleared for 2 h at 4 °C with protein G-agarose/salmon sperm DNA beads (Upstate-Millipore) and incubated with 5 μg of the appropriate antibody overnight at 4 °C. Immunoprecipitation was carried out with protein G-agarose/salmon sperm DNA beads for 2 h at 4 °C. DNA·protein·antibody·bead complexes were washed out with low salt buffer (150 mm NaCl, 20 mm Tris-HCl, pH 8, 2 mm EDTA, 1% Triton X-100, 0.1% SDS), high salt buffer (500 mm NaCl, 20 mm Tris-HCl, pH 8, 2 mm EDTA, 1% Triton X-100, 0.1% SDS), LiCl buffer (250 mm LiCl, 10 mm Tris-HCl, pH 8, 1 mm EDTA, 1% Igepal, 1% sodium deoxycholate), and TE buffer (10 mm Tris-HCl pH 8, 1 mm EDTA). Proteins were eluted with elution buffer (100 mm NaHCO3, 1% SDS). Cross-linking was reversed incubating samples with 200 mm NaCl overnight at 65 °C. Before DNA purification (phenol-chloroform-isoamilic alcohol), proteins were digested with 20 μg of proteinase K (Roche Diagnostics) for 2 h at 45 °C. Immunoprecipitated DNA was used as template in the PCR reactions. The primers are listed in supplemental Table 1.
Luciferase Reporter Assays
Cells were transiently co-transfected with pGL3-basic-SMC2 promoter (1 μg/106 cells) alone or in combination with VP16-TCF4 (3 μg/106 cells) or pBCAT (3 μg/106 cells) using Lipofectamine 2000 (Invitrogen), according to the manufacturer's instructions. pRL-TK Renilla (0.2 μg/106 cells) was introduced in all samples to allow data normalization. pTOPFLASH and pFOPFLASH were used as positive and negative luciferase reporter controls, respectively. 24 h post-transfection, cells were lysed, and luciferase activity was measured according to the Dual-Luciferase reporter assay using a Clarity Luminescence Microplate Reader (BioTek Instruments).
SMC2 Knockdown
Cells were transiently transfected with 20 μm siRNA using HiPerfect Transfection Reagent (Qiagen®) according to the manufacturer's instructions. SMC2 and scrambled siRNA were purchased from Qiagen® (catalog no. SI02654260 and 1027281, respectively). Cells used in the xenograft assays were cultured for 48 h and subjected to a second round of transfection. For stable knockdown, cells were transduced with lentiviral particles containing five different shRNAs targeting SMC2 (MISSION shRNA, Sigma-Aldrich, clone IDs NM_006444.1-3720s1c1, -1295s1c1, -1961s1c1, -3173s1c1, and -3300s1c1) prior to puromycin selection.
Assessment of Cell Cycle Profile
Cells transiently silenced for 24, 48, 72, or 96 h were trypsinized, washed with cold PBS, fixed with 70% ethanol, and stained with propidium iodide (40 μg/ml). DNA content was assessed using a FACSCalibur instrument and CellQuest software (BD Biosciences).
Xenograft Study
Female athymic nude mice (Hsd:athymic nude-Foxn1 nu/nu; Harlan Interfauna Iberica) were maintained in pathogen-free conditions and used at 5–6 weeks of age. Animal care was handled in accordance with the Guide for the Care and Use of Laboratory Animals of the Vall d'Hebron Hospital Animal Experimentation Ethical Committee. 1.5 × 106 silenced DLD1 cells were injected subcutaneously in the rear flanks of mice. Tumor growth was monitored three times per week for 5 weeks by conventional caliper measurements (tumor volume = D × d2/2, where D is the major diameter and d is the minor diameter).
Statistical Analysis
Unless stated differently, descriptive data were expressed as mean ± S.D. The GraphPad Prism statistical package was used to investigate group differences by unpaired Student's t test. p values are indicated for statistically different means.
RESULTS
The Core Subunit of Human Condensin Complex, SMC2, Is Overexpressed in CRC
SMC2 protein expression was evaluated in clinical samples from 29 patients that had undergone surgery for colon carcinoma. Protein detection by WB showed that SMC2 was up-regulated in 20 of the 29 tumor samples (69%) compared with the matched normal controls (subset shown in Fig. 1A). SMC2 overexpression in CRC was further confirmed by quantitative PCR of 16 clinical samples, showing also a clear up-regulation of SMC2 in the tumor counterpart samples in 11 cases (68.5%) (Fig. 1B). As SMC4 is the natural partner in the core of the condensin complex, its levels were also studied in 27 clinical samples and found to be overexpressed in 13 tumor counterparts (48.1%) (Fig. 1C). Further analysis of non-SMC subunits in patient samples confirmed the trend for increased expression of all condensin complex members in tumor samples versus normal tissue (supplemental Fig. 2). A strong positive correlation between the protein levels of SMC2 and SMC4 in clinical samples and also in CRC cell lines was identified (Fig. 1E). Interestingly, levels of SMC2/SMC4 protein negatively correlated with population doubling times in CRC cell lines (supplemental Fig. 3). Nevertheless, neither SMC2 nor SMC4 overexpression could be correlated to any clinicopathological variables (age, sex, tumor stage, or tumor location; supplemental Table 2) in the clinical samples studied.
FIGURE 1.

SMC2 is up-regulated in human CRC. A, WB analysis of SMC2 in human CRC. A representative subset of 29 cases studied is shown. Actin was used as loading control. B, quantitative real-time PCR for SMC2 in 16 pairs of colon adenocarcinoma tumors and matched adjacent normal colonic tissues. Data are representative of three independent experiments. The mean values of SMC2 levels were compared using Student's t test (upper boxplot). C and D, SMC2, SMC4, and β-catenin levels were evaluated by WB in both colorectal cancer cell lines (n = 14) and samples from CRC patients (n = 27, a representative subset is shown). Actin was used as loading control. E and F, SMC2, SMC4, and β-catenin protein levels on WB were determined by gel band quantification and normalized to the corresponding actin levels. Values were used to perform correlation studies following Spearman test. G, immunohistochemistry of SMC2 in paraffin-embedded tissue. A representative specimen is shown. Magnified regions of the normal and tumor mucosa are shown on the right. N, normal tissue; T: tumor tissue (adenocarcinoma).
Furthermore, additional immunohistochemistry studies were performed in paraffin embedded sections of normal colon mucosa and tumor tissue. SMC2 protein was up-regulated in tumor cells (Fig. 1G), both in the cytoplasmic compartment and nuclei. Normal tissue staining confirmed β-catenin, SMC2, and NCAPH (a non-SMC subunit of the condensin complex) accumulation in cells located in the lower part of the intestinal crypts, where WNT signaling is active and cells proliferate actively to maintain the normal epithelial homeostasis (supplemental Fig. 4A) (26). Our observation that SMC2 is naturally expressed in cells where WNT pathway is engaged, prompted us to examine whether there was a correlation between the levels of SMC2/SMC4 and β-catenin in a panel of 14 CRC cell lines, and also in a subset of 14 pairs (normal/tumor) of clinical samples by WB (Fig. 1, C and D). It was confirmed that there was a strong positive correlation between the protein levels of β-catenin and SMC2 and SMC4 (Fig. 1F). Furthermore, immunohistochemical analysis of tumor samples confirmed that membrane-localized β-catenin corresponded to low levels of SMC2 and NCAPH, predominantly localized in the cytoplasm, whereas nuclear β-catenin staining was found in conjunction with increased levels of SMC2 and NCAPH expression, predominantly in the nucleus (supplemental Fig. 4, B–I). Because SMC2 was up-regulated in cells actively proliferating in response to WNT signaling and correlated with β-catenin levels in CRC cell lines and clinical samples, we were interested to determine whether SMC2 expression could be directly regulated by the WNT/β-catenin pathway.
SMC2 Is Down-regulated in Cellular Models for WNT Pathway Inhibition
First, we wanted to determine whether disruption of WNT/β-catenin signaling could affect SMC2/SMC4 transcription. For this purpose, we used two in vitro systems, Ls174T/dnTCF4 and Ls174T/pTER-β-catenin cell lines, for WNT pathway inhibition. Ls174T/dnTCF4 cells carry a doxycycline-inducible expression plasmid encoding N-terminally truncated version of TCF4, which acts as a dominant negative form of TCF4 (dnTCF4). Even though dnTCF4 protein binds to DNA it does not bind B-catenin acting as a potent inhibitor of endogenous β-catenin·TCF4 complexes (26). Induction of dnTCF4 after 96 h of doxycycline treatment resulted in a decrease in SMC2 protein levels in a dnTCF4 protein dose-dependent manner (Fig. 2A). Longer inductions were not tested as these cells rapidly undergo G1 arrest (26, 27). To confirm inhibition of β-catenin·TCF4 activity, the levels of c-Myc, a well characterized WNT target gene, were evaluated (28). The same effect could be observed in the SMC4 protein levels under the same conditions (Fig. 2B).
FIGURE 2.

SMC2 protein is down-regulated upon WNT signaling inhibition. Ls174T/dnTCF4 (A and B) and Ls174T/pTER-β-catenin (C and D) cell lines were cultured in absence or presence of 5 μg/μl doxycycline (Dox) during the indicated time points. Cells were lysed and analyzed by WB using the indicated antibodies. Representative data from three replicates/independent experiments are shown.
To substantiate the dnTCF4 result, we examined the levels of SMC2/4 in Ls174T/pTER-β-catenin, a cellular model that expresses a doxycycline-inducible form of the RNA polymerase III H1 promoter to drive expression of an siRNA, directed to β-catenin. Addition of doxycycline to the growth medium induced rapid down-regulation of β-catenin messenger RNA (27) and protein (Fig. 2C) in these cells. In this context, following 96 h of doxycycline treatment, we observed a down-regulation in SMC2 (Fig. 2C) and SMC4 protein levels (Fig. 2D) that correlated to decreased β-catenin protein levels and implied a strong association between SMC subunit expression and β-catenin·TCF4 transcription factor.
SMC2 Promoter Responds to WNT Pathway Activation/Inhibition
To determine whether SMC2/4 could be targets of the β-catenin·TCF4 transcription factor complex, upstream sequences of the human SMC2 and SMC4 genes were obtained from the Ensembl database (30). Three different software packages were used for in silico prediction of the SMC2 promoter: Gene2Promoter recognized a very highly promoter-like region between the −308 bp and +420 bp region (considering 0 bp the transcription start site); promoter 2.0 predicted a promoter region starting in the −476 bp position; finally, promoterScan located two putative regulatory regions, from the −597 bp to −348 bp position, and from −313 bp to −64 bp, respectively. For subsequent studies, we compiled an SMC2 promoter based on the different predictions, which was determined to be from position −597 bp to the translation start site (+1059 bp) (Fig. 3A). In this region, two TATA boxes were identified at positions −591 and −12 bp, and three recognition sites for the Sp1 transcriptional factor were situated at −561 bp, −301 bp, and +219 bp positions. The predicted SMC2 promoter was subjected to an in silico screen for TCF binding elements (TBE). rVista (version 2.0, NCBI DCODE), TESS (Transcription Element Search System), and Matinspector software (Genomatix), which predicted four different elements: TBE1 (−389 bp), TBE3 (−20 bp), TBE4 (+57 bp), and TBE6 (+724 bp). Additionally, Matinspector located two further TBEs: TBE2 (−37 bp) and TBE5 (+98 bp) (Fig. 3A).
FIGURE 3.

Functional study of SMC2 promoter activity. A, schematic representation of human SMC2 promoter. Predicted TCF response elements are also indicated; arrows indicate target sequence for ChIP PCR amplification. B, Ls174T/dnTCF4 (left) and Ls174T/pTER-β-catenin (right) cell lines were transfected with SMC2 promoter-luciferase reporter construct together with control Renilla luciferase reporter pRL-TK for normalization (RLU, relative luciferase units). Where indicated, cells were doxycline (Doxy)-treated to induce the TCF4 dominant-negative form (left) or a siRNA targeting β-catenin (right). TOP-flash vector was used as positive control for WNT signaling activity/repression. A representative result out of at least three different experiments run in triplicates is shown. C, DLD-1 or HCT116 cell lines were co-transfected with SMC2 promoter luciferase construct and pcDNA (empty vector), β-catenin, or VP16-TCF4 expression vectors. D, PCR analyses of DNA pulled down by isotypic antibody (negative control) or anti-TCF-4 monoclonal antibody in ChIP assay. c-myc promoter sequence containing TBE1 element and APC promoter region 1B sequences were amplified as positive and negative controls, respectively. Error bars indicate S.D. (Student's t test; **, p < 0.01).
Promoter software (version 2.0) predicted that the region from the −1500 bp position to the transcription start site (0 bp) of SMC4 was highly likely to be a promoter region, in which one TATA box (−837 bp) and three Sp1 sites (−1066 bp, −21 bp, and −5 bp) could be identified. Two putative TBE were predicted in this region (−1270 bp and −1294 bp), but none of these were phylogenetically conserved in mammals (data not shown), so we continued our study by focusing on WNT pathway regulation of SMC2 expression.
The full-length promoter of SMC2 was cloned into a pGL3 firefly luciferase reporter vector, and its activity was assayed in Ls174T/dnTCF4 cells following transient transfection alone, or in combination with β-catenin expression vector (pBCAT). This promoter was active under normal conditions, even more than the positive control TOPFLASH (run in parallel). After doxycycline induction of the dnTCF4 form, luciferase activity was significantly reduced. Moreover, the promoter was able to respond to β-catenin transduction, but this capacity was lost when the dnTCF4 form was induced by doxycycline (Fig. 3B, left). SMC2 promoter was also tested in Ls174T/pTER-β-catenin cells. Doxycycline-induced down-regulation of β-catenin also diminished the luciferase activity of the full-length promoter of SMC2 (Fig. 3B, right).
To confirm that the SMC2 promoter was a target of activated WNT signaling, we tested the luciferase activity of the promoter in two colon carcinoma cell lines, DLD1 and HCT116, carrying an activating mutation in β-catenin or a deactivating mutation in APC (31), respectively. Cells were co-transfected with pGL3-SMC2 promoter and expression vectors for β-catenin (pBCAT) or VP16-TCF4 (a constitutively active form of TCF4). In both cell lines, a significant gene transactivation increase could be observed after the co-transfection (Fig. 3C), supporting the hypothesis that the activated WNT pathway can drive transcription from the SMC2 promoter via the β-catenin·TCF4 transcription complex.
TCF4 Transcription Factor Is Bound to the SMC2 Promoter in Vivo
We aimed to determine whether TCF4 interaction with the SMC2 promoter was direct or indirect. Therefore, ChIP experiments were used to test whether TCF4 could occupy the SMC2 promoter. Chromatin from DLD1 cells was cross-linked prior to anti-TCF4 antibody immunoprecipitation of DNA·protein complexes. The SMC2 promoter sequence that contains TBE 2, 3, 4, and 5 was present in the TCF4 eluate (Fig. 3D), confirming that this transcription factor can bind to the SMC2 promoter in vivo. Primers for c-myc and APC promoter amplification were used as positive and negative controls, respectively (28).
The Region Located between −389 bp and +98 bp in SMC2 Promoter Is Defined as the Minimal Regulatory Fragment of the SMC2 Gene
To define the minimal transcriptional regulatory region in the SMC2 promoter, we cloned a series of terminal deletions of the full-length sequence based on the position of the predicted TCF response elements (Fig. 4A). DLD1 and HCT116 cells were transfected with three different deletion mutants, and luciferase activity was measured.
FIGURE 4.
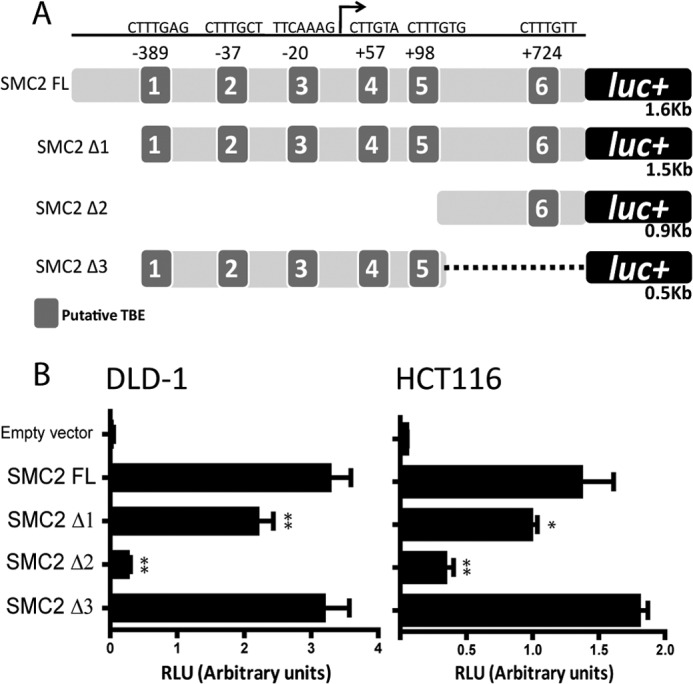
Determination of the minimal regulatory region of SMC2 promoter. A, relative position and sequences of the putative TBEs predicted in silico in the SMC2 promoter and deletion mutants for luciferase (luc) reporters performed. B, determination of fragment 3 as the minimal regulatory region of the SMC2 promoter. Luciferase activity of each deletion mutant was normalized to Renilla luciferase internal control (RLU, relative luciferase units) in DLD1 (left) or HCT116 (right) cell lines; a representative result is shown of at least three independent experiments. *, p < 0.05; **, p < 0.01; Student's t test (promoter activity versus full-length SMC2 promoter (SMC2 FL).
Deletion of the first 100 base pairs in the SMC2 promoter resulted in decreased luciferase activity, and the promoter activity was almost lost when the deletion removed all of the putative TBEs (except TBE6). It was also confirmed that the 0.5 kb (Δ3 sequence), which contains TBEs 1, 2, 3, 4, and 5, maintained the maximal activity in both cell lines. Fragment Δ3 showed a luciferase activity similar to the full length sequence. Thus, we defined Δ3 as the minimal regulatory region and used it for further mutational studies (Fig. 4B).
The TCF Response Element Located at −20 bp (TBE3) Is Susceptible to β-Catenin·TCF4 Transactivation
Interspecies conservation analysis showed that two of the six TBEs predicted, TBE2 and TBE3, were highly conserved in ortholog SMC2 promoters of mouse, rat, macaque, and chimpanzee (Fig. 5A), and both were present in the minimal regulatory region, Δ3. Interestingly, these two TBEs are the closest ones to the transcription start site.
FIGURE 5.

Elucidation of the TBE responsible for β-catenin·TCF4 transactivation in the SMC2 promoter. A, sequence alignment of SMC2 promoter in different species; Hs, Homo sapiens; Pt, Pan troglodytes; Mmt, Macaca mulatta; Rn, Rattus novergicus; Mms, Mus musculus. Conserved TBEs are highlighted in gray background. B, schematic representation of SMC2 promoter mutant variants. C, DLD1 (left) or HCT116 (right) cell lines were transfected with constructs above. Luciferase activity was normalized to Renilla activity (RLU, relative luciferase units); a representative result is shown out of at least three independent experiments. D, DLD-1 (left) or HCT116 (right) cell lines were co-transfected with Δ3 fragment mutational combinations and expression vectors for β-catenin, TCF4-VP16 (constitutively active form of TCF4), or the empty vector pcDNA3 (pcDNA); a representative result is shown out of at least three independent experiments (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
To study the functionality of those conserved TBEs, we performed site-directed mutagenesis to disrupt TCF4 binding ability (Fig. 5B). We detected a significant decrease in luciferase activity when TBE3, located at −20 bp, was mutated. However, mutations in all other TBEs did not affect luciferase activity driven by the SMC2 promoter (Fig. 5C).
To confirm TBE3 susceptibility to WNT signaling transactivation, we measured luciferase activity after co-transfection of β-catenin or VP16-TCF4 expression plasmids and different mutational combinations in Δ3 fragment. Enhancement of luciferase activity in response to WNT/β-catenin stimulation was lost when TBE3 was disrupted (Fig. 5D). As expected, mutations in TBE 1, 2, 4, and 5 did not affect promoter response to β-catenin or VP16-TCF4 stimulation. Thus, we identified the TCF response element located at −20 bp (TBE3) as the sole entity responsible for β-catenin·TCF4 transactivation of the SMC2 promoter.
SMC2 Knockdown Results in Decreased Tumor Growth in Vivo
Because we had established that the SMC2 promoter could be driven by WNT signaling, and SMC2 has a clear role in mitosis, we hypothesized that perturbing SMC2 expression may reduce WNT-induced cell proliferation. Therefore, we investigated the effect of SMC2 down-regulation in WNT-activated CRC cell lines. DLD1, HT29, and HCT116 cells were transiently transfected with an siRNA targeting SMC2 for 48, 72, and 96 h. SMC2 knockdown efficiency was assessed by WB. Furthermore, a decrease in SMC4 and NCAPH protein expression was also detected, implying a reduction in the condensin complex as a whole (supplemental Fig. 5). Cell cycle profile was studied by FACS determination of propidium iodide stained DNA. A significant increase in haplo-diploid (apoptosis), 4n (G2/M), and aneuploid (>4n) DNA content populations could be observed along treatments, whereas the 2n DNA content population (G1) decreased drastically (supplemental Fig. 6, A–C). For stable SMC2 knockdown, lentiviral particles containing an shRNA targeted to SMC2 were used to transduce HT29 cells. Five different sequences targeting different regions of SMC2 mRNA were tested, but only three regions were able to down-regulate SMC2 efficiently. As expected, stable knockdown of SMC2 impaired HT29 cell viability. Morphological changes in SMC2-down-regulated cells could be appreciated after 1 week in culture in terms of enlarged multinucleated and non-viable cells, a phenotype clearly associated to shRNA-SMC2 knockdown efficiency (supplemental Fig. 6, D and E). These results implied that decreasing SMC2 protein levels could attenuate cell division even in cells that are receiving strong proproliferation signals, such as the WNT/β-catenin pathway.
To test this concept in vivo, we investigated whether CRC tumor cells require SMC2 expression to proliferate in a xenograpft model of tumor progression. To prolong the knockdown effect, two rounds of transfection were performed in DLD1 cells before injection into athymic nude mice (Fig. 6A). To asses SMC2 knockdown durability, SMC2 protein was evaluated by WB of whole cell extracts 72, 96, and 120 h post-transfection, confirming that SMC2 levels remained down-regulated for at least 120 h under these experimental conditions (Fig. 6B). siRNA-SMC2 or scrambled siRNA transfected DLD1 cells were injected into the flanks of 11 nude mice and tumor growth was measured over 5 weeks. Transient knockdown of SMC2 was enough to significantly reduce tumor size compared with controls even at 12 days post-injection, and this difference became more pronounced after 35 days, the point at which the animals were sacrificed (Fig. 1, C and D). Although further investigation is required, the significantly tumor growth-retarding effect of SMC2 knockdown in vivo could make SMC2 an interesting novel chemotherapeutic target.
FIGURE 6.

siRNA knockdown of SMC2 impairs tumor growth in a xenograft mouse model. A, schematic representation of the experimental design. DLD1 cells were transiently transfected with an siRNA targeting SMC2 or a scrambled sequence. After 48 h, a second round of transfection was performed. 24 h later, 1.5 × 106 cells were injected subcutaneously in the dorsal flanks of athymic nude mice. B, SMC2 knockdown was assessed by WB using whole cell extracts from in vitro culture until 120 h post-tranfection (sc, scrambled siRNA). C, representative resected tumors from the same animal at day 40 post-injection. Scale bar, 1 cm. D, tumor growth curves. Tumor volume was measured every 2–3 days for 36 days. The graph is representative of two independent experiments. Error bars represent S.E. (n = 11). Differences were evaluated with paired Student's t test (p = 0.0201); (*, p < 0.05, t test in each time point).
DISCUSSION
It is becoming apparent that the WNT signaling pathway appears to be intimately linked with the mitotic machinery. In this study, we have demonstrated that the TCF4 transcription factor can bind to and drive the SMC2 promoter in vitro and that preventing β-catenin binding to TCF4 markedly reduces SMC2 protein levels. Our in vivo study suggests that depletion of SMC2 levels in human CRC cells expressing constitutively active β-catenin significantly affected tumor growth in an immunodeficient mouse model.
In this study, we observed SMC2 protein levels to correlate directly with SMC4 protein levels in a panel of colorectal cell lines and tumor lysates, in accordance with the heterodimeric structure of the condensin SMC2/4 core. We were unable to locate a conserved TCF4 transcription element within the SMC4 promoter; however, expression of either condensin SMC subunit appears to be very tightly linked to expression of its partner. Indeed, preliminary experiments in which SMC2 expression is depleted in DLD1 cells using siRNA to SMC2 show that there is a corresponding reduction in the levels of SMC4 and the non-SMC regulatory subunits (supplemental Fig. 5).
Chromatin is generally thought to be transcriptionally silent around the G2/M transition. Furthermore, Takemoto et al. (32) demonstrated that in unstimulated cells, SMC protein levels remained stable throughout the cell cycle. However, in WNT-activated cells, the situation may be different. WNT signaling is enhanced by cyclin γ and peaks around G2/M (16). Therefore, it is important to consider whether WNT target genes, such as SMC2, could be actively transcribed during this phase of the cell cycle. A recent study using conditional gene knock-out (KO) mice highlighted the link between cell cycle regulators and WNT signaling, and goes some way to answering the question. Deletion of all three members of the CDC25 protein phosphatase family led to a lethal reduction in enterocyte proliferation due to arrest at G2/M. Notably, in the same animals, WNT target gene expression was up-regulated in putative epithelial crypt progenitor cells, and there was a 50% increase in the total number of crypt cells staining positive for nuclear β-catenin (34). This result confirms the possibility that β-catenin·TCF4 could be actively driving transcription of SMC2 and other target genes during G2/M in vivo. The physiological significance of SMC2 transcription at this point in the cell cycle is unclear; however, it could be a method of ensuring that sufficient levels of DNA-condensing proteins are available at the juncture where they are required most.
Our initial immunohistological observations of normal human intestine confirmed that SMC2 protein expression was up-regulated in crypt cells staining positive for nuclear β-catenin. As WNT signaling drives cell proliferation, it is not particularly surprising that higher levels of condensins are required by tissues with elevated cell turnover such as the gut. However, it is exciting to note that SMC2 expression can be directly driven by TCF4 transcription factor. It is possible that WNT signaling can drive a positive feedback loop, whereby rapidly dividing cells are induced to produce elevated levels of proteins involved in the cell division machinery.
Previously, Ghiselli and co-workers (29) found that one of the cohesin SMC subunits, SMC3, was up-regulated in human colorectal adenocarcinomas and APCMin mouse adenomas. The SMC3 promoter also contains two conserved transcriptional binding sites for β-catenin·TCF4 in the human and mouse promoters, which could be driven by elevated β-catenin (29, 33). Our data confirms that the promoter of a condensin subunit, SMC2, can also be a target of β-catenin·TCF4 activation, and our in vivo knock-down experiment suggests that reducing SMC2 levels could be an effective way of retarding or ablating tumor growth.
Our analysis of a bank of human CRC cell lines showed that SMC2 and SMC4 proteins are highly expressed in many transformed cells. Interestingly, there appeared to be a correlation between the level of SMC protein expressed, and the rate of cell division (i.e. cells with higher levels of SMC2 tended to be the fastest growing; supplemental Fig. 3). Furthermore, SMC2 levels are significantly reduced in non-dividing senescent cells (data not shown), supporting the positive feedback hypothesis suggested above. Moreover, our analysis of human colorectal tissue samples implies that up-regulation of SMC2 and SMC4 is a common occurrence in human intestinal cancer, corroborating the idea that up-regulation of condensin can be linked to β-catenin-induced hyperplasia. Analysis of non-SMC condensin subunits at the mRNA and protein levels confirmed up-regulation of the condensin complex as a whole in tumor versus normal tissue samples (supplemental Figs. 2 and 4). Our observation that SMC2 expression is up-regulated in cells with nuclear β-catenin suggests that β-catenin·TCF4 may drive production of condensin, which might be required to allow rapid cell division.
Interestingly, knockdown of the SMC2 subunit alone was sufficient to cause a significant reduction in proliferation of an APC mutant colorectal cell line in vivo (confirmed by FACS, supplemental Fig. 6B). Upon further analysis, we found that two additional CRC tumor cell lines treated with SMC2 siRNA appeared to be undergoing aneuploid division and apoptosis, most likely as result of mitotic catastrophe (supplemental Fig. 6, B and C). Therefore, given that reducing SMC2 expression alone is enough to induce growth arrest and apoptosis of tumor cells, the SMC2 condensin subunit could be an attractive novel target for therapeutic intervention in cancer treatment. Of particular significance is the fact that SMC2 is highly expressed alongside nuclear β-catenin in a few cells located at the base of intestinal crypts, which are putative stem cells. This suggests that high expression of SMC2 may be a characteristic of stem cells in normal colon tissues.
In summary, this study has identified SMC2, one of the condensin ATPase subunits, as a novel, bone fide target of β-catenin·TCF4 transcription. Furthermore, overexpression of condensin appears to be a frequent feature of human CRC. Our data suggests that elevated levels of condensin may be required to allow WNT-driven cell proliferation and that reducing SMC2 expression can lead to tumor cell apoptosis. Therefore, modulation of condensin SMC protein expression may offer exciting novel therapeutic potential in the treatment of human neoplasia.
Acknowledgments
We thank Professor Hans Clevers and Dr. Antonio García de Herreros for kindly providing the vectors and cellular models. We also thank Dr. Águeda Martinez-Barriocanal for critical reading of the manuscript.
This work was supported in part by Grants EU2008-0170 from the Spanish Ministry of Science and Innovation, PI080771 from the “Fondo de Investigaciones Sanitarias,” and grants from the Spanish Ministry of Science and Innovation, the Networking Centre on Bioengineering, Biomaterials, and Nanomedicine (CIBER-BBN), Instituto de Salud Carlos III, and Spanish Ministry of Science and Innovation.

This article contains supplemental Tables 1 and 2 and Figs. S1–S6.
- APC
- adenomatous polyposis coli
- dnTCF4
- dominant-negative transcription factor 4
- CRC
- colorectal cancer
- WB
- Western blot
- TBE
- TCF-binding element
- NCAPH
- non-SMC condensin I complex, subunit H.
REFERENCES
- 1. Hirano T. (2006) At the heart of the chromosome: SMC proteins in action. Nat. Rev. Mol. Cell Biol. 7, 311–322 [DOI] [PubMed] [Google Scholar]
- 2. Losada A., Hirano T. (2005) Dynamic molecular linkers of the genome: the first decade of SMC proteins. Genes Dev. 19, 1269–1287 [DOI] [PubMed] [Google Scholar]
- 3. Oliveira R. A., Nasmyth K. (2010) Getting through anaphase: splitting the sisters and beyond. Biochem. Soc. Trans. 38, 1639–1644 [DOI] [PubMed] [Google Scholar]
- 4. Hudson D. F., Marshall K. M., Earnshaw W. C. (2009) Condensin: Architect of mitotic chromosomes. Chromosome. Res. 17, 131–144 [DOI] [PubMed] [Google Scholar]
- 5. Paliulis L. V., Nicklas R. B. (2004) Micromanipulation of chromosomes reveals that cohesion release during cell division is gradual and does not require tension. Curr. Biol. 14, 2124–2129 [DOI] [PubMed] [Google Scholar]
- 6. Bazile F., St.-Pierre J., D'Amours D. (2010) Three-step model for condensin activation during mitotic chromosome condensation. Cell Cycle 9, 3243–3255 [DOI] [PubMed] [Google Scholar]
- 7. St.-Pierre J., Douziech M., Bazile F., Pascariu M., Bonneil E., Sauvé V., Ratsima H., D'Amours D. (2009) Polo kinase regulates mitotic chromosome condensation by hyperactivation of condensin DNA supercoiling activity. Mol. Cell 34, 416–426 [DOI] [PubMed] [Google Scholar]
- 8. Takemoto A., Kimura K., Yanagisawa J., Yokoyama S., Hanaoka F. (2006) Negative regulation of condensin I by CK2-mediated phosphorylation. EMBO J. 25, 5339–5348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Takemoto A., Murayama A., Katano M., Urano T., Furukawa K., Yokoyama S., Yanagisawa J., Hanaoka F., Kimura K. (2007) Analysis of the role of Aurora B on the chromosomal targeting of condensin I. Nucleic Acids Res. 35, 2403–2412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Takemoto A., Maeshima K., Ikehara T., Yamaguchi K., Murayama A., Imamura S., Imamoto N., Yokoyama S., Hirano T., Watanabe Y., Hanaoka F., Yanagisawa J., Kimura K. (2009) The chromosomal association of condensin II is regulated by a noncatalytic function of PP2A. Nat. Struct. Mol. Biol. 16, 1302–1308 [DOI] [PubMed] [Google Scholar]
- 11. Ham M. F., Takakuwa T., Rahadiani N., Tresnasari K., Nakajima H., Aozasa K. (2007) Condensin mutations and abnormal chromosomal structures in pyothorax-associated lymphoma. Cancer Sci. 98, 1041–1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Strunnikov A. V. (2010) One-hit wonders of genomic instability. Cell Div. 5, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Logan C. Y., Nusse R. (2004) The WNT signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 20, 781–810 [DOI] [PubMed] [Google Scholar]
- 14. Chien A. J., Conrad W. H., Moon R. T. (2009) A WNT survival guide: from flies to human disease. J. Invest. Dermatol. 129, 1614–1627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rao T. P., Kühl M. (2010) An updated overview on WNT signaling pathways: a prelude for more. Circ. Res. 106, 1798–1806 [DOI] [PubMed] [Google Scholar]
- 16. Davidson G., Niehrs C. (2010) Emerging links between CDK cell cycle regulators and WNT signaling. Trends Cell Biol. 20, 453–460 [DOI] [PubMed] [Google Scholar]
- 17. Orford K., Orford C. C., Byers S. W. (1999) Exogenous expression of beta-catenin regulates contact inhibition, anchorage-independent growth, anoikis, and radiation-induced cell cycle arrest. J. Cell Biol. 146, 855–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Banks J. D., Heald R. (2004) Adenomatous polyposis coli associates with the microtubule-destabilizing protein XMCAK. Curr. Biol. 14, 2033–2038 [DOI] [PubMed] [Google Scholar]
- 19. Dikovskaya D., Newton I. P., Näthke I. S. (2004) The adenomatous polyposis coli protein is required for the formation of robust spindles formed in CSF Xenopus extracts. Mol. Biol. Cell 15, 2978–2991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kaplan D. D., Meigs T. E., Kelly P., Casey P. J. (2004) Identification of a role for β-catenin in the establishment of a bipolar mitotic spindle. J. Biol. Chem. 279, 10829–10832 [DOI] [PubMed] [Google Scholar]
- 21. Bahmanyar S., Kaplan D. D., Deluca J. G., Giddings T. H., Jr., O'Toole E. T., Winey M., Salmon E. D., Casey P. J., Nelson W. J., Barth A. I. (2008) β-Catenin is a Nek2 substrate involved in centrosome separation. Genes Dev. 22, 91–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ong Tone S., Dayanandan B., Fournier A. E., Mandato C. A. (2010) GSK3 regulates mitotic chromosomal alignment through CRMP4. PLoS One 5, e14345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bex A., Wullich B., Endris V., Otto T., Rembrink K., Stöckle M., Rübben H. (2001) Comparison of the malignant phenotype and genotype of the human androgen-independent cell line DU 145 and a subline derived from metastasis after orthotopic implantation in nude mice. Cancer Genet. Cytogenet. 124, 98–104 [DOI] [PubMed] [Google Scholar]
- 24. Korinek V., Barker N., Morin P. J., van Wichen D., de Weger R., Kinzler K. W., Vogelstein B., Clevers H. (1997) Constitutive transcriptional activation by a β-catenin-Tcf complex in APC−/− colon carcinoma. Science 275, 1784–1787 [DOI] [PubMed] [Google Scholar]
- 25. Arango D., Mariadason J. M., Wilson A. J., Yang W., Corner G. A., Nicholas C., Aranes M. J., Augenlicht L. H. (2003) c-Myc overexpression sensitises colon cancer cells to camptothecin-induced apoptosis. Br. J. Cancer 89, 1757–1765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. van de Wetering M., Sancho E., Verweij C., de Lau W., Oving I., Hurlstone A., van der Horn K., Batlle E., Coudreuse D., Haramis A. P., Tjon-Pon-Fong M., Moerer P., van den Born M., Soete G., Pals S., Eilers M., Medema R., Clevers H. (2002) The β-catenin·TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell 111, 241–250 [DOI] [PubMed] [Google Scholar]
- 27. van de Wetering M., Oving I., Muncan V., Pon Fong M. T., Brantjes H., van Leenen D., Holstege F. C., Brummelkamp T. R., Agami R., Clevers H. (2003) Specific inhibition of gene expression using a stably integrated, inducible small-interfering-RNA vector. EMBO Rep. 4, 609–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. He T. C., Sparks A. B., Rago C., Hermeking H., Zawel L., da Costa L. T., Morin P. J., Vogelstein B., Kinzler K. W. (1998) Identification of c-MYC as a target of the APC pathway. Science 281, 1509–1512 [DOI] [PubMed] [Google Scholar]
- 29. Ghiselli G., Coffee N., Munnery C. E., Koratkar R., Siracusa L. D. (2003) The cohesin SMC3 is a target the for β-catenin·TCF4 transactivation pathway. J. Biol. Chem. 278, 20259–20267 [DOI] [PubMed] [Google Scholar]
- 30. Hubbard T. J., Aken B. L., Beal K., Ballester B., Caccamo M., Chen Y., Clarke L., Coates G., Cunningham F., Cutts T., Down T., Dyer S. C., Fitzgerald S., Fernandez-Banet J., Graf S., Haider S., Hammond M., Herrero J., Holland R., Howe K., Howe K., Johnson N., Kahari A., Keefe D., Kokocinski F., Kulesha E., Lawson D., Longden I., Melsopp C., Megy K., Meidl P., Ouverdin B., Parker A., Prlic A., Rice S., Rios D., Schuster M., Sealy I., Severin J., Slater G., Smedley D., Spudich G., Trevanion S., Vilella A., Vogel J., White S., Wood M., Cox T., Curwen V., Durbin R., Fernandez-Suarez X. M., Flicek P., Kasprzyk A., Proctor G., Searle S., Smith J., Ureta-Vidal A., Birney E. (2007) Ensembl 2007. Nucleic Acids Res. 35, D610–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ilyas M., Tomlinson I. P., Rowan A., Pignatelli M., Bodmer W. F. (1997) β-Catenin mutations in cell lines established from human colorectal cancers. Proc. Natl. Acad. Sci. U.S.A. 94, 10330–10334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Takemoto A., Kimura K., Yokoyama S., Hanaoka F. (2004) Cell cycle-dependent phosphorylation, nuclear localization, and activation of human condensin. J. Biol. Chem. 279, 4551–4559 [DOI] [PubMed] [Google Scholar]
- 33. Ghiselli G., Iozzo R. V. (2000) Overexpression of bamacan/SMC3 causes transformation. J. Biol. Chem. 275, 20235–20238 [DOI] [PubMed] [Google Scholar]
- 34. Lee G., White L. S., Hurov K. E., Stappenbeck T. S., Piwnica-Worms H. (2009) Response of small intestinal epithelial cells to acute disruption of cell division through CDC25 deletion. Proc. Natl. Acad. Sci. U.S.A. 106, 4701–4706 [DOI] [PMC free article] [PubMed] [Google Scholar]