

Background: Agonist binds to an extracellular agonist-binding domain in AMPA receptors.
Results: Willardiines induce a range of cleft closure states in the agonist-binding domain of AMPA receptors.
Conclusion: The fraction of the agonist-binding domains in a closed cleft conformation correlates with the extent of activation.
Significance: The dynamics and extent of cleft closure in the agonist-binding domain control activation of AMPA receptors.
Keywords: Fluorescence Resonance Energy Transfer (FRET); Glutamate Receptors Ionotropic (AMPA, NMDA); Ion Channels; Ligand-binding Protein; Membrane Proteins; AMPA Receptors; Single-molecule FRET
Abstract
We have investigated the range of cleft closure conformational states that the agonist-binding domains of the α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptors occupy when bound to a series of willardiine derivatives using single-molecule FRET. These studies show that the agonist-binding domain exhibits varying degrees of dynamics when bound to the different willardiines with differing efficacies. The chlorowillardiine- and nitrowillardiine-bound form of the agonist-binding domain probes a narrower range of cleft closure states relative to the iodowillardiine bound form of the protein, with the antagonist (αS)-α-amino-3-[(4-carboxyphenyl)methyl]-3,4-dihydro-2,4-dioxo-1(2H)-pyrimidinepropanoic acid (UBP-282)-bound form exhibiting the widest range of cleft closure states. Additionally, the average cleft closure follows the order UBP-282 > iodowillardiine > chlorowillardiine > nitrowillardiine-bound forms of agonist-binding domain. These single-molecule FRET data, along with our previously reported data for the glutamate-bound forms of wild type and T686S mutant proteins, show that the mean currents under nondesensitizing conditions can be directly correlated to the fraction of the agonist-binding domains in the “closed” cleft conformation. These results indicate that channel opening in the AMPA receptors is controlled by both the ability of the agonist to induce cleft closure and the dynamics of the agonist-binding domain when bound to the agonist.
Introduction
Glutamate receptors mediate excitatory neurotransmission by forming cation selective transmembrane channels upon binding to the neurotransmitter glutamate (1–7). They are involved in learning and memory and are implicated in neurodegenerative disorders such as Huntington, Parkinson, and Alzheimer diseases and in neurodegeneration associated with stroke and amyotrophic lateral sclerosis (8–15). Glutamate receptors are classified into three subtypes: α-amino-5-methyl-3-hydroxy-4-isoxazole propionate (AMPA),3 N-methyl-d-aspartate (NMDA), and kainate receptors, based on their sequence, functionality, and pharmacological profiles. The recent full-length structure of the antagonist-bound form of the AMPA receptor shows that the receptor is a dimer of dimers, with each subunit being made up of modular segments of the N-terminal domain, agonist-binding domain, transmembrane segments, and intracellular C-terminal domain (16). This structure, along with FRET investigations of the full-length receptor, establishes that the isolated agonist-binding domain is a good model of the domain in the full-length receptor, thus validating its use in detailed structure and dynamics investigations (17–19).
There are currently numerous structures available for the agonist-binding domain of the AMPA receptor determined in complex with antagonists, as well as agonists of varying efficacy (20–25). Based on the x-ray structures, it was initially thought that the extent of cleft closure is the primary mechanism by which agonists mediate receptor activation, i.e., increased cleft closure leads to increased activation (23). However, there were several structures such as those of the glutamate-bound form of the T686A mutant and the structures of the AMPA bound form of the L650T mutant that do not follow this trend (22, 25–27). Ensemble FRET investigations with the wild type and L650T mutant were consistent with the x-ray structures, further validating these deviations from the cleft closure hypothesis (28, 29).
Recent single-molecule FRET (smFRET) investigations of wild type and T686S mutant receptors provide additional insight into the complete structural landscape in terms of cleft closure (30). These studies showed that both the most probable state and the average state probed by the T686S mutant showed a closed cleft conformation consistent with the x-ray structures. However, the T686S mutant protein also probed a wider range of cleft closure states that covered more open states than the glutamate bound wild type receptor. Thus, the probability that the T686S mutant was in the closed state was lower than that of the wild type protein when bound to glutamate, and this decrease in probability could lead to lower activation (27, 30). These smFRET investigations emphasized the need to study the overall cleft closure landscape and dynamics of the agonist-binding domain to obtain a better insight into the role of cleft closure in activation.
The smFRET results are also consistent with NMR investigations that show wide variation in the dynamics of the agonist-binding domain upon binding to different agonists. In particular, NMR studies investigating the methyl group dynamics indicate that at slower time scales, willardiines, which are partial agonists of the AMPA receptor, exhibit the largest flexibility in the agonist-binding domain relative to full agonists and antagonists (31, 32). Here we have performed smFRET investigations with substituted willardiines bound to the agonist-binding domain and compared the spectrum of states that the protein probes to the previously reported NMR-based dynamics and additionally correlated these results with activation of the receptor.
EXPERIMENTAL PROCEDURES
Purification and Labeling of the Agonist-binding Domain of GluA2 Subunit of the AMPA Receptor
The plasmid expressing the agonist-binding domain of the GluA2 subunit of AMPA receptors was provided by Dr. Eric Gouaux (Oregon Health and Science University). Mutations T394C and S652C were made by standard mutation reactions using Pfu Turbo DNA polymerase (Agilent Technologies). Plasmid expressing this double mutant was transformed, and the protein was expressed in Escherichia coli origami DE3 cells (EMD Chemicals) in LB broth, Miller (Fisher Scientific) containing ampicillin (Sigma Aldrich), kanamycin (Fisher Scientific), and tetracycline (Shelton Scientific) at concentrations of 50, 15, and 12.5 μg/ml, respectively. The protein expression was induced using isopropyl β-d-1-thiogalactopyranoside (Fisher Scientific) when cells reached an optical density of 0.8–0.9. After isopropyl β-d-1-thiogalactopyranoside addition, cells were grown at 20 °C for 24 h. The cells were harvested and lysed in 20 mm Tris buffer containing 150 mm NaCl, 1 mm glutamate, 5 mm MgSO4, 0.5 mm PMSF, 50 μg/ml lysozyme, 125 μg/ml sodium deoxycholate, 25 μg/ml DNaseI. The lysed cells were centrifuged at 35,000 rpm, 4 °C for 45 min. The supernatant was then purified using a HiTrap nickel column (GE Healthcare). The purified protein was dialyzed in phosphate-buffered saline containing 1 mm glutamate. The protein was labeled with a 1:3 ratio of maleimide derivatives of Alexa 555 (donor) and Alexa 647 (acceptor) (Invitrogen), respectively. The ratio was confirmed using absorbance measurements. Protein was allowed to conjugate with the fluorophores by incubating overnight at 4 °C in dark. The protein sample was dialyzed to remove the excess unbound fluorophores in phosphate-buffered saline without glutamate, and the appropriate willardiine derivative (Abcam Biochemicals) was added to the protein. This sample was then treated with sulfolink resin for 30 min. The resin was removed by centrifugation, and supernatant was conjugated with biotin-conjugated anti-His antibody (Rockland Immunochemicals) and used for smFRET investigations.
Sample Preparation for smFRET
Standard 22 × 22-mm glass microscope slides were plasma cleaned to remove the organic residues and other impurities from the surface. The surface was then functionalized with aminosilane groups through the Vectabond procedure. In this procedure, the slides were first treated with Vectabond-acetone 1% (w/v) solution (Vector Laboratories) for 5 min, rinsed with molecular biology grade water (HyClone) for 30 s, and dried with an N2 gas stream. The functionalized area was then exposed with 100:1 mixture of 5-kDa, methoxy-terminated, N-succinimidyl polyethylene glycol (Fluka; 33% w/w PEG in molecular biology grade water) and 5-kDa biotin-terminated PEG (NOF Corporation, 2.5% w/w in molecular biology grade water) in sodium bicarbonate (1% v/v, pH 8.0) buffer for ∼3 h. A sample chamber was assembled by placing a custom hybriwell chamber (Grace Bio-labs) with the help of two silicon ports (press fit tubing connectors; Grace Bio-labs) on top of the biotin-PEG glass slide (33). The silicon ports provided the inlet and outlet for the flow system. The biotin-PEG chamber was filled with 40 μl of 20% w/w streptavidin (Invitrogen) in PBS buffer (pH 7.4) and incubated in the dark for 10 min. The streptavidin acts as linker between the biotin-PEG slide and the biotin-conjugated anti-histidine antibody bound to GluA2 subunit. A PBS solution containing ∼250 nm protein tagged with biotin-conjugated anti-histidine monoclonal antibody was then added in ten 17-μl increments into the chamber and incubated for 20 min. The excess protein was then rinsed with PBS buffer.
Oxygen Scavenging System
To minimize the photobleaching and blinking of fluorophores, all of the experiments were performed in the presence of an oxygen scavenger system, consisting of 33% w/w β-d-(+)-glucose (Sigma-Aldrich), 1% w/w glucose oxidase, 0.1% v/v catalase (Sigma Aldrich), 1 mm methyl viologen (Sigma Aldrich), and 1 mm ascorbic acid (Sigma Aldrich) in molecular biology grade water saturated with phosphate buffer (34). In addition, 1 mm of the substituted willardiine was also added to the oxygen scavenging system, depending on the experimental conditions.
Experimental Setup for smFRET
All single-molecule fluorescence measurements were performed using a custom built confocal microscope (35, 36). A 532-nm diode-pumped solid state laser (Coherent, Compass 315M-100 SL) was used for sample excitation. The light was expanded to overfill the back aperture of a Fluar 100× 1.3 NA oil immersion microscope objective lens (Carl Zeiss), which resulted in the expansion of the laser light in a 1/e2 beam radius of ∼ 250 nm and height of ∼ 1 μm, respectively. The sample chamber with the flow system for oxygen scavenger was placed on top of a closed-loop x-y-z piezo stage (P-517.3CL; Physik Instrumente) with 100 × 100 × 20-μm travel range and 1-nm specificity (SPM 1000; RHK Technology, Maryville, TN). The power of the laser was controlled as necessary using neutral density filters. Fluorescence was collected and refocused by the same objective and was separated from the excited light by using a dichroic mirror (z532rdc; Chroma Technology). Fluorescence was collected and refocused by the same objective and the excited light was filtered via a notch filter (zet532nf, Chroma Technology). The refocused signal was further passed though a dichroic mirror (640dcxr, Chroma Technology) to split donor emission and acceptor emission by wavelength, and these fluorescence signals were collected by two avalanche photodiodes (SPCM-AQR-15; PerkinElmer Life Sciences). The signal to noise ratio was improved by the use of additional emission filters (NHPF-532.0, Kaiser Optical; and ET585, Chroma Technology) placed in front of the photodiode detectors.
Data Collection and Analysis
To obtain the smFRET trajectories for the individual protein molecules, a 10 × 10-μm area of the sample was scanned to spatially locate 20–25 molecules. The fluorescence signals of the donor and the acceptor were collected until the fluorophores were photobleached. The emission intensity trajectories were collected at 1-ms resolution and later binned up to 10-ms time steps to improve the signal to noise ratio. All of the data were analyzed with programs written in-house using MATLAB (R2009b; Mathworks). The corrected fluorescence signal trajectories were used directly to calculate the apparent FRET efficiency (EA) using the following equation,
 |
where IA and ID correspond to background corrected acceptor and donor fluorescence intensities, respectively. The distance between the two fluorophores was calculated with the following equation,
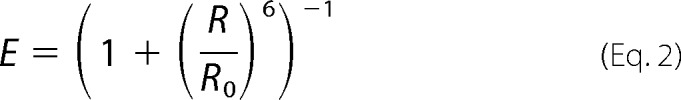 |
where: r = is the inter-dye distance, and R0 = is the Förster radius, which, for the Alexa 555-Alexa 647 pair, is ∼5.1 nm (Molecular Probes).
All the raw trajectories were analyzed by the above mentioned processing algorithm, and traces were automatically characterized into single step bleaching, multistep bleaching and high acceptor background. Traces were excluded if they met with the conditions of multistep bleaching and higher acceptor background. Details of the wavelet denoising technique have been described previously (30, 37, 38).
For experiments requiring multiple solution conditions on the same proteins, a secondary method of data collection and analysis was used. Single-molecule samples were prepared as described above along with two buffer solutions following the stated procedure for oxygen scavenging solutions and using glutamate (1 mm) and iodowillardiine (1 mm) as agonists. A dual syringe pump system initially delivered glutamate containing buffer with a flow rate of 0.001 ml/min followed by both the glutamate and iodowillardiine buffers and then finally iodowillardiine alone. A 30 × 30-μm area was repeatedly raster scanned using the same confocal setup as above, with a 30-s repetition rate between images. The images were analyzed using a custom-designed program that locates and tracks single molecules to correct for possible stage drift. Molecules were selected from the final frame of the acceptor channel images to avoid skewing of intensities by photobleaching and to ensure FRET occurred in all molecules during the imaging period.
Electrophysiology
The GluA2-flip plasmid was donated by Dr. Seeburg (Max Planck Institute, Heidelberg, Germany). Endogenous accessible cysteine residues were removed, and the T394C and S652C mutations were introduced into wild type GluA2 as described above. This construct was co-transfected with enhanced GFP into human embryonic kidney 293 tSA201 (HEK 293) cells (ATCC CRL 11268) using the calcium phosphate technique with 1–2 μg of GluA2/ml for 10–12 h. Electrophysiological experiments were performed 48–72 h later. Alexa 555 (100 nm) was added to the recording dish 10–20 min prior to recording. Whole cell recordings were obtained using thick walled borosilicate pipettes with resistances of 2–4 MΩ and filled with solution containing 135 mm CsF, 33 mm CsOH, 2 mm MgCl2, 1 mm CaCl2, 11 mm EGTA, and 10 mm HEPES (pH 7.4). The extracellular bath solution consisted of (in mm) 150 NaCl, 2.8 KCl, 1.8 CaCl2, 1 MgCl2, 5 mm glucose, and 10 mm HEPES (pH 7.4). All of the ligand solutions were prepared in extracellular buffer and kept at pH 7.4. Cyclothiazide at a concentration of 100 μm was used for all experiments. Cells expressing mutant GluA2 were voltage-clamped at −60 mV, and solutions were locally applied using computer controlled valve switcher (VC-6; Warner Instruments) and homemade application pipette. All of the recordings were performed using an Axon 200B amplifier (Molecular Devices), with data acquired at 10 kHz, low pass filtered at 3 kHz (8-pole Bessel, −3 dB) and under the control of pCLAMP 10.1 software (Molecular Devices). Two-tailed repeated measures t test was performed for statistical analysis of differences in the responses evoked by the different ligands.
RESULTS
Functional Characterization of Willardiines
Whole cell currents were obtained using saturating concentrations of glutamate (10 mm) and chlorowillardiine (1 mm), nitrowillardiine (1 mm), iodowillardiine (1 mm), and (2 mm) UBP-282, on GluA2-flip receptors under nondesensitizing conditions in the presence of 100 μm cyclothiazide. To compare our results directly with the smFRET experiments, agonist-evoked responses were recorded from the same agonist-binding domain mutations, i.e., T394C/S652C GluA2. The currents evoked by the willardiines were normalized to 10 mm glutamate responses and are shown in Fig. 1. The data shown in the bar graph are consistent with the numerous previous more detailed investigations and show that the willardiines are partial agonists with iodowillardiine have a lower response relative to chloro- and nitro-willardiines (31, 32, 39). A two-tailed repeated measures t test was performed for statistical analysis of differences in the measured currents, and a p value of ≤0.05 was considered significant in our experiments. The p values for chlorowillardiine, nitrowillardiine, and iodowillardiine were calculated to be less than 0.0005, 0.0025, and 0.0005, respectively, in comparison with mean currents with glutamate. As expected, the antagonist UBP-282 produced no response beyond the base-line noise. These willardiines, along with glutamate, thus provide a spectrum of ligands that allow us to draw correlations between the smFRET-based states and receptor activation.
FIGURE 1.

a, sample traces of electrophysiological responses evoked by glutamate (Glu), chlorowilladiine (Chloro), nitrowillardiine (Nitro), iodowillardiine (Iodo), and UBP-282 (UBP) under nondesensitizing conditions from cells expressing the T394C/S652C GluA2. Saturating concentrations of Glu (10 mm), chlorowilladiine (1 mm), nitrowillardiine (1 mm), iodowillardiine (1 mm), and UBP-282 (2 mm) were used in the continual presence of 100 μm cyclothiazide. b, summary data showing the electrophysiological response of each willardiine compared with glutamate in the same cell.
Conformational Changes in the Agonist-binding Domain
A representative smFRET trace of a single GluA2 subunit agonist-binding domain tagged with donor and acceptor fluorophores is shown in Fig. 2. The sites tagged with the donor and acceptor fluorophores are T394C and S652C, similar to those used in our previous smFRET investigations (30). The smFRET data from a number of such traces (170–210 traces) for each willardiine were then processed for background and cross-talk correction (35, 36) and denoised using wavelet decomposition as described previously (37). The data were then plotted as histograms of fraction of occurrence versus the FRET efficiency to determine the spread of states that the protein explores (Fig. 3). The denoised histograms for the chlorowillardiine, nitrowillardiine, iodowillardiine, and the antagonist UBP-282-bound forms are shown in Fig. 3. The distances calculated for the average state for the glutamate-, UBP-282-, and willardiine-bound forms, based on the FRET efficiencies are listed in Table 1. The average distances follow the rank order Glu < nitrowillardiine < chlorowilladiine < iodowillardiine < UBP-282. Although the correlation of cleft closure with activation is present for the extreme cases, this does not hold true for nitrowillardiine and cholorwillardiine. Nitrowillardiine and cholorwillardiine have similar electrophysiological responses; however, the average smFRET distances indicate that the nitrowillardiine-bound state is on average slightly more closed relative to chlorowillardiine bound state. A similar breakdown was observed in the crystal structures where no significant changes were observed in the distances at the positions labeled for the FRET measurements between these willardiines (shown in Table 1) (39).
FIGURE 2.

a, representative smFRET trace measured from a single agonist-binding domain of GluA2 subunit in complex with chlorowillardiine showing the raw trajectory for donor (blue) and acceptor (red) photons as a function of time. b, the resulting calculated FRET trajectory (green) and its denoised counterpart (black). c and d, histograms of calculated smFRET values from the raw data (c) and following denoising (d).
FIGURE 3.

Denoised ensemble FRET histograms for single agonist-binding domains of GluA2 subunit of AMPA receptors in complex with chlorowillardiine (a), nitrowillardiine (b), iodowillardiine (c), and UBP-282 (d).
TABLE 1.
Comparison of the distances between residues Thr-394 and Ser-652 in the x-ray structure and smFRET studies
The smFRET studies provide insight into possible reasons for this discrepancy because they are able to provide the spread of closed cleft states that the protein occupies (Fig. 3). Because the time resolution of the smFRET measurements is in milliseconds, the breadth of the histograms reflect the dynamics in the millisecond time scale. The histograms show that the chlorowillardiine-bound form probes a narrower range of cleft closures relative to the nitrowillardiine, whereas the iodowillardiine-bound form exhibits a wider range of cleft closure states (Fig. 3). The willardiine-based antagonist, UBP-282, showed the broadest distribution of closed cleft conformations (Fig. 3d). These data suggest that the cleft is more stabilized when bound to chlorowillardiine relative to nitrowillardiine, with the iodowillardiine- and UBP-282-bound forms being the most destabilized, exploring a wide range of cleft closures and consequently spending little time in conformations able to activate the receptor.
To confirm that tethered and tagged ligand-binding domains retained their function during imaging, we performed a displacement experiment. A collection of single ligand-binding domains were imaged in the presence of 1 mm glutamate alone for ∼20 min. Subsequently, the perfusing buffer was switched to a glutamate and iodowillardiine containing solution for 20 min and finally 1 mm iodowillardiine alone for the final 20 min. As seen in Fig. 4, the average FRET intensity from this experiment progressively decreases from 0.81 in glutamate alone to 0.76 in iodowillardiine. This demonstrates that the labeled and tethered single ligand-binding domains retain their ability to bind and unbind ligands. Single-molecule histograms from this experiment also exhibit a progressive shift from a narrow, focused distribution in glutamate to a broader distribution in iodowillardiine (Fig. 4) similar to the trends seen in the detailed histograms shown in Fig. 3.
FIGURE 4.

a, plot of average single-molecule FRET efficiency over time during a wash from glutamate to glutamate plus iodowillardiine to iodowillardiine alone. Note the progressive decrease in FRET efficiency as glutamate is displaced by iodowillardiine (IW). b, single-molecule histograms from the experiment in a.
Fraction in Closed Cleft Conformations versus Activation
Based on the histograms, the fraction of the protein exhibiting efficiencies higher than 0.76 was determined for each of the willardiine bound state. The fraction for the glutamate-bound forms of GluA2 and T686S mutant protein, as well as the apo state of the protein, was also determined from the previous report (30). The FRET efficiency of 0.76 was chosen because it corresponds to a FRET distance of 42 Å, which is less than the most probable (46 Å), as well as the average state (43 Å) of the apo state, and thus is expected to be the least distance at which the cleft closure could trigger channel opening. This fraction of the protein exhibiting efficiencies higher than 0.76 shows a strong correlation with the currents mediated by the agonist, as shown in Fig. 5. The activation shown for the glutamate-bound form of T686S mutant is relative to that obtained with quisqualate on the same mutant (22, 25, 26). Quisqualate and glutamate mediate similar currents in wild type receptors (22, 25, 26).
FIGURE 5.

Plot of the fraction of the agonist-binding domain of the GluA2 subunit exhibiting FRET efficiencies higher than 0.76 versus normalized mean currents obtained with the corresponding agonists activating the full-length receptors. The data for the glutamate bound and apo form of wild type, as well as glutamate-bound form of T686S mutant, were obtained from Landes et al. (30). ClW, chlorowilladiine; NO2W, nitrowillardiine; IW, iodowillardiine.
The linear dependence both for the willardiines and the T686S mutant indicates that the fraction of agonist-binding domains in the closed cleft conformation, which takes into account the ability of the agonist to induce cleft closure, as well as the range of states that the protein probes, is a good determinant of the ability of an agonist drive activation rather than the most probable state or the average state that the protein-agonist complex occupies.
DISCUSSION
The modular nature of the AMPA receptors has allowed for the study of the isolated agonist-binding domain by crystallography, NMR, FRET, and FTIR spectroscopy (4, 6, 20–25, 28, 31, 32, 39–49). The initial crystal structures indicated a correlation between cleft closure and activation (50). However, later investigations showed a number of partial agonists and mutant proteins that did not follow this trend.
NMR and more recently smFRET experiments, along with functional studies, showed that the stability of closed cleft states and the spread of conformations that the protein explores play a key role in translating cleft closure to activation, thus accounting for some of these discrepancies (30, 31, 49). Indeed, as seen in Fig. 5, the smFRET data allow us to explicitly determine the fraction of agonist-binding domains that exist in a “productive” closed cleft conformation, which correlates with the extent of activation even in previously “discrepant” cases such as the T686S mutation (30). The willardiines were initially thought to be classical examples for showing a graded cleft closure consistent with their activation (50). However, further studies using crystal structures (39) and NMR showed that the correlation is not as straightforward as first thought (31, 49). NMR studies investigating the exchange at the side chain methyl groups showed differences among the various willardiines and indicated that the dynamics must also play a role in the activation. Specifically, these studies show that the number of residues exhibiting exchange was higher for chlorowillardiine relative to iodowillardiine. The smFRET investigations reported here are consistent with this observation, with the chlorowillardiine-bound form exhibiting a narrower range of states than the protein probes relative to iodowillardiine-bound form (Fig. 4) and add to these studies by showing the whole range of conformations probed. Additionally, the crystal structures of GluA2 and GluA3 agonist-binding domain in complex with chlorowillardiine show that despite the small size of the chloro substituent, the chlorowillardiine-bound form is on average more open than the glutamate-bound form (39). The nitrowillardiine-bound form, despite nitro being a larger substituent, is more closed in six of the nine structures studied relative to the fluorowillardiine-bound form. The larger nitro group was accommodated by the change in the rotamer state of the β-carbon of M712 as well as elimination of a water group. Furthermore, the structures showed that the nitrowillardiine-bound form exhibited a wider range of lobe openings relative to the glutamate-bound form of the protein. The smFRET data presented here are in agreement with these structures and show that the chlorowillardiine-bound form is on average slightly more closed than the nitrowillardiine-bound form (Table 1) and that the nitrowillardiine-bound form has a broader range of cleft closure states relative to the chlorowillardiine-bound form.
The smFRET data are also consistent with recent crystal structure “lobe-locking” experiments, which found that ligand-binding domains could be trapped in closed conformations when bound by partial agonists or even antagonists (51). Taken together, these data provide strong support for recent proposals that agonist efficacy at AMPA receptors (27), and perhaps kainate receptors (52), is governed not by the extent of closure in a single state of the binding domain but by the relative stability of a range of variously productive closed cleft conformations.
Acknowledgments
C. F. Landes thanks the Norman Hackerman Welch Young Investigator Program at Rice University.
This work was supported, in whole or in part, by National Institutes of Health Grant GM094246. This work was also supported by Gulf Coast Consortia Grant/Welch Foundation Grant H-E-0035/R-11-0055 and American Heart Association Grant 11GRNT7890004.
- AMPA
- α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid
- UBP-282
- (αS)-α-Amino-3-[(4-carboxyphenyl)methyl]-3,4-dihydro-2,4-dioxo-1(2H)-pyrimidinepropanoic acid
- smFRET
- single-molecule FRET.
REFERENCES
- 1. Nakanishi S. (1992) Molecular diversity of glutamate receptors and implications for brain function. Science 258, 597–603 [DOI] [PubMed] [Google Scholar]
- 2. Nakanishi S., Masu M. (1994) Molecular diversity and functions of glutamate receptors. Annu. Rev. Biophys. Biomol. Struct. 23, 319–348 [DOI] [PubMed] [Google Scholar]
- 3. Madden D. R. (2002) The structure and function of glutamate receptor ion channels. Nat. Rev. Neurosci. 3, 91–101 [DOI] [PubMed] [Google Scholar]
- 4. McFeeters R. L., Oswald R. E. (2004) Emerging structural explanations of ionotropic glutamate receptor function. FASEB J. 18, 428–438 [DOI] [PubMed] [Google Scholar]
- 5. Valentine E. R., Palmer A. G., 3rd. (2005) Microsecond-to-millisecond conformational dynamics demarcate the GluR2 glutamate receptor bound to agonists glutamate, quisqualate, and AMPA. Biochemistry 44, 3410–3417 [DOI] [PubMed] [Google Scholar]
- 6. Oswald R. E., Ahmed A., Fenwick M. K., Loh A. P. (2007) Structure of glutamate receptors. Curr. Drug Targets 8, 573–582 [DOI] [PubMed] [Google Scholar]
- 7. Traynelis S. F., Wollmuth L. P., McBain C. J., Menniti F. S., Vance K. M., Ogden K. K., Hansen K. B., Yuan H., Myers S. J., Dingledine R. (2010) Glutamate receptor ion channels. Structure, regulation, and function. Pharmacol. Rev. 62, 405–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sommer B., Seeburg P. H. (1992) Glutamate receptor channels. Novel properties and new clones. Trends Pharmacol. Sci. 13, 291–296 [DOI] [PubMed] [Google Scholar]
- 9. Madsen U., Ebert B., Krogsgaard-Larsen P. (1994) Modulation of AMPA receptor function in relation to glutamatergic abnormalities in Alzheimer's disease. Biomed. Pharmacother. 48, 305–311 [DOI] [PubMed] [Google Scholar]
- 10. Zamanillo D., Sprengel R., Hvalby O., Jensen V., Burnashev N., Rozov A., Kaiser K. M., Köster H. J., Borchardt T., Worley P., Lübke J., Frotscher M., Kelly P. H., Sommer B., Andersen P., Seeburg P. H., Sakmann B. (1999) Importance of AMPA receptors for hippocampal synaptic plasticity but not for spatial learning. Science 284, 1805–1811 [DOI] [PubMed] [Google Scholar]
- 11. Kwak S., Kawahara Y. (2005) Deficient RNA editing of GluR2 and neuronal death in amyotropic lateral sclerosis. J. Mol. Med. 83, 110–120 [DOI] [PubMed] [Google Scholar]
- 12. Shepherd J. D., Huganir R. L. (2007) The cell biology of synaptic plasticity. AMPA receptor trafficking. Annu. Rev. Cell Dev. Biol. 23, 613–643 [DOI] [PubMed] [Google Scholar]
- 13. O'Neill M. J., Witkin J. M. (2007) AMPA receptor potentiators. Application for depression and Parkinson's disease. Curr. Drug Targets 8, 603–620 [DOI] [PubMed] [Google Scholar]
- 14. Johnson K. A., Conn P. J., Niswender C. M. (2009) Glutamate receptors as therapeutic targets for Parkinson's disease. CNS Neurol. Disord. Drug Targets 8, 475–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Keifer J., Zheng Z. (2010) AMPA receptor trafficking and learning. Eur. J. Neurosci. 32, 269–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sobolevsky A. I., Rosconi M. P., Gouaux E. (2009) X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature 462, 745–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Du M., Reid S. A., Jayaraman V. (2005) Conformational changes in the ligand-binding domain of a functional ionotropic glutamate receptor. J. Biol. Chem. 280, 8633–8636 [DOI] [PubMed] [Google Scholar]
- 18. Gonzalez J., Rambhadran A., Du M., Jayaraman V. (2008) LRET investigations of conformational changes in the ligand binding domain of a functional AMPA receptor. Biochemistry 47, 10027–10032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gonzalez J., Du M., Parameshwaran K., Suppiramaniam V., Jayaraman V. (2010) Role of dimer interface in activation and desensitization in AMPA receptors. Proc. Natl. Acad. Sci. U.S.A. 107, 9891–9896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Armstrong N., Sun Y., Chen G. Q., Gouaux E. (1998) Structure of a glutamate-receptor ligand-binding core in complex with kainate. Nature 395, 913–917 [DOI] [PubMed] [Google Scholar]
- 21. Armstrong N., Gouaux E. (2000) Mechanisms for activation and antagonism of an AMPA-sensitive glutamate receptor. Crystal structures of the GluR2 ligand binding core. Neuron 28, 165–181 [DOI] [PubMed] [Google Scholar]
- 22. Armstrong N., Mayer M., Gouaux E. (2003) Tuning activation of the AMPA-sensitive GluR2 ion channel by genetic adjustment of agonist-induced conformational changes. Proc. Natl. Acad. Sci. U.S.A. 100, 5736–5741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gouaux E. (2004) Structure and function of AMPA receptors. J. Physiol. 554, 249–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ahmed A. H., Wang Q., Sondermann H., Oswald R. E. (2009) Structure of the S1S2 glutamate binding domain of GLuR3. Proteins 75, 628–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Birdsey-Benson A., Gill A., Henderson L. P., Madden D. R. (2010) Enhanced efficacy without further cleft closure. Reevaluating twist as a source of agonist efficacy in AMPA receptors. J. Neurosci. 30, 1463–1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Robert A., Armstrong N., Gouaux J. E., Howe J. R. (2005) AMPA receptor binding cleft mutations that alter affinity, efficacy, and recovery from desensitization. J. Neurosci. 25, 3752–3762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang W., Cho Y., Lolis E., Howe J. R. (2008) Structural and single-channel results indicate that the rates of ligand binding domain closing and opening directly impact AMPA receptor gating. J. Neurosci. 28, 932–943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mankiewicz K. A., Rambhadran A., Wathen L., Jayaraman V. (2008) Chemical interplay in the mechanism of partial agonist activation in α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors. Biochemistry 47, 398–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ramanoudjame G., Du M., Mankiewicz K. A., Jayaraman V. (2006) Allosteric mechanism in AMPA receptors. A FRET-based investigation of conformational changes. Proc. Natl. Acad. Sci. U.S.A. 103, 10473–10478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Landes C. F., Rambhadran A., Taylor J. N., Salatan F., Jayaraman V. (2011) Structural landscape of isolated agonist-binding domains from single AMPA receptors. Nat. Chem. Biol. 7, 168–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fenwick M. K., Oswald R. E. (2008) NMR spectroscopy of the ligand-binding core of ionotropic glutamate receptor 2 bound to 5-substituted willardiine partial agonists. J. Mol. Biol. 378, 673–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Maltsev A. S., Oswald R. E. (2010) Hydrophobic side chain dynamics of a glutamate receptor ligand binding domain. J. Biol. Chem. 285, 10154–10162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Karam P., Ngo A. T., Rouiller I., Cosa G. (2010) Unraveling electronic energy transfer in single conjugated polyelectrolytes encapsulated in lipid vesicles. Proc. Natl. Acad. Sci. U.S.A. 107, 17480–17485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vogelsang J., Doose S., Sauer M., Tinnefeld P. (2007) Single-molecule fluorescence resonance energy transfer in nanopipets. Improving distance resolution and concentration range. Anal. Chem. 79, 7367–7375 [DOI] [PubMed] [Google Scholar]
- 35. Darugar Q., Kim H., Gorelick R. J., Landes C. (2008) Human T-cell lympotropic virus type 1 nucleocapsid protein-induced structural changes in transactivation response DNA measured by single molecule fluorescence resonance energy transfer. J. Virol. 82, 12164–12171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Taylor J. N., Darugar Q., Kourentzi K., Willson R. C., Landes C. F. (2008) Dynamics of an anti-VEGF aptamer. A single molecule study. Biochem. Biophys. Res. Commun. 373, 213–218 [DOI] [PubMed] [Google Scholar]
- 37. Taylor J. N., Makarov D. E., Landes C. F. (2010) Denoising single-molecule FRET trajectories with wavelets and Bayesian inference. Biophys. J. 98, 164–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Taylor J. N., Landes C. F. (2011) Improved resolution of complex single-molecule FRET systems via wavelet shrinkage. J. Phys. Chem. B 115, 1105–1114 [DOI] [PubMed] [Google Scholar]
- 39. Poon K., Ahmed A. H., Nowak L. M., Oswald R. E. (2011) Mechanisms of modal activation of GluA3 receptors. Mol. Pharmacol. 80, 49–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jayaraman V., Keesey R., Madden D. R. (2000) Ligand-protein interactions in the glutamate receptor. Biochemistry 39, 8693–8697 [DOI] [PubMed] [Google Scholar]
- 41. Jayaraman V., Thiran S., Madden D. R. (2000) Fourier transform infrared spectroscopic characterization of a photolabile precursor of glutamate. FEBS Lett. 475, 278–282 [DOI] [PubMed] [Google Scholar]
- 42. Madden D. R., Thiran S., Zimmermann H., Romm J., Jayaraman V. (2001) Stereochemistry of quinoxaline antagonist binding to a glutamate receptor investigated by Fourier transform infrared spectroscopy. J. Biol. Chem. 276, 37821–37826 [DOI] [PubMed] [Google Scholar]
- 43. Madden D. R., Cheng Q., Thiran S., Rajan S., Rigo F., Keinänen K., Reinelt S., Zimmermann H., Jayaraman V. (2004) Stereochemistry of glutamate receptor agonist efficacy. Engineering a dual-specificity AMPA/kainate receptor. Biochemistry 43, 15838–15844 [DOI] [PubMed] [Google Scholar]
- 44. Jayaraman V. (2004) Spectroscopic and kinetic methods for ligand-protein interactions of glutamate receptor. Methods Enzymol. 380, 170–187 [DOI] [PubMed] [Google Scholar]
- 45. Cheng Q., Du M., Ramanoudjame G., Jayaraman V. (2005) Evolution of glutamate interactions during binding to a glutamate receptor. Nat. Chem. Biol. 1, 329–332 [DOI] [PubMed] [Google Scholar]
- 46. Mankiewicz K. A., Jayaraman V. (2007) Glutamate receptors as seen by light. Spectroscopic studies of structure-function relationships. Braz. J. Med. Biol. Res. 40, 1419–1427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. McFeeters R. L., Oswald R. E. (2002) Structural mobility of the extracellular ligand-binding core of an ionotropic glutamate receptor. Analysis of NMR relaxation dynamics. Biochemistry 41, 10472–10481 [DOI] [PubMed] [Google Scholar]
- 48. Ahmed A. H., Loh A. P., Jane D. E., Oswald R. E. (2007) Dynamics of the S1S2 glutamate binding domain of GluR2 measured using 19F NMR spectroscopy. J. Biol. Chem. 282, 12773–12784 [DOI] [PubMed] [Google Scholar]
- 49. Maltsev A. S., Ahmed A. H., Fenwick M. K., Jane D. E., Oswald R. E. (2008) Mechanism of partial agonism at the GluR2 AMPA receptor. Measurements of lobe orientation in solution. Biochemistry 47, 10600–10610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jin R., Banke T. G., Mayer M. L., Traynelis S. F., Gouaux E. (2003) Structural basis for partial agonist action at ionotropic glutamate receptors. Nat. Neurosci. 6, 803–810 [DOI] [PubMed] [Google Scholar]
- 51. Ahmed A. H., Wang S., Chuang H. H., Oswald R. E. (2011) Mechanism of AMPA receptor activation by partial agonists. Disulfide trapping of closed lobe conformations. J. Biol. Chem. 286, 35257–35266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Maclean D. M., Wong A. Y., Fay A. M., Bowie D. (2011) Cations but not anions regulate the responsiveness of kainate receptors. J. Neurosci. 31, 2136–2144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ahmed A. H., Thompson M. D., Fenwick M. K., Romero B., Loh A. P., Jane D. E., Sondermann H., Oswald R. E. (2009) Mechanisms of antagonism of the GluR2 AMPA receptor. Structure and dynamics of the complex of two willardiine antagonists with the glutamate binding domain. Biochemistry 48, 3894–3903 [DOI] [PMC free article] [PubMed] [Google Scholar]