

Background: PCSK9 regulates low-density lipoprotein receptor levels in the liver. The importance of the M1, M2, and M3 modules within the C terminus of PCSK9 is unknown.
Results: The M2 module is needed for the extracellular, but not intracellular, activity of PCSK9.
Conclusion: The integrity of the M2 module is essential for the extracellular function of PCSK9.
Significance: Targeting the M2 module should neutralize circulating PCSK9 and reduce LDL-cholesterol.
Keywords: Atherosclerosis; Cholesterol Regulation; Convertases; Receptor Regulation; Trafficking; LDLR Degradation; M2 Module, CHRD; PCSK9; Structure-Function; Hypercholesterolemia
Abstract
PCSK9 enhances the cellular degradation of the LDL receptor (LDLR), leading to increased plasma LDL cholesterol. This multidomain protein contains a prosegment, a catalytic domain, a hinge region, and a cysteine-histidine rich domain (CHRD) composed of three tightly packed modules named M1, M2, and M3. The CHRD is required for the activity of PCSK9, but the mechanism behind this remains obscure. To define the contribution of each module to the function of PCSK9, we dissected the CHRD structure. Six PCSK9 deletants were generated by mutagenesis, corresponding to the deletion of only one (ΔM1, ΔM2, ΔM3) or two (ΔM12, ΔM13, ΔM23) modules. Transfection of HEK293 cells showed that all deletants were well processed and expressed compared with the parent PCSK9 but that only those lacking the M2 module were secreted. HepG2 cells lacking endogenous PCSK9 (HepG2/shPCSK9) were used for the functional analysis of the extracellular or intracellular activity of PCSK9 and its deletants. To analyze the ability of the deletants to enhance the LDLR degradation by the intracellular pathway, cellular expressions revealed that only the ΔM2 deletant retains a comparable total LDLR-degrading activity to full-length PCSK9. To probe the extracellular pathway, HepG2/shPCSK9 cells were incubated with conditioned media from transfected HEK293 or HepG2/shPCSK9 cells, and cell surface LDLR levels were analyzed by FACS. The results showed no activity of any secreted deletant compared with PCSK9. Thus, although M2 is dispensable for secretion, its presence is required for the extracellular activity of PCSK9 on cell surface LDLR.
Introduction
Proprotein convertase subtilisin/kexin type 9 (PCSK9) is the latest and ninth member of the mammalian subtilisin-like serine proprotein convertases (PC)2 family (1, 2). Its high expression in liver, localization of its gene PCSK9 on human chromosome 1p32, and characterization of two gain-of-function (GOF) mutants in families with high levels of circulating low density lipoprotein cholesterol (LDLc) led to its identification as the third locus implicated in autosomal dominant hypercholesterolemia, with the low-density lipoprotein receptor (LDLR), apolipoprotein B (3), and apolipoprotein E (4) as the other three.
Early studies demonstrated that, similar to the other PC family members, PCSK9 is first synthesized as a zymogen (proPCSK9) that undergoes an autocatalytic cleavage of its prosegment (1) at VFAQ152↓ (5, 6) within the endoplasmic reticulum (ER). This cleavage is a prerequisite for the exit and secretion of the prosegment PCSK9 complex from the ER, as the zymogen is not secreted (1, 5). It became apparent that, different from the other eight PC family members, PCSK9 never gets rid of its inhibitory prosegment (2) and is thus secreted as a catalytically inactive protease. This tight prosegmentPCSK9 interaction was confirmed by the crystal structure of PCSK9 that revealed multiple points of contact between the prosegment and the catalytic subunit of PCSK9 (7–9). These data further showed that following the catalytic domain, the PCSK9 structure exhibits the presence of an exposed hinge region (residues 422–439) (10) followed by a C-terminal Cys/His-rich domain (CHRD) composed of three repeat modules termed M1 (amino acids 453–531), M2 (amino acids 530–605), and M3 (amino acids 604–692) (supplemental Fig. S1) (7).
PCSK9 complex from the ER, as the zymogen is not secreted (1, 5). It became apparent that, different from the other eight PC family members, PCSK9 never gets rid of its inhibitory prosegment (2) and is thus secreted as a catalytically inactive protease. This tight prosegmentPCSK9 interaction was confirmed by the crystal structure of PCSK9 that revealed multiple points of contact between the prosegment and the catalytic subunit of PCSK9 (7–9). These data further showed that following the catalytic domain, the PCSK9 structure exhibits the presence of an exposed hinge region (residues 422–439) (10) followed by a C-terminal Cys/His-rich domain (CHRD) composed of three repeat modules termed M1 (amino acids 453–531), M2 (amino acids 530–605), and M3 (amino acids 604–692) (supplemental Fig. S1) (7).
Soon after its discovery, it became clear that PCSK9 is implicated in the degradation of the LDLR itself, as its overexpression in mice (11), primary hepatocytes (12), and/or cell lines including its natural GOF mutants (5) resulted in decreased levels of this receptor. It was also shown that the degradation of the PCSK9LDLR complex occurs in acidic compartments (5) likely to be endosomes/lysosomes (13). Cellular studies showed that PCSK9 targets the LDLR for degradation by two pathways: an intracellular one from the trans Golgi network directly to lysosomes, implicating clathrin light chains (14), and an extracellular one (15) requiring clathrin heavy chain-mediated endocytosis of the cell surface PCSK9LDLR complex (13).
Biochemical, cell biological, and structural studies demonstrated that the catalytic subunit of PCSK9 binds the epidermal growth factor-like repeat A domain of the LDLR (16–18). As suspected from the exclusive secretion of the catalytically inactive prosegmentPCSK9 complex, it was formally shown that the catalytic activity of PCSK9 was not required for its ability to enhance the degradation of the LDLR (19), suggesting that the latter is performed by undefined endogenous endosomal/lysosomal proteases. This was further confirmed for the other two receptors that PCSK9 also targets, namely Very Low Density Receptor and Apolipoprotein E receptor 2 (20).
The cell surface endocytosis of the PCSK9LDLR complex is a dominant degradation pathway that shunts the typical LDLR recycling route (21, 22). As a major consequence of PCSK9 action, levels of hepatocyte cell surface LDLR decrease, as confirmed in mouse knockout models (23, 24), leading to the accumulation of LDLc in mouse and human plasma (23–25). In cases of high levels or GOF mutants of PCSK9, this can result in inflammation and the formation of atherosclerotic plaques (26), ultimately leading to cardiovascular disease (27). Accordingly, inhibition or silencing of PCSK9 function is a novel powerful therapeutic approach to lower LDLc, as attested by multiple ongoing phase II clinical trials (2, 28).
In contrast to major advances in PCSK9-based therapies, the molecular mechanisms regulating the sorting of the prosegmentPCSK9LDLR complex to endosomes/lysosomes by the extracellular or intracellular pathways are poorly defined. Our present understanding of the subcellular trafficking of the PCSK9LDLR complex leading to its degradation is that it requires the presence of the CHRD (13, 29) but not the cytosolic tail of the LDLR (30). Furthermore, removal of the acidic N-terminal sequence of the prosegment significantly enhances the degradation efficacy of the complex (17, 31, 32).
To expand our understanding of the contribution of the CHRD in the enhanced degradation of LDLR by PCSK9, we present structure-function studies in which we dissected the M1, M2, and M3 modules of the CHRD and analyzed the activity of resulting deletants in the extracellular and intracellular pathways of LDLR degradation. The data demonstrated the critical requirement for the M2 domain in the extracellular pathway but not for the intracellular one.
EXPERIMENTAL PROCEDURES
Plasmids and Reagents
Human PCSK9 and its mutant cDNAs (hPCSK9 L455X and PCSK9 CHRD) were cloned into pIRES2-EGFP (Clontech, Mountain view, CA) as described (1, 13). Human HepG2/shPCSK9 cells essentially lacking endogenous PCSK9 (14) and HEK293 cells (ATCC) were cultivated in DMEM (Invitrogen) supplemented with 10% FBS (Wisent). Puromycin (2 μg/ml, Invitrogen) was added only to HepG2/shPCSK9 cells as a selection antibiotic. Lipoprotein-deficient serum was from Biomedical Technologies.
cDNA Constructs
The construct pIRES2-EFGP-human PCSK9-V5 (C-terminal V5-tag) (1) was used as a template to generate cDNAs coding for human PCSK9 mutants and module deletants. The oligonucleotides used are listed in supplemental Table S1. Two-step PCRs were used to introduce deletion mutants as described previously (5). All constructs contain a V5 tag at the C terminus. All constructs were confirmed by DNA sequencing.
Cell Culture and Transfections
HepG2/shPCSK9 cells were seeded at 1 × 105cells/well in a 12-well microplate (Greiner Bio-One). After 24 h, the cells were transfected with 1 μg of cDNAs using FuGENE HD (Roche Applied Science), and 24 h post-transfection, the cells were washed and then incubated with fresh DMEM without serum for an additional 24 h before recovering media and cells.
Preparation of Conditioned Media
2 × 106 HEK293 or HepG2/shPCSK9 cells in a 100-mm Petri dish coated with poly-l-lysine (Invitrogen) were transfected with a total of 4 μg of cDNA using Effectene (Qiagen). At 24 h post-transfection, the cells were washed and incubated with serum-free media. Conditioned media were recovered 72 h post-transfection. The spent media were then concentrated on an Amicon Ultra-15 centrifugal filter unit with a 10-kDa membrane cutoff (Millipore). Concentrated V5-tagged PCSK9 and its derivatives in conditioned media were quantitated by enzyme-linked immunosorbent assay detecting V5-tagged proteins (ELISA-V5, Invitrogen, catalog no. R960-25).
Media Transfer Experiments
HepG2/shPCSK9 cells were seeded in a 12-well microplate at 3 × 105 cells/well (Greiner Bio-One). After an overnight incubation, cells were washed and incubated in LPDS media (Dulbecco's phosphate-buffered saline (Invitrogen), 0.1% sodium pyruvate (Invitrogen), and 5% lipoprotein-deficient serum (Biomedical Technologies)). Following 24-h incubation, media were replaced by conditioned media containing PCSK9 or its mutants/deletants at a final concentration of 1.2 μg/ml. After 4 h of incubation at 37 °C, cells were lysed in 1× radioimmune precipitation assay buffer (RIPA: 150 mm NaCl, 50 mm Tris-HCl (pH 8.0)) containing 1% Nonidet P-40, 0.5% sodium deoxycholate, and 0.1% SDS supplemented with 1× complete protease inhibitor mixture (Roche Applied Science)) and then analyzed.
Western Blot Analyses
Media were recovered 48 h post-transfection, and the cells were lysed in 1× radioimmune precipitation assay buffer. Proteins in the cell lysates and media were resolved by 10% Tris-glycine SDS-PAGE. The gels were blotted onto PVDF (PerkinElmer Life Sciences) membranes (GE Healthcare), blocked for 1 h in TBS-T (50 mm Tris-HCl (pH 7.5), 150 mm NaCl, 0.1% Tween 20) containing 5% nonfat milk, and immunoblotted with a homemade polyclonal human PCSK9 antibody (1:1000) (13), human LDLR antibody (1:1000, R&D Systems), β-actin (1:5000; Sigma), and mAb V5-HRP (1:5000, Sigma). Appropriate horseradish peroxidase-conjugated secondary antibodies (1:10000, Sigma) were used for detection with enhanced chemiluminescence using the ECL Plus kit (GE Healthcare). Quantitation of protein bands was obtained using ImageJ software.
FACS Analysis
HepG2/shPCSK9 cells were incubated at 37 °C for the indicated times (0, 30, 60 min) with various conditioned media containing WT PCSK9, single-point mutants, or module deletants. The cells were then washed three times with solution A (calcium/magnesium-free Dulbecco's PBS (Invitrogen) containing 0.5% bovine serum albumin (Sigma) and 1g/liter glucose). The cells were then incubated for 10 min at room temperature with 1× Versene solution (Invitrogen) followed by the addition of 5 ml of solution A. The cells were then incubated for 40 min in solution A containing a human LDLR mAb-C7 (1:100, Santa Cruz Biotechnology). Following washes, the cells were then incubated for 20 min in solution A containing a secondary antibody (Alexa Fluor 647 donkey anti-mouse antibody, 1:250, Molecular Probes). Following suspension in PBS containing 0.2% of propidium iodide, the cells were analyzed by FACS for both propidium iodide (dead cells) and LDLR in live cells with Alexa Fluor 647 using the FACS Becton Dickinson LSR (BD Biosciences).
Immunocytochemistry
Cells were washed three times with PBS, fixed with a solution of 4% paraformaldehyde, 4% sucrose in PBS for 15 min, and permeabilized with a solution of 25% BSA, 10% fetal bovine serum, 0.3 m glycine, and 0.1% Tween/PBS for 10 min. The cells were then incubated for 30 min with 5% BSA (Fraction V, Sigma), followed by an overnight incubation at 4 °C with selected antibodies (1:100 goat polyclonal anti-hLDLR, R&D Systems; 1:500 mouse mAb-V5, Invitrogen; 1:200 human TGN46, Serotech; 1:200 human protein disulfide isomerase, Santa Cruz Biotechnology). The cells were then incubated for 60 min with the corresponding Alexa Fluor-conjugated secondary antibodies (Molecular Probes) and mounted in ProLong Gold antifade reagent (Molecular Probes, Invitrogen). Immunofluorescence analyses were performed with a Zeiss LSM 710 confocal microscope coupled with a Nikon Eclipse TE2000-U laser scanning microscope with 405-, 488-, 543-, and 633-nm laser lines. Images were processed with Zen 2009 software. For internalization experiments, HepG2/shPCSK9 cells were preincubated for 15 min at room temperature followed by 30 min of incubation with conditioned media at room temperature, and then transferred to 37 °C for 0, 60, or 180 min before preparation for immunofluorescence analysis as described above.
RESULTS
Expression, Zymogen Processing, and Secretion of CHRD Module Deletants
The reported crystal structures of PCSK9 revealed that its C-terminal CHRD contains three distinct Cys/His-rich modules named M1, M2, and M3 (7–9) (supplemental Fig. S1). The CHRD was shown to be critical for targeting the PCSK9LDLR complex to endosomes/lysosomes for degradation (13, 29). Thus, it was of interest to define the contribution of each module in this process and whether they could equally affect the extracellular (33) and intracellular (14) pathways of PCSK9-induced LDLR degradation. Accordingly, six C-terminally V5-tagged CHRD module deletants were generated by mutagenesis, resulting in human PCSK9 forms lacking only one (ΔM1, ΔM2, or ΔM3) or two (ΔM12, ΔM23, or ΔM13) modules (Fig. 1A). To analyze the fate of each deletant and its ability to fold and be secreted, among other cell lines, we first selected HEK293 cells, which are efficiently transfected, allowing the collection of media with enough material for multiple extracellular incubations. Accordingly, HEK293 (or HepG2/shPCSK9) cells were transiently transfected with cDNAs coding for these constructs. Cell lysates and media were then analyzed by Western blot analysis using mAb-V5. The data show that, similar to WT PCSK9 (1, 5) and its construct lacking the CHRD (L455X) (31), each CHRD module deletant is synthesized as a zymogen (pPCSK9), which is cleaved into a mature form, similar to WT PCSK9 (Fig. 1B, cell lysates, left panel). The CHRD only shows one protein product because it lacks both the prosegment and catalytic domain of PCSK9. However, only PCSK9, L455X, and CHRD and PCSK9 ΔM23, ΔM12, and ΔM2 are secreted (Fig. 1B, media, right panel). This suggested that loss of the M2 domain does not negatively affect folding and/or secretion (Fig. 1B, media, right panel). In contrast, in the presence of M2, the loss of M1, M3, or both (ΔM1, ΔM3, ΔM13) prevents secretion into the medium (Fig. 1B, media, right panel) but not zymogen cleavage (A, cells, left panel). In agreement, immunocytochemistry (supplemental Fig. S2) confirmed that ΔM1, ΔM3, and ΔM13 are retained in the ER and colocalize with the ER marker protein disulfide isomerase. In contrast, the secreted ΔM2, ΔM12, and ΔM23 colocalize with the trans Golgi network marker protein TGN46. We also noted that all secreted constructs containing the catalytic domain exhibit small amounts of the product of furin-cleavage at Arg218, resulting in the loss of the N-terminal residues 152–218 (ΔN218) (34, 35).
FIGURE 1.

Expression, autocleavage, and secretion of CHRD module deletants. A, schematic representation of V5-tagged WT human PCSK9, PCSK9 domain deletants (L455X, lacking CHRD, and CHRD, lacking the prosegment (PRO) and catalytic domain but retaining the hinge region (10)), and PCSK9 CHRD module deletants (ΔM1, ΔM2, ΔM3, ΔM12, ΔM23, and ΔM13). SP: Signal Peptide. B, HEK293 cells were transiently transfected with cDNAs encoding the above constructs. 48 h post-transfection, proteins in cell lysates and media were resolved by SDS-PAGE and analyzed by Western blot analysis using mAb-V5. The migration positions of reference molecular weight proteins, as well as those of the precursor and mature PCSK9 products are emphasized. The secreted CHRD module deletants are labeled with a star. Ctl, control.
LDLR Degradation by Overexpressed PCSK9 and Its Domain Deletants
Because PCSK9 is mostly expressed in hepatocytes (1, 24) it was pertinent to study its ability to degrade LDLR in these cells. We previously reported the activity of PCSK9 on LDLR in the human hepatocyte-derived HepG2 cell line (5, 31). Furthermore, because of the endogenous expression of PCSK9 in naïve HepG2 cells, we engineered cells lacking its expression by stable shRNA silencing. The resultant HepG2/shPCSK9 cells showed a more robust activity of added PCSK9 (14). Accordingly, we used these cells to further study the various PCSK9 constructs. Western blot analysis of the expression of endogenous PCSK9 in HepG2 cells expressing non-functional shRNA (Control) versus HepG2/shPCSK9 cells revealed that in the latter, the total protein levels (cells and media) of endogenous PCSK9 are reduced by > 90% (Fig. 2A).
FIGURE 2.

Effect of intracellular expression of CHRD module deletants on total LDLR levels in HepG2/shPCSK9 cells. Western blot analysis of total LDLR in HepG2/shPCSK9 cells transiently transfected with either an empty vector control, WT PCSK9, L455X, CHRD, and CHRD module deletants (ΔM23, ΔM12, ΔM2, ΔM3, ΔM1, and ΔM13). A, intracellular and secretion levels of endogenous PCSK9 in HepG2 cells expressing non-target shRNA (Control) compared with HepG2/shPCSK9 cells. The migration positions of proPCSK9 and PCSK9 forms are emphasized. IB, immunoblot. B, total LDLR reducing activity of WT PCSK9, L455X, CHRD, and CHRD domain deletants compared with cells expressing an empty vector control (upper panels). The levels of cellular and secreted PCSK9 forms are shown (center panels) as well as those of the β-actin cellular loading controls (lower panels). Endogenous LDLR was detected using a polyclonal antibody recognizing its extracellular domain, and all PCSK9 constructs, except the control empty vector one, were detected with a V5 HRP-conjugated mAb. C, total LDLR levels normalized to β-actin. As negative controls we used either L455X (a construct lacking the CHRD) or the secreted CHRD domain, which had no effect on the LDLR (31). The black bars emphasize the secreted CHRD module deletants. These data are representative of at least three independent experiments. D, intracellular degradation of LDLR by PCSK9 and PCSK9 ΔM2 occurs in acidic compartment(s) and is blocked upon 7h incubation of HepG2/shPCSK9 cells by 10 mm NH4Cl but not by 50 μm acetyl-Leu-Leu-norleucinal (ALLN).
We next transiently transfected cDNAs coding for PCSK9 and its various V5-tagged deletion constructs in HepG2/shPCSK9 cells and analyzed their intracellular production and effects on the levels of LDLR 48 h post-transfection (Fig. 2B). All the data obtained with HepG2/shPCSK9 cells were confirmed in naïve HepG2 cells (supplemental Fig. S3A). The expression and intracellular processing of PCSK9 and its deletants were very similar to those observed in HEK293 cells (Fig. 1B, cells, left panel). Western blot analysis of total LDLR in these cells (Figs. 2B, upper panel, and C) showed that only PCSK9, its GOF D374Y mutant, and PCSK9 ΔM2 effectively enhance the degradation of endogenous LDLR in these HepG2/shPCSK9 cells. Furthermore, WT PCSK9 and its ΔM2 deletant are equipotent in this respect (Fig. 2C). Surprisingly, even though PCSK9 ΔM23 and ΔΜ12 are secreted (Fig. 1B), they do not enhance LDLR degradation, similar to the PCSK9 L455X and the CHRD (Fig. 3C). In accordance with the observed absence of secretion of the module deletants PCSK9 ΔM13, ΔM3, and ΔM1 (Fig. 1B, media, right panel), these constructs are completely inactive on the LDLR (Fig. 2, B and C).
FIGURE 3.

Lack of modulation of LDLR levels following extracellular incubation of HepG2/shPCSK9 cells with CHRD module deletants. A, ELISA-V5 assay of secreted PCSK9, L455X, CHRD, D374Y, and CHRD deletants (ΔM2, ΔM12, and ΔM23) from transiently transfected HEK293 cells. B and C, HepG2/shPCSK9 cells were incubated with equal amounts of PCSK9 constructs (1.2 μg/ml) for 4 h at 37 °C. Cells were then lysed, and total LDLR levels were analyzed by Western blot analysis (B) and normalized to β-actin (C). Total LDLR was detected using a polyclonal antibody recognizing its extracellular domain, and all PCSK9 constructs were detected with a V5 HRP-conjugated antibody. D, HepG2/shPCSK9 cells were incubated for 0, 2, and 4 h at 37 °C with equal amounts (1.2 μg/ml) of secreted PCSK9 constructs, including ΔM2. Cells were detached as described under “Experimental Procedures,” and the levels of cell surface LDLR were measured by FACS analysis. As negative controls we used either a construct lacking the CHRD (L455X) or that expressing only the CHRD, both of which had no effect on LDLR levels, similar to the PCSK9 ΔM2 construct. These data are representative of at least three independent experiments. Only PCSK9 and its D374Y mutant significantly (p < 0.001) enhance the degradation of the LDLR.
The similar activity of PCSK9 and its M2 deletant begged the question of whether they both act in a similar fashion to induce LDLR degradation in acidic endosomal/lysosomal compartments. Similar to what was previously observed for WT PCSK9 in HEK293 cells (5), incubation of HepG2/shPCSK9 cells with 10 mm of the alkalinizing agent NH4Cl completely abrogated the effects of both PCSK9 and its ΔM2 deletant (Fig. 2D), as also observed in control cells. Note that the proteasome inhibitor acetyl-Leu-Leu-norleucinal does not significantly affect the ability of either PCSK9 or PCSK9 ΔM2 to induce the degradation of LDLR. Thus, we can conclude that similar to WT PCSK9, the PCSK9 ΔM2 construct also enhances the degradation of the PCSK9LDLR complex in acidic compartment(s).
LDLR Degradation in HepG2/shPCSK9 Cells by Extracellular PCSK9 and its Domain Deletants
We next proceeded to gauge the effects of PCSK9 and its domain deletants on the extracellular pathway of LDLR degradation. Accordingly, we concentrated approximately ten times the media of HEK293 cells obtained 72 h post-transfection with each cDNA construct. The concentration of each V5-tagged protein in these media was assessed by an ELISA-V5 (Fig. 3A). As expected, no protein is secreted from the ΔM1, ΔM3, and ΔM13 constructs. We also observed that the D374Y mutant and the deletants analyzed are all less secreted than WT PCSK9, with ΔM23 and ΔM12 being the least (five times less) secreted ones (Fig. 3A, Ratio column).
All protein constructs were adjusted to a final 1.5 μg/ml concentration in LPDS media and then incubated for 4 h at 37 °C with HepG2/shPCSK9 cells that were preincubated for 24 h in LPDS media to stimulate LDLR expression (5). Unexpectedly, Western blot analysis of total LDLR revealed that only WT PCSK9 and its GOF D374Y are active in this assay. These data confirm the observed inactivity of extracellular L455X and CHRD on LDLR (31) and extends this observation to constructs lacking M23 and M12 modules, expected from their inactivity in the intracellular pathway (Fig. 2, B and C). However, in contrast to the intracellular expression of PCSK9 ΔM2 (Fig. 2, B and C), its extracellular incubation with HepG2/shPCSK9 cells had no significant effect on LDLR levels (Fig. 3, B and C). This unexpected observation was repeated many times and further confirmed in naïve HepG2 cells (supplemental Fig. S3B) as well as by FACS analysis of cell surface LDLR following 0, 2-h, and 4-h incubations (Fig. 3D).
To test whether the lack of activity observed upon incubation of cells with 1.5 μg/ml may be due to a lower intrinsic activity that may be detectable at higher concentrations, HepG2/shPCSK9 cells were incubated for 4 h at 37 °C with increasing concentrations (0, 0.5, 1, 3, and 5 μg/ml) of either PCSK9 or its ΔM2 deletant (Fig. 4A). The data show that 5 μg/ml PCSK9 can reduce total LDLR levels by ∼60% but that PCSK9 ΔM2 is still completely inactive at the same concentration. Finally, to eliminate the possibility that post-translational modifications in hepatocytes would be different from HEK293 cells, we also confirmed the loss of extracellular function of PCSK9 ΔM2, even when it is produced in and tested on HepG2/shPCSK9 cells (Fig. 4B). Thus, we can conclude that the M2 domain is critical for the activity of PCSK9 on LDLR in the extracellular pathway but not in the intracellular one.
FIGURE 4.
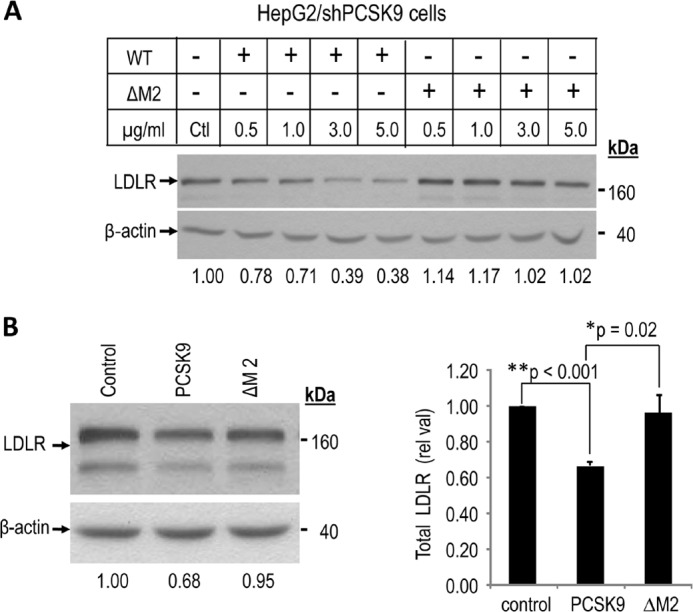
Loss of extracellular degradation activity on LDLR by PCSK9 ΔM2 produced in and/or tested on HepG2/shPCSK9 cells. Wild type PCSK9 and PCSK9 ΔM2 obtained from HEK293 or HepG2/shPCSK9 cells incubated for 4 h at (A) increasing concentrations (0 [control, Ctl], 0.5, 1.0, 3.0, and 5.0 μg/ml, quantified by ELISA assays) with HepG2/shPCSK9 cells, or (B) 1.5 μg/ml (quantified by ELISA assays) with HepG2/shPCSK9 cells. Cells were then lysed and total LDLR was analyzed by Western blot. Total LDLR was detected using a polyclonal antibody recognizing its extracellular domain and its levels were normalized to β-actin cellular loading controls. These data are representative of three independent experiments. Statistical values were estimated by Student's t-test and considered significant when p values are < 0.05. The data show that PCSK9 significantly reduces LDLR (**, p < 0.001) and that the LOF of PCSK9 ΔM2 versus PCSK9 is significant with *, p = 0.02, but not versus control, supporting its complete LOF.
Internalization of Extracellular PCSK9, ΔM2, and L455X in HepG2/shPCSK9 Cells
In view of the inactivity of either PCSK9 L455X or PCSK9 ΔM2 on LDLR, it was of interest to define the critical step that these deletants cannot achieve and that is required to enhance the degradation of the PCSK9LDLR complex. To avoid degradation in acidic compartments, HepG2/shPCSK9 cells were preincubated overnight with 5 mm NH4Cl and maintained in these conditions until cell fixation. To slow down cellular metabolism, cells were placed for 15 min at room temperature. To allow cell surface binding, HepG2/shPCSK9 cells were incubated for 30 min with each of the three constructs. Subsequently, incubations were shifted to 37 °C for 0, 60, or 180 min to follow the possible cellular internalization of the proteins by confocal microscopy under permeabilizing conditions (31).
As expected, analysis of confocal microscopy images showed that PCSK9 colocalized with cell surface LDLR and, after 60 and 180 min, was internalized into the cell with the LDLR (Fig. 5A). Confocal images revealed that at t = 0 min, L455X (supplemental Fig. S4) and the module deletant ΔM2 (Fig. 5B) also colocalized with the LDLR, indicative of their binding to this receptor. Subsequently, at t = 60 and 180 min, both deletant proteins were also found to increasingly colocalize with intracellular LDLR, suggesting that their internalization also occurred (Fig. 5B and supplemental Fig. S4).
FIGURE 5.

Internalization of PCSK9 ΔM2. HepG2/shPCSK9 cells were incubated at the indicated times with HEK293 media containing 1.5 μg/ml of PCSK9 (A) and PCSK9 ΔM2 (B), both V5-tagged. After 0, 60, or 180 min of incubation, cells were prepared for immunocytochemistry under permeabilizing conditions, and the internalization of V5-tagged proteins was analyzed by confocal microscopy. Scale bar = 10 μm.
Role of the Basic Residues Containing Loops within the Hinge Region and M2
The hinge region of PCSK9, linking the catalytic subunit to the CHRD, is not a disordered sequence as it is visible in all crystal structures and contains an exposed loop, possibly representing a binding region to a partner protein. The latter could regulate the PCSK9-induced degradation of LDLR. Alternatively, the hinge region could be necessary for the correct folding of PCSK9 and the spatial positioning of the CHRD versus the rest of the molecule to optimize its function. Indeed, we showed previously that a natural mutation of the basic residue at Arg-434 present within an exposed loop within the hinge region (supplemental Fig. S5, A and B) into Trp (R434W) results in an ∼70% reduction in the extracellular pathway function of PCSK9 (10). In this report, we extend this observation and show that following transfection of HepG2/shPCSK9 cells, the R434W mutant lacks intracellular activity on the LDLR (Fig. 6B). These data point to the critical importance of the hinge region in regulating both the intracellular and extracellular activity of PCSK9 on the LDLR. We thus analyzed the effects of constructs lacking the hinge region (PCSK9 ΔH, ΔH ΔM2, and ΔH CHRD; supplemental Fig. S5) on the overall intracellular activity of PCSK9. Overexpression of these V5-tagged constructs (supplemental Fig. S6) in HEK293 cells revealed that removal of the hinge region does not affect the expression or secretion of the CHRD. Similarly, zymogen processing of PCSK9 and its ΔM2 deletant are not affected, but the lack of the hinge region results in a significantly reduced secretion of PCSK9 and less so of the ΔM2 construct (Fig. 6A). Lack of the hinge region in the latter proteins results in a complete loss of their intracellular activity on LDLR (Fig. 6B), supporting the notion that the hinge region is critical to maintain an active PCSK9 conformation.
FIGURE 6.

Impact of basic residues in the hinge region and M2 module on the activity of PCSK9. Hinge region: A, HepG2/shPCSK9 cells were transiently transfected with various V5-tagged constructs containing or lacking the hinge region of PCSK9 (aa 405–420) and/or lacking the M2 module. At 48 h post-transfection, the proteins in the media and cell lysates were separated by SDS-PAGE and analyzed by Western blot using mAb-V5. The migration positions of proPCSK9 (pPCSK9) and mature PCSK9 are emphasized. B, the total LDLR-degrading activity of each construct was analyzed 48 h post transfection by Western blot of cell lysates using a polyclonal LDLR antibody, and the levels of the ∼160 kDa were normalized to β-actin cellular loading controls. The ∼110 kDa LDLR represents the immature form of the receptor. The black triangles point to the abrogation of the intracellular activity in WT and ΔM2 constructs upon removal of the hinge region. M2 module: C, HepG2/shPCSK9 cells were transiently transfected with various V5-tagged constructs expressing PCSK9 ΔM2, WT PCSK9 or some of its single point mutant (GOF D374Y) and the charge modified double point mutants (HQ553,554RR; HQ553,554AA; HQ553,554EE). The total LDLR-degrading activity of each construct was analyzed in B. D, cell surface LDLR levels were evaluated by FACS analysis following 4 h incubation of HepG2/shPCSK9 cells with most of the constructs in C. The star emphasizes the GOF of the HQ553,554RR mutant in this assay. These data are representative of at least three independent experiments. Statistical significance is emphasized (*, p < 0.05).
A survey of known PCSK9 natural mutations modifying the charge around basic residues within an exposed loop of the CHRD M2 module (residues 530–605, supplemental Fig. S5C) led us to concentrate on the only reported natural GOF H553R and LOF Q554E (36) mutants found within the sequence RVHCH553QQGH. Accordingly, we doubly mutated residues 553 and 554 to Arg, Ala, or Glu (HQ-RR, HQ-AA, and HQ-EE). The resulting double mutants did not affect the zymogen processing or secretion of PCSK9 (Fig. S7) or its capacity to enhance the degradation of the LDLR via the intracellular pathway (Fig. 6C). However, we note that the HQ-RR mutant results in a 1.7-fold enhanced extracellular activity of PCSK9, as measured by quantitative FACS analysis of cell surface LDLR (Fig. 6D). This result agrees with the observation that the H533R is a GOF but also circumscribes this enhanced activity to the extracellular pathway and not the intracellular one. We conclude that the critical importance of the M2 domain in the extracellular and intracellular pathways can be enhanced by specific mutations that increase the basicity of this exposed loop, such as the selected GOF HQ-RR mutation. This finding further emphasizes the major role played by the M2 module, and specific exposed residues within, in regulating the activity of PCSK9.
DISCUSSION
Hypercholesterolemia, which is characterized by elevated blood LDLc, is one of the main risk factors for cardiovascular disease worldwide (37) and is observed in > 20% of the Western population. Among existing treatments, “statins,” which target HMG-CoA reductase, are by far the most efficient drugs. However, at least 15% of patients either do not respond to statin treatment, encounter serious side effects (38), or do not reach ideal target levels of circulating LDLc to control their dyslipidemia. Consequently, new drugs to treat elevated LDLc and tools to identify responders versus non-responders to LDLc-lowering treatments are needed urgently.
Studies performed over the past 9 years have indicated that the proprotein convertase PCSK9 plays a central role in regulating blood LDLc levels (2, 21). Indeed, the mature, active form of PCSK9 binds to the LDLR at the external face of the plasma membrane to enhance its degradation in endosomes/lysosomes, thus preventing internalization of circulating LDLc by liver hepatocytes. Most notably, PCSK9 has been shown to be an efficient therapeutic target in humans (2, 21, 39). Indeed, injections of PCSK9-silencing siRNAs or anti-PCSK9 mAbs, currently in phase II and III clinical trials, have been shown to efficiently reduce LDLc in the blood circulation of patients (2, 28).
Natural mutations within the hinge region and CHRD were reported to modify the function of PCSK9 and, thus, circulating LDLc levels. For example, the R434W hinge mutation results in a LOF of PCSK9 (10). The five significant LOF mutations in the CHRD are in the M1 (S462P), M2 (Q554E), and M3 (P616L, S668R, and C679X) modules, and the reported eight GOF mutations are in the M1 (R469W, E482G, R496W, A514T, F515L, and A522T), M2 (H553R), and M3 (V624M) modules. Even though the hinge region and M2 module exhibit the highest prevalence of structural mobility, the latter possibly because of the lack of disulfide stabilization (7), they are the structural units with the least reported natural mutations, with only two juxtaposed mutations (GOF H553R and LOF Q554E) within the exposed loop of the M2 module (supplemental Fig. S5).
On the basis of the above observations, the requirement of the CHRD for the activity of PCSK9 (13, 29) and the demonstration of the presence of intracellular and extracellular pathways by which PCSK9 enhances the degradation of the LDLR (14, 15), we aimed to define the contribution of the hinge region and the three CHRD modules in the regulation of the PCSK9 activity.
Accordingly, we generated six CHRD module deletants (ΔM1, ΔM2, ΔM3, ΔM12, ΔM23, or ΔM13) (Fig. 1A). We next evaluated their role in the PCSK9 intracellular processing, secretion, subcellular localization, and the two LDLR degradation pathways. Interestingly, intracellular expression of the CHRD domain module deletants in HEK293, HepG2/shPCSK9 (cells in Figs. 1B and 2B) or HepG2 cells (not shown) revealed that the lack of one or two modules in the CHRD domain did not affect the ability of proPCSK9 to undergo autocatalytic processing into mature PCSK9. Surprisingly, among all six CHRD module deletants, only PCSK9 ΔM2, ΔM12, and ΔM23 were secreted (media in Fig. 1A), indicating that for secretion, M2 was the only dispensable module.
Our previous data showed that except for the GOF D374Y, cellular overexpression of WT PCSK9 mostly results in lowering LDLR levels by the intracellular pathway (14). This is due to the relatively low secretion levels of PCSK9 from transfected cells (∼0.1 μg/ml), versus the ten times higher concentration necessary to effectively enhance LDLR degradation from the extracellular pathway (31). We conclude that the intracellular overexpression of PCSK9 and its ΔM2 deletant exhibit a comparable ability to enhance the degradation of the LDLR, likely by the intracellular pathway.
We next assessed the extracellular activity of the three secreted CHRD module deletants (Fig. 3, B and C, and supplemental Fig. S3). Unexpectedly, none of them showed any activity on the LDLR, even the ΔM2 at a concentration up to 5 μg/ml, which results in an ∼60% reduction of LDLR levels by WT PCSK9 (Fig. 4A). This conclusion was further confirmed by FACS analysis, which also revealed that similar incubations resulted in no detectable reduction of cell surface LDLR levels (Fig. 3D). We analyzed the cellular association of each construct by incubating them with HepG2/shPCSK9 cells followed by extensive washes. Western blot analysis of cell lysates demonstrated that all of them bound to cells (data not shown). Thus, the lack of activity of the PCSK9 L455X and CHRD module deletants is not due to their inability to bind and likely internalize into cells.
To rationalize the inactivity of the extracellular ΔM2 protein, immunocytochemistry was used to analyze its fate. Confocal images showed that PCSK9, L455X, and even the ΔM2 deletant colocalize with LDLR at all time points, revealing their cell surface binding and internalization (Fig. 5 and supplemental Fig. S4). However, these steps were not accomplished with the same efficiency, as evidenced by the higher binding and faster internalization of WT PCSK9 versus L455X or ΔM2. On the basis of these observations, it is clear that the inability of PCSK9 ΔM2 to enhance LDLR degradation does not reside in its lack of LDLR binding or its cointernalization with the LDLR, suggesting that the M2 module carries structural requirements to achieve the critical and late LDLR sorting and degradation steps within endosomes/lysosomes.
The hinge region is tightly linked to the CHRD domain, even if the hinge is only 17 amino acids long, its sequence seems to be important to maintain the full activity of WT PCSK9. Notably, the only natural mutation (R434W) reported (10) in the hinge region is a loss of function. This evidence led us to more closely examine the role of the hinge region in the folding, secretion, and, ultimately, in PCSK9 activity. On the basis of PCSK9 crystal structures, we selected to delete the exposed loop (residues 422–439) corresponding to the hinge region (supplemental Fig. S5). Interestingly, although autocatalytically processed, PCSK9 lacking amino acids 422–439 is very poorly secreted (PCSK9 ΔH, supplemental Fig. S6A). In contrast, removal of the same sequence from either the N terminus of the CHRD or the ΔM2 construct (PCSK9 ΔH ΔM2) had much less effect. Overexpression of either construct in HepG2/shPCSK9 cells revealed that none of these constructs is active on the LDLR (Fig. 6B). Thus, although the PCSK9 ΔM2 is active intracellularly, the loss of the hinge region abrogates its activity. Hence, the integrity of the hinge region is critical for the intracellular activity of PCSK9 (Fig. 6B). This is likely due to its structural requirement as a linker providing flexibility to the CHRD. Amazingly, replacement of only one basic residue (Arg-434) by a Trp within the hinge region results in an almost complete intracellular loss of function (R434W in Fig. 6B) and a reduction by ∼70% of its extracellular activity on LDLR (10).
The gain of a basic residue at neutral pH in the H553R mutant within the M2 domain does not affect the intracellular activity (data not shown) and, yet, results in a GOF (36), as also observed for the double mutant HQ554-RR554 (Fig. 6D). Interestingly, the intracellular activity is either not affected when HQ554 is replaced by neutral residues (HQ554-AA554) or decreased if replaced by acidic residues (HQ554-EE554; Fig. 6C). The only natural mutations reported in the M2 module (36) are in the exposed His-rich HCHQQGH557 loop (supplemental Fig. S5C), resulting in either an increased basicity at neutral pH (GOF, H553R) or increased acidity (relatively weak LOF, Q554E). We thus conclude that the whole M2 module is only critical for the extracellular activity of PCSK9 and that natural mutations within it affect this pathway. This is relevant, because neutralizing mAb injections are being presently tested in phases II and III of various clinical trials to reduce hyper-cholesterolemia by inhibiting the function of PCSK9 on LDLR (2, 28). All these mAbs target the catalytic subunit of PCSK9 and prevent its extracellular binding to cell surface LDLR. The only attempts to obtain CHRD-directed interfering mAbs against the extracellular activity of PCSK9 targeted the M3 domain, achieving a maximum of 50% reduction in PCSK9 activity (40). According this study, targeting the M2 module of the CHRD could represent a novel strategy to inhibit the extracellular activity of PCSK9.
This concept of increased basicity favoring the CHRD contribution to the extracellular activity of PCSK9 has also been proposed recently (41). In agreement with the present results, the authors show that replacements of up to 7 His, mostly located in the M2 module, do not affect the intracellular activity of PCSK9. Unfortunately, the authors have not measured the extracellular activity of their mutants, which according to our study should have been the predominantly affected pathway. Furthermore, the authors suggest that replacement of the CHRD by an unrelated, similarly charged basic domain, results in a comparable activity to WT PCSK9 (41). Although the total basicity of the CHRD may well be important, our data further zoom in on a specific site within the M2 domain that is a hot spot for natural mutations. We show that, indeed, the higher basicity of the HQ554-RR554 mutant within an exposed loop results in a robust ∼2-fold higher extracellular activity on LDLR (Fig. 6D).
A recent report also suggested that the M2 module is dispensable for the intracellular activity of PCSK9 (42). However, the authors did not measure the extracellular function of their PCSK9 ΔM2 construct, which is abrogated according to this work (Figs. 3 and 4).
The mechanism governing the critical role of the M2 module, and possibly its exposed HCHQQGH557 loop (supplemental Fig. S5), in regulating the extracellular activity of PCSK9 is not yet defined. One possibility would be that the M2 module interacts with a critical, as yet undefined, protein that would result in the efficient targeting of the PCSK9LDLR complex to lysosomes for degradation by endogenous hydrolases (13, 29). This would fit with the observation that an excess of CHRD could compete extracellularly with PCSK9 for LDLR degradation (42). Even though it was reported that in vitro high concentrations of the CHRD could bind the N-terminal R1-R7 repeat domain of the LDLR, the crystal structure of PCSK9 bound to the ectodomain (18) or full-length (PDB codes 3M0C and 3P5B) LDLR do not support this model, as the CHRD is far away in space from the β-barrel repeat domains of this receptor. Thus, the hepatocyte CHRD-binding protein remains to be identified.
In conclusion, the structural requirements of the hinge and M2 domains are not the same for the intracellular and extracellular pathways of PCSK9-induced LDLR degradation. Although the R434W mutation in the hinge region is absolutely deleterious for the intracellular activity of PCSK9, it reduces by ∼70% the extracellular one. In contrast, the loss of M2 is tolerated for the intracellular activity of PCSK9 but not for the extracellular one. This information should lead to the development of more targeted approaches, possibly against the M2 domain, to inhibit the extracellular function of PCSK9 and reduce circulating LDLc.
Acknowledgments
We thank all the members of the Seidah laboratory for helpful discussions and Brigitte Mary for efficacious editorial assistance.
This research was supported by Canadian Institute of Health Research Grants MOP-102741 and CTP 82946, a Bristol-Myers Squibb collaborative agreement grant, a Strauss Foundation grant, and Canada Chair no. 216684.

This article contains supplemental Figs. S1–S7 and Table S1.
- PC
- proprotein convertase
- GOF
- gain of function
- LDLc
- LDL cholesterol
- LDLR
- LDL receptor
- ER
- endoplasmic reticulum
- CHRD
- cysteine/histidine-rich domain.
REFERENCES
- 1. Seidah N. G., Benjannet S., Wickham L., Marcinkiewicz J., Jasmin S. B., Stifani S., Basak A., Prat A., Chretien M. (2003) The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1). Liver regeneration and neuronal differentiation. Proc. Natl. Acad. Sci. U.S.A. 100, 928–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Seidah N. G., Prat A. (2012) The biology and therapeutic targeting of the proprotein convertases. Nat. Rev. Drug Discov. 11, 367–383 [DOI] [PubMed] [Google Scholar]
- 3. Abifadel M., Varret M., Rabès J. P., Allard D., Ouguerram K., Devillers M., Cruaud C., Benjannet S., Wickham L., Erlich D., Derré A., Villéger L., Farnier M., Beucler I., Bruckert E., Chambaz J., Chanu B., Lecerf J. M., Luc G., Moulin P., Weissenbach J., Prat A., Krempf M., Junien C., Seidah N. G., Boileau C. (2003) Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 34, 154–156 [DOI] [PubMed] [Google Scholar]
- 4. Marduel M., Ouguerram K., Serre V., Bonnefont-Rousselot D., Marques-Pinheiro A., Berge K. E., Devillers M., Luc G., Lecerf J. M., Tosolini L., Erlich D., Peloso G. M., Stitziel N., Nitchke P., Jais J. P., Abifadel M., Kathiresan S., Leren T. P., Rabes J. P., Boileau C., Varret M. (2012) Description of a large family with autosomal dominant hypercholesterolemia associated with the APOE p.Leu167del mutation. Hum. Mutat., doi: 10.1002/humu.22215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Benjannet S., Rhainds D., Essalmani R., Mayne J., Wickham L., Jin W., Asselin M. C., Hamelin J., Varret M., Allard D., Trillard M., Abifadel M., Tebon A., Attie A. D., Rader D. J., Boileau C., Brissette L., Chretien M., Prat A., Seidah N. G. (2004) NARC-1/PCSK9 and its natural mutants: zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. J. Biol. Chem. 279, 48865–48875 [DOI] [PubMed] [Google Scholar]
- 6. Naureckiene S., Ma L., Sreekumar K., Purandare U., Lo C. F., Huang Y., Chiang L. W., Grenier J. M., Ozenberger B. A., Jacobsen J. S., Kennedy J. D., DiStefano P. S., Wood A., Bingham B. (2003) Functional characterization of Narc 1, a novel proteinase related to proteinase K. Arch. Biochem. Biophys. 420, 55–67 [DOI] [PubMed] [Google Scholar]
- 7. Cunningham D., Danley D. E., Geoghegan K. F., Griffor M. C., Hawkins J. L., Subashi T. A., Varghese A. H., Ammirati M. J., Culp J. S., Hoth L. R., Mansour M. N., McGrath K. M., Seddon A. P., Shenolikar S., Stutzman-Engwall K. J., Warren L. C., Xia D., Qiu X. (2007) Structural and biophysical studies of PCSK9 and its mutants linked to familial hypercholesterolemia. Nat. Struct. Mol. Biol. 14, 413–419 [DOI] [PubMed] [Google Scholar]
- 8. Piper D. E., Jackson S., Liu Q., Romanow W. G., Shetterly S., Thibault S. T., Shan B., Walker N. P. (2007) The crystal structure of PCSK9. A regulator of plasma LDL-cholesterol. Structure 15, 545–552 [DOI] [PubMed] [Google Scholar]
- 9. Hampton E. N., Knuth M. W., Li J., Harris J. L., Lesley S. A., Spraggon G. (2007) The self-inhibited structure of full-length PCSK9 at 1.9 A reveals structural homology with resistin within the C-terminal domain. Proc. Natl. Acad. Sci. U.S.A. 104, 14604–14609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dubuc G., Tremblay M., Paré G., Jacques H., Hamelin J., Benjannet S., Boulet L., Genest J., Bernier L., Seidah N. G., Davignon J. (2010) A new method for measurement of total plasma PCSK9. Clinical applications. J. Lipid Res. 51, 140–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Maxwell K. N., Breslow J. L. (2004) Adenoviral-mediated expression of Pcsk9 in mice results in a low-density lipoprotein receptor knockout phenotype. Proc. Natl. Acad. Sci. U.S.A. 101, 7100–7105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Park S. W., Moon Y. A., Horton J. D. (2004) Post-transcriptional regulation of low density lipoprotein receptor protein by proprotein convertase subtilisin/kexin type 9a in mouse liver. J. Biol. Chem. 279, 50630–50638 [DOI] [PubMed] [Google Scholar]
- 13. Nassoury N., Blasiole D. A., Tebon Oler A., Benjannet S., Hamelin J., Poupon V., McPherson P. S., Attie A. D., Prat A., Seidah N. G. (2007) The cellular trafficking of the secretory proprotein convertase PCSK9 and its dependence on the LDLR. Traffic 8, 718–732 [DOI] [PubMed] [Google Scholar]
- 14. Poirier S., Mayer G., Poupon V., McPherson P. S., Desjardins R., Ly K., Asselin M. C., Day R., Duclos F. J., Witmer M., Parker R., Prat A., Seidah N. G. (2009) Dissection of the endogenous cellular pathways of PCSK9-induced LDLR degradation. Evidence for an intracellular route. J. Biol. Chem. 284, 28856–28864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Holla Ø. L., Cameron J., Berge K. E., Ranheim T., Leren T. P. (2007) Degradation of the LDL receptors by PCSK9 is not mediated by a secreted protein acted upon by PCSK9 extracellularly. BMC Cell Biol. 8, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang D. W., Lagace T. A., Garuti R., Zhao Z., McDonald M., Horton J. D., Cohen J. C., Hobbs H. H. (2007) Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat a of low density lipoprotein receptor decreases receptor recycling and increases degradation. J. Biol. Chem. 282, 18602–18612 [DOI] [PubMed] [Google Scholar]
- 17. Kwon H. J., Lagace T. A., McNutt M. C., Horton J. D., Deisenhofer J. (2008) Molecular basis for LDL receptor recognition by PCSK9. Proc. Natl. Acad. Sci. U.S.A. 105, 1820–1825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lo Surdo P., Bottomley M. J., Calzetta A., Settembre E. C., Cirillo A., Pandit S., Ni Y. G., Hubbard B., Sitlani A., Carfí A. (2011) Mechanistic implications for LDL receptor degradation from the PCSK9/LDLR structure at neutral pH. EMBO Rep. 12, 1300–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. McNutt M. C., Lagace T. A., Horton J. D. (2007) Catalytic activity is not required for secreted PCSK9 to reduce low density lipoprotein receptors in HepG2 cells. J. Biol. Chem. 282, 20799–20803 [DOI] [PubMed] [Google Scholar]
- 20. Poirier S., Mayer G., Benjannet S., Bergeron E., Marcinkiewicz J., Nassoury N., Mayer H., Nimpf J., Prat A., Seidah N. G. (2008) The proprotein convertase PCSK9 induces the degradation of low density lipoprotein receptor (LDLR) and its closest family members VLDLR and ApoER2. J. Biol. Chem. 283, 2363–2372 [DOI] [PubMed] [Google Scholar]
- 21. Horton J. D., Cohen J. C., Hobbs H. H. (2009) PCSK9. A convertase that coordinates LDL catabolism. J. Lipid Res. 50, S172-S177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Seidah N. G. (2009) PCSK9 as a therapeutic target of dyslipidemia. Expert Opin. Ther. Targets 13, 19–28 [DOI] [PubMed] [Google Scholar]
- 23. Rashid S., Curtis D. E., Garuti R., Anderson N. N., Bashmakov Y., Ho Y. K., Hammer R. E., Moon Y. A., Horton J. D. (2005) Decreased plasma cholesterol and hypersensitivity to statins in mice lacking PCSK9. Proc. Natl. Acad. Sci. U.S.A. 102, 5374–5379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zaid A., Roubtsova A., Essalmani R., Marcinkiewicz J., Chamberland A., Hamelin J., Tremblay M., Jacques H., Jin W., Davignon J., Seidah N. G., Prat A. (2008) Proprotein convertase subtilisin/kexin type 9 (PCSK9). Hepatocyte-specific low-density lipoprotein receptor degradation and critical role in mouse liver regeneration. Hepatology 48, 646–654 [DOI] [PubMed] [Google Scholar]
- 25. Cohen J. C., Boerwinkle E., Mosley T. H., Jr., Hobbs H. H. (2006) Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N. Engl. J. Med. 354, 1264–1272 [DOI] [PubMed] [Google Scholar]
- 26. Denis M., Marcinkiewicz J., Zaid A., Gauthier D., Poirier S., Lazure C., Seidah N.G., Prat A. (2012) Gene inactivation of PCSK9 reduces atherosclerosis in mice. Circulation 125, 894–901 [DOI] [PubMed] [Google Scholar]
- 27. Davignon J., Dubuc G., Seidah N. G. (2010) The influence of PCSK9 polymorphisms on serum low-density lipoprotein cholesterol and risk of atherosclerosis. Curr. Atheroscler. Rep. 12, 308–315 [DOI] [PubMed] [Google Scholar]
- 28. Stein E. A., Gipe D., Bergeron J., Gaudet D., Weiss R., Dufour R., Wu R., Pordy R. (2012) Effect of a monoclonal antibody to PCSK9, REGN727/SAR236553, to reduce low-density lipoprotein cholesterol in patients with heterozygous familial hypercholesterolaemia on stable statin dose with or without ezetimibe therapy. A phase 2 randomised controlled trial. Lancet 380, 29–36 [DOI] [PubMed] [Google Scholar]
- 29. Zhang D. W., Garuti R., Tang W. J., Cohen J. C., Hobbs H. H. (2008) Structural requirements for PCSK9-mediated degradation of the low-density lipoprotein receptor. Proc. Natl. Acad. Sci. U.S.A. 105, 13045–13050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Strøm T. B., Holla Ø. L., Tveten K., Cameron J., Berge K. E., Leren T. P. (2010) Disrupted recycling of the low density lipoprotein receptor by PCSK9 is not mediated by residues of the cytoplasmic domain. Mol. Genet. Metab. 101, 76–80 [DOI] [PubMed] [Google Scholar]
- 31. Benjannet S., Saavedra Y. G., Hamelin J., Asselin M. C., Essalmani R., Pasquato A., Lemaire P., Duke G., Miao B., Duclos F., Parker R., Mayer G., Seidah N. G. (2010) Effects of the prosegment and pH on the activity of PCSK9. Evidence for additional processing events. J. Biol. Chem. 285, 40965–40978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Holla Ø. L., Laerdahl J. K., Strøm T. B., Tveten K., Cameron J., Berge K. E., Leren T. P. (2011) Removal of acidic residues of the prodomain of PCSK9 increases its activity towards the LDL receptor. Biochem. Biophys. Res. Commun. 406, 234–238 [DOI] [PubMed] [Google Scholar]
- 33. Cameron J., Holla Ø. L., Ranheim T., Kulseth M. A., Berge K. E., Leren T. P. (2006) Effect of mutations in the PCSK9 gene on the cell surface LDL receptors. Hum. Mol. Genet. 15, 1551–1558 [DOI] [PubMed] [Google Scholar]
- 34. Benjannet S., Rhainds D., Hamelin J., Nassoury N., Seidah N. G. (2006) The proprotein convertase PCSK9 is inactivated by furin and/or PC5/6A. Functional consequences of natural mutations and post-translational modifications. J. Biol. Chem. 281, 30561–30572 [DOI] [PubMed] [Google Scholar]
- 35. Essalmani R., Susan-Resiga D., Chamberland A., Abifadel M., Creemers J. W., Boileau C., Seidah N. G., Prat A. (2011) In vivo evidence that furin from hepatocytes inactivates PCSK9. J. Biol. Chem. 286, 4257–4263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kotowski I. K., Pertsemlidis A., Luke A., Cooper R. S., Vega G. L., Cohen J. C., Hobbs H. H. (2006) A spectrum of PCSK9 Alleles contributes to plasma levels of low-density lipoprotein cholesterol. Am. J. Hum. Genet. 78, 410–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Davignon J., Dufour R. (2007) Primary Hyperlipidemias: An Atlas of Investigation and Diagnosis, pp. 1–128, Clinical Publishing, Oxford [Google Scholar]
- 38. Sikka P., Kapoor S., Bindra V. K., Sharma M., Vishwakarma P., Saxena K. K. (2011) Statin intolerance. Now a solved problem. J. Postgrad. Med. 57, 321–328 [DOI] [PubMed] [Google Scholar]
- 39. Cariou B., Le May C., Costet P. (2011) Clinical aspects of PCSK9. Atherosclerosis 216, 258–265 [DOI] [PubMed] [Google Scholar]
- 40. Ni Y. G., Condra J. H., Orsatti L., Shen X., Di Marco S., Pandit S., Bottomley M. J., Ruggeri L., Cummings R. T., Cubbon R. M., Santoro J. C., Ehrhardt A., Lewis D., Fisher T. S., Ha S., Njimoluh L., Wood D. D., Hammond H. A., Wisniewski D., Volpari C., Noto A., Lo Surdo P., Hubbard B., Carfí A., Sitlani A. (2010) A proprotein convertase subtilisin-like/kexin type 9 (PCSK9) C-terminal domain antibody antigen-binding fragment inhibits PCSK9 internalization and restores low density lipoprotein uptake. J. Biol. Chem. 285, 12882–12891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Holla Ø. L., Cameron J., Tveten K., Strøm T. B., Berge K. E., Laerdahl J. K., Leren T. P. (2011) Role of the C-terminal domain of PCSK9 in degradation of the LDL receptors. J. Lipid Res. 52, 1787–1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Du F., Hui Y., Zhang M., Linton M. F., Fazio S., Fan D. (2011) Novel domain interaction regulates secretion of proprotein convertase subtilisin/kexin type 9 (PCSK9) protein. J. Biol. Chem. 286, 43054–43061 [DOI] [PMC free article] [PubMed] [Google Scholar]