

Background: Mutagenesis was used to gain insights into the structure/function of IP3R channels.
Results: Only 5 of 22 residues mutated proved essential for channel function.
Conclusion: We propose that Ile-2588 and Ile-2589 are at the channel gate. Arg-2596, His-2630, and His-2635 maintain the structure of the pore and facilitate contacts critical for channel gating.
Significance: A revised model of channel gating is proposed.
Keywords: Calcium; Calcium Intracellular Release; Endoplasmic Reticulum (ER); Inositol 1,4,5-Trisphosphate; Ion Channels
Abstract
We have combined alanine mutagenesis and functional assays to identify amino acid residues in the channel domain that are critical for inositol 1,4,5-trisphosphate receptor (IP3R) channel function. The residues selected were highly conserved in all three IP3R isoforms and were located in the cytosolic end of the S6 pore-lining helix and proximal portion of the C-tail. Two adjacent hydrophobic amino acids (Ile-2588 and Ile-2589) at the putative cytosolic interface of the S6 helix inactivated channel function and could be candidates for the channel gate. Of five negatively charged residues mutated, none completely eliminated channel function. Of five positively charged residues mutated, only one inactivated the channel (Arg-2596). In addition to the previously identified role of a pair of cysteines in the C-tail (Cys-2610 and Cys-2613), a pair of highly conserved histidines (His-2630 and His-2635) were also essential for channel function. Expression of the H2630A and H2635A mutants (but not R2596A) produced receptors with destabilized interactions between the N-terminal fragment and the channel domain. A previously unrecognized association between the cytosolic C-tail and the TM 4,5-loop was demonstrated using GST pulldown assays. However, none of the mutations in the C-tail interfered with this interaction or altered the ability of the C-tail to assemble into dimers. Our present findings and recent information on IP3R structure from electron microscopy and crystallography are incorporated into a revised model of channel gating.
Introduction
The inositol 1,4,5-trisphosphate receptor (IP3R)2 Ca2+ channel is the principal mechanism involved in the release of Ca2+ from internal stores elicited by hormones, growth factors, and neurotransmitters (1–3). A family of three IP3R isoforms with multiple splice variants and different tissue distributions have been identified. All of these isoforms share the basic domain architecture consisting of an N-terminal ligand-binding domain (LBD) and a C-terminal channel domain containing six transmembrane domains. The intervening ∼2000 amino acids is referred to as the regulatory or coupling domain. Structural studies on IP3Rs have principally used single particle analysis of EM images (4–7) or x-ray crystal structures of isolated domains (8, 9). The LBD is composed of an inner IP3-binding core (IBC, amino acids 226–604) and a suppressor domain (SD, amino acids 1–225), which decreases the affinity of IP3 binding to the IBC. The IBC consists of two domains: a β-trefoil domain and an α-helical domain. Both the IP3-bound IBC and the SD have been crystallized separately (8, 9), and more recently the crystal structures of the bound and apo forms of the LBD have been reported (10, 11).
A rudimentary picture of how IP3 binding at the N-terminal region may lead to channel opening in the C-terminal region has emerged based on the observations of a direct association of these two domains (12, 13). A consensus model proposes that conformational changes occurring upon IP3 binding to the N-terminal IBC are transmitted via the SD to a helical loop located in the C-terminal domain between transmembrane domains 4 and 5 (S4S5 loop) (14–16). By analogy with other channels, the S4S5 loop is believed to be orientated parallel to the membrane (17) and to compress the cytosolic aspect of the S6 pore-lining helix maintaining the channel in a closed state. IP3 binding is hypothesized to move the S4S5 loop away from the central axis of the channel, thereby allowing the S6 helixes to separate and leading to channel opening. Many aspects of this simple model require additional support, and it is clear that our knowledge of the gating and permeation mechanism of this channel are limited.
Mutagenesis has proved a valuable approach to our understanding of the structure function relationships in many ion channels, but this approach has received only limited application in the case of the IP3R channel (2). The C-terminal end of the S6 pore-lining helix is a particularly important region of the receptor for further investigation. In many other channels critical hydrophobic residues serve to restrict ion flow through the narrow aperture formed at the intersection of the S6 helixes on the cytosolic face of the membrane (18–21). The putative residues at the channel gate in IP3Rs remain to be identified. In the crystal structure of the Kv1.2 channel, it is apparent that several residues at the cytosolic end of the S6 helix make contact with residues from the S4S5 loop of the same subunit (17). Such interactions have now been documented in other voltage-gated ion channels (23–25) but have not been investigated in IP3Rs. Finally, an additional 160 amino acid residues project into the cytoplasm beyond the S6 helix (C-tail). In ryanodine receptors (RyRs), it has been suggested that clusters of negatively charged residues in the proximal portion of the C-tail facilitate the transit and exit of Ca2+ from the ion channel (26). However, the distribution of charged residues in the C-tail of IP3Rs and RyRs are different, and their role in IP3R channel function remains to be evaluated. In the present study, we have used a mutagenesis approach to examine the functional role of highly conserved residues in the distal S6 helix and the proximal region of the C-tail in IP3Rs. The data identify putative gating residues, highlight a critical role for histidines at positions 2630 and 2635, and reveal an interaction between the S4S5 loop and C-tail. Mutation of most of the negatively and positively charged residues in the C-tail had minimal impact on channel function, with the exception of the Arg-2596 residue, which is highly conserved in all IP3Rs and RyRs. Our findings provide information on critical residues in the channel domain and are incorporated into a modified model of channel gating.
EXPERIMENTAL PROCEDURES
Cloning of Mutations in Full-length Type 1 IP3R
The cDNA encoding the IP3R type 1 in pCMV3 was the kind gift of Dr. Thomas Sudof (University of Texas Southwestern Medical Center). All amino acid numbering is with reference to the rat type 1 IP3R (27). The splice variant used in this study was SI(−), SII(+), and SIII(−). All point mutations were made using a QuikChange site-directed mutagenesis kit (Stratagene) utilizing a cassette encompassing the BstBI/XbaI fragment of the type 1 IP3R in pBluescript (Invitrogen). The mutants were confirmed by sequencing, and the BstBI/XbaI-digested inserts were subcloned into the full-length IP3R.
Cloning and Expression of Fusion Proteins Encoding the C-tail and S4S5 Loop
The wild-type and C-tail mutants prepared in pBluescript as described above were used as templates for PCR amplification of amino acids Asp-2590 to Ala-2749 of the C-tail. The NdeI/XhoI-digested PCR fragment was ligated into a similarly digested pET-24b vector (Novagen), which allowed the expression of the C-tail with a C-terminally attached His6 tag. The S4S5 loop corresponding to amino acids Asp-2418 to Arg-2438 was PCR-amplified with primers encoding EcoRI and BamHI. The purified, digested PCR product was ligated into EcoRI/BamHI-digested pGEX-2TK, which allowed expression of the S4S5 loop as an N-terminally tagged GST fusion protein. The plasmid encoding the GST-RyR1 S4S5 loop (28) was a kind gift of Dr. Eun Hui Lee (Catholic University of Korea, Seoul, South Korea). All the constructs were transformed into BL21-DE3 bacteria. The cultures were grown to an optical density of ∼0.6 and induced with 0.2 mm isopropyl β-d-thiogalactopyranoside for 4 h at 37 °C. Frozen and thawed bacterial pellets were resuspended in PBS containing 1 mg/ml lysozyme, EDTA-free protease inhibitor mixture, and 1 mm β-mercaptoethanol. The pellet was disrupted by sonication, and bacterial lysates were used for pulldown experiments. However, in experiments using the cross-linking agents, the His6 C-tail constructs were partially purified by incubation of the bacterial lysates for 1 h at 4 °C with 0.5 ml of Talon resin (Clontech). The Talon beads were washed in PBS containing 50 mm imidazole and eluted in the same buffer containing 250 mm imidazole. The imidazole was removed by passing the eluate through a PD-10 Sephadex G-25 column (GE Healthcare) equilibrated in PBS with 10% glycerol.
GST Pulldown Assays
Bacterial lysates (0.5 mg of protein) containing GST, GST-IP3R1 loop, or GST-RyR1 loop were incubated with 50 μl of glutathione-Sepharose-4B (GE Biosciences) for 2 h. The beads were washed twice in 150 mm NaCl, 20 mm Tris-HCl, pH 8.3, and then incubated for a further 2 h with 0.5 mg of bacterial lysate expressing the C-terminal tail. The beads were washed once in PBS containing an additional 250 mm NaCl and then twice in PBS alone. The amount of C-tail retained on the beads was estimated by immunoblotting with C-tail Ab. The blots were stripped and reprobed with anti-GST Ab to estimate the relative levels of the GST constructs.
Transfection of Cells and Preparation of Microsomal Vesicles
COS-7 cells were grown in 150-mm plates and were cotransfected with DNA encoding IP3R (10 μg) and SERCA2b (10 μg) using LT-1 (Mirus) and Novafector (VennNova, Inc.) transfection reagents as described previously (29). Transfected cells were washed twice in PBS and scraped into isolation buffer (320 mm sucrose, 20 mm K-HEPES, pH 7.8, 0.5 mm EGTA, and 0.5 mm DTT). The cells were lysed by five passes through a 26.5-gauge needle. The lysates were spun at 750 × g for 5 min, and the supernatant was then centrifuged at 115,000 × g for 45 min. The microsomal pellet was resuspended in a buffer containing 320 mm sucrose, 20 mm K-HEPES, pH 7.8, and 50 μm EGTA and stored at −80 °C. In more recent experiments, we have switched to using HEK-293 cells because these cells can be transfected economically and with high efficiency using 10% polyethyleneimine/DNA (ratio 3:1) (30). The flux properties of the isolated HEK-293 cell microsomes are similar to COS cell microsomes (see Fig. 3).
FIGURE 3.

Two histidines in the C-terminal tail are also critical for function. Two highly conserved histidines in the C-tail (His-2630 and His-2635) and the immediately adjacent residues were mutated to alanine and expressed in HEK-293 cells together with SERCA2b. Representative immunoblots of microsomal expression and IP3-sensitive flux responses are shown in A and B, respectively. *, not significantly different from wild type (p > 0.05). In C, the indicated constructs were transiently transfected into IP3R triple knock-out DT40 cells together with dsRed, and single-cell Ca2+ responses to anti-IgM were measured as described in Fig. 2. Representative responses of three cells are shown, and the total number of responding cells is shown in the figure.
45Ca2+ Flux Assays
Assays were performed as previously described (31). Briefly, microsomal vesicles prepared from cells expressing SERCA2b and IP3R were incubated for 25 min at 30 °C in a 200 nm free [Ca2+] buffer supplemented with ATP and an ATP regenerating system, 45Ca2+, 2.5 mm potassium oxalate, and 2 μm ruthenium red. 45Ca2+ uptake was estimated in the absence of any addition or in the presence of 10 μm IP3 or 1 μm A23187. After incubation, microsomes were vacuum-filtered over a 0.3-μm filter (Millipore Corp.) and washed (150 mm KCl). Filters were counted in Budget Solve complete counting mixture (Research Products International Corp.). An unpaired Student's t test was used to assess statistical significance of differences in flux assays of the various mutant constructs. GraphPad QuickCalcs software was used for all statistical analyses.
[3H]IP3 Binding Assays
Ligand binding at a subsaturating concentration (3 nm [3H]IP3) (NEN Radiochemicals, Waltham, MA) was measured with 30 μg of microsomal protein in a final volume of 120 μl in a buffer containing 120 mm KCl, 20 mm Tris-HCl, pH 8.3, 1 mm DTT, and 1 mm EDTA. The samples were incubated on ice for 5 min, and the binding reaction was terminated by centrifugation at 15,000 × g for 10 min. The pellet was solubilized in 0.5 ml of 0.1 m NaOH, and radioactivity was determined in a scintillation counter. Each condition was measured in triplicate, and nonspecific binding was measured in the presence of 10 μm unlabeled IP3.
Digital Imaging of [Ca2+]i in Intact Cells
Transient transfection of DT40 TKO cells was carried out using electroporation as described previously (32) using both IP3R DNA and a plasmid encoding dsRed. The transfected DT40 cells were washed once in a HEPES-buffered physiological saline solution containing 5.5 mm glucose, 137 mm NaCl, 0.56 mm MgCl2, 4.7 mm KCl, 1 mm Na2HPO4, 10 mm HEPES, pH 7.4, 1.2 mm CaCl2, and 1% (w/v) bovine serum albumin. The cells were loaded with 5 μm fura2/acetoxymethyl ester for 20 min in the presence of 100 μm sulfinpyrazone and 0.003% pluronic acid at room temperature. The cells attached to coverslips were placed in 1 ml of buffer on the heated stage (35 °C) of an inverted epifluorescence microscope (40× oil objective) connected to a cooled CCD camera. Ratiometric imaging of fura2 in dsRed-positive cells was used to monitor [Ca2+] as described previously (33).
RESULTS
Gating Residues and Charged Residues in the Distal Portion of the S6 Helix
Fig. 1A shows the primary sequence of the S6 pore-lining helix of type I IP3R and a portion of the subsequent 160 amino acids constituting the C-tail that projects into the cytosol. Analysis of the sequence by several secondary structure programs predicts that the S6 helix may extend as a continuous helix considerably beyond the membrane. There is experimental support for this conclusion from high resolution analysis of EM structures of RyR1 and IP3R1 (7, 34). For comparison, the primary sequence of RyR1 is also shown in Fig. 1A, and the residues that are conserved between all three IP3R and RyR isoforms are indicated. To examine effects on channel function, we mutated selected residues to alanine. All mutants expressed at levels comparable with, or in excess of, the wild-type receptor (Fig. 1B). The channel function of recombinant receptors was evaluated using two independent experimental methods. First, we measured the effect of IP3 in a 45Ca2+ uptake assay using microsomes prepared from cells cotransfected with SERCA2b as described previously (31). In this assay the presence of a functional IP3R channel decreases the SERCA-mediated 45Ca2+ accumulation into oxalate-loaded microsomes (Fig. 1C). Second, we transiently transfected the mutants into a chicken DT40 cell line with a targeted disruption of all three IP3R isoforms (35, 36). Responses to ligation of the B-cell receptor with anti-IgM were measured by Ca2+ imaging (Fig. 2).
FIGURE 1.

The effects of point mutations in the S6 pore-lining helix and C-tail on IP3R channel function. A shows the segment of the rat type I IP3R sequence investigated in the present study. The predicted transmembrane domain and secondary structure elements within the sequence were obtained from TMpred and PSIpred (both available at online). For comparison, the RyR1 sequence and the consensus identical residues in all three IP3R and RyR sequences are also shown. Shaded residues in the IP3R1 sequence indicate sites mutated in the present study or in previously published work (underlined) (29, 31). Residues mutated in RyRs are from Refs. 22, 26, 42, and 55. B shows the expression of IP3R constructs in microsomes determined by immunoblotting. Previous studies have established the absence of endogenous type I IP3R in COS cells (31). C shows that IP3R channel function of wild-type and mutant receptors was measured by quantitating the inhibitory effect of 1 μm IP3 on 45Ca2+ uptake into microsomal vesicles prepared from SERCA2b cotransfected cells as described under “Experimental Procedures.” The data shown are the means ± S.E. of three to eight measurements made on a minimum of two independent microsomal preparations. *, not significantly different from wild type (p > 0.05).
FIGURE 2.

Functional activity of mutants transiently transfected into IP3R null DT40 cells. DT40 TKO cells were transiently transfected with the indicated IP3R constructs by electroporation. Cotransfection with dsRed was used to identify transfected cells. After 48 h the cells were used for Ca2+ imaging experiments as described under “Experimental Procedures.” The cells were stimulated with anti-IgM (1 μg/ml added at arrows). Representative traces from three cells are shown or each mutant (panels a–n), and the total number of responding cells in a minimum of two coverslips from separate transfections is indicated in each panel.
We initially selected to mutate highly conserved residues including two hydrophobic residues at the predicted membrane/cytosol interface and a series of positive and negatively charged residues in the distal portion of S6. Hydrophobic residues located at the narrowest point of the channel where the S6 helices cross over each other are known to play important roles in the gating of other ion channels (18, 20, 21, 37). In a previous study we found that mutation of a candidate highly conserved residue (Phe-2592) had little effect on channel function (29). Figs. 1C and 2 (b and c) show that mutation of either Ile-2588 or Ile-2589 led to complete inactivation of the channel. The proposed location of these residues at the membrane/cytosol interface of the S6 helix suggests that potentially one (or both) residues could function as the channel gate. Of the five negative charges mutated (Asp-2590, Asp-2594, Glu-2598, Glu-2603, and Glu-2604), none resulted in complete inactivation of the channel. When assayed by the 45Ca2+ flux method, significant inhibitory effects were observed particularly for the D2590A mutant (Fig. 1C). However, all the negative charged mutants displayed Ca2+ responses in the imaging measurements (Fig. 2, d–h). The greater heterogeneity of single-cell Ca2+ signals elicited by anti-IgM did not allow any systematic differences to be identified. Although individual mutation of the residues had little effect on channel function, the mutation of all five negative charges led to loss of channel activity (Fig. 2i). Although D2590A gave the largest inhibition (54%) in the 45Ca2+ flux assay, there was no evidence for a critical combination of negative charges based on the analysis of multiple negative charge mutants (data not shown). Of the five positively charged residues mutated (Arg-2596, Lys-2599, Lys-2601, Lys-2602, and Lys-2607), only the R2596A mutant led to loss of channel function (Figs. 1C and 2, j–n). This effect was not simply due to a loss of charge at this position because the R2596K mutation was also inactive (Fig. 1C). It is interesting to note that although the distribution of charged residues are quite different in the distal portion of S6 in RyRs, the Arg-2596 position is highly conserved in all three IP3R and RyR isoforms (Fig. 1A).
Conserved Histidines in the C-tail
The proximal portion of the C-tail, just beyond the S6 helix, contains two highly conserved cysteine residues (Cys-2610 and Cys-2613) that have previously been shown to be essential for IP3R channel function (38). A pair of highly conserved histidines (His-2630 and His-2635) are present C-terminal to these cysteines (Fig. 1A). We chose to mutate these and their immediately adjacent residues. The 45Ca2+ flux data in Fig. 3B and the Ca2+ imaging data in Fig. 3C indicate that the mutation H2630A or H2635A inactivated channel function. However, mutation of the adjacent residues Glu-629, Ile-2631, and Glu-2634 had no significant effects. Mutation of the Asn-2636 residue, adjacent to His-2635, also retained function but had inhibited activity in the 45Ca2+ flux assay that was not apparent in the imaging measurements. The data suggest that in addition to the cysteines, the histidines are also essential residues in the C-tail. Pairs of cysteine followed by a pair of histidines are a common motif in zinc finger domains. However, the spacing of the sequence CX2CX16HX4H is atypical, and TPEN (a zinc chelator) had no effects on the in vitro oligomerization of the C-tail (data not shown).
Binding of the C-tail to S4S5 Loop
A key role for the S4S5 loop has been proposed in the gating mechanism of the IP3R channel (14). In addition to making direct (or indirect) contacts with the N-terminal ligand-binding region of the channel, it has been speculated that the loop may also interact with the C-tail (14) as observed in several other channels (17, 21–25). This possibility was experimentally tested using fusion proteins encoding the C-tail and S4S5 loop. The sequence of the predicted S4S5 loop is shown in Fig. 4A and can be seen to be highly conserved in all three IP3R isoforms. By contrast the S4S5 loop of the RyR is shorter (16 versus 21 amino acids) and contains several residues that are dissimilar from IP3Rs. Fig. 4B shows that the GST fusion protein encoding the IP3R 4,5-loop bound specifically to the IP3R C-tail but not to the GST-RyR 4,5-loop. Five of the mutants shown to be nonfunctional in the flux assays (R2596A, C2610A, C2613A, H2630A, and H2635A) were transposed individually into the fusion protein encoding the C-tail and tested for binding to the GST IP3R 4,5-loop. Fig. 4C shows that none of the point mutations interfered significantly with the binding.
FIGURE 4.

Interaction between the 4,5-loop and the C-tail. A, putative sequences of the highly conserved loop between transmembrane domains 4 and 5 from all three IP3R isoforms and RyR1 are shown. The indicated sequences for IP3R1 and RyR1 were expressed as GST fusion proteins. B, GST alone or GST-IP3R1 loop and GST-RyR1 loop were immobilized on GSH-agarose and incubated with bacterial lysate expressing the C-tail as described under “Experimental Procedures.” Bound C-tail was detected with CT-1 Ab. The input lane was 10 μg of the C-tail lysate. The blots were stripped and reprobed with anti-GST Ab. The data from three experiments were quantitated. C, binding of immobilized GST-IP3R1 loop to bacterial lysates expressing wild-type and mutant C-tails was carried out as described for B. The amount of C-tail bound was estimated by immunoblotting with CT-1 Ab after normalization for differences in expression levels of the constructs determined by analyzing 10-μg aliquots of the input lysates. The data shown are the means ± S.E. of three experiments. D, bacterial lysates expressing His6-tagged wild-type or mutant receptors were partially purified on Talon resin as described under “Experimental Procedures.” Aliquots (3 μg of protein) were incubated for 30 min in a final volume of 50 μl with 125 μm of the noncleavable cross-linker SMPH. The reaction was terminated by the addition of 5 mm Tris-HCl and SDS sample buffer. The reaction was run on 17.5% gels, and cross-linked bands were detected with CT-1 Ab. At lower exposure, the cross-linked bands appear as a closely spaced doublet.
A previously documented property of the C-tail is the ability to self-associate to form dimers (39). A coiled-coil domain has also been identified in the C-tail (amino acids 2694–2721), which forms tetramers in vitro (14). We examined the self-association properties of the wild-type and mutant C-tails in the presence of a noncleavable heterobifunctional cross-linker (SMPH) that cross-links -NH2 and -SH groups in close proximity. As observed previously, a small fraction of dimers survives the denaturing conditions of SDS-PAGE, and the formation of the dimer bands is enhanced in the presence of the cross-linking agent (39) (Fig. 4D). The amount of dimer formed in the presence or absence of cross-linker was not different between the wild-type and mutant C-tails. The accumulation of small amounts of an additional band corresponding to the expected molecular weight of a tetramer was not reproducibly observed.
We tested the hypothesis that gross structural changes may underlie the loss of channel function of the inactivating mutations identified in the present study (R2596A, H2630A, and H2635A). However, the trypsin digestion patterns of the mutants were not different from wild-type receptors (data not shown). Previous studies have found that N-terminal trypsin fragment (fragment I) remains attached to the membrane after cleavage and that mutations that impair the interaction between the LBD and channel domains cause an increased release of fragment I into the supernatant (14, 40). Membrane attachment assays (Fig. 5A) indicate that although the R2596A mutant behaves like wild-type receptors, the attachment of fragment I to membranes is destabilized in the H2630A and H2635A mutants. A similar finding was made by Yamazaki et al. (40) with the C2613S mutant. Previous studies have shown higher [3H]IP3 binding in mutants with defective coupling between ligand binding and channel domains (14). [3H]IP3 binding to microsomes was comparable with wild-type receptor for the R2596A mutant but was significantly higher in the H2630A and H2635A mutants (Fig. 5B).
FIGURE 5.
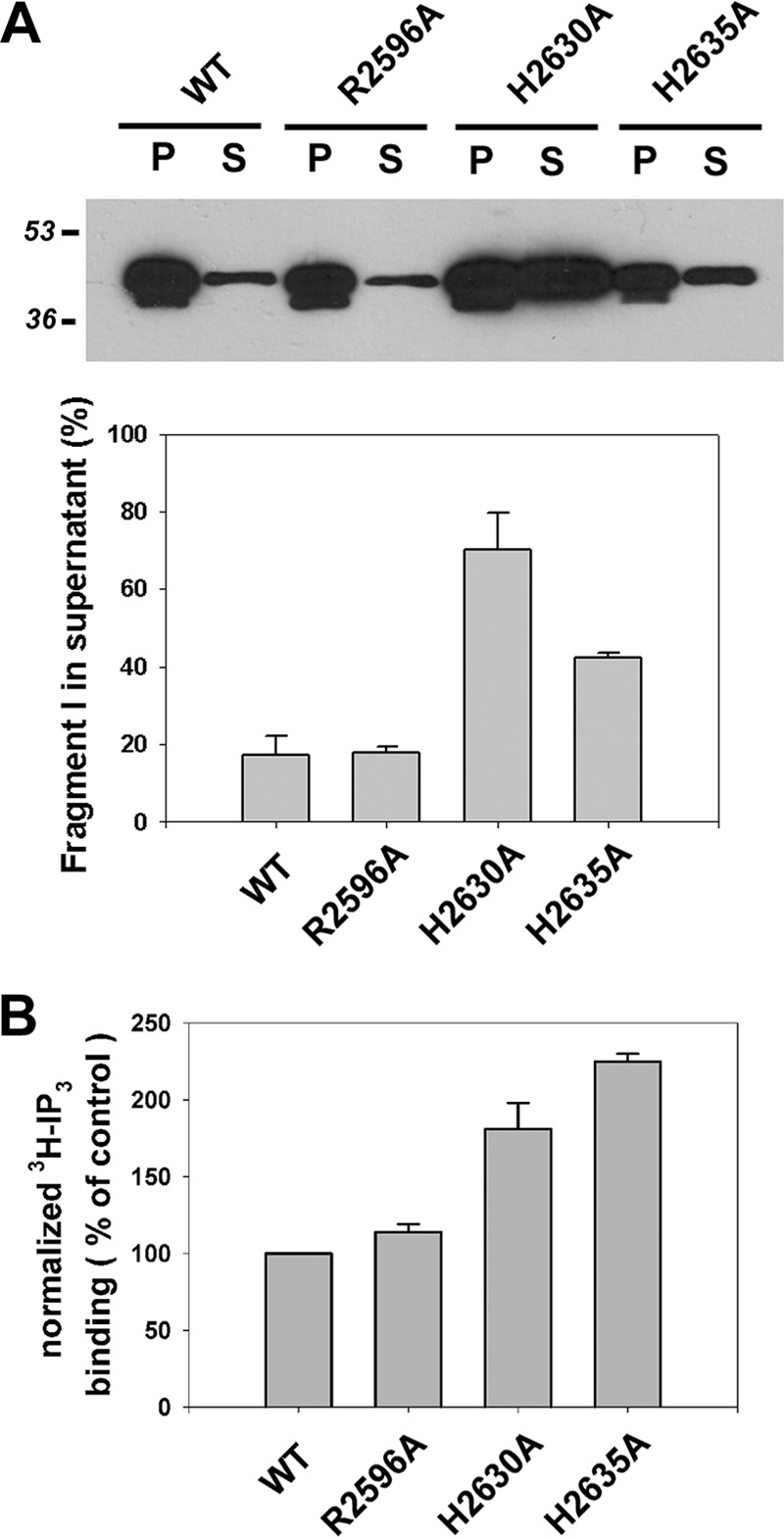
Quantitating membrane attachment of N-terminal trypsin fragment and [3H]IP3 binding in three channel inactivating mutants. A, HEK-293 microsomes (40 μg of protein) expressing wild-type, R2596A, H2630A, and H2635A constructs were incubated with 1 μg of trypsin for 5 min, and cleavage was stopped by adding 10 μg of soybean trypsin inhibitor and 1 mm PMSF. The samples were spun at 100,000 × g in a TLA-100 rotor. SDS sample buffer was added to the pellet (P) and supernatant (S) fractions, and the amount of the N-terminal trypsin fragment in the fractions was determined by immunoblotting with NT-1 Ab. The percentage of N-terminal fragment released into the supernatant fraction was calculated for three experiments. B, [3H] IP3 binding (3 nm) was measured in 30-μg aliquots of wild-type and mutant microsomal membranes as described under “Experimental Procedures.” Western blots of the microsomes (not shown) were used to determine the relative expression of the IP3R constructs, and this was used to normalize the binding data. The data are the means ± S.E. of three experiments.
DISCUSSION
A key aspect of IP3R channel function relates to the operation of the pore-lining S6 helix during channel gating. In previous studies we mutated the Gly-2586 and Phe-2592 in the S6 helix to test the possibility that these residues function respectively as a “hinge” during gating or may be present at the gate itself. Neither mutation had significant effects on channel function (29). Based on this we proposed that the transmembrane portion of S6 may function as a rigid helix and that other hydrophobic residues in addition to Phe-2592 may be present at the channel gate. The present study shows that I2588A and I2589A, two hydrophobic residue mutations preceding Phe-2592, both inactivate IP3R channel function. Secondary structure prediction programs place Ile-2588 and Ile-2589 at the cytosolic end of the transmembrane portion of S6 (Fig. 1A). A homology model of the IP3R pore domain built using the closed state structure of KirBac1.1 is shown in Fig. 6A. This model also places these residues at the narrowest point where the helixes emerge into the cytosol, and therefore these residues could be located at the critical “gate” region of the channel. A cluster of adjacent hydrophobic residues is known to stabilize S6-S6 interaction interfaces in the gate of other channels (19, 20, 41). The S6 helix of IP3Rs and RyRs are highly conserved, and both Ile residues are present in all isoforms of this family of channels. Surprisingly previous mutagenesis studies have shown that RyR2 is inactivated by mutation of the Ile corresponding to 2588 but not 2589, although the latter mutation does suppress caffeine responses (43, 44). This result could indicate differences in the molecular architecture of the RyR and IP3R pore.
FIGURE 6.

Model for the gating mechanism of the IP3R. A, homology model of the S5S6 region of the IP3R was built using the closed state structure of KirBac1.1 as template as described previously (29). Three of the four subunits are shown with the putative gating residues at Ile-2588 and Ile-2599 as indicated. B, a cartoon model of the receptor with only the S4, S5, and S6 helices of two subunits of the tetramer is shown. The crystal structure of the N-terminal LBD in the apo and bound forms are placed at the cytosolic surface of the receptor based on docking of the crystal structure into the cryo-EM model (50). The SD, β-trefoil (β), and α-helical domains (α) are separately colored. CC, coiled-coil domain. The three main features of the model are: 1) the LBD does not make direct contact with the S4S5 loop but retains interactions with the C-tail, which extends to the cytosol within the central portion of the receptor; 2) the S4S5 loop associates with the proximal C-tail; and 3) conformational changes in the LBD are transmitted to the channel domain by alterations in multiple interactions involving the C-tail and an interior coupling domain (ICD), which interacts with the S4S5 loop and whose position is maintained by interactions with key residues in the proximal C-tail. For additional details see text.
Multiple secondary structure programs predict that the S6 helix extends for a considerable distance beyond the membrane. This would be in agreement with the long inner helix proposed for RyRs (34, 45). It is also apparent that in some channels the S6 helix bends sharply as it exits the membrane. In the case of the Kv1.2 channel, the trajectory approaches the S4S5 loop sufficiently closely to make interactions between these two regions (17). The present study provides biochemical evidence for an association between the S4S5 loop and the C-tail of IP3Rs in vitro. This association appears to be specific, because the S4S5 loop of RyR1 did not bind to the IP3R C-tail, although it has been found previously to bind to the RyR1 C-tail (28). In our experiments none of the single point mutations that inactivated channel function in the S6 helix or C-tail influenced the interaction with the S4S5 loop. This is not entirely unexpected because in other voltage-gated K+ and Ca2+ channels, the interaction interface is complex and involves multiple residues from both domains (25, 46, 47). Although further studies will be required to resolve the molecular basis of the S4S5 loop interaction with the C-tail, the data suggest that the S4S5 loop does not simply function as a mechanical lever during channel gating.
Eight negatively charged residues located in the cytosolic extension of the S6 helix and extending to residue 2634 were mutated in this study. None of the single mutations inactivated channel function, although one residue (Asp-2590) did cause significant inhibition. This is in marked contrast to RyR1 where two negatively charged residues have been identified as inactivating mutations (E4942Q and D4953Q) (26). It has been argued that rings of negative charges on both the luminal and cytosolic side of the transmembrane channel would aid in the high rates of cation flux seen in RyRs (26, 48). The Asp-2590 residue is completely conserved in RyRs, and mutation of this residue did substantially reduce K+ conductance of the channel but did not eliminate caffeine responses or high affinity ryanodine binding (26, 49). The higher impact of this residue presumably stems from its location just beyond the putative gate where residues from each subunit of the tetramer are sufficiently close to form a negatively charged ring. Negatively charged residues further away would not be expected to contribute to this effect if, as predicted, the S6 helixes bend away from each other in the cytosolic vestibule.
Of the five positively charged residues mutated, only one inactivated the channel completely (R2596A) with the remaining four showing smaller inhibitory effects. The Arg-2596 residue is located within the cytosolic extension of the S6 helix and is conserved in all three IP3R and RyR isoforms. The substitution of the smaller positively charged lysine (R2596K) did not restore activity, indicating that charge alone is not the sole determinant for inactivation. Some mutations that lead to loss of microsomal 45Ca2+ fluxes are nevertheless able to conduct K+ ions in nuclear patch clamp measurements (29): this was not the case for the R2596A mutant.3 There was no evidence for gross structural defects in the R2596A mutant as indicated by unchanged C-tail/4,5-loop interactions, C-tail assembly, interactions with the LBD, or altered [3H]IP3 binding. In addition to forming intra- or intersubunit salt bridges, the arginine side chain has the potential to form as many as five H-bonds. Thus, the Arg-2596 residue may be critical for maintaining the structure of the cytosolic region of the S6 helix. Further insights into the role of this residue will require more detailed structural studies on the IP3R channel domain.
Two studies describing the crystal structures of the LBD in the presence and absence of IP3 have recently published (10, 50). Both report relatively small conformational changes after IP3 binding that principally affect the interface between the SD and the β-helical domain. In addition, Seo et al. (50) docked their crystal structure into the high resolution cryo-EM model of the receptor in the closed state (7). The docking study reveals that none of the N-terminal domains are likely to penetrate sufficiently into the IP3R to make direct contact with the channel domain, supporting previous conclusions derived from cysteine accessibility studies (51). By contrast NMR experiments and pulldown assays have provided evidence for a direct interaction of sequences between the β8 and β9 helices of the SD and the 4,5-loop in the channel domain (15, 40). However, the docking model of Seo et al. suggests that the β8β9 loop is more likely to be critical for maintaining intersubunit contacts within the tetramer (50). If the SD does not make direct contact with the channel domain, then how is the channel gated? One model that would be consistent with the available experimental data is shown in Fig. 6B. The main assumption of the model, based on previous cysteine accessibility studies (52), is that the C-terminal tail projects all the way to the cytosolic surface within the central portion of the receptor assembly. In this model we suggest that one mechanism by which IP3 induced conformational changes in the LBD could be transmitted to the channel domain is by altering the conformation of the C-terminal tail. The model remains consistent with the experimental finding that the N-terminal and C-terminal trypsin fragments cross-link and coimmunoprecipitate (13, 53), despite the SD and 4,5-loop not making direct contact. Rotational or other conformational changes of the tetrameric LBD ring induced by IP3 binding could influence the S6 helix bundle crossing directly by altering interactions with the distal portions of the C-tail or indirectly via interior domains of the receptor that interact with the proximal portion of the C-tail and/or the 4,5-loop.
In the present study we find that two highly conserved histidines (His-2630 and His-2635) in the C-tail are critical for channel function. These residues, together with the previously identified cysteine residues (Cys-2610 and Cys-2613), share the common feature that they destabilize interactions of the N-terminal trypsin fragment with the remaining membrane attached fragments of the receptor (Fig. 5 and Ref. 40). It is important to emphasize that it is the specific residues, rather than the surrounding domains, that are critical. Mutation of adjacent residues, even when highly conserved, have no effect on channel function (Fig. 3B and Ref. 14). The distance constraints imposed by structural studies would seem to rule out a direct interaction of these residues with the N-terminal fragment, suggesting that the effects of mutation on membrane association of this fragment are likely to be secondary to other structural perturbations. We hypothesize that the residues could be important in maintaining proper interactions of the C-tail with the interior coupling domains of the receptor and for positioning these domains appropriately to make contacts with the S4S5 loop (Fig. 6B).
An important role for the C-tail is supported by the finding that deletion of 43 residues from the C-tail of IP3R1 (14) or 15 residues from the C-tail of RyR1 (54) is sufficient to disrupt channel function. However, it is difficult to reconcile these observations with the finding that a chimera of the IP3R with the channel domain of the RyR1 continues to be gated by IP3, albeit with a 20-fold lower efficacy (50). This chimeric IP3R construct contains the shorter RyR1 C-tail, which is 100 residues compared with 160 residues in IP3R1 C-tail. These data would suggest that interactions with the highly conserved residues in the proximal portion of the C-tail may play the dominant role in channel gating. At present the available data are consistent with a model in which multiple domain interactions are involved in transmitting the concerted motion of the tetrameric ring of LBDs at the receptor surface to the channel domain. Further studies are needed to probe the interior architecture and nearest neighbor domain relationships of the receptor to clarify the molecular mechanism of IP3R channel gating.
Acknowledgments
We thank the following students for help in the initial stages of the project: Annika Barber, Ben Woodall, and Ron Vagnozzi. We also thank Drs. David Yule and Gyorgy Hajnozcky for assistance.
This work was supported, in whole or in part, by National Institutes of Health Grant DK34804.
D. Yule, personal communication.
- IP3R
- inositol 1,4,5-trisphosphate receptor
- LBD
- ligand-binding domain
- IBC
- IP3-binding core
- SD
- suppressor domain
- RyR
- ryanodine receptor.
REFERENCES
- 1. Berridge M. J. (2009) Inositol trisphosphate and calcium signalling mechanisms. Biochim. Biophys. Acta 1793, 933–940 [DOI] [PubMed] [Google Scholar]
- 2. Foskett J. K., White C., Cheung K. H., Mak D. O. (2007) Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev. 87, 593–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Taylor C. W., Tovey S. C. (2010) IP3 receptors. Toward understanding their activation. Cold Spring Harb. Perspect. Biol. 2, a004010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. da Fonseca P. C., Morris S. A., Nerou E. P., Taylor C. W., Morris E. P. (2003) Domain organization of the type 1 inositol 1,4,5-trisphosphate receptor as revealed by single-particle analysis. Proc. Natl. Acad. Sci. U.S.A. 100, 3936–3941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Serysheva I. I., Bare D. J., Ludtke S. J., Kettlun C. S., Chiu W., Mignery G. A. (2003) Structure of the type 1 inositol 1,4,5-trisphosphate receptor revealed by electron cryomicroscopy. J. Biol. Chem. 278, 21319–21322 [DOI] [PubMed] [Google Scholar]
- 6. Sato C., Hamada K., Ogura T., Miyazawa A., Iwasaki K., Hiroaki Y., Tani K., Terauchi A., Fujiyoshi Y., Mikoshiba K. (2004) Inositol 1,4,5-trisphosphate receptor contains multiple cavities and L-shaped ligand-binding domains. J. Mol. Biol. 336, 155–164 [DOI] [PubMed] [Google Scholar]
- 7. Ludtke S. J., Tran T. P., Ngo Q. T., Moiseenkova-Bell V. Y., Chiu W., Serysheva I. I. (2011) Flexible architecture of IP3R1 by cryo-EM. Structure. 19, 1192–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bosanac I., Alattia J. R., Mal T. K., Chan J., Talarico S., Tong F. K., Tong K. I., Yoshikawa F., Furuichi T., Iwai M., Michikawa T., Mikoshiba K., Ikura M. (2002) Structure of the inositol 1,4,5-trisphosphate receptor binding core in complex with its ligand. Nature 420, 696–700 [DOI] [PubMed] [Google Scholar]
- 9. Bosanac I., Yamazaki H., Matsu-Ura T., Michikawa T., Mikoshiba K., Ikura M. (2005) Crystal structure of the ligand binding suppressor domain of type 1 inositol 1,4,5-trisphosphate receptor. Mol. Cell 17, 193–203 [DOI] [PubMed] [Google Scholar]
- 10. Lin C. C., Baek K., Lu Z. (2011) Apo and InsP3-bound crystal structures of the ligand-binding domain of an InsP3 receptor. Nat. Struct. Mol. Biol. 18, 1172–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sanderson M. J., Delmotte P., Bai Y., Perez-Zogbhi J. F. (2008) Regulation of airway smooth muscle cell contractility by Ca2+ signaling and sensitivity. Proc. Am. Thorac. Soc. 5, 23–31 [DOI] [PubMed] [Google Scholar]
- 12. Joseph S. K., Pierson S., Samanta S. (1995) Trypsin digestion of the inositol trisphosphate receptor. Implications for the conformation and domain organization of the protein. Biochem. J. 307, 859–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Boehning D., Joseph S. K. (2000) Direct association of ligand-binding and pore domains in homo- and heterotetrameric inositol 1,4,5-trisphosphate receptors. EMBO J. 19, 5450–5459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schug Z. T., Joseph S. K. (2006) The role of the S4-S5 linker and C-terminal tail in inositol 1,4,5-trisphosphate receptor function. J. Biol. Chem. 281, 24431–24440 [DOI] [PubMed] [Google Scholar]
- 15. Chan J., Yamazaki H., Ishiyama N., Seo M. D., Mal T. K., Michikawa T., Mikoshiba K., Ikura M. (2010) Structural studies of inositol 1,4,5-trisphosphate receptor. Coupling ligand binding to channel gating. J. Biol. Chem. 285, 36092–36099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rossi A. M., Riley A. M., Tovey S. C., Rahman T., Dellis O., Taylor E. J., Veresov V. G., Potter B. V., Taylor C. W. (2009) Synthetic partial agonists reveal key steps in IP3 receptor activation. Nat. Chem. Biol. 5, 631–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Long S. B., Campbell E. B., Mackinnon R. (2005) Voltage sensor of Kv1.2. Structural basis of electromechanical coupling. Science 309, 903–908 [DOI] [PubMed] [Google Scholar]
- 18. Jin T., Peng L., Mirshahi T., Rohacs T., Chan K. W., Sanchez R., Logothetis D. E. (2002) The βγ subunits of G proteins gate a K+ channel by pivoted bending of a transmembrane segment. Mol. Cell 10, 469–481 [DOI] [PubMed] [Google Scholar]
- 19. Rojas A., Wu J., Wang R., Jiang C. (2007) Gating of the ATP-sensitive K+ channel by a pore-lining phenylalanine residue. Biochim. Biophys. Acta 1768, 39–51 [DOI] [PubMed] [Google Scholar]
- 20. Stepanovic S. Z., Potet F., Petersen C. I., Smith J. A., Meiler J., Balser J. R., Kupershmidt S. (2009) The evolutionarily conserved residue A653 plays a key role in HERG channel closing. J. Physiol 587, 2555–2566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sackin H., Nanazashvili M., Palmer L. G., Krambis M., Walters D. E. (2005) Structural locus of the pH gate in the Kir1.1 inward rectifier channel. Biophys. J. 88, 2597–2606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang R., Bolstad J., Kong H., Zhang L., Brown C., Chen S. R. (2004) The predicted TM10 transmembrane sequence of the cardiac Ca2+ release channel (ryanodine receptor) is crucial for channel activation and gating. J. Biol. Chem. 279, 3635–3642 [DOI] [PubMed] [Google Scholar]
- 23. Decher N., Chen J., Sanguinetti M. C. (2004) Voltage-dependent gating of hyperpolarization-activated, cyclic nucleotide-gated pacemaker channels. Molecular coupling between the S4-S5 and C-linkers. J. Biol. Chem. 279, 13859–13865 [DOI] [PubMed] [Google Scholar]
- 24. Ferrer T., Rupp J., Piper D. R., Tristani-Firouzi M. (2006) The S4-S5 linker directly couples voltage sensor movement to the activation gate in the human ether-á-go-go-related gene (hERG) K+ channel. J. Biol. Chem. 281, 12858–12864 [DOI] [PubMed] [Google Scholar]
- 25. Labro A. J., Boulet I. R., Choveau F. S., Mayeur E., Bruyns T., Loussouarn G., Raes A. L., Snyders D. J. (2011) The S4-S5 linker of KCNQ1 channels forms a structural scaffold with the S6 segment controlling gate closure. J. Biol. Chem. 286, 717–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xu L., Wang Y., Gillespie D., Meissner G. (2006) Two rings of negative charges in the cytosolic vestibule of type-1 ryanodine receptor modulate ion fluxes. Biophys. J. 90, 443–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mignery G. A., Südhof T. C. (1990) The ligand binding site and transduction mechanism in the inositol-1,4,5-triphosphate receptor. EMBO J. 9, 3893–3898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lee E. H., Allen P. D. (2007) Homo-dimerization of RyR1 C-terminus via charged residues in random coils or in an alpha-helix. Exp. Mol. Med. 39, 594–602 [DOI] [PubMed] [Google Scholar]
- 29. Schug Z. T., da Fonseca P. C., Bhanumathy C. D., Wagner L., 2nd, Zhang X., Bailey B., Morris E. P., Yule D. I., Joseph S. K. (2008) Molecular characterization of the inositol 1,4,5-trisphosphate receptor pore-forming segment. J. Biol. Chem. 283, 2939–2948 [DOI] [PubMed] [Google Scholar]
- 30. Buguliskis J. S., Casta L. J., Butz C. E., Matsumoto Y., Taraschi T. F. (2007) Expression and biochemical characterization of Plasmodium falciparum DNA ligase I. Mol. Biochem. Parasitol. 155, 128–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Boehning D., Joseph S. K. (2000) Functional properties of recombinant type I and type III inositol 1, 4,5-trisphosphate receptor isoforms expressed in COS-7 cells. J. Biol. Chem. 275, 21492–21499 [DOI] [PubMed] [Google Scholar]
- 32. Khan M. T., Joseph S. K. (2010) Role of inositol trisphosphate receptors in autophagy in DT40 cells. J. Biol. Chem. 285, 16912–16920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Csordás G., Hajnóczky G. (2001) Sorting of calcium signals at the junctions of endoplasmic reticulum and mitochondria. Cell Calcium 29, 249–262 [DOI] [PubMed] [Google Scholar]
- 34. Samsó M., Feng W., Pessah I. N., Allen P. D. (2009) Coordinated movement of cytoplasmic and transmembrane domains of RyR1 upon gating. PLoS Biol. 7, e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sugawara H., Kurosaki M., Takata M., Kurosaki T. (1997) Genetic evidence for involvement of type 1, type 2 and type 3 inositol 1,4,5-trisphosphate receptors in signal transduction through the B-cell antigen receptor. EMBO J. 16, 3078–3088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Taylor C. W., Rahman T., Tovey S. C., Dedos S. G., Taylor E. J., Velamakanni S. (2009) IP3 receptors. Some lessons from DT40 cells. Immunol. Rev. 231, 23–44 [DOI] [PubMed] [Google Scholar]
- 37. Chatelain F. C., Alagem N., Xu Q., Pancaroglu R., Reuveny E., Minor D. L., Jr. (2005) The pore helix dipole has a minor role in inward rectifier channel function. Neuron 47, 833–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Uchida K., Miyauchi H., Furuichi T., Michikawa T., Mikoshiba K. (2003) Critical regions for activation gating of the inositol 1,4,5-trisphosphate receptor. J. Biol. Chem. 278, 16551–16560 [DOI] [PubMed] [Google Scholar]
- 39. Galvan D. L., Mignery G. A. (2002) Carboxyl-terminal sequences critical for inositol 1,4,5-trisphosphate receptor subunit assembly. J. Biol. Chem. 277, 48248–48260 [DOI] [PubMed] [Google Scholar]
- 40. Yamazaki H., Chan J., Ikura M., Michikawa T., Mikoshiba K. (2010) Tyr-167/Trp-168 in type 1/3 inositol 1,4,5-trisphosphate receptor mediates functional coupling between ligand binding and channel opening. J. Biol. Chem. 285, 36081–36091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kuo A., Gulbis J. M., Antcliff J. F., Rahman T., Lowe E. D., Zimmer J., Cuthbertson J., Ashcroft F. M., Ezaki T., Doyle D. A. (2003) Crystal structure of the potassium channel KirBac1.1 in the closed state. Science 300, 1922–1926 [DOI] [PubMed] [Google Scholar]
- 42. Hurne A. M., O'Brien J. J., Wingrove D., Cherednichenko G., Allen P. D., Beam K. G., Pessah I. N. (2005) Ryanodine receptor type 1 (RyR1) mutations C4958S and C4961S reveal excitation-coupled calcium entry (ECCE) is independent of sarcoplasmic reticulum store depletion. J. Biol. Chem. 280, 36994–37004 [DOI] [PubMed] [Google Scholar]
- 43. Szado T., Vanderheyden V., Parys J. B., De Smedt H., Rietdorf K., Kotelevets L., Chastre E., Khan F., Landegren U., Söderberg O., Bootman M. D., Roderick H. L. (2008) Phosphorylation of inositol 1,4,5-trisphosphate receptors by protein kinase B/Akt inhibits Ca2+ release and apoptosis. Proc. Natl. Acad. Sci. U.S.A. 105, 2427–2432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Steinmann C., Landsverk M. L., Barral J. M., Boehning D. (2008) Requirement of inositol 1,4,5-trisphosphate receptors for tumor-mediated lymphocyte apoptosis. J. Biol. Chem. 283, 13506–13509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ramachandran S., Serohijos A. W., Xu L., Meissner G., Dokholyan N. V. (2009) A structural model of the pore-forming region of the skeletal muscle ryanodine receptor (RyR1). PLoS Comput. Biol. 5, e1000367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Labro A. J., Raes A. L., Grottesi A., Van Hoorick D., Sansom M. S., Snyders D. J. (2008) Kv channel gating requires a compatible S4-S5 linker and bottom part of S6, constrained by non-interacting residues. J. Gen. Physiol. 132, 667–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wall-Lacelle S., Hossain M. I., Sauvé R., Blunck R., Parent L. (2011) Double mutant cycle analysis identified a critical leucine residue in the IIS4S5 linker for the activation of the CaV2.3 calcium channel. J. Biol. Chem. 286, 27197–27205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gillespie D., Xu L., Wang Y., Meissner G. (2005) (De)constructing the ryanodine receptor. Modeling ion permeation and selectivity of the calcium release channel. J. Phys. Chem. B 109, 15598–15610 [DOI] [PubMed] [Google Scholar]
- 49. Du G. G., Guo X., Khanna V. K., MacLennan D. H. (2001) Functional characterization of mutants in the predicted pore region of the rabbit cardiac muscle Ca2+ release channel (ryanodine receptor isoform 2). J. Biol. Chem. 276, 31760–31771 [DOI] [PubMed] [Google Scholar]
- 50. Seo M. D., Velamakanni S., Ishiyama N., Stathopulos P. B., Rossi A. M., Khan S. A., Dale P., Li C., Ames J. B., Ikura M., Taylor C. W. (2012) Structural and functional conservation of key domains in InsP3 and ryanodine receptors. Nature 483, 108–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Anyatonwu G., Joseph S. K. (2009) Surface accessibility and conformational changes in the N-terminal domain of type I inositol trisphosphate receptors. Studies using cysteine substitution mutagenesis. J. Biol. Chem. 284, 8093–8102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Anyatonwu G., Khan M. T., Schug Z. T., da Fonseca P. C., Morris E. P., Joseph S. K. (2010) Calcium-dependent conformational changes in inositol trisphosphate receptors. J. Biol. Chem. 285, 25085–25093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yoshikawa F., Iwasaki H., Michikawa T., Furuichi T., Mikoshiba K. (1999) Trypsinized cerebellar inositol 1,4,5-trisphosphate receptor. Structural and functional coupling of cleaved ligand binding and channel domains. J. Biol. Chem. 274, 316–327 [DOI] [PubMed] [Google Scholar]
- 54. Stewart R., Zissimopoulos S., Lai F. A. (2003) Oligomerization of the cardiac ryanodine receptor C-terminal tail. Biochem. J. 376, 795–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang R., Zhang L., Bolstad J., Diao N., Brown C., Ruest L., Welch W., Williams A. J., Chen S. R. (2003) Residue Gln4863 within a predicted transmembrane sequence of the Ca2+ release channel (ryanodine receptor) is critical for ryanodine interaction. J. Biol. Chem. 278, 51557–51565 [DOI] [PubMed] [Google Scholar]