

Background: Two forms of PCSK9, an intact and a furin cleaved form, circulate in blood.
Results: Both forms, as highly purified recombinant proteins, are able to bind to and trigger degradation of LDL receptors and elevate serum cholesterol levels.
Conclusion: Furin cleavage is not associated with a loss of PCSK9 polypeptides or a significant loss of function.
Significance: LDL-c levels are controlled by both forms of PCSK9.
Keywords: Antibodies, Cholesterol, Lipoprotein Receptor, Low Density Lipoprotein (LDL), Serine Protease, LDL Receptor, PCSK9, Furin, Hepsin, Proprotein Convertases
Abstract
Proprotein convertase subtilisin/kexin 9 (PCSK9) regulates plasma LDL cholesterol levels by regulating the degradation of LDL receptors. Another proprotein convertase, furin, cleaves PCSK9 at Arg218-Gln219 in the surface-exposed “218 loop.” This cleaved form circulates in blood along with the intact form, albeit at lower concentrations. To gain a better understanding of how cleavage affects PCSK9 function, we produced recombinant furin-cleaved PCSK9 using antibody Ab-3D5, which binds the intact but not the cleaved 218 loop. Using Ab-3D5, we also produced highly purified hepsin-cleaved PCSK9. Hepsin cleaves PCSK9 at Arg218-Gln219 more efficiently than furin but also cleaves at Arg215-Phe216. Further analysis by size exclusion chromatography and mass spectrometry indicated that furin and hepsin produced an internal cleavage in the 218 loop without the loss of the N-terminal segment (Ser153–Arg218), which remained attached to the catalytic domain. Both furin- and hepsin-cleaved PCSK9 bound to LDL receptor with only 2-fold reduced affinity compared with intact PCSK9. Moreover, they reduced LDL receptor levels in HepG2 cells and in mouse liver with only moderately lower activity than intact PCSK9, consistent with the binding data. Single injection into mice of furin-cleaved PCSK9 resulted in significantly increased serum cholesterol levels, approaching the increase by intact PCSK9. These findings indicate that circulating furin-cleaved PCSK9 is able to regulate LDL receptor and serum cholesterol levels, although somewhat less efficiently than intact PCSK9. Therapeutic anti-PCSK9 approaches that neutralize both forms should be the most effective in preserving LDL receptors and in lowering plasma LDL cholesterol.
Introduction
Proprotein convertase subtilisin/kexin type 9 (PCSK9)2 is a member of the proprotein convertase family and a ligand of hepatic LDL receptors. PCSK9 prevents LDL receptor recycling by directing the ligand·receptor complex for lysosomal degradation, resulting in reduced LDL cholesterol (LDL-c) clearance and increased plasma LDL-c levels. The importance of PCSK9 in lipid metabolism is strongly supported by human genetics and by physiologic studies (reviewed in Refs. 1–4). Moreover, the strong reduction in coronary heart disease caused by loss of function mutations in the PCSK9 gene (5) has provided a strong rationale for the development of PCSK9 inhibitors for the treatment of dyslipidemia.
Mature PCSK9 is composed of the prodomain that is noncovalently attached to the subtilisin-like catalytic domain, which is followed by the C-terminal domain (see Fig. 1A) (6–8). PCSK9 binds to the EGF(A) domain of LDL receptor in a calcium-dependent fashion (9–11). Enzymatic activity of PCSK9 is required for autocatalytic processing of the single-chain PCSK9 precursor in the endoplasmic reticulum but not for LDL receptor binding and LDL receptor degradation (12, 13). Although the C-terminal domain is not directly involved in binding to cell surface LDL receptor, it interacts with cell surface proteins (14, 15) and engages in LDL receptor interactions in the endosomal compartment (16, 17).
FIGURE 1.

PCSK9 domains and protease cleavage sites. A, cartoon indicating the three main PCSK9 domains: the prodomain, catalytic (Cat), and C-terminal (CT) domains. The N-segment (Ser153–Arg218) comprises the N-terminal portion of the catalytic domain up to the furin cleavage site residue Arg218. The molecular masses determined by MS (see Fig. 5) are indicated for the N-segment, ΔN-Cat_CT, and Cat_CT. B, the 218 loop comprising the P4-P4′ amino acid sequence according to the Schechter and Berger nomenclature (29). The primary cleavage site for furin and hepsin (Arg218-Gln219) is indicated by a solid arrow, and the secondary cleavage site that is specific for hepsin (Arg215-Phe216) is indicated by a dashed arrow.
The membrane-bound proprotein convertase furin was shown to cleave PCSK9 at the Arg218-Gln219 peptide bond (18). Another proprotein convertase PC5/6A was also found to hydrolyze this bond, albeit with much reduced efficiency compared with furin (18). This cleavage site is located in a surface loop, the “218 loop,” which appears to have inherent flexibility. In vivo studies with liver-targeted furin knock-out mice corroborated the in vitro findings and demonstrated that furin is the main PCSK9 processing protease in vivo (19). When analyzed by SDS-PAGE, the cleaved PCSK9 shows a shift of the ∼60-kDa band to a lower ∼50-kDa species that lacks the Ser153–Arg218 amino acid stretch of the catalytic domain, designated the N-segment (see Fig. 1A). Results from cellular assays suggested that cleaved PCSK9 is no longer able to degrade LDL receptor (18). This complete lack of activity was attributed to the loss of the N-segment, as well as the prodomain from PCSK9 (18).
The cleaved PCSK9 form circulates in human plasma (18–21) and constitutes 15–40% of total circulating PCSK9 (18, 19). Cleaved PCSK9 was also found in mouse plasma contributing 30–50% to total PCSK9 levels (19, 22). The furin cleavage sequence 215RFHRQ219 harbors naturally occurring gain of function mutations, R215H, F216L, and R218S, associated with hypercholesterolemia (23–25). Analysis of patient plasma samples and in vitro mutagenesis studies indicated that these mutations impair furin-mediated PCSK9 cleavage (18, 19, 23), suggesting that furin resistance is the underlying molecular mechanism for the gain of function phenotype.
The question of whether furin cleavage of PCSK9 affects receptor binding or post-ligation events has not yet been formally addressed. For example, if cleaved PCSK9 is biologically inactive but would retain LDL receptor binding, then the circulating cleaved PCSK9 could act as a competitive inhibitor of the intact form. To gain insight into these questions, we generated highly purified cleaved PCSK9 protein for functional studies in biochemical assays and for in vivo LDL receptor degradation. We took advantage of monoclonal antibody Ab-3D5 that differentially recognizes furin-cleaved and intact PCSK9, allowing us to obtain highly purified cleaved PCSK9. Functional studies revealed that cleavage only moderately affected LDL receptor binding and physiologic functions. These findings were consistent with biophysical measurements indicating that cleaved PCSK9 remained intact and without loss of fragments. The unexpected functional competence of cleaved PCSK9 can be rationalized on the basis of the PCSK9 structure, and its ramifications on biological and therapeutic aspects of PCSK9 function are discussed.
EXPERIMENTAL PROCEDURES
Reagents
Soluble human furin and soluble LDL receptor ectodomain were from R & D Systems, factor Xa, activated protein C, and thrombin from Hematologic Technologies and factor XIIa from Enzyme Research Laboratories. Soluble hepsin, hepatocyte growth factor activator, and matriptase were expressed and purified as described (26, 27). The neutralizing anti-hepsin antibody Ab25 was described recently (26).
Construction, Expression, and Purification of Wild Type and Mutant PCSK9 Proteins
Human PCSK9 cDNA containing a His8 C-terminal tag was cloned into a mammalian expression vector (pRK5). Human PCSK9 R215A/R218A mutant was made by site-directed mutagenesis using QuikChange Lightning (Aligent Technologies, Santa Clara, CA). Human PCSK9 R218A mutant was constructed by ACTG, Inc. (Wheeling, IL) using site-directed mutagenesis. Mutants were confirmed by DNA sequencing. The recombinant human PCSK9 proteins (wild type and mutants) were transiently expressed in CHO cells and purified from conditioned media by affinity chromatography using a nickel nitrilotriacetic agarose column (Qiagen) followed by gel filtration on a Sephacryl S 200 column (GE Healthcare).
Production of Monoclonal Anti-PCSK9 Antibodies
8–12-week-old PCSK9−/− mice (22) were immunized with recombinant human PCSK9. Three days after the final boost, lymphocytes from spleens and lymph nodes were harvested for fusion with SP2/0 myeloma cells (American Type Culture Collection). After 7–10 days, single hybridoma clones were picked by ClonePix (Genetix) and transferred into 96-well cell culture plates (Becton Dickinson) with 200 μl/well ClonaCell-HY medium E (Stem Cell Technologies). After at least two rounds of single cell subcloning by limiting dilution, the final clones, including Ab-3D5 and Ab-7G7, were scaled up, and supernatants were collected for antibody purification. The hybridoma supernatants were purified by protein A affinity chromatography, then sterile filtered (Nalge Nunc International), and stored at 4 °C in PBS.
PCSK9 Cleavage Assays
Unless otherwise indicated, all of the cleavage reactions were performed in PCSK9 buffer (50 mm Tris, pH 8.0, 150 mm NaCl), and reaction products were analyzed by SDS-PAGE under nonreducing conditions. PCSK9 (1.9 μm) was incubated with 40 nm of furin, factor Xa, factor XIIa, activated protein C, or thrombin in PCSK9 buffer supplemented with 4 mm CaCl2, or with hepsin, hepatocyte growth factor activator, matriptase in PCSK9 sample without CaCl2, for 6 h at 20 °C. In hepsin blocking experiments, 40 nm of hepsin was preincubated with 3 μm of hepsin-specific antibody Ab25 (26) for 20 min prior to the addition of PCSK9.
Experiments with PCSK9 mutants R218A and R215A/R218A (2.6 μm) were carried out in PCSK9 buffer supplemented with 4 mm CaCl2 by incubation with either 40 nm hepsin for 6 h or with 80 nm furin for 20 h. Proteins were analyzed by SDS-PAGE followed by staining with SimplyBlue SafeStain (Invitrogen).
The relative PCSK9-processing activities of hepsin and furin were assessed by incubation of PCSK9 (1.9 μm) with increasing concentrations of hepsin or furin for 6 h in PCSK9 buffer supplemented with 4 mm CaCl2, followed by SDS-PAGE and densitometry of intact PCSK9 bands by the NIH ImageJ software. The results are the averages ± S.D. of three experiments.
Inhibition assays with Ab-3D5 or Ab-7G7 were performed by incubating PCSK9 (1.9 μm) with 3 μm of either antibody for 20 min prior to the addition of 80 nm furin or 40 nm hepsin in PCSK9 buffer supplemented with 4 mm CaCl2 for 6 h. The gel pictures of the cleavage experiments shown in Figs. 2 and 3 are representative of at least three independently performed experiments.
FIGURE 2.

PCSK9 cleavage by serine proteases furin and hepsin. A, PCSK9 (1.9 μm) was incubated with 40 nm of various serine proteases for 6 h at room temperature, and the reaction products were analyzed by SDS-PAGE (nonreducing conditions) followed by gel staining with SimplyBlue SafeStain. The individual bands are indicated according to the nomenclature in Fig. 1A, and the determined N-terminal sequences are in parentheses. The far right lane shows hepsin that was pretreated with the inhibitory anti-hepsin antibody Ab25 before incubation with PCSK9. Ctrl, untreated PCSK9; APC, activated protein C; HGFA, hepatocyte growth factor activator. B, comparison of proteolytic activities of furin and hepsin toward PCSK9. PCSK9 (1.9 μm) was incubated for 6 h with increasing protease concentrations (soluble hepsin and soluble furin) and analyzed by SDS-PAGE (right panel). Disappearance of the ∼60-kDa PCSK9 band (Cat_CT domain) was quantified and plotted against protease concentration (left panel). The calculated concentrations for 50% PCSK9 cleavage were 11.6 ± 0.6 nm for hepsin and 80.7 ± 29.1 nm for furin (average ± S.D. of three experiments).
FIGURE 3.

Inhibition of protease-mediated PCSK9 cleavage by mutagenesis of 218 loop residues and by antibody 3D5. A, PCSK9 WT and PCSK9 mutants R218A and R215A/R218A (2.6 μm each) were incubated for 6 h with 40 nm of hepsin (upper panel) or for 20 h with 80 nm of furin (lower panel). The results indicate that hepsin has two cleavage sites, Arg218-Gln219 and Arg215-Phe216, whereas furin only cleaves at Arg218-Gln219 (see also Fig. 1B). B, PCSK9 (1.9 μm) was preincubated with antibodies 3D5 or 7G7 for 20 min before treatment with 40 nm hepsin (H) or with 80 nm furin (F) for 6 h. The results indicate that antibody 3D5 completely inhibited PCSK9 cleavage by either protease, whereas antibody 7G7 did not.
Purification of Cleaved PCSK9
PCSK9 was treated with 80 nm furin in PCSK9 buffer plus 4 mm CaCl2 for 20 h or with 40 nm hepsin in PCSK9 buffer for 6 h at room temperature. Then Ab-3D5 (IgG) and anti-furin antibody (IgG) (H-220; Santa Cruz Biotechnology) were added to the furin-treated samples to remove intact PCSK9 and furin, respectively. Ab-3D5 (IgG) and the hepsin antibody Ab25 (IgG) were added to the hepsin-treated samples to remove intact PCSK9 and hepsin, respectively. After 30 min of incubation, the mixtures were applied to S-200 size exclusion columns (HiLoadTM 16/60 SuperdexTM prep grade; GE Healthcare), and elution fractions were collected and analyzed by SDS-PAGE. Fractions containing pure cleaved PCSK9 (PCSK9c_fu and PCSK9c_hep, respectively) were pooled and stored at −80 °C for subsequent functional studies, analysis by N-terminal sequencing, and mass spectrometry.
Affinity Measurements by Biolayer Interferometry
LDL receptor binding to cleaved PCSK9 was measured on an Octet Red384 system (ForteBio). LDL receptor (ectodomain; R & D Systems) was biotinylated according to the manufacturer's instructions (Thermo Scientific) and immobilized on streptavidin biosensors (ForteBio). Measurements of association and dissociation constants were carried out in the presence of increasing concentrations of PCSK9c_hep or PCSK9c_fu in 50 mm Tris, pH 7.5, 300 mm NaCl, 2 mm CaCl2, 1 mg/ml BSA, 0.1% Tween 20. Kinetic parameters kon, koff, and KD were calculated from a nonlinear fit of the data using the Octet software Version 6.1 (ForteBio). Each reported value represents an average ± S.D. of at least three independent experiments.
Mass Spectrometry
PCSK9, PCSK9c_hep, and PCSK9c_fu were analyzed by reverse phase liquid chromatography-electrospray quadrupole time of flight mass spectrometry (6520; Agilent Technologies). Raw spectra were deconvoluted using Mass Hunter software (v.B.04.00; Agilent Technologies).
Cell Surface LDL Receptor Assay with HepG2 Cells
HepG2 cells (American Type Culture Collection) were seeded into 48-well plates (Corning) at 1 × 105 cells/well in high glucose medium (DMEM; Invitrogen) containing 2 mm glutamine (Sigma), penicillin/streptomycin (Invitrogen), and 10% FBS (Sigma) and incubated overnight. The medium was changed to DMEM containing 10% lipoprotein-deficient serum (Intracel), and after 24 h various concentrations of PCSK9, PCSK9c_fu or PCSK9c_hep were added to the cells and incubated at 37 °C for 4 h. The cells were rinsed with PBS and detached using cell dissociation buffer (Invitrogen). The cells were collected, centrifuged, and incubated with 1:20 anti-LDL receptor antibody (Progen Biotechnik) in PBS, 1% BSA on ice for 10 min. The samples were then washed with PBS and incubated with 1:200 goat anti-mouse IgG (H + L) Alexa Fluor 488 (Invitrogen) on ice for 5 min. After two PBS washes, the cells were resuspended in PBS containing 10 μg/ml of propidium iodide and analyzed on a dual laser flow cytometer (FACScan; Becton Dickinson). Relative fluorescence units were used to quantify LDL receptor expression levels on the HepG2 cell surface. The results were expressed as percentages of LDL receptor levels measured in the absence of PCSK9 ( = control) and are shown as the averages ± S.D. of three independent experiments.
Mouse Model of Liver LDL Receptor Degradation
8-week-old male C57BL/6 mice were purchased from Jackson Laboratory and housed for 2 weeks before starting the experiment. The mice were randomized into three groups (three mice/group) based on body weight and were injected intravenously with either PBS (vehicle control) or four doses (3, 15, 45, and 90 μg) of PCSK9 or of PCSK9c_fu. Experiments with PCSK9c_hep followed the same protocol. After 1 h, the livers were harvested and snap frozen. Approximately 200 mg of each liver was homogenized in extraction buffer I supplemented with protease inhibitor mixture (ProteoExtract native membrane protein extraction kit; Calbiochem) using the TissueLyser (Qiagen). The lysates were centrifuged, and the cell pellet resuspended in extraction buffer II supplemented with protease Inhibitor mixture (Calbiochem). After 30 min of gentle agitation at 4 °C, the samples were centrifuged, and the membrane protein concentrations of the supernatants were quantified using the Bradford assay. For each group (n = 3), liver proteins were pooled for a total of 100 μg of protein and separated by SDS-PAGE on a 4–12% Bis-Tris Midi gel (Invitrogen). Proteins were transferred to nitrocellulose membranes using iBlot (Invitrogen). The membranes were blocked with 5% nonfat milk and incubated with anti-LDL receptor (Abcam) in 5% nonfat milk. After washing with 10 mm Tris, pH 8.0, 150 mm NaCl, 0.1% Tween 20, blots were incubated with 1:5000 anti-rabbit horseradish peroxidase (GE Healthcare) in 5% nonfat milk, washed, and treated with ECL-Plus (GE Healthcare) followed by exposure to XAR film (Kodak). After washing with 10 mm Tris, pH 8.0, 150 mm NaCl, 0.1% Tween 20, membranes were incubated with anti-transferrin receptor antibody (Invitrogen) overnight at 4 °C. The membranes were incubated in 1:5000 anti-mouse horseradish peroxidase (GE Healthcare) for 1 h, washed, and then treated with ECL Plus and exposed to XAR film.
Determination of Serum Cholesterol Levels in Mice
8-week-old female C57BL/6 mice were purchased from Jackson Laboratory and housed for 2 weeks before starting the experiment. The mice were randomized into three groups (five mice/group) based on body weight and total cholesterol level and were injected intravenously with either PBS (vehicle group) or 60 μg of furin-cleaved PCSK9 or intact PCSK9. Blood samples were collected by orbital bleed under anesthesia from each group of mice at 0 h and 6, 9, and 48 h after intravenous dosing. Serum total cholesterol was measured by use of an enzymatic assay (InfinityTM; Thermo Scientific). Statistical analysis was performed using JMP v.9.0.2 software (SAS Institute, Inc.). Multiple comparisons with the pre-dose control (0 h) were performed using Dunnett's method. p values of <0.05 were considered as significant.
RESULTS
The Serine Protease Hepsin Cleaves PCSK9 at the Furin Cleavage Site
In contrast to membrane-localized furin, the soluble form of furin was shown to be rather inefficient in cleaving PCSK9 at the Arg218-Gln219 site (18, 28). To identify a more efficient protease, we tested a panel of trypsin-like serine proteases having preference for an arginine at the P1 position (nomenclature according to Schechter and Berger (29) (Fig. 1B). The results showed that none of the examined proteases appreciably cleaved PCSK9 during a 6-h incubation period, except for soluble furin (referred to as furin) and the soluble form of hepsin (referred to as hepsin), a type II transmembrane serine protease (Fig. 2A). The cleavage by hepsin was specific, because pretreatment with the neutralizing anti-hepsin antibody Ab25 completely inhibited PCSK9 cleavage (Fig. 2A). Hepsin appeared to be more efficient than furin, because most of the intact ∼60-kDa PCSK9 band (Cat_CT; Fig. 1A) was converted into the ∼50-kDa cleaved form (ΔN-Cat_CT; Fig. 1A), whereas less than 50% was converted by furin. N-terminal sequencing showed that for both furin and hepsin, the ΔN-Cat_CT domain (∼50 kDa) started with 219QAS and the ∼10-kDa segment (N-segment; Figs. 1 and 2A) started with 153SIP. Therefore, hepsin cleaved at the same site as furin, i.e., at the Arg218-Gln219 peptide bond (Fig. 1B). The relative cleavage efficiencies of hepsin and furin were quantified by measuring the disappearance of the intact 60-kDa band by densitometry. The results showed that during a 6-h reaction period, hepsin cleaved 50% of PCSK9 at a concentration of 11.6 nm compared with 80.7 nm for furin, indicating that hepsin was ∼7-fold more efficient (Fig. 2B).
To find out whether hepsin specifically cleaved at the Arg218-Gln219 site, we mutated the Arg218 residue to Ala, which should abolish cleavage. However, hepsin still cleaved PCSK9, although at a reduced rate, whereas furin was no longer able to cleave PCSK9 (Fig. 3A). An additional mutation of the nearby Arg215 residue was made to generate the double mutant R215A/R218A. This mutant was completely resistant to cleavage by hepsin (Fig. 3A), suggesting that hepsin cleaved at the two sites Arg215-Phe216 and Arg218-Gln219, whereas furin only cleaved at the latter site (Fig. 1B).
Identification of an Antibody That Differentiates Intact and Furin-cleaved PCSK9
To generate highly purified cleaved PCSK9, we wished to identify an antibody that only recognized intact but not cleaved PCSK9 for use in affinity purification of the cleaved form. Monoclonal antibodies derived from PCSK9−/− mice immunized with intact PCSK9 were analyzed for their ability to inhibit PCSK9 cleavage. One of these antibodies, Ab-3D5, completely prevented the cleavage by both furin and hepsin, whereas Ab-7G7 did not (Fig. 3B). This suggested that Ab-3D5 might bind to the intact 218 loop. This was examined by two different epitope mapping approaches. First, in an epitope excision experiment, PCSK9 alone or in complex with Ab-3D5 was digested with trypsin for various time periods. The peptide segments were identified by mass spectrometry and mapped to the PCSK9 sequence (supplemental Fig. S1A). The results indicated that Ab-3D5 (in the PCSK9·Ab-3D5 complex) protected trypsin-mediated cleavage at the Arg215-Phe216 and Arg218-Gln219 sites, which were readily cleaved when PCSK9 alone was digested. Therefore, the Ab-3D5 epitope encompassed the 214TRFHRQ219 sequence. Second, a panel of overlapping synthetic peptides covering the entire PCSK9 amino acid sequence was prepared (supplemental Fig. S2). ELISA experiments measuring binding of Ab-3D5 to the biotinylated peptides captured on streptavidin-coated plates showed that Ab-3D5 only bound to two peptides spanning the 218 loop, with the consensus sequence 211EDGTRFHRQA220 (supplemental Fig. S1B). The results from both approaches were in good agreement and clearly indicated that the Ab-3D5 epitope encompassed the PCSK9 218 loop, which harbors the hepsin and furin cleavage sites.
The recent crystal structure of the LDL receptor·PCSK9 complex revealed that the EGF(B) domain of LDL receptor is located close to the 218 loop, suggesting that Ab-3D5 may interfere with LDL receptor binding. Indeed, biolayer interferometry binding experiments demonstrated that Ab-3D5 completely inhibited LDL receptor binding to PCSK9 (supplemental Fig. S3A). In contrast, Ab-7G7, whose binding site did not overlap with that of Ab-3D5 according to competition binding experiments (data not shown), did not inhibit LDL receptor binding to PCSK9 (supplemental Fig. S3A). Additional studies with HepG2 cells further showed that Ab-3D5 inhibited PCSK9-mediated LDL receptor degradation in a concentration-dependent manner, whereas Ab-7G7 did not (supplemental Fig. S3B). Therefore, it is very probable that by binding to the 218 loop, Ab-3D5 sterically interfered with the LDL receptor binding.
The binding of Ab-3D5 to intact PCSK9 and to cleaved PCSK9, obtained by PCSK9 treatment with hepsin (6 h) or furin (20 h), was determined by biolayer interferometry. Hepsin and furin were removed in a repurification step, but the preparations still contained residual intact PCSK9. The results showed that Ab-3D5 bound with high affinity to intact PCSK9 (KD 3.5 nm) but had 170-fold and >280-fold reduced affinities for furin- and hepsin-cleaved PCSK9, respectively (supplemental Table S1). In contrast, the control antibody Ab-7G7 bound to intact and cleaved forms with similar affinities (supplemental Table S1). These findings suggested that Ab-3D5 differentially recognized intact and cleaved PCSK9 and thus could be utilized for affinity purification of the cleaved form.
Purification and Characterization of Cleaved PCSK9
The residual intact PCSK9 in the furin-treated preparation was removed by the addition of Ab-3D5. The formed complexes of intact PCSK9 and Ab-3D5 (high molecular weight peak) could be easily separated from the cleaved (uncomplexed) form by size exclusion chromatography (supplemental Fig. S4). SDS-PAGE analysis of the elution fractions showed a clear separation of intact PCSK9 co-eluting with Ab-3D5 IgG ( = intact PCSK9·Ab-3D5 complex) and the cleaved form eluting in later fractions (Fig. 4A). The pooled fractions of the cleaved PCSK9 contained the 50-kDa ΔN-Cat_CT (starting with Gln219), the ∼15-kDa prodomain, and the N-segment (Ser153–Arg218). However, these fractions did not contain any detectable intact PCSK9. Similar results were obtained with hepsin-cleaved PCSK9 (data not shown).
FIGURE 4.
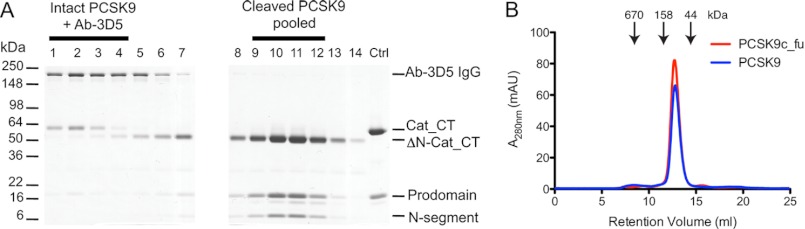
Purification and analysis of furin-cleaved PCSK9. A, PCSK9 was treated with furin for 20 h, after which an anti-furin antibody and Ab-3D5 were added, and the protein mixture was applied to a quantitative S-200 size exclusion column. The first elution peak contained the complex formed of Ab-3D5 with residual intact PCSK9 (∼60-kDa Cat_CT; lanes 1–4) and was separated from the later eluting pure cleaved PCSK9 (∼50-kDa ΔN-Cat_CT; lanes 9–12) (see also Suppl. Fig. 4). B, pooled fractions (9–12) of pure furin-cleaved PCSK9 (PCSK9c_fu; red elution profile) were compared with intact PCSK9 (PCSK9; blue elution profile) by analytical size exclusion chromatography. The elution volumes for PCSK9c_fu and PCSK9 were identical (12.72 and 12.78 ml), both having a deduced mass of 77 kDa. Molecular mass markers are indicated by arrows.
The pooled fractions of the purified furin-cleaved PCSK9 (referred to as PCSK9c_fu) were reapplied to a S-200 analytical size exclusion column and compared with intact PCSK9. Both proteins eluted at the same elution volume (12.72 and 12.78 ml) with a deduced mass of ∼77 kDa (Fig. 4B). This suggested that furin cleavage did not result in the loss of any substantial PCSK9 fragment. This was further investigated by electrospray mass spectrometry of PCSK9c_fu and of hepsin-cleaved purified PCSK9 (referred to as PCSK9c_hep). Intact PCSK9, which was used as reference material, gave a single peak of Mr 60,516 corresponding to the Cat_CT domain, several prodomain peaks (Mr 13,756–14,000; data not shown), but no peaks in the mass range of the N-segment (Fig. 5). For PCSK9c_fu and PCSK9c_hep, we observed peaks that corresponded the prodomain similar to intact PCSK9 (data not shown) and to two complementary portions of the Cat_CT domain, the ΔN-Cat_CT segment (Mr ∼52,800 starting at Gln219) and the N-segment (Mr 7,731; Ser153–Arg218) (Fig. 5). For PCSK9c_hep, there was an additional peak (Mr 7,290) identified as the C-terminally truncated N-segment Ser153–Arg215 (Fig. 5). This result was in excellent agreement with the mutagenesis experiments (Fig. 3A), demonstrating that furin cleaved only at the Arg218-Gln219 site, whereas hepsin additionally cleaved at the Arg215-Phe216 site. Therefore, the presence of prodomain, N-segment and ΔN-Cat_CT domain of the PCSK9c_fu and PCSK9c_hep preparations indicated that cleavage by furin or hepsin did not result in the loss of any PCSK9 fragments, except for the partial excision and probable loss of the three-amino acid stretch Phe216–Arg218 in the case of PCSK9c_hep. This is consistent with the co-elution of ΔN-Cat_CT, prodomain, and N-segment observed by SDS-PAGE analysis and the identical masses of intact and cleaved PCSK9 by size exclusion chromatography (Fig. 4). Thus, hepsin and furin only produced internal cleavages at the 218 loop, and the Ser153–Arg218 segment (and Ser153–Arg215 segment for PCSK9c_hep) remained noncovalently attached to the catalytic domain. These segments, like the noncovalently attached prodomain, only dissociate from the catalytic domain under denaturing conditions used for SDS-PAGE or for electrospray mass spectrometry measurements.
FIGURE 5.

Mass spectra of furin- and hepsin-cleaved PCSK9. The left panels show the peaks of Cat_CT and ΔN-Cat_CT, and the right panels show the peaks of the N-segment for intact (top panels), hepsin-cleaved (middle panels), and furin-cleaved PCSK9 (bottom panels) (see Fig. 1A for domain nomenclature). The peaks are labeled with relative molecular masses (Mr) deconvoluted from electrospray time of flight mass spectra and their assigned amino acid residues in PCSK9. The N-segment accounts for the reduced Mr of ΔN-Cat_CT compared with Cat_CT.
Binding of Cleaved PCSK9 to LDL Receptor
Competition binding ELISA experiments showed that both cleaved PCSK9 forms retained much of the LDL receptor binding function, having only slightly reduced binding affinities compared with intact PCSK9 (supplemental Table S2). Compared with the respective intact PCSK9 controls, the affinity losses (IC50 intact/IC50 cleaved) of PCSK9c_fu and PCSK9c_hep were 1.1- and 1.4-fold. Moreover, biolayer interferometry experiments showed that the intact PCSK9 controls bound to immobilized soluble LDL receptor with KD values of 130 and 177 nm (Table 1). These values are similar to those reported by Cunningham et al. (6) but significantly lower than reported by other studies (8, 30). The determined KD values for PCSK9c_fu and PCSK9c_hep were 357 and 268 nm, respectively (Table 1), and the corresponding affinity losses (KD cleaved/KD intact) were 2.0- and 2.1-fold. The results indicated that cleavage at the 218 loop had only a moderate effect on LDL receptor binding.
TABLE 1.
Binding affinities of LDL receptor interaction with intact and cleaved PCSK9
Kinetic constants were determined by biolayer interferometry using immobilized biotinylated LDL receptor ectodomain. Furin- and hepsin-cleaved PCSK9 (PCSK9c_fu and PCSK9c_hep) were purified by size exclusion chromatography using Ab-3D5. PCSK9 indicates intact PCSK9. The values are the averages ± S.D. of at least three independent experiments.
| Analyte | kon | koff | KD |
|---|---|---|---|
| 104m−1 s−1 | 10−4 s−1 | 10−9 m | |
| PCSK9 | 4.7 ± 1.0 | 61.2 ± 13.1 | 129.7 ± 17.4 |
| PCSK9c_hep | 3.2 ± 0.6 | 85.8 ± 18.0 | 267.7 ± 10.9 |
| PCSK9 | 3.6 ± 0.5 | 62.4 ± 12.4 | 177.0 ± 38.9 |
| PCSK9c_fu | 3.9 ± 0.5 | 138.5 ± 2.7 | 356.5 ± 50.7 |
LDL Receptor Degradation by Cleaved PCSK9
The LDL receptor degradation by cleaved PCSK9 was measured in two systems, in a cellular HepG2 assay and in a mouse model of liver LDL receptor degradation. In the HepG2 assays, intact PCSK9 reduced surface LDL receptor levels in a concentration-dependent fashion with a half-maximal reduction at 3.7–11 μg/ml (Fig. 6). PCSK9c_fu and PCSK9c_hep also reduced LDL receptor surface levels in a concentration-dependent fashion. Both cleaved forms were equally potent with half-maximal activity at 11–33 μg/ml (Fig. 6). Thus, the cleaved forms were ∼3-fold less potent compared with intact PCSK9 in reducing LDL receptor surface levels on HepG2 cells.
FIGURE 6.

Cleaved PCSK9 forms reduce LDL receptor levels on HepG2 surface. A, HepG2 cells were treated for 4 h with increasing concentrations of intact PCSK9 (PCSK9) or furin-cleaved PCSK9 (PCSK9c_fu), and cell surface LDL receptor (LDLR) was quantified by FACS analysis using an anti-LDL receptor antibody. B, same experimental protocol as in Fig. 6A for the comparison of intact PCSK9 with hepsin-cleaved PCSK9 (PCSK9c_hep). The results are the averages ± S.D. of at least three independent experiments.
In a mouse liver LDL receptor degradation model, the liver LDL receptor levels were determined by immunoblotting after mice were injected with increasing doses of intact or cleaved PCSK9 forms. Both PCSK9c_fu and intact PCSK9 almost completely reduced liver LDL receptor levels at the 45-μg dose (Fig. 7A). Similarly, PCSK9c_hep and intact PCSK9 almost completely reduced LDL receptor levels at 45 and 15 μg, respectively (Fig. 7B). Therefore, the cleaved forms efficiently degraded mouse liver LDL receptor with activities that were only slightly reduced compared with intact PCSK9.
FIGURE 7.

Cleaved PCSK9 degrades liver LDL receptor and increases serum cholesterol levels. A, C57BL/6 mice in groups of three mice were injected intravenously with PBS or with the indicated doses of intact PCSK9 (PCSK9) or with furin-cleaved PCSK9 (PCSK9c_fu). After 1 h, livers were harvested, and LDL receptor (LDLR) levels of pooled liver lysates were visualized by immunoblotting. B, same experimental protocol as in Fig. 7A for the comparison of intact PCSK9 with hepsin-cleaved PCSK9 (PCSK9c_hep). C, C57BL/6 mice in groups of n = 5 were injected intravenously with PBS (Vehicle) or with 60 μg of intact PCSK9 (PCSK9) or furin-cleaved PCSK9 (PCSK9c_fu). Blood samples were drawn at the indicated time points, and the serum total cholesterol levels were measured. Individual values for each group of mice (n = 5) are shown for each time point. The black bars indicate the means ± S.E. for each group. Multiple comparisons with the pre-dose control (0 h) were performed using Dunnett's method. p values of <0.05 were considered statistically significant and are denoted by asterisks.
Furin-cleaved PCSK9 Increases Cholesterol Levels in Mice
To find out whether reduction in liver LDL receptor by furin-cleaved PCSK9 led to increased serum cholesterol levels, the mice were injected intravenously with a single bolus of 60 μg of furin-cleaved or intact PCSK9. The results demonstrated that furin-cleaved PCSK9 elevated cholesterol levels significantly at 6 and 9 h (by 27% and 23%) in comparison with the corresponding pre-dose levels (Fig. 7C). These increases were slightly lower compared with those produced by intact PCSK9 (35% and 24%). After 48 h the cholesterol levels returned to base-line levels for the furin-cleaved PCSK9 group but were still slightly elevated for the intact PCSK9 group. These results demonstrated that furin-cleaved PCSK9 is competent to modulate physiologic processes, albeit with what is likely a moderately reduced activity.
DISCUSSION
There is ample experimental evidence that circulating PCSK9 is an important regulator of plasma LDL-c levels. Blocking antibodies that target circulating PCSK9 very effectively lower plasma LDL-c levels in mice, monkeys, and humans (31–33). Although there is evidence that furin-cleaved PCSK9 circulates in blood at significant levels, it has been assumed that the LDL-c levels were regulated only by the intact PCSK9 form because furin-cleaved PCSK9 was considered an inactive species (18). However, our study provides evidence that furin-cleaved PCSK9 retains substantial activity in vitro and in vivo. Therefore, liver LDL receptor and blood cholesterol levels appear to be regulated by both species of circulating PCSK9, the intact and the cleaved forms. The relative contribution of the two forms to LDL receptor degradation is difficult to assess. Considering that the cleaved PCSK9 is less abundant in circulation and has somewhat lower activity, it may only have a relatively modest impact on cholesterol levels. It is important to point out, however, that efforts to increase the levels of cleaved PCSK9 for therapeutic purposes are unlikely to produce any significant reduction in LDL-c levels.
The herein described functional characterization of cleaved PCSK9 was contingent upon the identification of Ab-3D5, which, based on its differential recognition of intact and cleaved PCSK9 forms, was used to produce highly purified cleaved PCSK9. Therefore, experimentally determined activities of cleaved PCSK9 could unambiguously be assigned to the cleaved form without any contribution from intact PCSK9 to confound interpretation. The molecular basis for the differential recognition of the antibody was provided by epitope mapping and binding experiments, which showed that Ab-3D5 bound to the intact 218 loop but lost its binding once the loop was cleaved by furin or hepsin. Based on the surmised functional incompetence of cleaved PCSK9 (18), it was unexpected that both furin- and hepsin-cleaved PCSK9 were able to degrade LDL receptor on HepG2 cells and in mouse liver, resulting in elevated serum cholesterol levels.
The moderate 2-fold reduced LDL receptor binding affinity of cleaved PCSK9 is best understood in the context of localized structural effects on this interaction. Biochemical and biophysical experiments established that furin-mediated cleavage at Arg218-Gln219 is an internal cleavage event without the loss of any fragment. The N-segment (Ser153–Arg218), as well as the prodomain, remained noncovalently attached to the catalytic domain. The same is true for hepsin cleavage, except that the additional cleavage at Arg215-Phe216 likely resulted in the partial loss of the 216FHR218 tripeptide. In addition, structural considerations favor attachment rather than loss of the N-segment and prodomain upon cleavage at the 218 loop. An inspection of published PCSK9 crystal structures indicated that the N-segment is unlikely to dissociate without a major structural reorganization of the PCSK9 catalytic domain because of extensive interactions between the N-segment and the catalytic domain (supplemental Fig. S5). Analysis of this interface (Protein Data Bank code 3H42) by PISA (34) revealed that the N-terminal segment and catalytic domain share 6283 Å2 of combined buried surface area, 9 salt bridges, and 58 main chain and side chain hydrogen bonds. Particularly notable is the N-segment β-strand Glu181–Asp186 that is part of a five-stranded β-sheet and engages in numerous β-strand type H-bond interactions (supplemental Fig. S5).
The recently published structure of the LDL receptor·PCSK9 complex (28) provides a basis for our attempt to rationalize the moderate effect of cleavage on LDL receptor binding. Both the EGF(B) and EGF(A) domains are in proximity of the 218 loop, which is only partially resolved in the structure and is missing the stretch Gln219–Lys222 (Protein Data Bank code 3P5C). Therefore, one possibility is that cleavage results in the loss of interactions between this region and EGF(B) and/or EGF(A). Alternatively, the positively charged N terminus (Gln219) and the negatively charged C terminus (Arg218) generated by furin cleavage are no longer constrained in a loop conformation and may now extend toward EGF(A,B) to disturb LDL receptor binding.
Our findings are incongruent with the reported lack of LDL receptor degradation by furin-cleaved PCSK9 in cellular assays (18). A significant difference was the use of a variant protein, engineered to be highly sensitive to furin cleavage, in the prior studies (18, 35), whereas we used purification to produce a fully cleaved preparation of the wild-type protein. Because the variant protein was not purified or characterized in detail, the possibility that the engineered furin cleavage site had effects on protein function beyond increasing the rate of furin cleavage is a likely explanation for the lack of LDL receptor down-regulation in those studies versus ours. For example, it is possible that cleavage of the altered amino acid sequence, 215RRRREL220 (18) versus the wild-type 215RFHRQA220, has detrimental effects on the interaction of PCSK9 with EGF(B) and/or EGF(A) domains or with the ability of PCSK9 to direct sorting to the lysosome following internalization.
The surface-exposed 218 loop is resistant to cleavage by most of the tested proteases, such as the proprotein convertases (18) and various trypsin-like serine proteases (this study). Only furin, PC5/6 and the herein reported hepsin can cleave it. Although both furin and hepsin are serine proteases, their catalytic domains have completely different folds (subtilisin-like and trypsin-like, respectively) and different active site conformations. Therefore, it is intriguing that they both cleave PCSK9 at the Arg218-Gln219 site. The additional partial cleavage at the Arg215-Phe216 site by hepsin had no further impact on PCSK9 activity as demonstrated by the equivalent activities of furin-and hepsin-cleaved forms. Of note, both PCSK9 and hepsin are mainly expressed in the liver. However, so far there is no evidence to suggest that hepsin is involved in lipid metabolism and, particularly in PCSK9 biology. Hepsin deficiency causes deafness in mice (36), whereas hepsin overexpression is associated with prostate cancer progression (37–39). On the other hand, there is compelling evidence from in vivo studies that furin is the major protease that cleaves PCSK9 and regulates the plasma levels of the cleaved form (19). PCSK9 gain of function mutations located in the 218 loop, R215H, F216L, and R218S, are resistant to furin cleavage (18, 19, 23) and predisposed to increased LDL-c levels, which was interpreted by the inability of furin to inactivate PCSK9 (18, 19). This interpretation was made under the assumption that cleaved PCSK9 is completely devoid of any LDL receptor degradation activity. Our findings do call this interpretation into question because the reduction in activity of cleaved PCSK9 is only ∼3-fold or less. However, it is very possible that in a highly regulated and complex physiologic system, changes of this magnitude could have significant effects. Heterozygosity for a loss of function mutation substantially alters both LDL-c levels and cardiovascular risk, demonstrating that PCSK9 levels and activity have significant physiological effects within a narrow range of values near the norm.
The responsiveness of LDL-c plasma levels to the level and activity of circulating PCSK9 highlights the potential for therapeutic intervention, and initial results (33) have borne this out. Our study highlights the importance of considering the antibody epitope that is targeted for therapeutic intervention. It is likely that an antibody whose binding site encompasses the 218 loop region, such as the 3D5 antibody described here or mAb1 (31), will have much reduced ability to neutralize cleaved PCSK9 as compared with an antibody whose epitope is distant from this site. The increased understanding of the role of cleaved PCSK9 provided in this study could improve the probability of success of therapeutic intervention and the likelihood that further reductions in cardiovascular risk will be achieved in the near future.
Acknowledgment
We thank Wilson Phung for expert help with mass spectrometry analysis of PCSK9 samples.

This article contains supplemental text, Tables S1 and S2, and Figs. S1–S5.
- PCSK9
- proprotein convertase subtilisin/kexin type 9
- PCSK9c_fu
- purified furin-cleaved PCSK9
- PCSK9c_hep
- purified hepsin-cleaved PCSK9
- Cat_CT
- catalytic and C-terminal domain of PCSK9
- ΔN-Cat_CT
- catalytic and C-terminal domain of PCSK9 without the N-segment
- LDL-c
- LDL cholesterol.
REFERENCES
- 1. Horton J. D., Cohen J. C., Hobbs H. H. (2009) PCSK9. A convertase that coordinates LDL catabolism. J. Lipid Res. 50, (suppl.) S172–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Seidah N. G. (2009) PCSK9 as a therapeutic target of dyslipidemia. Expert. Opin. Ther. Targets 13, 19–28 [DOI] [PubMed] [Google Scholar]
- 3. Costet P., Krempf M., Cariou B. (2008) PCSK9 and LDL cholesterol. Unravelling the target to design the bullet. Trends Biochem. Sci. 33, 426–434 [DOI] [PubMed] [Google Scholar]
- 4. Duff C. J., Hooper N. M. (2011) PCSK9. An emerging target for treatment of hypercholesterolemia. Expert. Opin. Ther. Targets 15, 157–168 [DOI] [PubMed] [Google Scholar]
- 5. Cohen J. C., Boerwinkle E., Mosley T. H., Jr., Hobbs H. H. (2006) Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N. Engl. J. Med. 354, 1264–1272 [DOI] [PubMed] [Google Scholar]
- 6. Cunningham D., Danley D. E., Geoghegan K. F., Griffor M. C., Hawkins J. L., Subashi T. A., Varghese A. H., Ammirati M. J., Culp J. S., Hoth L. R., Mansour M. N., McGrath K. M., Seddon A. P., Shenolikar S., Stutzman-Engwall K. J., Warren L. C., Xia D., Qiu X. (2007) Structural and biophysical studies of PCSK9 and its mutants linked to familial hypercholesterolemia. Nat. Struct. Mol. Biol. 14, 413–419 [DOI] [PubMed] [Google Scholar]
- 7. Hampton E. N., Knuth M. W., Li J., Harris J. L., Lesley S. A., Spraggon G. (2007) The self-inhibited structure of full-length PCSK9 at 1.9 A reveals structural homology with resistin within the C-terminal domain. Proc. Natl. Acad. Sci. U.S.A. 104, 14604–14609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Piper D. E., Jackson S., Liu Q., Romanow W. G., Shetterly S., Thibault S. T., Shan B., Walker N. P. (2007) The crystal structure of PCSK9. A regulator of plasma LDL-cholesterol. Structure 15, 545–552 [DOI] [PubMed] [Google Scholar]
- 9. Kwon H. J., Lagace T. A., McNutt M. C., Horton J. D., Deisenhofer J. (2008) Molecular basis for LDL receptor recognition by PCSK9. Proc. Natl. Acad. Sci. U.S.A. 105, 1820–1825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang D. W., Lagace T. A., Garuti R., Zhao Z., McDonald M., Horton J. D., Cohen J. C., Hobbs H. H. (2007) Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat A of low density lipoprotein receptor decreases receptor recycling and increases degradation. J. Biol. Chem. 282, 18602–18612 [DOI] [PubMed] [Google Scholar]
- 11. Malby S., Pickering R., Saha S., Smallridge R., Linse S., Downing A. K. (2001) The first epidermal growth factor-like domain of the low-density lipoprotein receptor contains a noncanonical calcium binding site. Biochemistry 40, 2555–2563 [DOI] [PubMed] [Google Scholar]
- 12. McNutt M. C., Lagace T. A., Horton J. D. (2007) Catalytic activity is not required for secreted PCSK9 to reduce low density lipoprotein receptors in HepG2 cells. J. Biol. Chem. 282, 20799–20803 [DOI] [PubMed] [Google Scholar]
- 13. Li J., Tumanut C., Gavigan J. A., Huang W. J., Hampton E. N., Tumanut R., Suen K. F., Trauger J. W., Spraggon G., Lesley S. A., Liau G., Yowe D., Harris J. L. (2007) Secreted PCSK9 promotes LDL receptor degradation independently of proteolytic activity. Biochem. J. 406, 203–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ni Y. G., Condra J. H., Orsatti L., Shen X., Di Marco S., Pandit S., Bottomley M. J., Ruggeri L., Cummings R. T., Cubbon R. M., Santoro J. C., Ehrhardt A., Lewis D., Fisher T. S., Ha S., Njimoluh L., Wood D. D., Hammond H. A., Wisniewski D., Volpari C., Noto A., Lo Surdo P., Hubbard B., Carfí A., Sitlani A. (2010) A proprotein convertase subtilisin-like/kexin type 9 (PCSK9) C-terminal domain antibody antigen-binding fragment inhibits PCSK9 internalization and restores low density lipoprotein uptake. J. Biol. Chem. 285, 12882–12891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mayer G., Poirier S., Seidah N. G. (2008) Annexin A2 is a C-terminal PCSK9-binding protein that regulates endogenous low density lipoprotein receptor levels. J. Biol. Chem. 283, 31791–31801 [DOI] [PubMed] [Google Scholar]
- 16. Yamamoto T., Lu C., Ryan R. O. (2011) A two-step binding model of PCSK9 interaction with the low density lipoprotein receptor. J. Biol. Chem. 286, 5464–5470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Holla Ø. L., Cameron J., Tveten K., Strøm T. B., Berge K. E., Laerdahl J. K., Leren T. P. (2011) Role of the C-terminal domain of PCSK9 in degradation of the LDL receptors. J. Lipid Res. 52, 1787–1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Benjannet S., Rhainds D., Hamelin J., Nassoury N., Seidah N. G. (2006) The proprotein convertase (PC) PCSK9 is inactivated by furin and/or PC5/6A. Functional consequences of natural mutations and post-translational modifications. J. Biol. Chem. 281, 30561–30572 [DOI] [PubMed] [Google Scholar]
- 19. Essalmani R., Susan-Resiga D., Chamberland A., Abifadel M., Creemers J. W., Boileau C., Seidah N. G., Prat A. (2011) In vivo evidence that furin from hepatocytes inactivates PCSK9. J. Biol. Chem. 286, 4257–4263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dubuc G., Tremblay M., Paré G., Jacques H., Hamelin J., Benjannet S., Boulet L., Genest J., Bernier L., Seidah N. G., Davignon J. (2010) A new method for measurement of total plasma PCSK9. Clinical applications. J. Lipid Res. 51, 140–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Troutt J. S., Alborn W. E., Cao G., Konrad R. J. (2010) Fenofibrate treatment increases human serum proprotein convertase subtilisin kexin type 9 levels. J. Lipid Res. 51, 345–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zaid A., Roubtsova A., Essalmani R., Marcinkiewicz J., Chamberland A., Hamelin J., Tremblay M., Jacques H., Jin W., Davignon J., Seidah N. G., Prat A. (2008) Proprotein convertase subtilisin/kexin type 9 (PCSK9). Hepatocyte-specific low-density lipoprotein receptor degradation and critical role in mouse liver regeneration. Hepatology 48, 646–654 [DOI] [PubMed] [Google Scholar]
- 23. Cameron J., Holla O. L., Laerdahl J. K., Kulseth M. A., Ranheim T., Rognes T., Berge K. E., Leren T. P. (2008) Characterization of novel mutations in the catalytic domain of the PCSK9 gene. J. Intern. Med. 263, 420–431 [DOI] [PubMed] [Google Scholar]
- 24. Abifadel M., Varret M., Rabès J. P., Allard D., Ouguerram K., Devillers M., Cruaud C., Benjannet S., Wickham L., Erlich D., Derré A., Villéger L., Farnier M., Beucler I., Bruckert E., Chambaz J., Chanu B., Lecerf J. M., Luc G., Moulin P., Weissenbach J., Prat A., Krempf M., Junien C., Seidah N. G., Boileau C. (2003) Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 34, 154–156 [DOI] [PubMed] [Google Scholar]
- 25. Allard D., Amsellem S., Abifadel M., Trillard M., Devillers M., Luc G., Krempf M., Reznik Y., Girardet J. P., Fredenrich A., Junien C., Varret M., Boileau C., Benlian P., Rabes J. P. (2005) Novel mutations of the PCSK9 gene cause variable phenotype of autosomal dominant hypercholesterolemia. Hum. Mutat. 26, 497. [DOI] [PubMed] [Google Scholar]
- 26. Ganesan R., Zhang Y., Landgraf K. E., Lin S. J., Moran P., Kirchhofer D. (2012) An allosteric anti-hepsin antibody derived from a constrained phage display library. Protein Eng. Des. Sel. 25, 127–133 [DOI] [PubMed] [Google Scholar]
- 27. Kirchhofer D., Peek M., Li W., Stamos J., Eigenbrot C., Kadkhodayan S., Elliott J. M., Corpuz R. T., Lazarus R. A., Moran P. (2003) Tissue expression, protease specificity, and Kunitz domain functions of hepatocyte growth factor activator inhibitor-1B (HAI-1B), a new splice variant of HAI-1. J. Biol. Chem. 278, 36341–36349 [DOI] [PubMed] [Google Scholar]
- 28. Lo Surdo P., Bottomley M. J., Calzetta A., Settembre E. C., Cirillo A., Pandit S., Ni Y. G., Hubbard B., Sitlani A., Carfí A. (2011) Mechanistic implications for LDL receptor degradation from the PCSK9/LDLR structure at neutral pH. EMBO Rep. 12, 1300–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schechter I., Berger A. (1967) On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 27, 157–162 [DOI] [PubMed] [Google Scholar]
- 30. Fisher T. S., Lo Surdo P., Pandit S., Mattu M., Santoro J. C., Wisniewski D., Cummings R. T., Calzetta A., Cubbon R. M., Fischer P. A., Tarachandani A., De Francesco R., Wright S. D., Sparrow C. P., Carfi A., Sitlani A. (2007) Effects of pH and low density lipoprotein (LDL) on PCSK9-dependent LDL receptor regulation. J. Biol. Chem. 282, 20502–20512 [DOI] [PubMed] [Google Scholar]
- 31. Chan J. C., Piper D. E., Cao Q., Liu D., King C., Wang W., Tang J., Liu Q., Higbee J., Xia Z., Di Y., Shetterly S., Arimura Z., Salomonis H., Romanow W. G., Thibault S. T., Zhang R., Cao P., Yang X. P., Yu T., Lu M., Retter M. W., Kwon G., Henne K., Pan O., Tsai M. M., Fuchslocher B., Yang E., Zhou L., Lee K. J., Daris M., Sheng J., Wang Y., Shen W. D., Yeh W. C., Emery M., Walker N. P., Shan B., Schwarz M., Jackson S. M. (2009) A proprotein convertase subtilisin/kexin type 9 neutralizing antibody reduces serum cholesterol in mice and nonhuman primates. Proc. Natl. Acad. Sci. U.S.A. 106, 9820–9825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liang H., Chaparro-Riggers J., Strop P., Geng T., Sutton J. E., Tsai D., Bai L., Abdiche Y., Dilley J., Yu J., Wu S., Chin S. M., Lee N. A., Rossi A., Lin J. C., Rajpal A., Pons J., Shelton D. L. (2012) Proprotein convertase substilisin/kexin type 9 antagonism reduces low-density lipoprotein cholesterol in statin-treated hypercholesterolemic nonhuman primates. J. Pharmacol. Exp. Ther. 340, 228–236 [DOI] [PubMed] [Google Scholar]
- 33. Stein E. A., Mellis S., Yancopoulos G. D., Stahl N., Logan D., Smith W. B., Lisbon E., Gutierrez M., Webb C., Wu R., Du Y., Kranz T., Gasparino E., Swergold G. D. (2012) Effect of a monoclonal antibody to PCSK9 on LDL cholesterol. N. Engl. J. Med. 366, 1108–1118 [DOI] [PubMed] [Google Scholar]
- 34. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 35. Duff C. J., Scott M. J., Kirby I. T., Hutchinson S. E., Martin S. L., Hooper N. M. (2009) Antibody-mediated disruption of the interaction between PCSK9 and the low-density lipoprotein receptor. Biochem. J. 419, 577–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Guipponi M., Tan J., Cannon P. Z., Donley L., Crewther P., Clarke M., Wu Q., Shepherd R. K., Scott H. S. (2007) Mice deficient for the type II transmembrane serine protease, TMPRSS1/hepsin, exhibit profound hearing loss. Am. J. Pathol. 171, 608–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Magee J. A., Araki T., Patil S., Ehrig T., True L., Humphrey P. A., Catalona W. J., Watson M. A., Milbrandt J. (2001) Expression profiling reveals hepsin overexpression in prostate cancer. Cancer Res. 61, 5692–5696 [PubMed] [Google Scholar]
- 38. Klezovitch O., Chevillet J., Mirosevich J., Roberts R. L., Matusik R. J., Vasioukhin V. (2004) Hepsin promotes prostate cancer progression and metastasis. Cancer Cell 6, 185–195 [DOI] [PubMed] [Google Scholar]
- 39. Li W., Wang B. E., Moran P., Lipari T., Ganesan R., Corpuz R., Ludlam M. J., Gogineni A., Koeppen H., Bunting S., Gao W. Q., Kirchhofer D. (2009) Pegylated kunitz domain inhibitor suppresses hepsin-mediated invasive tumor growth and metastasis. Cancer Res. 69, 8395–8402 [DOI] [PubMed] [Google Scholar]