

Background: HuR regulates expression of many oncogenic proteins by modulating mRNA stability.
Results: Glycolysis inhibition facilitates HuR degradation through a novel β-TrCP-mediated mechanism.
Conclusion: This mechanism underlies the complexity in the regulation of HuR turnover under different stress stimuli.
Significance: The ability of glycolysis inhibitors to target expression of oncogenic proteins by promoting HuR degradation might foster novel strategies for cancer therapy.
Keywords: Cancer Therapy, E3 Ubiquitin Ligase, Glycolysis, Protein Degradation, Signal Transduction, HuR, β-TrCP, Energy Restriction, Glucose Transporter Inhibitor, Metabolic Stress
Abstract
The mRNA-stabilizing protein HuR acts a stress response protein whose function and/or protein stability are modulated by diverse stress stimuli through posttranslational modifications. Here, we report a novel mechanism by which metabolic stress facilitates proteasomal degradation of HuR in cancer cells. In response to the glucose transporter inhibitor CG-5, HuR translocates to the cytoplasm, where it is targeted by the ubiquitin E3 ligase β-TrCP1 for degradation. The cytoplasmic localization of HuR is facilitated by PKCα-mediated phosphorylation at Ser-318 as the Ser-318 → alanine substitution abolishes the ability of the resulting HuR to bind PKCα and to undergo nuclear export. The mechanistic link between β-TrCP1 and HuR degradation was supported by the ability of ectopically expressed β-TrCP1 to mimic CG-5 to promote HuR degradation and by the protective effect of dominant negative inhibition of β-TrCP1 on HuR ubiquitination and degradation. Substrate targeting of HuR by β-TrCP1 was further verified by coimmunoprecipitation and in vitro GST pull-down assays and by the identification of a β-TrCP1 recognition site. Although HuR does not contain a DSG destruction motif, we obtained evidence that β-TrCP1 recognizes an unconventional motif, 296EEAMAIAS304, in the RNA recognition motif 3. Furthermore, mutational analysis indicates that IKKα-dependent phosphorylation at Ser-304 is crucial to the binding of HuR to β-TrCP1. Mechanistically, this HuR degradation pathway differs from that reported for heat shock and hypoxia, which underlies the complexity in the regulation of HuR turnover under different stress stimuli. The ability of glycolysis inhibitors to target the expression of oncogenic proteins through HuR degradation might foster novel strategies for cancer therapy.
Introduction
Substantial evidence has demonstrated the clinical relevance of targeting the mRNA-binding protein HuR in cancer therapy in light of its pivotal role in regulating the expression of a wide array of proteins implicated in oncogenesis and tumor progression by conferring stability and/or altering translation rates of target mRNAs (1–3). HuR is primarily localized in the nucleus and translocates to the cytoplasm in response to various stress stimuli, including oxidative stress (4), heat shock (5), hypoxia (6), UV light (7), amino acid starvation (7), polyamine depletion (8), and staurosporine (9). This nuclear export is integral to the function of HuR to control the stability and translation of mRNA. HuR interacts with target mRNAs through the recognition of AU-rich elements in the 5′ and 3′ untranslated regions via its RNA recognition motifs (RRMs)2. Although the mechanism by which HuR stabilizes mRNAs remains undefined, it is rationalized that this HuR binding antagonizes AU-rich element-directed mRNA destabilization mediated by other RNA binding proteins (10).
Consistent with its oncogenic role, HuR protein is highly abundant in many types of malignancies (11–14). Recent evidence indicates that cancer cells increase HuR expression, in part, through posttranslational modifications (15) or by inhibiting miRNA-mediated translational repression (16, 17). For example, a recent report indicates that oncogenic murine double minute (MDM) 2-mediated neddylation protected HuR from ubiquitin-dependent degradation in cancer cells (15). Although the machinery that regulates HuR proteolysis is not fully elucidated, HuR was reported to undergo ubiquitin-dependent degradation following heat shock (5). However, the identity of the responsible E3 ubiquitin ligase remains unclear.
In this study, we demonstrate that HuR was subjected to ubiquitin-dependent degradation via a novel mechanism in prostate cancer cells in response to metabolic stress induced by glucose depletion or glycolysis inhibitors, including CG-5, a glucose transporter inhibitor (18), and 2-deoxyglucose (2-DG). Moreover, we obtained evidence that HuR was targeted for ubiquitination by the E3 ubiquitin ligase β-transducin repeat-containing protein (β-TrCP) 1 in response to glycolysis inhibition, and that PKCα and IκB kinase (IKK) α were involved in facilitating cytoplasmic translocation and β-TrCP1 recognition of HuR, respectively, in the course of ubiquitin-dependent degradation. It is noteworthy that, although HuR lacks the DSGXnS destruction motif, mutational analysis indicates that HuR is recognized by β-TrCP1 through an unconventional motif, 296EEAAMAIAS304. From a mechanistic perspective, the ability of glycolysis inhibitors to target a broad range of HuR-regulated oncogenic proteins by facilitating HuR degradation might foster novel strategies for cancer therapy.
EXPERIMENTAL PROCEDURES
Cell Culture and Reagents
LNCaP, PC3, and DU-145 prostate cancer cells were obtained from the ATCC. Cells were maintained and treated with individual test agents in 10% FBS-supplemented RPMI 1640 medium (Invitrogen) at 37 °C in a humidified incubator containing 5% CO2. CG-5 was synthesized according to a published procedure (18). 2-DG, MG132, cycloheximide, and leptomycin B were purchased from Sigma-Aldrich (St. Louis, MO). The kinase inhibitors SB216763, GF109203X, SB203580, and BAY11-7082 were purchased from Cayman Chemical (Ann Arbor, MI), and AT7519 was obtained from Selleckchem (Houston, TX). Antibodies against the following proteins were used: HuR, cyclin B1, cyclin E1, VEGF, cyclin D1, β-catenin, PKCα, PKCδ, and p-Thr-507-PKCδ (Santa Cruz Biotechnology, Santa Cruz, CA); MMP-9, Myc, HA, IKKα, p-Ser-180-IKKα, and p-Ser-PKC substrate (Cell Signaling Technology, Beverly, MA); Snail and MDM2 (Abcam, Cambridge, MA); p-Ser-657-PKCα (Millipore, Billerica, MA); β-TrCP1 (Invitrogen); β-TrCP2 (Rockland Immunochemicals, Gilbertsville, PA); FLAG (Sigma-Aldrich); GST (GE Healthcare); and β-actin (MP Biomedicals, Irvine, CA). The IκB kinase dominant-negative mutant (pIKK2M) was obtained as described (19).
Plasmid Construction and Site-directed Mutagenesis
The HuR human cDNA clone was purchased from Origene Technologies (Rockville, MD, catalog no. SC119271, pCMV6-XL5) and subcloned into the HindIII/BglII sites of the p3XFLAG-CMV26 expression vector (Sigma-Aldrich). Using the FLAG-tagged full-length HuR plasmid as a template, a series of truncated or mutated HuR constructs were created. The ΔHNS-RRM3-HuR-FLAG, ΔRRM1-HuR-FLAG, and ΔRRM1/2-HuR-FLAG plasmids were constructed by PCR amplification and subsequently cloned into the p3XFLAG-CMV26 vector. Plasmids encoding various HuR mutations were generated by site-directed mutagenesis from the FLAG-tagged full-length HuR by using the QuikChange II XL site-directed mutagenesis kit (Stratagene, Santa Clara, CA). The forward and reverse primers used to generate individual HuR mutations were as follows, respectively: S158A, 5′-CGGTTTGACAAACGGGCGGAGGCAGAAGAGG-3′ and 5′-CCTCTTCTGCCTCCGCCCGTTTGTCAAACCG-3′; S221A, 5′-AGAGATTCAGGTTCGCCCCCATGGGCGTC-3′ and 5′-GACGCCCATGGGGGCGAACCTGAATCTCT-3′; S318A, 5′-GGGGGACAAAATCTTACAGGTTGCCTTCAAAACCAACAA-3′ and 5′-TTGTTGGTTTTGAAGGCAACCTGTAAGATTTTGTCCCCC-3′; E296/297A, 5′-TTGTGACCATGACAAACTATGCAGCAGCCGCGATGGCC-3′ and 5′-GGCCATCGCGGCTGCTGCATAGTTTGTCATGGTCACAA-3′; S304A, 5′-CGATGGCCATAGCCGCCCTGAACGGCTACC-3′ and 5′-GGTAGCCGTTCAGGGCGGCTATGGCCATCG-3′; and S304E, 5′-GCGATGGCCATAGCCGAGCTGAACGGCTACCGC-3′ and 5′-GCGGTAGCCGTTCAGCTCGGCTATGGCCATCGC-3′. The Myc-tagged full-length β-TrCP1 and ΔF-β-TrCP1 were generated from a β-TrCP1 clone as described previously (20).
RNA Isolation and Quantitative Real-time PCR (qRT-PCR)
Total RNA was isolated and reverse-transcribed to cDNA using TRIzol reagent (Invitrogen) and the iScript cDNA synthesis kit (Bio-Rad), respectively, according to the instructions of the manufacturer. qRT-PCR was carried out in the Bio-Rad CFX96 real-time PCR detection system with iQ SYBR Green Supermix (Bio-Rad). Relative gene expression was normalized to 18 S rRNA and calculated by using the 2-▵▵Ct method. PCR primers used were as follows: HuR, 5′-TGTCTAATGGTTATGAAGACC-3′ and 5′-AGGAAACCTGTAAGATTTTGTC-3′ and GAPDH, 5′-AGGGGTCTACATGGCAACTG-3′ and 5′-CGACCACTTTGTCAAGCTCA-3′.
Cell Fractionation and Immunoblotting
Cytoplasmic and nuclear protein extracts were prepared by using the NE-PER nuclear and cytoplasmic extraction kit (Thermo Scientific, Rockford, IL) according to the instructions of the manufacturer. Immunoblotting was performed as described previously (18).
Transient Transfection, RNA Interference, and Coimmunoprecipitation Analysis
Transfections were performed by electroporation using Nucleofector kit R of the Amaxa nucleofector system (Lonza, Walkersville, MD) according to the protocol of the manufacturer. For siRNA experiments, cells were electroporated with 100 nm scrambled, PKCα, PKCδ (Santa Cruz Biotechnology), or β-TrCP1 (Dharmacon, Lafayette, CO) siRNA and incubated for 48 h. Two independent siRNAs were used for the knockdown of β-TrCP1 with the following sequences: 1, 5′-CACAUAAACUCGUAUCUUA-3′ and 2, 5′-GCAGAGAGAUUUCAUAACU-3′. After exposure to 5 μm CG-5, cells were harvested and subjected to immunoblotting analyses. For coimmunoprecipitation analysis, cells were lysed in lysis buffer (20 mm Tris-HCl (pH 7.5), 150 mm NaCl, 1% Triton X-100) containing a protease inhibitor mixture (Sigma-Aldrich) on ice for 30 min. After centrifugation at 13,000 rpm for 10 min, one-tenth volume of supernatant was reserved as input, and the remainder was incubated with anti-FLAG or anti-HuR antibodies in the presence of protein A/G-agarose (Santa Cruz Biotechnology) at 4 °C for 12 h. After a brief centrifugation, immunoprecipitates were washed four times with lysis buffer, resuspended in 2× SDS sample buffer (100 mm Tris-HCl (pH 6.8), 4% SDS, 5% β-mercaptoethanol, 20% glycerol, and 0.1% bromphenol blue), boiled for 10 min, resolved by 10% SDS-polyacrylamide gel, and subjected to immunoblotting.
Immunocytochemical Analysis
LNCaP cells growing on slides in 6-well plates and were fixed with 3.7% formaldehyde at room temperature for 20 min. Cells were then permeabilized with 0.1% Triton X-100 in PBS for 15 min and blocked for 15 min with 1% BSA. After washing the cells twice with PBS, immunostaining was performed by incubating cells with mouse anti-HuR (1:200 dilution in 1% BSA) or anti-FLAG (1:500 dilution in 1% BSA) primary antibody at 4 °C for 12 h. After washing with PBS, the bound primary antibodies were labeled with Alexa Fluor 555 goat anti-mouse antibody (Invitrogen, 1:400 dilution in 1% BSA) at room temperature for 1 h. Nuclear counterstaining was performed using a DAPI-containing mounting medium (Vector Laboratories, Burlingame, CA), and images were acquired on a confocal microscope (FV1000, Olympus) using a ×40 UPLFLN oil objective (1.30 NA) at room temperature. Scanning was set with 1024 × 1024 frame size, and ×2 zoom. FV10-ASW software (Olympus) was used for data acquisition, and images were imported and processed with Photoshop (Adobe).
GST Fusion Protein Preparation and Pull-down Assay
The full-length β-TrCP1 and Skp2 cDNA were subcloned into the BamHI/EcoRI and EcoRI/HindIII sites, respectively, of the pGEX vector for GST fusion protein expression. GST-β-TrCP1 and GST-Skp2 fusion proteins were expressed in Escherichia coli strain BL21 (DE3) by isopropyl 1-thio-β-d-galactopyranoside induction for 3 h at 37 °C, and bacteria were lysed in STE buffer (10 mm Tris (pH 8.0), 150 mm NaCl, 1 mm EDTA, 5 mm DTT, and 1 mm PMSF) containing 1 mg/ml lysozyme (Sigma-Aldrich) and sonicated on ice for 5 min. The lysates were centrifuged for 20 min at 16,000 rpm, and the pellets were dissolved in 10 ml of 1.5% N-laurylsarcosine-containing STE buffer at 4 °C for 1 h. After centrifugation at 16,000 rpm for 20 min, supernatants were neutralized by adding 2% Triton X-100. Recombinant GST and GST fusion proteins were purified by incubation with glutathione-Sepharose beads (GE Healthcare) with gentle rocking at 4 °C for 2 h. The fusion proteins immobilized onto glutathione beads were washed with ice-cold PBS buffer and used for the GST pull-down assay. For the GST pull-down assay, LNCaP cell lysates were prepared by incubation in lysis buffer (20 mm Tris-HCl (pH 7.5), 150 mm NaCl, 1% Triton X-100) containing a protease inhibitor mixture on ice for 30 min and then incubated with equal amounts of GST-, GST-β-TrCP1-, or GST-Skp2-immobilized glutathione beads at 4 °C for 2 h. After washing three times with lysis buffer, the precipitates were subjected to immunoblotting analysis with antibodies against HuR and GST.
RESULTS
HuR Is Degraded in Response to Glycolysis Inhibition
As the stability and cellular distribution of HuR protein are modulated by diverse stress signals, we investigated the effect of glycolysis inhibition-induced metabolic stress, induced by glucose deprivation or glycolysis inhibitors, on HuR expression in prostate cancer cells. As shown, exposure of LNCaP, PC-3, and DU-145 cells to glucose-depleted medium resulted in substantial decreases in HuR protein levels, whereas qRT-PCR analysis indicates that the mRNA expression was undisturbed (Fig. 1A). Similarly, treatment of LNCaP cells with CG-5, a glucose transporter inhibitor (18), or 2-DG resulted in a dose- and/or time-dependent reduction in HuR protein expression, whereas the mRNA levels remained unaffected (Fig. 1B). Together, these findings suggest that glycolysis inhibition-induced down-regulation of HuR was mediated at the posttranscriptional level. This CG-5-mediated ablation of HuR was also noted in PC-3 and DU-145 cells (Fig. 1C), indicating that this drug effect was not a cell line-specific cellular response.
FIGURE 1.

Glycolysis inhibition suppresses HuR expression at the protein level in prostate cancer cells. A, Western blot (left panel) and qRT-PCR (right panel) analyses of the effect of glucose deprivation on the protein and mRNA expression levels of HuR in LNCaP, PC-3, and DU-145 cells in 10% FBS-supplemented, glucose-free RPMI 1640 medium for 72 h. B, Western blot (upper panel) and qRT-PCR (lower panel) analyses of dose- and/or time-dependent effects of CG-5 and 2-DG on the protein and mRNA expression levels of HuR in LNCaP cells. C, dose-dependent effects of CG-5 on HuR protein expression in PC-3 and DU-145 cells after 48 h of exposure.
Moreover, this CG-5-induced reduction in HuR abundance was accompanied by parallel decreases in the levels of various tumor-promoting signaling proteins encoded by HuR-targeted mRNAs, including cyclin B1, cyclin E1, matrix metallopeptidase 9, Snail, and VEGF, that, however, could be rescued by ectopic expression of HuR (Fig. 2A). Together, these findings verify that glycolysis inhibition reduced HuR protein levels, leading to suppression of HuR signaling.
FIGURE 2.
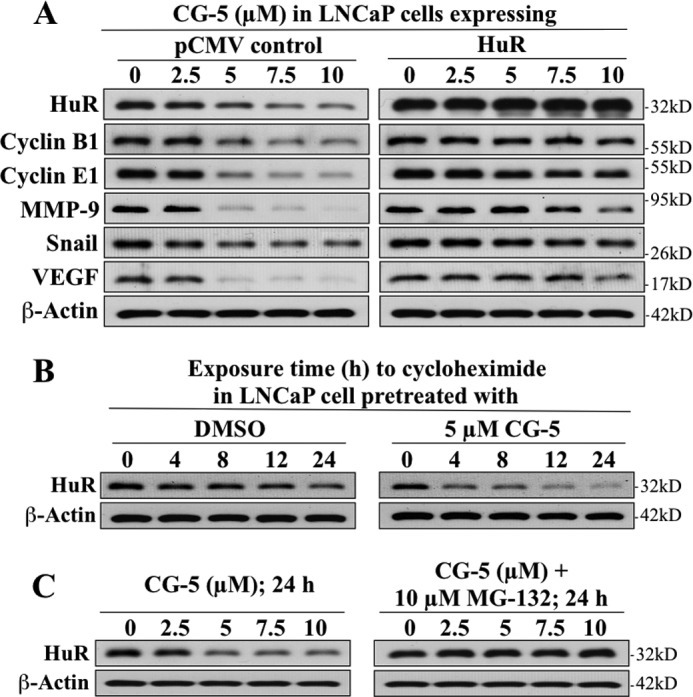
Effects of CG-5 on the expression of HuR-targeted signaling proteins and HuR protein stability. A, validation of the role of HuR in mediating the suppressive effect of CG-5 on various tumor-promoting signaling proteins encoded by HuR-targeted mRNAs, including cyclin B1, cyclin E1, MMP9, Snail, and VEGF in LNCaP cells. Ectopic expression of full-length HuR protected cells from CG-5-mediated suppression of these proteins. B, cycloheximide chase assays of the effect of CG-5 on the half-life of HuR protein in LNCaP cells. Cells were treated with 5 μm CG-5 for 12 h, followed by exposure to 100 μg/ml cycloheximide for the indicated time intervals. C, protective effect of proteasome inhibitor MG132 on CG-5-mediated HuR degradation. LNCaP cells were treated with CG-5 at the indicated concentration, alone or in combination with 10 μm MG132, for 24 h.
Evidence indicates that this CG-5-induced down-regulation of HuR was attributable to reduced protein stability through proteasomal degradation. As shown, CG-5 significantly shortened the half-life of HuR relative to the dimethyl sulfoxide (DMSO) control in LNCaP cells cotreated with cycloheximide (Fig. 2B), and the proteasome inhibitor MG132 effectively blocked the drug-induced ablation of HuR protein (Fig. 2C).
As earlier reports indicated that heat shock- and hypoxia-induced HuR proteolysis is preceded by its cytoplasmic translocation (5, 6), we examined the effect of leptomycin B, an inhibitor of CRM1-dependent nuclear export, on CG-5-mediated HuR ablation. As shown in Fig. 3A, cotreatment of LNCaP cells with leptomycin B blocked the ability of CG-5 to facilitate HuR depletion, indicating the dependence of this cellular response on nuclear export.
FIGURE 3.

CG-5-induced HuR degradation is preceded by nuclear export in LNCaP cells. A, the nuclear export inhibitor, leptomycin B (LMB), protected cells from CG-5-mediated HuR degradation. B, dose- and time-dependent suppressive effects of CG-5 on HuR expression in the cytoplasm versus nucleus. β-Actin and histone H3 signals indicate the quality of the cytoplasmic and nuclear fractionations, respectively. C, dose-dependent effect of 2-DG on the cytoplasmic and nuclear expression of HuR protein after 48 h of treatment. D, left panel, Western blot analysis of the expression of the two paralogs of β-TrCP, β-TrCP1 and β-TrCP2, in LNCaP cells. Right panel, dose-dependent effects of CG-5 on cytoplasmic and nuclear expression levels of putative HuR-targeted E3 ligases, including β-TrCP1, β-TrCP2, and MDM2.
Pursuant to this finding, we further investigated the spatial and temporal dynamics of this glycolysis inhibition-induced HuR degradation by examining the dose- and time-dependent suppressive effect of CG-5 on HuR expression in the cytoplasm versus the nucleus of LNCaP cells. Exposure to CG-5 led to dose- and time-dependent reductions in HuR that were more rapid in the cytoplasm than in the nucleus (Fig. 3B). This rapid clearance of cytoplasmic HuR was also noted in 2-DG-treated cells (Fig. 3C). Together, these findings suggest that CG-5-induced proteolysis of HuR occurred in the cytoplasm and that nuclear export represented a rate-limiting step for this cellular event.
β-TrCP1 Targets HuR for Degradation in the Cytoplasm
It has been reported that HuR stability correlates with MDM2 expression levels in hepatocellular carcinoma cells as MDM2 stabilizes HuR through neddylation (15). However, exposure of LNCaP cells to CG-5 had no appreciable effect on MDM2 expression in either the cytoplasmic or nuclear fraction, refuting the involvement of MDM2 in CG-5-mediated HuR degradation (Fig. 3D). We thus turned our attention to the potential involvement of the E3 ubiquitin ligase β-TrCP in CG-5-mediated HuR proteolysis in light of our previous finding that β-TrCP-facilitated protein degradation represents a cellular response to energy restriction in cancer cells (21). Mechanistic evidence indicates that the effect of CG-5 on β-TrCP up-regulation was attributable to its ability to promote the neddylation-dependent destabilization of Skp2, the E3 ligase responsible for β-TrCP degradation (22).
β-TrCP consists of two orthologs. β-TrCP1 contains 569 amino acid residues, and β-TrCP2 comprises 542 amino acid residues (23), with estimated molecular masses of 69 kDa and 62 kDa, respectively. As shown, the LNCaP cell expressed both orthologs, and equally important, although there is a high degree of similarity in sequences, no cross-reactivity between β-TrCP1 and β-TrCP2 was noted with either antibody, indicating the high degree of specificity of these antibodies (Fig. 3D, left panel). Western blotting indicates that CG-5 exhibited differential effects on the expression and/or cellular localization of the two β-TrCP paralogs, β-TrCP1 and β-TrCP2, in LNCaP cells. As shown, CG-5 treatment led to a concentration-dependent accumulation of β-TrCP1 in the cytoplasm, accompanied by gradual decreases in nuclear β-TrCP1, whereas the expression of β-TrCP2, which was exclusively cytoplasmic, was unaffected (Fig. 3D, right panel). Moreover, CG-5-induced increases in cytoplasmic β-TrCP1 expression were inversely correlated with the concomitant reduction in HuR expression (Fig. 3D). This selective up-regulation of β-TrCP1 suggests its unique role in CG-5-mediated HuR down-regulation.
The role of β-TrCP1 in targeting HuR for degradation in response to CG-5 in LNCaP cells was borne out by several lines of evidence. First, ectopic expression of β-TrCP1, as manifested by reduced expression levels of the β-TrCP substrates β-catenin and cyclin D1, promoted the ablation of HuR in a manner similar to that of CG-5 (Fig. 4A). Second, both dominant-negative inhibition and siRNA-mediated knockdown of β-TrCP1 protected against the suppressive effect of CG-5 on HuR expression. As ΔF-β-TrCP1, an F-box-deleted, dominant-negative mutant form of β-TrCP1 (24), competes with endogenous β-TrCP for substrate binding in a nonproductive manner, enforced expression of ΔF-β-TrCP1 abolished CG-5-facilitated HuR degradation (Fig. 4B). This protective effect was also noted with β-TrCP1 knockdown using two independent siRNAs (Fig. 4C), suggesting the critical role of the physical interaction between β-TrCP1 and HuR in CG-5-induced down-regulation of HuR. Cycloheximide chase assays revealed that silencing of β-TrCP1 increased the protein stability of HuR in CG-5-treated LNCaP cells (Fig. 4D).
FIGURE 4.

Evidence that β-TrCP1 is involved in CG-5-mediated HuR degradation in LNCaP cells. A, effect of ectopically expressed full-length β-TrCP1 versus pCMV vector control on the expression of HuR and known β-TrCP substrates, cyclin D1 and β-catenin. B, protective effect of ΔF-β-TrCP1-Myc, a dominant-negative mutant form of β-TrCP1, against CG-5-mediated HuR degradation. C, protective effects of siRNA-mediated knockdown of β-TrCP1 by using two independent siRNAs (#1 and #2) on CG-5-mediated HuR degradation. D, cycloheximide chase analysis of the effect of siRNA-mediated knockdown of β-TrCP1 on HuR protein stability in response to DMSO versus 5 μm CG-5 LNCaP cells. Cells were electroporated with scrambled or β-TrCP1 siRNA. After incubation for 48 h, these cells were treated with 5 μm CG-5 for 12h, followed by exposure to 100 μg/ml cycloheximide for the indicated time intervals.
Coimmunoprecipitation analysis in LNCaP cells ectopically expressing HA-tagged ubiquitin (HA-Ub) and/or FLAG-tagged HuR (FLAG-HuR) indicates that CG-5 facilitated HuR ubiquitination (Fig. 5, A and B, left panel) and that ΔF-β-TrCP1-mediated HuR stabilization was attributable to the inhibition of CG-5-induced HuR ubiquitination (B, right panel). Third, physical interactions between β-TrCP1 and HuR were verified in two ways: co-immunoprecipitation and in vitro GST pull-down assays. LNCaP cells were doubly transfected with plasmids expressing either HuR-FLAG or β-TrCP1-Myc, and exposed to DMSO or 5 μm CG-5 in the presence of 10 μm MG132 for 24 h. Cell lysates were immunoblotted with anti-FLAG or anti-Myc antibodies (input) or immunoprecipitated by anti-FLAG affinity matrix. Equal amounts of the immunoprecipitated proteins were subjected to Western blotting with anti-Myc antibodies. As shown, relative to the DMSO control, CG-5 increased binding of β-TrCP1 to HuR (Fig. 6A). Moreover, the direct association of endogenous HuR and β-TrCP1 was demonstrated by using both anti-HuR (Fig. 6B, left panel) and anti-β-TrCP1 (right panel) antibodies to perform the coimmunoprecipitation analysis of the protein complex. As shown, in both cases, CG-5 enhanced the β-TrCP-HuR complex formation.
FIGURE 5.

CG-5 facilitated β-TrCP1-mediated HuR ubiquitination in LNCaP cells. A, coimmunoprecipitation analysis of the time-dependent effect of CG-5 (5 μm) on HuR ubiquitination. LNCaP cells ectopically expressing HA-Ubiquitin (HA-Ub) were treated with 5 μm CG-5 for 12 or 36 h, followed by cotreatment with proteasome inhibitor MG132 for an additional 12 h. Equal amounts of cell lysates were immunoprecipitated (IP) with anti-HuR antibody and protein A/G-agarose followed by immunoblotting (WB) with anti-HA antibodies. B, ectopic expression of ΔF-β-TrCP1-Myc blocked CG-5-mediated HuR ubiquitination. LNCaP cells cotransfected with plasmids encoding HA-Ub, HuR-FLAG, and ΔF-β-TrCP1-Myc or empty vector (pCMV) were treated with 5 μm CG-5 for 12 or 36 h, followed by cotreatment with proteasome inhibitor MG132 for an additional 12 h. Equal amounts of cell lysates were immunoprecipitated with anti-FLAG antibody and protein A/G-agarose followed by immunoblotting with anti-HA and anti-FLAG antibodies.
FIGURE 6.

Evidence that HuR physically interacts with β-TrCP1. A, coimmunoprecipitation analysis revealed the association of HuR with β-TrCP1 in response to CG-5 treatment. LNCaP cells ectopically expressing both FLAG-tagged HuR and Myc-tagged β-TrCP1 were treated with 5 μm CG-5 for 12 h, followed by cotreatment with 10 μm MG132 for an additional 12 h. Equal amounts of cell lysates were immunoprecipitated (IP) with anti-FLAG antibody and protein A/G-agarose followed by immunoblotting (WB) with anti-Myc and anti-FLAG antibodies. B, coimmunoprecipitation analysis showed the association of endogenous HuR and β-TrCP1 in response to CG-5 treatment. Cells were treated according to the aforementioned procedure. Equal amounts of cell lysates were immunoprecipitated with anti-HuR (left panel) and anti-β-TrCP1 (right panel) antibodies and protein A/G-agarose, followed by immunoblotting with anti-β-TrCP1 and anti-HuR antibodies, respectively. C, in vitro pull-down of HuR by bacterially expressed GST-β-TrCP1. Equal amounts of LNCaP cell lysates were incubated with recombinant GST, GST-β-TrCP1, or GST-Skp2 immobilized onto glutathione beads. The resulting complexes were washed, centrifuged, and subjected to immunoblotting analysis with HuR antibody (right panel). One-tenth volume of cell lysates were collected as input and probed with HuR and β-actin antibodies, and recombinant GST-fusion proteins were purified and probed with GST antibody (left panel).
For the GST pull-down assays, GST, GST-β-TrCP1, and a negative control, GST-Skp2, were expressed in E. coli cells and purified by immobilization onto glutathione beads that were then incubated with LNCaP cell lysates. The resulting complexes were washed and immunoblotted with HuR antibodies. The results show that HuR was pulled down by bacterially expressed GST-β-TrCP1 but not by GST or GST-Skp2 (Fig. 6C).
β-TrCP1 Targets the HuR Nucleocytoplasmic Shuttling (HNS)-RRM3 motifs to Facilitate HuR Degradation
HuR contains three RRMs and a hinge region encompassing the HNS motif, each of which plays a role in regulating the biological function, cellular localization, and/or protein stability of HuR through posttranslational modifications by distinct kinases (2). To understand the mode of HuR recognition by β-TrCP1, we investigated the interaction of ectopically expressed β-TrCP1-Myc with a series of FLAG-tagged, truncated HuR mutants, including those with deleted HNS-RRM3 (ΔHNS-RRM3), deleted RRM1 (ΔRRM1), and deleted RRM1 and RRM2 (ΔRRM1/2) versus wild-type HuR by coimmunoprecipitation. As shown, deletion of the HNS-RRM3 motif, but not that of RRM1 or RRM1/2, abrogated the ability of HuR to bind β-TrCP1 (Fig. 7A). Furthermore, ΔHNS-RRM3 was resistant to CG-5-mediated degradation, whereas ΔRRM1 and ΔRRM1/2 were as sensitive as endogenous HuR to proteolysis in CG-5-treated cells (Fig. 7B).
FIGURE 7.
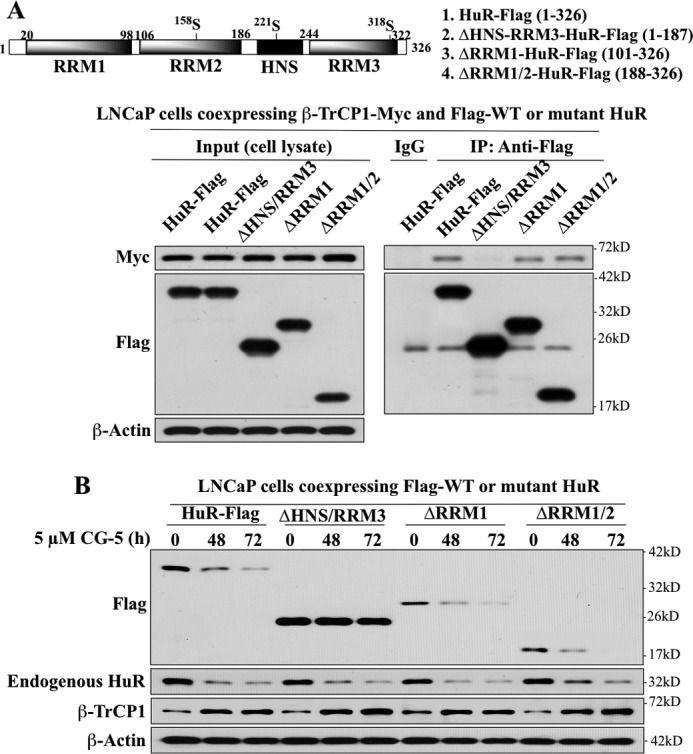
Evidence that the HNS-RRM3 motif is involved in the recognition and degradation of HuR by β-TrCP1. A, upper panel, schematic representation of the structures of wild-type and various truncated mutant forms of HuR-FLAG. Lower panel, coimmunoprecipitation analysis of the interaction of wild-type HuR versus various truncated HuR mutants with β-TrCP1. LNCaP cells were transiently cotransfected with Myc-tagged β-TrCP1 and FLAG-tagged wild-type HuR or individual truncated mutants for 48 h. Immunoprecipitation (IP) with anti-FLAG-agarose conjugates and immunoblotting with anti-Myc and anti-FLAG antibodies were performed as described in Fig. 6. B, time-dependent effect of CG-5 on the degradation of FLAG-tagged HuR and various truncated HuR mutants versus endogenous HuR.
Kinase Dependence of CG-5-mediated HuR Proteolysis
The above findings demonstrated a pivotal role for the HNS-RRM3 motif in regulating the metabolic fate of HuR in response to CG-5. This finding, however, contrasts with a previous report showing that Lys-182 within the RRM2 was involved in promoting heat shock-mediated HuR degradation (5). The HNS-RRM3 motif has been reported to contain multiple phosphorylation sites for controlling the cellular localization and RNA binding of HuR (2), each of which is targeted by a distinct kinase, including Ser-202, which is phosphorylated by cyclin-dependent kinase (CDK) 1 (25), Ser-221 by PKCα and δ (26, 27), Ser-242 by an undetermined kinase (28), and Ser-318 by PKCδ (27) (Fig. 8A). To gain insight into how the HNS-RRM3 motif regulates HuR degradation, we examined the effect of various kinase inhibitors on the protein stability of HuR in CG-5-treated LNCaP cells, including those of the aforementioned CDK (AT7519) and PKC (GF109203X) as well as p38 MAP kinase (SB203580), glycogen synthase kinase (GSK)-3β (SB216763), and IKK (BAY11–7028). Among these kinases, CDK, PKC, and p38 have been reported to regulate the cellular distribution of HuR in response to different stress stimuli (4, 25–27), whereas GSK-3β and IKKα are involved in β-TrCP recognition of various target proteins (20, 29). As shown, GF109203X and BAY11–7082 were effective in blocking CG-5-mediated HuR proteolysis, whereas the other three inhibitors examined showed no appreciable protection (Fig. 8B). Pursuant to this finding, we examined the effect of CG-5 on the phosphorylation of PKCα, PKCδ, and IKKα. Western blotting indicates a dose-dependent increase in the phosphorylation levels of PKCα and IKKα, whereas no significant change in PKCδ phosphorylation, which was constitutively high, was noted (Fig. 8C). Together, these findings suggest the involvement of PKC and IKKα in facilitating CG-5-induced HuR degradation, presumably through the regulation of the cytoplasmic translocation and β-TrCP1 recognition of HuR, respectively.
FIGURE 8.

Evidence that PKCα and IKKα play pivotal roles in CG-5-facilitated HuR degradation in LNCaP cells. A, the amino acid sequence of the HNS-RRM3 motif of HuR. B, effects of various kinase inhibitors, including SB216763 (GSK3β), GF109203X (PKC), SB203580 (p38), AT7519 (CDK), and BAY11–7082 (IKKα) on CG-5-mediated HuR degradation. Cells were treated with CG-5 in combination with individual kinase inhibitors at the indicated concentrations for 24 h. C, dose-dependent effect of CG-5 on the phosphorylation status of PKCα, PKCδ, and IKKα in LNCaP cells. D, coimmunoprecipitation analysis of the time-dependent effect of 5 μm CG-5 on PKC-mediated serine phosphorylation of HuR. Cells were treated with 5 μm CG-5 for 12 or 24 h followed by cotreatment with 10 μm MG132 for an additional 12 h. Equal amounts of cell lysates were immunoprecipitated (IP) with anti-HuR antibody and protein A/G-agarose followed by immunoblotting (WB) with anti-p-Ser PKC substrate and anti-HuR antibodies. E, effects of siRNA-mediated silencing of PKCα versus PKCδ on CG-5-mediated HuR degradation. Cells were transfected with 100 nm scrambled (Scrb), PKCα (left panel), or PKCδ (right panel) siRNA for 48 h and were then treated with 5 μm CG-5 for 24 h. F, dominant-negative inhibition of IKKα by IKK2M protected HuR from CG-5-mediated degradation.
To demonstrate that HuR was phosphorylated by PKC in response to CG-5, HuR was immunoprecipitated from LNCaP cells after treatment with 5 μm CG-5 in the presence of the proteasome inhibitor MG132 for different time intervals and was immunoblotted with a p-Ser-PKC substrate antibody that recognizes phosphorylated PKC consensus motifs. The results indicate that exposure of cells to CG-5 led to a time-dependent increase in PKC-dependent HuR phosphorylation (Fig. 8D). To discern the role of PKCα versus PKCδ, we examined the effects of siRNA-mediated knockdown of these two isoforms on CG-5-mediated HuR degradation. The silencing of PKCα blocked the ability of CG-5 to ablate HuR, whereas that of PKCδ had no appreciable effect (Fig. 8E). To validate the role of IKKα, we transfected LNCaP cells with a plasmid encoding the dominant-negative IKKα mutant IKK2M (19) or the empty pCMV vector as control to examine the effects on CG-5-facilitated HuR degradation. As shown, enforced expression of IKK2M blocked drug-induced reduction in HuR protein expression (Fig. 8F). Together, these findings implicate PKCα and IKKα in regulating the metabolic fate of HuR in response to CG-5.
Role of PKCα in CG-5-induced HuR Nuclear Export
Two PKC phosphorylation sites have been reported within the HNS-RRM3 motif of HuR, i.e. Ser-221 for PKCα/δ and Ser-318 for PKCδ (26, 27). To identify which of these two sites was targeted by PKCα in response to CG-5, these serine residues were individually replaced by an alanine by site-directed mutagenesis to generate two mutant forms of FLAG-HuR, S221A and S318A. In addition, Ser-158, a PKCα phosphorylation site in RRM2 (26), was also mutated to provide a negative control, S158A. Analysis of the metabolic fate of these mutants versus wild-type FLAG-HuR in CG-5-treated LNCaP cells revealed that S318A was resistant to CG-5-induced HuR degradation, whereas S158A and S221A were as susceptible to the drug effect as their wild-type counterpart and endogenous HuR (Fig. 9A). Coimmunoprecipitation analysis indicates that mutation of Ser-318 to Ala abrogated the ability of HuR to bind PKCα (Fig. 9B). Moreover, in contrast to the wild-type HuR, S318A was localized exclusively to the nucleus (Fig. 9C, DMSO control). This nuclear sequestration thus protected the mutant HuR from β-TrCP1-mediated degradation in the cytoplasm. Together, these findings demonstrate that PKCα-mediated Ser-318 phosphorylation plays a crucial role in CG-5-mediated HuR turnover by facilitating HuR nuclear export.
FIGURE 9.

Mutational analyses demonstrating the crucial role of Ser-318 in CG-5-mediated HuR cytoplasmic translocation and degradation in LNCaP cells. A, effect of alanine substitution at Ser-158, Ser-221, and Ser-318 on CG-5-mediated HuR degradation. B, coimmunoprecipitation analysis of the effect of the Ser-318 → Ala substitution on HuR binding to PKCα. LNCaP cells ectopically expressing FLAG-tagged WT or S318A mutant form of HuR were treated with 5 μm CG-5 for 24 h. Immunoprecipitation (IP) with anti-FLAG-agarose conjugates and immunoblotting (WB) with anti- PKCα and anti-FLAG antibodies were performed as described in Fig. 6. C, immunocytochemical analysis of the cellular distribution and turnover of FLAG-tagged WT versus the S318A mutant of HuR in CG-5-treated cells. Scale bars = 10 μm.
Identification of the β-TrCP1 Recognition Site in HuR
Pursuant to our finding that HNS-RRM3 is critical for β-TrCP1-facilitated degradation of HuR, we sought to identify the β-TrCP1 recognition site within the HNS-RRM3 motif. In many of its target proteins, β-TrCP recognizes the consensus sequence of DSGXnS (X is any amino acid, n = 2–4) after phosphorylation of both Ser residues by different kinases (29). Although the HNS-RRM3 motif lacks this DSG destruction domain (Fig. 6A), it contains a partial sequence, 296EEAMAIAS304, that resembles an unconventional β-TrCP recognition sequence reported in the Cdc2 inhibitory kinase Wee1 (116EEGFGS121) (30) and cyclin D1 (279EEVDLACT286) (20), in which the upstream Ser of the recognition motif is replaced by a phosphomimetic amino acid, Glu.
The involvement of 296EEAMAIAS304 in regulating β-TrCP recognition was corroborated by a series of mutational analyses. As shown by coimmunoprecipitation, the phosphomimetic substitution of Ser-304 by Glu (S304E) mimicked the ability of CG-5 to enhance HuR binding to β-TrCP1-Myc in the presence of MG132 (Fig. 10A), indicating the obligatory role of Ser-304 phosphorylation in mediating the interaction between HuR and β-TrCP1. In contrast, replacement of Glu-296/297 or Ser-304 by alanine (E296/297A and S304A, respectively) abolished CG-5-mediated HuR binding to β-TrCP1 (Fig. 10B), and, subsequently, CG-5-facilitated HuR degradation (Fig. 10C).
FIGURE 10.

Identification of the β-TrCP1 recognition motif in HuR in LNCaP cells via mutational analyses. A, phosphomimetic substitution of Ser-304 by Glu promotes the binding of HuR to β-TrCP1. LNCaP cells were cotransfected with Myc-tagged β-TrCP1 and FLAG-tagged WT or S304E mutant of HuR in the presence of 10 μm MG132 for 48 h. Immunoprecipitation (IP) with anti-FLAG-agarose conjugates and immunoblotting (WB) with anti-Myc and anti-FLAG antibodies were performed as described in Fig. 6. B, coimmunoprecipitation analysis revealed that the E296A/E297A and S304A mutants were incapable of binding to β-TrCP1. LNCaP cells ectopically expressing FLAG-tagged WT, E296A/E297A, or S304A mutants of HuR were treated with 5 μm CG-5 for 12 h followed by a cotreatment with 10 μm MG132 for an additional 12 h. C, the E296A/E297A and S304A mutants of HuR are resistant to CG-5-mediated proteolysis. D, immunocytochemical analysis of the cellular distribution and turnover of FLAG-tagged WT HuR versus the S304A mutant in CG-5-treated cells. Scale bars = 10 μm. E, coimmunoprecipitation analysis revealed that the S304A mutant was incapable of binding to IKKα.
Moreover, the effects of the Ser-304 → Ala substitution on the cellular distribution and metabolic fate of the resulting mutant HuR was examined by immunocytochemistry (Fig. 10D). The data show that, in untreated cells (DMSO control), S304A was located in the nucleus and, to a lesser extent, the cytoplasm in a manner similar to that of the wild-type HuR. In contrast to wild-type HuR, however, treatment with CG-5 led to a cytoplasmic accumulation of S304A accompanied by its disappearance from the nucleus, indicating its ability to be exported from the nucleus, and its resistance to CG-5-mediated degradation. This behavior of S304A was distinctly different from that of S318A, which was protected from CG-5-induced degradation by nuclear sequestration.
In light of the proposed role of IKKα in β-TrCP recognition of its target proteins and our finding that IKKα is important for CG-5-facilitated HuR degradation, we validated the role of IKKα in mediating Ser-304 phosphorylation by coimmunoprecipitation analysis. The results show that, although CG-5 increased the association of wild-type HuR with IKKα, S304A did not respond to CG-5 treatment with increased IKKα binding (Fig. 10E).
DISCUSSION
In this study, we report a novel mechanism by which glycolysis inhibition facilitates ubiquitin-dependent HuR degradation. We obtained evidence that, in response to the glucose transporter inhibitor CG-5, HuR translocates to the cytoplasm, where it is targeted by β-TrCP1 for degradation. This nuclear export of HuR was facilitated by PKCα-mediated phosphorylation at Ser-318. Although Ser-318 was identified previously as a PKCδ phosphorylation site in angiotensin II-induced HuR export (27), our data indicate that PKCα was responsible for Ser-318 phosphorylation in CG-5-facilitated HuR shuttling on the basis of the findings that silencing of PKCδ had no appreciable effect on the drug-induced HuR degradation (Fig. 8E) and that replacement of Ser-318 with alanine abolished the ability of the resulting HuR to bind PKCα and to undergo nuclear export (Fig. 9, B and C).
Up-regulation of β-TrCP expression represents a cellular response to energy restriction in cancer cells (21), and our data indicate that increased β-TrCP1 expression led to proteasomal degradation of HuR in CG-5-treated LNCaP cells. The differential effect of CG-5 on the expression and cellular distribution of β-TrCP1 versus β-TrCP2 was noteworthy (Fig. 3D). It has been reported that β-TrCP1 and β-TrCP2 are predominantly localized to the nucleus and cytoplasm, respectively (31, 32). In this study, we found that CG-5 facilitated the cytoplasmic accumulation of β-TrCP1 with concomitant decreases in the nuclear counterpart. It is noteworthy that as β-TrCP1 is devoid of a canonical nuclear localization signal. Its transport in and out of the nucleus is regulated by the nuclear phosphoprotein hnRNP-U, which serves as a pseudosubstrate for β-TrCP1 to prevent untoward engagements of this E3 ligase with target proteins (32). From a mechanistic perspective, the effect of CG-5 on β-TrCP1 expression and localization is in line with the previous finding that in the course of β-TrCP1-mediated degradation of cytoplasmic substrates such as IκBa and β-catenin, hnRNP-U would shuttle β-TrCP1 from the nucleus to the cytoplasm to facilitate the targeted degradation (32).
This mechanistic link was supported by the ability of ectopically expressed β-TrCP1 to mimic CG-5 to promote HuR degradation (Fig. 4A) and by the protective effect of dominant-negative inhibition and siRNA-mediated knockdown of β-TrCP1 on CG-5-induced HuR ubiquitination and/or degradation (Fig. 4, B and C, and Fig. 5). Substrate targeting of HuR by β-TrCP1 was further verified by coimmunoprecipitation and in vitro GST pull-down assays to demonstrate direct physical interactions (Fig. 6) and by the identification of a β-TrCP1 recognition site in the HNS-RRM3 via mutational analyses (Fig. 10). Although HuR does not contain a DSG destruction motif common to many β-TrCP substrates, there exists an unconventional motif, 296EEAMAIAS304, near the C terminus in which Glu-297 acts as a phosphomimetic of the upstream serine residue. Furthermore, we obtained evidence that IKKα-dependent phosphorylation at Ser-304 is crucial to the binding of HuR to β-TrCP1, as the S304A mutant lost its ability to bind β-TrCP1 and IKKα.
As a stress response protein, HuR is subjected to posttranslational modifications at different sites in response to diverse stress stimuli (33). Many of these stress signals, including oxidative stress (4), UV light (7), amino acid starvation (7), and polyamine depletion (8), result in the cytoplasmic accumulation of HuR and the subsequent stabilization of target mRNAs, suggesting the functional role of HuR in adaptive response to environmental changes to maintain cell homeostasis. However, other stress signals, such as heat shock (5), hypoxia (6), and staurosporine (9), were reported to reduce HuR expression through proteasomal degradation or caspase-mediated cleavage. The function of this HuR proteolysis, however, differs in the context of different types of stress. For example, the transient decrease in HuR expression through proteasomal degradation in response to heat shock was thought to confer cytoprotection against thermal stress by slowing down cell proliferation to allow cells to repair heat damage (5). In contrast, caspase-facilitated cleavage of HuR was shown to amplify the apoptotic response to staurosporine by enhancing apoptosome activity (9). In light of the role of this mRNA-stabilizing protein in regulating the expression of many oncogenic proteins (Fig. 2A), the ability of CG-5 to facilitate β-TrCP1-mediated degradation of HuR, along with other β-TrCP targets such as cyclin D1, β-catenin, and Sp1, might contribute to the antiproliferative activity of the drug.
Mechanistically, CG-5-mediated proteasomal degradation of HuR differs from that of heat shock in the mode of regulation. Under thermal stress, HuR degradation was facilitated by ubiquitination at Lys-182 located within the RRM2 (5). Although this degradation could be blocked by checkpoint kinase 2-mediated phosphorylation at Ser-88, Ser-100, and/or Thr-118, the kinase and E3 ligase responsible for this ubiquitination remain undefined. In contrast, in the course of β-TrCP1-facilitated degradation in response to CG-5, HuR is subjected to posttranslational modifications at Ser-318 and Ser-304, both located within the RRM3, by PKCα and IKKα, respectively. It has been reported previously that HuR function is regulated by a series of DNA damage response kinases (33). As IKKα is also involved in mediating DNA damage response downstream of ataxia-telangiectasia mutated (34), this finding adds IKKα to the list of DNA damage response kinases involved in HuR regulation.
In light of its tumorigenic role, HuR represents a therapeutically relevant target (1). Several natural product agents have been identified to inhibit HuR function through interference with HuR dimerization or HuR binding (35) to the AU-rich element of TNF-α mRNA (36). From a therapeutic perspective, the ability of CG-5 to ablate HuR through proteasomal degradation underlies the translational potential of this novel glucose transporter inhibitor as a cancer preventive/therapeutic agent, which is currently under investigation in various tumor models.
This work was supported by National Cancer Institute Public Health Service Grant R01CA112250.
- RRM
- RNA recognition motif
- 2-DG
- 2-deoxyglucose
- IKKα
- IκB kinase α
- qRT PCR
- quantitative real-time PCR
- DMSO
- dimethyl sulfoxide
- HNS
- HNS, HuR nucleocytoplasmic shuttling
- CDK
- cyclin-dependent kinase.
REFERENCES
- 1. Srikantan S., Gorospe M. (2012) HuR function in disease. Front. Biosci. 17, 189–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Abdelmohsen K., Gorospe M. (2010) Posttranscriptional regulation of cancer traits by HuR. Wiley Interdiscip. Rev. RNA 1, 214–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hinman M. N., Lou H. (2008) Diverse molecular functions of Hu proteins. Cell Mol. Life Sci. 65, 3168–3181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Song I. S., Tatebe S., Dai W., Kuo M. T. (2005) Delayed mechanism for induction of γ-glutamylcysteine synthetase heavy subunit mRNA stability by oxidative stress involving p38 mitogen-activated protein kinase signaling. J. Biol. Chem. 280, 28230–28240 [DOI] [PubMed] [Google Scholar]
- 5. Abdelmohsen K., Srikantan S., Yang X., Lal A., Kim H. H., Kuwano Y., Galban S., Becker K. G., Kamara D., de Cabo R., Gorospe M. (2009) Ubiquitin-mediated proteolysis of HuR by heat shock. EMBO J. 28, 1271–1282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Talwar S., Jin J., Carroll B., Liu A., Gillespie M. B., Palanisamy V. (2011) Caspase-mediated cleavage of RNA-binding protein HuR regulates c-Myc protein expression after hypoxic stress. J. Biol. Chem. 286, 32333–32343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang W., Furneaux H., Cheng H., Caldwell M. C., Hutter D., Liu Y., Holbrook N., Gorospe M. (2000) HuR regulates p21 mRNA stabilization by UV light. Mol. Cell Biol. 20, 760–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zou T., Mazan-Mamczarz K., Rao J. N., Liu L., Marasa B. S., Zhang A. H., Xiao L., Pullmann R., Gorospe M., Wang J. Y. (2006) Polyamine depletion increases cytoplasmic levels of RNA-binding protein HuR leading to stabilization of nucleophosmin and p53 mRNAs. J. Biol. Chem. 281, 19387–19394 [DOI] [PubMed] [Google Scholar]
- 9. Mazroui R., Di Marco S., Clair E., von Roretz C., Tenenbaum S. A., Keene J. D., Saleh M., Gallouzi I. E. (2008) Caspase-mediated cleavage of HuR in the cytoplasm contributes to pp32/PHAP-I regulation of apoptosis. J. Cell Biol. 180, 113–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen C. Y., Xu N., Shyu A. B. (2002) Highly selective actions of HuR in antagonizing AU-rich element-mediated mRNA destabilization. Mol. Cell Biol. 22, 7268–7278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Heinonen M., Bono P., Narko K., Chang S. H., Lundin J., Joensuu H., Furneaux H., Hla T., Haglund C., Ristimäki A. (2005) Cytoplasmic HuR expression is a prognostic factor in invasive ductal breast carcinoma. Cancer Res. 65, 2157–2161 [DOI] [PubMed] [Google Scholar]
- 12. Denkert C., Koch I., von Keyserlingk N., Noske A., Niesporek S., Dietel M., Weichert W. (2006) Expression of the ELAV-like protein HuR in human colon cancer: association with tumor stage and cyclooxygenase-2. Mod. Pathol. 19, 1261–1269 [DOI] [PubMed] [Google Scholar]
- 13. Erkinheimo T. L., Lassus H., Sivula A., Sengupta S., Furneaux H., Hla T., Haglund C., Butzow R., Ristimäki A. (2003) Cytoplasmic HuR expression correlates with poor outcome and with cyclooxygenase 2 expression in serous ovarian carcinoma. Cancer Res. 63, 7591–7594 [PubMed] [Google Scholar]
- 14. Niesporek S., Kristiansen G., Thoma A., Weichert W., Noske A., Buckendahl A. C., Jung K., Stephan C., Dietel M., Denkert C. (2008) Expression of the ELAV-like protein HuR in human prostate carcinoma is an indicator of disease relapse and linked to COX-2 expression. Int. J. Oncol. 32, 341–347 [PubMed] [Google Scholar]
- 15. Embade N., Fernández-Ramos D., Varela-Rey M., Beraza N., Sini M., Gutiérrez de Juan V., Woodhoo A., Martínez-López N., Rodríguez-Iruretagoyena B., Bustamante F. J., de la Hoz A. B., Carracedo A., Xirodimas D. P., Rodríguez M. S., Lu S. C., Mato J. M., Martínez-Chantar M. L. (2012) Murine double minute 2 regulates Hu antigen R stability in human liver and colon cancer through NEDDylation. Hepatology 55, 1237–1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Abdelmohsen K., Srikantan S., Kuwano Y., Gorospe M. (2008) miR-519 reduces cell proliferation by lowering RNA-binding protein HuR levels. Proc. Natl. Acad. Sci. U.S.A. 105, 20297–20302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Guo X., Wu Y., Hartley R. S. (2009) MicroRNA-125a represses cell growth by targeting HuR in breast cancer. RNA Biol. 6, 575–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang D., Chu P. C., Yang C. N., Yan R., Chuang Y. C., Kulp S. K., Chen C. S. (2012) Development of a novel class of glucose transporter inhibitors. J. Med. Chem. 55, 3827–3836 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 19. Hung J. H., Su I. J., Lei H. Y., Wang H. C., Lin W. C., Chang W. T., Huang W., Chang W. C., Chang Y. S., Chen C. C., Lai M. D. (2004) Endoplasmic reticulum stress stimulates the expression of cyclooxygenase-2 through activation of NF-κB and pp38 mitogen-activated protein kinase. J. Biol. Chem. 279, 46384–46392 [DOI] [PubMed] [Google Scholar]
- 20. Wei S., Yang H. C., Chuang H. C., Yang J., Kulp S. K., Lu P. J., Lai M. D., Chen C. S. (2008) A novel mechanism by which thiazolidinediones facilitate the proteasomal degradation of cyclin D1 in cancer cells. J. Biol. Chem. 283, 26759–26770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wei S., Kulp S. K., Chen C. S. (2010) Energy restriction as an antitumor target of thiazolidinediones. J. Biol. Chem. 285, 9780–9791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wei S., Chu P. C., Chuang H. C., Hung W. C., Kulp S. K., Chen C. S. (2012) Targeting the oncogenic e3 ligase skp2 in prostate and breast cancer cells with a novel energy restriction-mimetic agent. PLoS ONE 7, e47298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Suzuki H., Chiba T., Suzuki T., Fujita T., Ikenoue T., Omata M., Furuichi K., Shikama H., Tanaka K. (2000) Homodimer of two F-box proteins βTrCP1 or βTrCP2 binds to IκBα for signal-dependent ubiquitination. J. Biol. Chem. 275, 2877–2884 [DOI] [PubMed] [Google Scholar]
- 24. Latres E., Chiaur D. S., Pagano M. (1999) The human F box protein β-Trcp associates with the Cul1/Skp1 complex and regulates the stability of β-catenin. Oncogene 18, 849–854 [DOI] [PubMed] [Google Scholar]
- 25. Kim H. H., Abdelmohsen K., Lal A., Pullmann R., Jr., Yang X., Galban S., Srikantan S., Martindale J. L., Blethrow J., Shokat K. M., Gorospe M. (2008) Nuclear HuR accumulation through phosphorylation by Cdk1. Genes Dev. 22, 1804–1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Doller A., Huwiler A., Müller R., Radeke H. H., Pfeilschifter J., Eberhardt W. (2007) Protein kinase C α-dependent phosphorylation of the mRNA-stabilizing factor HuR. Implications for posttranscriptional regulation of cyclooxygenase-2. Mol. Biol. Cell 18, 2137–2148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Doller A., Akool el-S, Huwiler A., Müller R., Radeke H. H., Pfeilschifter J., Eberhardt W. (2008) Posttranslational modification of the AU-rich element binding protein HuR by protein kinase CΔ elicits angiotensin II-induced stabilization and nuclear export of cyclooxygenase 2 mRNA. Mol. Cell Biol. 28, 2608–2625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim H. H., Yang X., Kuwano Y., Gorospe M. (2008) Modification at HuR(S242) alters HuR localization and proliferative influence. Cell Cycle 7, 3371–3377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Frescas D., Pagano M. (2008) Deregulated proteolysis by the F-box proteins SKP2 and β-TrCP. Tipping the scales of cancer. Nat. Rev. Cancer 8, 438–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Watanabe N., Arai H., Nishihara Y., Taniguchi M., Watanabe N., Hunter T., Osada H. (2004) M-phase kinases induce phospho-dependent ubiquitination of somatic Wee1 by SCFβ-TrCP. Proc. Natl. Acad. Sci. U.S.A. 101, 4419–4424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lassot I., Ségéral E., Berlioz-Torrent C., Durand H., Groussin L., Hai T., Benarous R., Margottin-Goguet F. (2001) ATF4 degradation relies on a phosphorylation-dependent interaction with the SCF(βTrCP) ubiquitin ligase. Mol. Cell Biol. 21, 2192–2202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Davis M., Hatzubai A., Andersen J. S., Ben-Shushan E., Fisher G. Z., Yaron A., Bauskin A., Mercurio F., Mann M., Ben-Neriah Y. (2002) Pseudosubstrate regulation of the SCF(β-TrCP) ubiquitin ligase by hnRNP-U. Genes Dev. 16, 439–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kim H. H., Abdelmohsen K., Gorospe M. (2010) Regulation of HuR by DNA damage response kinases. J. Nucleic Acids 2010, pii: 981487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yoshida K., Ozaki T., Furuya K., Nakanishi M., Kikuchi H., Yamamoto H., Ono S., Koda T., Omura K., Nakagawara A. (2008) ATM-dependent nuclear accumulation of IKK-α plays an important role in the regulation of p73-mediated apoptosis in response to cisplatin. Oncogene 27, 1183–1188 [DOI] [PubMed] [Google Scholar]
- 35. Meisner N. C., Hintersteiner M., Mueller K., Bauer R., Seifert J. M., Naegeli H. U., Ottl J., Oberer L., Guenat C., Moss S., Harrer N., Woisetschlaeger M., Buehler C., Uhl V., Auer M. (2007) Identification and mechanistic characterization of low-molecular-weight inhibitors for HuR. Nat. Chem. Biol. 3, 508–515 [DOI] [PubMed] [Google Scholar]
- 36. Chae M. J., Sung H. Y., Kim E. H., Lee M., Kwak H., Chae C. H., Kim S., Park W. Y. (2009) Chemical inhibitors destabilize HuR binding to the AU-rich element of TNF-α mRNA. Exp. Mol. Med. 41, 824–831 [DOI] [PMC free article] [PubMed] [Google Scholar]