

Background: KLF8 is a cancer-promoting transcription factor.
Results: KLF8 inhibits DNA damage in breast cancer cells.
Conclusion: KLF8 is a novel effector of the PARP-1 and DNA-PK DNA damage response pathways.
Significance: KLF8 could be targeted for chemosensitizing therapy.
Keywords: Breast Cancer, DNA Damage Response, DNA Repair, Signal Transduction, SUMOylation, DNA-PK, KLF8, Krüppel-like factor, PARP-1
Abstract
Krüppel-like factor 8 (KLF8) regulates critical gene transcription and cellular events associated with cancer. However, the role of KLF8 in cancer remains largely unknown. Here, we report a surprisingly novel role for KLF8 in DNA repair in breast cancer cells. Comet, clonogenic, and WST-1 assays showed that KLF8 expression is required for protecting human breast cancer cells from doxorubicin-induced DNA damage and cell death. Western blotting indicated that overexpression of ectopic KLF8 attenuated the levels of the DNA damage marker γH2A.X in doxorubicin-treated PARP-1+/+ but not PARP-1−/− mouse embryonic fibroblasts, whereas the PARP-1-binding-defective KLF8 mutant failed to do so. Interestingly, in response to the DNA damage, KLF8 was phosphorylated by the DNA-dependent protein kinase catalytic subunit and, subsequently, SUMOylated by SUMO E3 ligases protein inhibitors of activated STAT (PIASs), which depends upon the interaction of KLF8 with DNA-dependent protein kinase catalytic subunit, PIASs, and PARP-1 as well as their enzymatic activities. Lastly, we show evidence that KLF8 was recruited to the DNA damage site. These results suggest a novel role and mechanism for KLF8 in the regulation of DNA repair and therapeutic resistance in breast cancer cells.
Introduction
In actively dividing normal cells, the genomic integrity is constantly threatened by DNA damage, resulting in DNA lesions such as single-strand breaks (SSBs)2 and double-strand breaks (DSBs) (1). To maintain the genomic integrity and thus avoid cell death or transformation, normal cells have inherited a variety of DNA repair mechanisms that fix SSBs or DSBs by the master sensors and regulators of DNA damage such as poly(ADP-ribose) polymerase 1 (PARP-1), BRCA1, BRCA2, and DNA-dependent protein kinase (DNA-PK) (2–5). In addition, many types of posttranslational modifications of proteins, such as SUMOylation, have been shown to play a critical role in the DNA repair processes (6, 7).
Because of their genotoxic and, thus, cytotoxic effect on cancer cells, DNA-damaging agents have been the mainstay of cancer therapy for decades. However, DNA damage responses by suboptimal or inaccurate DNA repair mechanisms not only arm cancer cells with therapeutic resistance but also contribute to aggressive progression of tumors by causing genomic instability (1). For this reason, inhibitors targeting DNA repair proteins such as PARP-1 and DNA-PK have been actively developed and tested to improve the cancer therapy (1). Understanding how cancer cells improperly respond to DNA damage is critical for designing novel anti-DNA repair therapeutic strategies for cancer.
Krüppel-like transcription factor 8 (KLF8) is aberrantly overexpressed in a variety of human cancers and promotes transformation or tumor progression of these cancers, including breast cancer (8–10), ovarian cancer (9, 11), hepatocellular carcinoma (12), renal cancer (11, 13), gastric cancer (14), and glioma (15–17). Interestingly, the nuclear function of KLF8 is tightly regulated by its posttranslational modifications, including SUMOylation (18, 19) and PARP-1-catalyzed PARylation (20, 21) to regulate the transcription of genes such as cyclin D1 (10, 18, 19, 22, 23), E-cadherin (10, 12), and MMP9 (8) that are essential for tumor progression.
In this work, we report the discovery of a novel role of KLF8 in promoting DNA repair and drug resistance in breast cancer cells and the underlying molecular mechanisms involving interaction with PARP-1, the DNA-PK catalytic subunit (DNA-PKcs), and PIASs and their catalyzed PARylation, phosphorylation, and sumoylation. Our results suggest that KLF8 could be a critical factor contributing to therapeutic resistance in breast cancer through improper DNA damage response.
EXPERIMENTAL PROCEDURES
Antibodies and Reagents
Antibodies against the HA tag (F-7), Myc tag (9E10), PARP-1, H2A.X (sc-54607), and DNA-PKcs (sc-5282) were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Anti-γ-H2A.X (9718S) antibody was purchased from Cell Signaling Technology, Inc. (Danvers, MA). Anti-phosphoserine was purchased from Chemicon (EMD Millipore Corp., Billerica, MA). Anti-HA and anti-Myc antibody-conjugated agarose beads and PJ-34 were purchased from Sigma. IgG was purchased from Jackson ImmunoResearch, Inc. (West Grove, PA). Doxorubicin (324380) was purchased from Calbiochem (EMD Millipore Corp.). Olaparib and ABT-888 (ABT) were purchased from Selleck Chemical LLC (Houston, TX). Nu7026 was purchased from Cayman Chemical Co. (Ann Arbor, MI).
Cell Culture, Transfection, and Plasmid Construction
HEK293T was purchased from the ATCC. The PARP-1+/+ and PARP-1−/− primary mouse embryonic fibroblast (MEF) cells (20, 24, 25), the MCF-10A line that expresses inducible KLF8 (10A-iK8), and the MDA-MB-231 line that expresses inducible KLF8 short hairpin RNA (231-K8ikd) (26) were described previously. Transfections of the plasmid DNAs were performed using Lipofectamine 2000 (Invitrogen) according to the instructions of the manufacturer. The mammalian expression plasmids pKH3, pKH3-KLF8, pHAN-PARP-1, -PIAS1, -PIAS2, -PIAS4, -SUMO-1, and the ZF1,2mCs and K67R mutants of KLF8 were described previously (18, 20–22). The pKH3-KLF8S80A serine-to-alanine mutant was generated by site-directed mutagenesis PCR and overlapping PCR using pKH3-KLF8 as the template and the mutation-specific primers of 5′-tag tga ttt cgc cct gcc c-3′ (forward) and 5′-tgg ggc agg gcg aaa tca c-3′ (reverse) paired with one of the master primers of 5′-CCC AAG CTT CTG CAG GTC G-3′ (forward) and 5′-GGA CAA ACC ACA ACT AGA ATG CAG-3′ (reverse).
RNA Interference, Quantitative Real-time PCR, Western Blotting, Coimmunoprecipitation (Co-IP), and SUMOylation Assays
These assays were performed as described previously (10, 18, 19, 23). PARP-1 and GFP ON-TARGETplus siRNAs were purchased from Dharmacon (J006656-05, 5′-GAU UUC AUC UGG UGU GAU A-3′; J006656-06, 5′-GAA AAC AGG UAU UGG AUA U-3′; J006656-07, 5′-GUU CUU AGC GCA CAU CUU G-3′; and J006656-08, 5′-CCA AUA GGC UUA AUC CUG U-3′). DNA-PKcs siRNAs (sc-35200) were purchased from Santa Cruz Biotechnology, Inc. Oligofectamine was used to transfect siRNAs according to the instructions of the manufacturer (Invitrogen). Primers for quantitative real-time PCR (forward/reverse) are as follows: GAPDH, 5′-tcg tac gtg gaa gga ctc a-3′ (forward) and 5′-cca gta gag gca ggg atg at-3′(reverse) and H2A.X, 5′-aga tcc tgg agc tgg c-3′ (forward) and 5′-acg gcc tgg atg ttg g-3′ (reverse).
ChIP Assays
The cyclin D1 gene promoter reporter assays were performed as described previously (23, 27). DNA damage site-targeted ChIP assays were carried out (28) to determine the recruitment of KLF8 to the DNA damage sites. Briefly, the DNA DSB reporter pDR-GFP that contains an I-SceI endonuclease recognition site was stably transfected into the 10A-iK8 cells. The established 10A-iK8/DR-GFP cells were treated with 1 μm tetracycline for 24 h for the induction of KLF8 expression. Then, the site-specific DNA DSB was induced by transfection of the I-SceI expression plasmid pCβASce. After 24 h, the ChIP assays were performed using the DNA damage site-specific PCR primer set DR-1 (adjacent to the DNA DSBs) (29).
Clonogenic Assay
Cell survival rates after doxorubicin treatment were performed as described (30). Briefly, the 10A-iK8 cells or 231-K8ikd cells were grown under induced or uninduced conditions for 2 days. MCF-10A cells were transfected with wild-type or mutant KLF8 for 24 h. The cells were then treated with doxorubicin, reseeded at low density, and allowed to grow for 2 weeks to allow colonies to form. Colonies were fixed with 3.7% paraformaldehyde, stained with 0.1% crystal violet, and counted. Colonies containing ∼50 or more cells were counted.
WST-1 Assay
The cell sensitivity to the cytotoxic effect of doxorubicin was assayed using Clontech WST-1 cell proliferation reagent (catalog no. 630118) according to the instructions of the manufacturer (PT3946-1) (Clontech Laboratories, Inc., Mountain View, CA). Briefly, cells of appropriate density were seeded and treated similarly as in a clonogenic assay. After being grown for 48 h, the cells were incubated with the WST-1 reagent for 2 h before optical density (A) values at 440-nm wavelength were measured using a multiplate bioreader (PerkinElmer Life Sciences). The OD values represent the cell viability.
DNA Damage Induction, γH2A.X Detection, and Comet Assay
Cellular DNA damage was induced by treating the cells in culture with the DNA-intercalating agent doxorubicin. Overall DNA damage levels were assessed by Comet assay using alkaline single-cell agarose gel electrophoresis and the Trevigen Comet assay kit (4250-050-K, Gaithersburg, MD) according to the instructions of the manufacturer. Briefly, cells were treated with doxorubicin with certain doses or at certain time points, harvested, and subjected to Comet analysis. After staining with SYBR Green, Comet images were captured by fluorescence microscopy. Tail moments (percentage of DNA in tail × tail length) were quantitated for 100 cells/slide using CometScore software. Western blotting was performed to determine the levels of γH2A.X, a marker for double-strand breaks of damaged DNA. Western blot analyses were quantified by chemiluminescence blot of Image Lab 3.0 (Bio-Rad). To control the exposure time between blots, for every panel, all membranes were exposed for the same duration. Then the quantitative data from three independent experiments were statistically analyzed. The reduction in the levels of DNA damage during the period of cell recovery post-doxorubicin removal was considered an indicator for DNA repair.
Statistical Analysis
All the data were summarized and presented as mean ± S.D. with a minimum of three observations per group. Unpaired, paired or single sample Student's t test with the Bonferroni correction for the multiple comparisons or Qi-Square test was applied as appropriate. Significance was determined by the α level of 0.05.
RESULTS
The Aberrantly Overexpressed KLF8 Protects Breast Cancer Cells from Doxorubicin-induced Cell Death by Reducing the Levels of DNA Damage
To test whether KLF8 expression protects breast cancer cells from death induced by DNA-damaging agent, we first treated our 231-K8ikd cells (an MDA-MB-231 variant expressing aberrantly high levels of endogenous KLF8 and Tet-on inducible short hairpin RNA against KLF8 (26)) with the DNA-damaging drug doxorubicin and examined the correlation between cell survival and KLF8 expression levels in the cells using clonogenic assays (Fig. 1, A and C, left panels). We found that when KLF8 knockdown was induced (I), the cells became more sensitive to doxorubicin-induced cell death than when KLF8 knockdown was uninduced (U). Conversely, in our 10A-iK8 cells (a MCF-10A cell line expressing little or no endogenous KLF8 and Tet-on inducible ectopic KLF8 (26)), the cells became more resistant to doxorubicin-induced death when the ectopic KLF8 expression was induced (I) than when the ectopic KLF8 expression was uninduced (U) (Fig. 1, A and C, right panels). Similar results were reproduced by the independent cell viability analysis using WST-1 assays (Fig. 1, B and D).
FIGURE 1.

KLF8 protects breast epithelial and cancer cells from doxorubicin-induced cell death by reducing DNA damage levels. A–D, overexpression of KLF8 confers resistance to doxorubicin-induced cell death. Cells were pretreated with 1 μm tetracycline for 48 h to induce KLF8 knockdown in the 231-K8ikd cells or overexpression in 10A-ik8 cells. After 4 h of treatment at the indicated doses (A and B) or 1 μm treatment for indicated periods of time (C and D) with doxorubicin and 2 h of recovery, the cells were prepared for clonogenic assay (A and C) or WST-1 assay (B and D). E and F, KLF8 promotes repair of doxorubicin-induced DNA damage. Cells were treated as in A. DNA damage levels were assessed using a Comet assay after the 4-h induction of DNA damage by doxorubicin of various doses, followed by 2 h of recovery (E) or after various time periods of DNA damage induction by 1 μm doxorubicin (F). G, KLF8 attenuates the levels of DSBs induced by doxorubicin. The cells were treated as in A. Whole cell lysates were collected as in E (top) or F (bottom) for assessing the expression of the DSB marker γ-H2A.X by Western blotting. The data represent the mean ± S.E. of at least three independent triplicate experiments. *, p < 0.05. I, induced; U, uninduced.
To test whether the expression levels of KLF8 in the cells affect the levels of DNA damage induced by doxorubicin, the cells were treated with doxorubicin similarly, and the levels of DNA damage were determined by Comet assays. The results showed that in both the 231-K8ikd and 10A-iK8 cells, the levels of KLF8 expression and the levels of doxorubicin-induced DNA damage were inversely correlated (Fig. 1, E and F). These results were further verified by an independent analysis of the induction of phosphorylated form of histone 2A.X, i.e. γ-H2A.X, a marker for DNA DSBs, using Western blotting (Fig. 1G).
These data clearly indicate that the aberrant overexpression of KLF8 in breast cancer cells is critical for the prevention of DNA damage, promotion of DNA repair, or both, and the subsequent resistance to the DNA damage-induced cell death.
KLF8 Attenuates DNA Damage in a PARP-1-dependent Manner
We have recently identified the KLF8 interaction with and regulation by PARP-1, a master regulator of DNA repair (1, 20). To determine whether PARP-1 plays a role in KLF8-induced reduction in DNA damage caused by doxorubicin, we first transfected the PARP-1+/+ primary MEF cells (20, 24, 25) with either the wild-type KLF8 or its PARP-1 binding-defective mutant ZF1,2mCs and compared the sensitivity between the wild-type and mutant KLF8-transfected cells to doxorubicin-induced DNA damage. We found that the cells transfected with the wild-type KLF8 showed a much lower level of γH2A.X than that shown in the vector-transfected cells (Fig. 2A, compare lanes or columns 4 and 2), consistent with the observations in the breast cells described above in Fig. 1. In contrast, the cells transfected with the mutant KLF8 maintained a level of γH2A.X that was comparable with that in the vector-transfected control cells and much higher than that in the wild-type KLF8-transfected cells (Fig. 2A, compare lane or column 6 to lanes 2 or 4). These results indicate that the interaction of KLF8 and PARP-1 is critical for the KLF8-induced decrease in the levels of DNA damage in the cells.
FIGURE 2.

KLF8 attenuates DNA damage-induced phosphorylation of H2A.X in a PARP-1-dependent manner. A, KLF8-mediated decrease in γH2A.X levels upon DNA damage requires its interaction with PARP-1. PARP-1+/+ cells were transfected with HA-KLF8, HA-ZF1,2mCs (the PARP-1 interaction-defective mutant of KLF8 (20)), or vector alone for 24 h. DNA damage was induced by doxorubicin (Doxo) (1 μm) treatment for 4 h. Whole cell lysates were prepared for Western blotting of γH2A.X. Left panel, representative image of Western blot analysis. Right panel, quantitative analysis of related γH2A.X levels. B, KLF8-mediated reduction of γH2A.X requires catalytically active PARP-1. PARP-1−/− cells were transfected with HA-KLF8, PARP-1, E988K (the auto-PARylation-defective mutant of PARP-1 (20)), or vector alone for 24 h and treated with doxorubicin (1 μm) for 4 h. Whole cell lysates were prepared for Western blotting (left panel) and quantitative analysis (right panel) of related γH2A.X levels. C, KLF8 does not impact H2A.X expression at mRNA levels. PARP-1+/+ cells were transfected with HA-KLF8 or vector alone for 24 h. Total RNA was prepared and used for examination of H2A.X mRNA expression levels by RT-PCR (left panels) and qRT-PCR (right panel). Data represent the mean ± S.E. of at least three independent duplicate experiments. *, p < 0.05.
To test whether the catalytic activity of PARP-1 is also involved, we transfected the PARP-1−/− MEFs with the wild-type PARP-1 or its auto-PARylation-defective mutant E988K (20, 25) along with KLF8 for 24 h, treated the cells with doxorubicin (1 μm) for 4 h, and then determined the difference in the levels of DNA damage between the wild-type and mutant PARP-1-transfected cells by Western blotting of γH2A.X. The result showed that without PARP-1 re-expression in the cells, overexpression of KLF8 did not cause a reduction in the levels of DNA damage (Fig. 2B, compare lanes or columns 4 and 2). Coexpression of wild-type PARP-1 restored the KLF8-induced reduction in the DNA damage levels, but the mutant PARP-1 failed to do so (Fig. 2A, compare lanes or columns 5 or 6 to 2 or 4), indicating that PARP-1-catalyzed auto-PARylation or trans-PARylation also plays an important role in KLF8 regulation of the DNA damage response.
These results suggest that it is important for KLF8 to interact with catalytically active PARP-1 to antagonize DNA damage. The results also suggest that KLF8 reduces the levels of DNA damage primarily by promoting DNA repair rather than preventing DNA damage in the cells, given that PARP-1 plays a critical role in DNA repair and that KLF8 does not appear to have an impact on the expression of H2A.X (Fig. 2C).
SUMOylation of KLF8 Is Induced by DNA Damage and This Modification is PARP-1-dependent and Essential for KLF8-mediated DNA Repair and Protection against Cell Death
It has been reported that KLF8 is regulated by SUMOylation (18, 19, 31), and SUMOylation plays an important role in DNA repair (6). To test whether doxorubicin treatment causes any change in the SUMOylation of KLF8 and possible involvement of PARP-1, we cotransfected the PARP-1+/+ and PARP-1−/− MEFs with wild-type KLF8 or its ZF1,2mCs mutant and SUMO-1 and examined the SUMOylation state of the KLF8 proteins. We found that in the presence of PARP-1, the SUMOylation of the wild-type KLF8 was highly stimulated by doxorubicin treatment (Fig. 3A, compare lane 3 to lane 1), and the SUMOylation remained at a high level within 2 h after doxorubicin was removed (Fig. 3A, compare lane 5 to lane 1, and B). In contrast, in the absence of PARP-1, doxorubicin failed to stimulate the SUMOylation of the wild-type KLF8 (Fig. 3A, compare lane 9 to lane 7). Little or no SUMOylation of the ZF1,2mCs mutant took place regardless of the expression of PARP-1 (Fig. 3A, compare lane 4 to lane 2 or lane 10 to lane 8). These results clearly indicate that KLF8 responds to doxorubicin-induced DNA damage through the SUMOylation of it and that the SUMOylation depends upon its interaction with PARP-1.
FIGURE 3.

KLF8 responds to DNA damage through PARP-1-dependent SUMOylation. A, SUMOylation of KLF8 is stimulated by DNA damage, requiring its interaction with PARP-1. PARP-1+/+ or PARP-1−/− MEFs were cotransfected with HA-KLF8 or HA-ZF1,2mCs with Myc-SUMO-1 for 24 h, treated with 1 μm doxorubicin for 4 h, and followed by 2 h of recovery. Whole cell lysates were prepared at the end of mock treatment (lanes 1, 2, 7, and 8), doxorubicin treatment (lanes 3, 4, 9, and 10) or recovery (lanes 5, 6, 11, and 12) for Western blotting (IB) and co-IP (IP). B, dynamic changes of SUMOylation on KLF8 during recovery from DNA damage. 293T cells were cotransfected with HA-KLF8 and Myc-SUMO-1 at a ratio of 5:1 for 24 h and then treated with 1 μm doxorubicin for 4 h. Whole cell lysates were prepared at indicated times post-doxorubicin removal for Western blotting and co-IP. C, the SUMOylation of KLF8 is required for the DNA repair. PARP-1+/+ cells were transfected with HA-KLF8, HA-K67R (the SUMOylation-defective mutant of KLF8 (18)), or vector alone for 24 h, treated with indicated concentrations of doxorubicin (Doxo) for 4 h, and followed by 2 h of recovery. Whole cell lysates were prepared for Western blotting and quantification of γH2A.X levels. D, SUMOylation of KLF8 and its interaction with PARP-1 are both essential for cell viability post-DNA damage. PARP-1+/+ and PARP-1−/− MEFs were transfected with HA-KLF8, HA-ZF1,2mCs, HA-K67R, or vector alone for 24 h, treated for 4 h with 1 μm doxorubicin, and collected for WST-1 assay. Data represent the mean ± S.E. of at least three independent duplicate experiments. *, p < 0.05. The arrows indicate the major band of SUMOylated KLF8 protein.
To test whether the SUMOylation of KLF8 plays a role in DNA repair, we transfected the PARP-1+/+ MEFs with the wild-type KLF8 or its K67R SUMOylation-defective mutant (18) and compared their effect on the expression levels of γH2A.X. As expected, the wild-type KLF8 reduced γH2A.X back to the basal level (Fig. 3C, compare lanes 3 or 4 to lane 1 or columns 5 or 6 to 4). The K67R mutant, however, failed to do so (Fig. 3C, compare lanes 6 or 7 to lane 1 or columns 8 or 9 to column 7). These results suggest that the SUMOylation of KLF8 is critical for the DNA repair.
We then transfected the PARP-1+/+ or PARP-1−/− MEFs with the wild-type KLF8 and the ZF1,2mCs or K67R mutant and determined their role in protecting the cells from doxorubicin-induced cell death. We found that the wild-type KLF8 enhanced the viability of the PARP-1+/+ but not the PARP-1−/− cells (Fig. 3D, compare column 3 to column 2 or column 8 to column 7), whereas both of the KLF8 mutants failed to do the same (Fig. 3D, compare lanes 4 or 5 to lanes 2 or 3). Taken together, these results suggest that the PARP-1-dependent SUMOylation of KLF8 is critical for DNA repair and resistance to DNA-damage-mediated cell death.
The DNA Damage-induced SUMOylation of KLF8 Is Mediated via Interaction between KLF8, PIASs, and Catalytic PARP-1
To determine the molecular mechanisms by which the SUMOylation of KLF8 is induced during doxorubicin-induced DNA damage, we first examined the role of PARP-1 for KLF8 interaction with the SUMO E3 ligases PIASs. Coexpression and co-IP analyses demonstrated that, unlike the wild-type KLF8 and the K67R mutant, the HA-ZF1,2mCs mutant hardly interacted with the PIASs (Fig. 4A, lanes 2, 5, and 8). On the other hand, the wild-type KLF8 barely interacted with PIASs either in the absence of PARP-1 (Fig. 4B). Treatment with doxorubicin dramatically enhanced the interaction between KLF8 and PIASs in the presence of PARP-1 (Fig. 4B). These results suggest that KLF8 interaction with PARP-1 is critical for the interaction between KLF8 and PIASs and subsequent sumoylation of KLF8 by PIASs during DNA damage response.
FIGURE 4.

SUMOylation of KLF8 is mediated by its interaction with PIASs and catalytic active PARP-1. A, the PARP-1 binding-defective mutant of KLF8 fails to interact with PIASs. 293T cells were cotransfected with HA-KLF8, HA-ZF1,2mCs, or HA-K67R with Myc-PIAS-1, -2, or -4 for 24 h. Whole cell lysates were prepared for Western blotting (IB) and co-IP (IP). B, KLF8 interaction with PIASs depends on PARP-1 expression and is enhanced by DNA damage. PARP-1+/+ MEFs and PARP-1−/− MEFs were cotransfected with HA-KLF8 and Myc-PIAS-1, -2, or -4, for 24 h. Whole cell lysates were prepared for Western blotting and co-IP. C, DNA damage-induced SUMOylation of KLF8 requires catalytically active PARP-1. PARP-1+/+ MEFs were cotransfected with HA-KLF8 and Myc-SUMO-1 for 24 h and treated first with the PARP-1 inhibitor PJ-34 for 24 h and then with doxorubicin (Doxo) for another 4 h. Whole cell lysates were prepared for Western blotting and co-IP. HA-K67R was used as a SUMOylation-negative control. The arrowheads indicate SUMOylated KLF8. D, recovery from the DNA damage releases PARP-1 and KLF8 from PIAS1. PARP-1+/+ cells were cotransfected with Myc-PIAS1 and HA-KLF8 for 24 h and treated with doxorubicin for 4 h. Whole cell lysates were collected at indicated time points after doxorubicin was removed for Western blotting and co-IP. E, the catalytic activity is required for PARP-1 interaction with PIAS1. PARP-1+/+ cells were transfected with Myc-PIAS1 for 24 h and treated similarly as in C prior to Western blotting and co-IP. F, KLF8 interaction with PARP-1 is independent of PARP-1 catalytic activity or PIAS1 levels. PARP-1+/+ cells were transfected with HA-KLF8 for 24 h and treated with PJ-34 for another 24 h prior to Western blotting and co-IP.
To test whether the PARylation activity of PARP-1 is involved in the SUMOylation of KLF8, we cotransfected the PARP-1+/+ MEFs with KLF8 and SUMO-1, treated the cells with both doxorubicin and the PARP-1 inhibitor PJ-34, and then examined the change in the SUMOylation state of KLF8. The results showed that the levels of SUMOylated KLF8 were markedly reduced when PARP-1 activity was inhibited (Fig. 4C, compare lane 1 to lane 2), suggesting that the PARylation activity of PARP-1 is required for DNA damage-induced SUMOylation of KLF8. Time course observation revealed that the PIAS-associated KLF8 and PARP-1 began to be released from PIASs around 1 h post-doxorubicin removal (Fig. 4D), suggesting that the KLF8-mediated DNA repair process may be finished quite quickly.
To determine the role of the PARylation activity of PARP-1 in the interaction between PARP-1, PIAS1, and KLF8, we first treated PIAS1-transfected PARP-1+/+ MEFs with both doxorubicin and PJ-34 and examined the interaction between PARP-1 and PIAS1 by co-IP. We found that inhibition of PARP-1 activity prevented DNA damage-enhanced interaction between PARP-1 and PIAS1 (Fig. 4E), suggesting that the PARylation activity of PARP-1 is critical for the interaction between PARP-1 and PIAS1 during DNA damage. We found that neither inhibition of PARP-1 activity nor overexpression of PIAS1 affected the interaction between PARP-1 and KLF8 in the cells (Fig. 4F), suggesting that KLF8 interaction with PARP-1 is independent of PARP-1 catalytic activity or PIAS1 expression levels. Taken together, these results support the notion that, during DNA damage, the catalytically active PARP-1 recruits KLF8 and PIASs together to facilitate PIASs catalyzed SUMOylation of KLF8 and subsequent DNA repair.
Phosphorylation of KLF8 at Ser-80 by DNA-PKcs Is Required for the DNA Damage-induced SUMOylation of KLF8
The kinase-specific phosphorylation sites on the KLF8 sequence were analyzed using NetPhosK software online, resulting in an interesting clue that the Ser-80 site of KLF8 is a potential phosphorylation target site for the serine kinase DNA-PKcs, another master sensor of DNA damage and regulator of DNA repair (32–34). To test whether Ser-80 plays a role in the DNA damage-induced SUMOylation of KLF8, the wild-type KLF8 or its S80A mutant were compared for their SUMOylation states in the PARP-1+/+ MEFs treated with doxorubicin. Unlike the wild-type KLF8 that was well SUMOylated, the S80A mutant showed no sign of being SUMOylated (Fig. 5A). This result suggests that the potential phosphorylation of KLF8 at Ser-80 by a serine kinase plays a critical role in the DNA damage-induced SUMOylation of KLF8.
FIGURE 5.

The phosphorylation of KLF8 at Ser-80 by DNA-PKcs is required for the SUMOylation of KLF8 upon DNA damage. A, the KLF8 S80A mutant fails to be SUMOylated during DNA damage. PARP-1+/+ MEFs were transfected with HA-KLF8, HA-S80A, or vector alone along with Myc-SUMO-1. The cells were treated with doxorubicin for 4 h followed by 2 h of recovery. Whole cell lysates (WCL) were prepared for Western blotting (IB) and co-IP (IP). The arrowhead, arrow, and asterisk indicate the SUMOylated KLF8, KLF8, and IgG heavy chain, respectively. B, KLF8 interacts with DNA-PKcs in a PARP-1-dependent fashion. PARP-1+/+ cells and PARP-1−/− cells were cotransfected with HA-KLF8, HA-ZF1,2mCs, or HA-S80A, treated similarly as in A, and prepared for Western blotting and co-IP. C, DNA-PKcs is required for the phosphorylation, and SUMOylation of KLF8. PARP-1+/+ cells were transfected with indicated HA-tagged WT KLF8 or its mutants along with siRNA against DNA-PKcs (siDPK) for 48 h or with 2.5 μm of the DNA-PKcs inhibitor Nu7026 (Nu) for 10 h. The cells were then treated with doxorubicin for 4 h. Whole cell lysates were prepared for IP with HA antibodies followed by Western blotting with phosphoserine (pSer) antibodies or DNA-PKcs (DPK) antibodies. The arrowhead indicates SUMOylated KLF8. D, the S80A and ZF1,2mCs mutants are less serine-phosphorylated than the wild-type KLF8 during DNA damage. PARP-1+/+ MEFs were transfected with indicated HA-tagged KLF8 WT or mutants for 48 h and treated with doxorubicin or vehicle control for 4 h. Whole cell lysates (WCL) were prepared for indicated Western blotting and co-IP (top panel). The ratio of serine-phosphorylated KLF8 to total KLF8 was normalized to the doxorubicin-treated wild-type KLF8-expressing group (bottom panel). Data represent the mean ± S.E. of at least three independent duplicate experiments. *, p < 0.05. E, KLF8 is serine-phosphorylated in response to DNA damage in MDA-MB-231 cells. The cells were treated with doxorubicin or vehicle control for 4 h, followed by 2 h recovery. Whole cell lysates were prepared for Western blotting and co-IP. IgG, IP negative control. The arrows indicate SUMOylated KLF8.
Co-IP experiments demonstrated an interaction between KLF8 and DNA-PKcs that depends on the expression of PARP-1 and KLF8 interaction with PARP-1 (Fig. 5B).
Silencing DNA-PKcs caused a significant decrease in the levels of DNA damage-induced SUMOylation of KLF8, and inhibiting DNA-PKcs catalytic activity with Nu7026 almost completely abolished the SUMOylation of KLF8 1 h after doxorubicin removal (Fig. 5C, top panel, compare lanes 5 or 6 to lane 1). This decrease in the SUMOylation of KLF8 was well correlated with the reduction in the overall serine phosphorylation of KLF8 (Fig. 5C, center panel, compare lanes 5 or 6 to lane 1). At the end of doxorubicin treatment, the levels of phosphorylation of KLF8 at serine residues were induced highly on the wild-type KLF8 and the K67R mutant but to a much less extent on the S80A and ZF1,2mCs mutants (Fig. 5D). Taken together, these results suggest that phosphorylation of KLF8 at Ser-80, presumably by DNA-PKcs, could possibly play a priming role for the PARP-1-dependent SUMOylation of KLF8 by PIASs during the cell response to doxorubicin-induced DNA damage.
The Phosphorylation and SUMOylation of KLF8 and Its Interaction with PARP-1, DNA-PKcs, and PIASs Are Critical for Its Role in Promoting DNA Repair and Cell Survival in Breast Cancer Cells
To test if the aforementioned multiple posttranslational modifications and protein-protein interactions also take place in breast cancer cells encountering DNA damage, we first treated the MDA-MB-231 cells and examined the serine-phosphorylation state of KLF8. We found that, as seen in the PARP-1+/+ MEFs, the levels of the KLF8 phosphorylation was markedly increased in response to doxorubicin treatment (Fig. 5E).
We then determined whether PARP-1 activity and expression are important in the KLF8-promoted DNA repair and cell survival against DNA damage. We grew the 231-K8ikd cells under uninduced (U) or induced (I) conditions, treated the cells simultaneously with both doxorubicin and the PARP-1 inhibitor or siRNAs, and examined the impact of the treatments on the levels of DNA damage and cell viability. We found that the doxorubicin-induced DNA damage was markedly enhanced when KLF8 expression in the cancer cells was silenced (I) (Fig. 6A, compare I and U). When PARP-1 activity was inhibited in the cells, the levels of DNA damage were increased similarly, regardless of the expression (U + ABT) or knockdown (I + ABT) of KLF8 in the cells (Fig. 6A, compare the ABT-treated and untreated groups). The increase in DNA damage levels was inversely correlated with cell viability (Fig. 6B). Silencing PARP-1 expression in the cells had an impact on cell viability similar to that caused by KLF8 knockdown (Fig. 6C). In MCF-10A cells, on the other hand, overexpression of the wild-type KLF8 but not the ZF1,2mCs, S80A, or K67R mutant, could significantly increase the cell resistance to doxorubicin-induced DNA damage (Fig. 6D) and cell death (E). These results strongly suggest that the multiple forms of protein modifications and interactions centering on KLF8 are critical for DNA repair and therapeutic resistance to DNA damage-induced death of human breast cancer cells.
FIGURE 6.

Multiple posttranslational modifications are involved in KLF8-mediated, PARP-1-dependent DNA repair and cell survival in breast cancer cells. A, PARP-1 catalytic activity is required for KLF8-promoted DNA repair in breast cancer cells. 231-K8iKd cells were grown with or without 1 μg/ml doxycycline to induce (I) or uninduce (U) the knockdown of KLF8. After 48 h, 2.5 μm of the PARP-1 inhibitor ABT-888 (ABT) was added for 8 h. The cells were then treated with 1 μm doxorubicin (Doxo) for indicated periods of time and processed for Western blotting (IB). B and C, PARP-1 catalytic activity is required for KLF8-dependent cell survival. 231-K8iKd cells were grown as in A. After 4 h of treatment with doxorubicin and 2 h of recovery, the cells were processed for WST-1 assay (B), or 231-K8iKd cells were transfected with indicated siRNAs for 24 h under U conditions and then grown in U or I conditions for 48 h. After 4 h of treatment with doxorubicin and 2 h of recovery, the cells were processed for WST-1 assay (C). D and E, disruption of PARP-1 interaction, phosphorylation on Ser-80 or SUMOylation on Lys-67 reduces the ability of KLF8 to promote DNA repair and cell survival. MCF-10A cells were transfected with HA-tagged wild-type KLF8 (WT), indicated mutants, or vector alone for 24 h and treated with doxorubicin for 4 h followed by 2 h of recovery. Cells were then prepared for Western blotting (D) or clonogenic assay (E). *, p < 0.05 compared with any one of the other three groups. Data represent the mean ± S.E. of three independent triplicate experiments.
The Phosphorylation and SUMOylation of KLF8 Redirects It to the DNA Damage Site during DNA Repair
To determine whether KLF8 is recruited to the DNA damage site during DNA damage response, the transcriptional activity of KLF8 on the promoter of cyclin D1, a known target of KLF8, and the presence of KLF8 at the DNA DSBs were examined. We found that in response to DNA damage, the activity of KLF8 on the cyclin D1 promoter was reduced in a Ser-80- and Lys-67-dependent manner (Fig. 7A) and that KLF8 was recruited to the DNA DSBs (B). These results suggest that the KLF8 protein was redirected from a target gene locus not associated with DNA damage to DNA damage site to facilitate DNA repair and that the phosphorylation and SUMOylation of KLF8 is critical for this redirection process.
FIGURE 7.
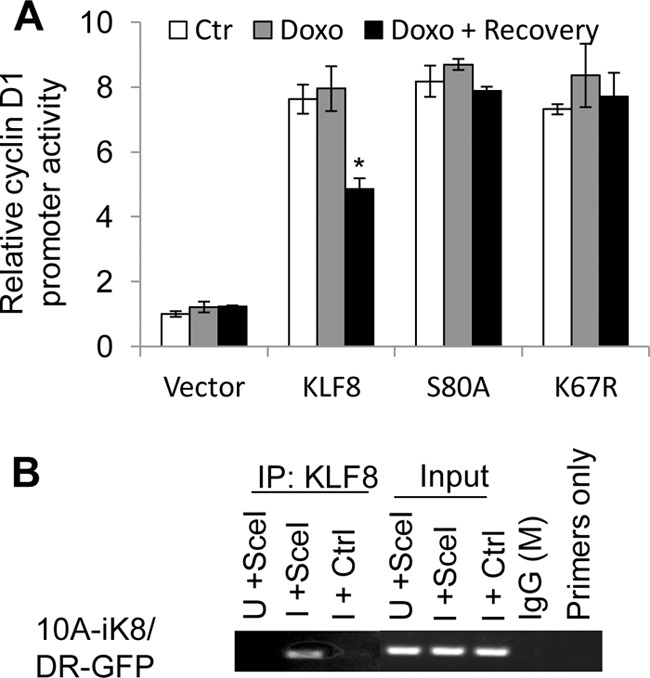
The phosphorylation at Ser-80 and the SUMOylation at Lys-67 of KLF8 are responsible for its reduced activity on the cyclin D1 promoter and its recruitment to the DNA damage site during DNA repair. A, the S80A and K67R mutants failed to respond to DNA damage in activating the cyclin D1 promoter. NIH3T3 cells were transfected with the cyclin D1 promoter reporter plasmid along with the indicated KLF8 DNAs or vector alone for 30 h. DNA damage was induced by 1 μm doxorubicin (Doxo) for 4 h of treatment, followed by 1 h of recovery. Dural luciferase assays were performed as described under “Experimental Procedures.” The data in the graph represent the mean ± S.E. of at least three independent duplicate experiments. *, p < 0.05. B, recruitment of KLF8 to double-stranded DNA breaks. The DNA damage site-targeted ChIP was performed as described under “Experimental Procedures.”
DISCUSSION
This study has identified a novel role and mechanisms for KLF8 in promoting DNA repair in breast cancer cells. This function of KLF8 depends on PARP-1 expression and activity, DNA-PKcs-mediated phosphorylation of KLF8 at Ser-80, and PIAS-catalyzed SUMOylation of KLF8. These findings suggest a potentially important mechanism underlying therapeutic resistance of breast cancer cells. Inhibitors of both PARP-1 and DNA-PKcs have been under active clinical trials for treatment of cancers of particularly BRCA-deficient types (1). Protein SUMOylation is generally accepted as a cellular process favoring tumor progression, and inhibitors of SUMOylation have also been explored as anti-cancer drugs (6). This work sheds new light on the drug-resistant mechanisms of breast cancer and advances the knowledge for designing novel strategies for breast cancer treatment.
Genotoxic anti-cancer drugs such as doxorubicin, proflavine, and daunomycin cause SSBs in treated cells. Failure to fix the SSBs will subsequently result in DSBs. SSBs and DSBs are primarily repaired by PARP-1 and BRCAs or DNA-PK, respectively (2, 4, 5). Our finding that KLF8 interacts with and is regulated by both PARP-1 and DNA-PKcs in the cells exposed to doxorubicin suggests that KLF8 may participate in both SSB and DSB repair processes.
Many interesting questions emerge regarding exactly what role KLF8 plays in protecting DNA. We have shown recently shown that KLF8 actively regulates the expression of genes that are associated with cell cycle progression and epithelial-to-mesenchymal transition such as cyclin D1, E-cadherin, and MMP9 in actively proliferating cells, including breast cancer cells (10, 11, 23, 26). We have also demonstrated that PARP-1 binding and modification of KLF8 are critical for KLF8 regulation of these genes and cellular processes (20) and that SUMOylation of KLF8 delimits such roles of KLF8 (18). In addition, one of the primary changes in SUMOylated proteins is their subcellular relocation to interact with distinct binding partner molecules (6, 7). Therefore, it appears likely that the cellular response to the DNA damage signals KLF8 away from these proliferation-promoting genes. PARP-1 appears to work as a “switchman” using SUMOylation of KLF8 as the “switch” to control the process. In other words, there appears to be a PARP-1-associated pool of KLF8 proteins. Depending upon the cell need, the KLF8 proteins are dispatched from this pool to distinct functional sites of KLF8. We speculate that in response to the DNA damage call, PARP-1 first aids in the SUMOylation of KLF8 and then redispatches it away from loci of proliferation-promoting genes. From there, PARP-1 further takes the SUMOylated KLF8 to the DNA damage sites and they work there together to fix the damage, or it sends KLF8 to loci of a different group of KLF8 target genes or microRNAs responsible for cell cycle arrest, DNA repair or protection, or anti-apoptosis (Fig. 8). Experiments are in progress to uncover the details of these very interesting possibilities. It is less plausible that all that happens to KLF8 during DNA damage response is to help brake the cell cycle wheel only.
FIGURE 8.

A model of redirection of KLF8 to DNA damage site during DNA repair. In the absence of DNA damage, KLF8 primarily targets target gene locus A (TGL A) not related to DNA repair, such as the cyclin D1 gene. In response to DNA damage, KLF8 is redirected to the DNA damage sites where it contributes to DNA repair in an unknown way. KLF8 could be redirected to a different TGL such as TGL B outside of the DNA damage site to regulate expression of genes associated with DNA repair. PARP-1-dependent phosphorylation and SUMOylation of KLF8 is critical for the redirection of KLF8.
Whatever the molecular action of KLF8 is on the DNA damage response, its prior SUMOylation, which is achieved by a combined effect from PARP-1, DNA-PKcs, and PIASs, seems to be essential. It has been suggested that phosphorylation plays an important role in initiating or promoting the SUMOylation of the same protein molecule (6). Although the amino acid sequence spanning the Lys-67 SUMOylation site and the Ser-80 site of KLF8 does not exactly match the predicted consensus sequence of the so-called phosphorylation-dependent SUMOylation motif (35, 36), the negative charges brought to the proximity to the Lys-67 by DNA-PKcs-catalyzed phosphorylation of the Ser-80 site could turn the region into a motif very similar to another SUMOylation consensus motif named negatively charged amino acid-dependent SUMOylation motif (37). It will be interesting to test whether it is essential for the phosphorylation of KLF8 at the Ser-80 by DNA-PKcs to prime the SUMOylation of KLF8 at Lys-67.
We have demonstrated recently that KLF8 interaction with PARP-1 in the nucleus does not depend upon PARP-1 activity, whereas KLF8 transactivation of its target gene promoters, such as cyclin D1, requires the PARylation of PARP-1 itself and of KLF8 (20). Our results here indicate that both the interaction of PARP-1 with KLF8, DNA-PKcs, and PIASs and the PARylation activity of PARP-1 are required for the SUMOylation of KLF8 during DNA damage. To the best of our knowledge, there has not been any report on PARylation of DNA-PKCS or PAISs. Further studies are worth doing to determine whether or not PARP-1 needs to catalyze the PARylation of any of the three interacting proteins to ensure the SUMOylation of KLF8.
DNA damage-based chemotherapies depend on the failure of cancer cells to repair the DNA damage and subsequent cell death. Aberrant high levels of DNA repair function in the cells likely increase not only the resistance of the cells to such therapies but also the malignancy of the cells because of improper DNA repair-mediated genomic and chromosomal instability. Indeed, our results have clearly linked the KLF8-promoted DNA repair to the cell resistance to doxorubicin-induced cell death. It remains to be determined whether KLF8 plays a similar role in repairing DNA damage caused by other types of genotoxic agents, such as DNA alkylating agents and ionizing radiation. Nevertheless, our results suggest that, in addition to enhancing the drug resistance of the cancer cells, KLF8 could play a role in disturbing genomic integrity through its aberrant DNA repair function to subsequently contribute to aggressive progression of cancer.
In summary, this work has identified KLF8 as a novel regulator of DNA repair in human breast cancer cells. Given its barely detectable expression in normal human breast epithelial cells and tissue (10, 11), KLF8 may represent a novel target for DNA repair-targeted therapy against breast cancer drug resistance.
This work was supported by NCI Grant CA132977 and Susan G. Komen for the Cure Grants KG090444 and KG080616 (to J. Z.).
- SSB
- single-strand break
- DSB
- double-strand break
- DNA-PK
- DNA-dependent protein kinase
- DNA-PKcs
- DNA-dependent protein kinase catalytic subunit
- MEF
- mouse embryonic fibroblast
- IP
- immunoprecipitation
- BRCA
- breast cancer.
REFERENCES
- 1. Lord C. J., Ashworth A. (2012) The DNA damage response and cancer therapy. Nature 481, 287–294 [DOI] [PubMed] [Google Scholar]
- 2. Cleaver J. E., Lam E. T., Revet I. (2009) Disorders of nucleotide excision repair. The genetic and molecular basis of heterogeneity. Nat. Rev. Genet. 10, 756–768 [DOI] [PubMed] [Google Scholar]
- 3. David S. S., O'Shea V. L., Kundu S. (2007) Base-excision repair of oxidative DNA damage. Nature 447, 941–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lieber M. R., Gu J., Lu H., Shimazaki N., Tsai A. G. (2010) Nonhomologous DNA end joining (NHEJ) and chromosomal translocations in humans. Subcell. Biochem. 50, 279–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Moynahan M. E., Jasin M. (2010) Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell Biol. 11, 196–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhao J. (2007) Sumoylation regulates diverse biological processes. Cell. Mol. Life Sci. 64, 3017–3033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Huen M. S., Chen J. (2008) The DNA damage response pathways. At the crossroad of protein modifications. Cell Res. 18, 8–16 [DOI] [PubMed] [Google Scholar]
- 8. Cao Q., Qin C., Meng X., Ju X., Ding Q., Wang M., Zhu J., Wang W., Li P., Chen J., Zhang Z., Yin C. (2011) Genetic polymorphisms in APE1 are associated with renal cell carcinoma risk in a Chinese population. Mol. Carcinog. 50, 863–870 [DOI] [PubMed] [Google Scholar]
- 9. Wang X., Urvalek A. M., Liu J., Zhao J. (2008) Activation of KLF8 transcription by focal adhesion kinase in human ovarian epithelial and cancer cells. J. Biol. Chem. 283, 13934–13942 [DOI] [PubMed] [Google Scholar]
- 10. Wang X., Zheng M., Liu G., Xia W., McKeown-Longo P. J., Hung M. C., Zhao J. (2007) Krüppel-like factor 8 induces epithelial to mesenchymal transition and epithelial cell invasion. Cancer Res. 67, 7184–7193 [DOI] [PubMed] [Google Scholar]
- 11. Wang X., Zhao J. (2007) KLF8 transcription factor participates in oncogenic transformation. Oncogene 26, 456–461 [DOI] [PubMed] [Google Scholar]
- 12. Li J. C., Yang X. R., Sun H. X., Xu Y., Zhou J., Qiu S. J., Ke A. W., Cui Y. H., Wang Z. J., Wang W. M., Liu K. D., Fan J. (2010) Up-regulation of Krüppel-like factor 8 promotes tumor invasion and indicates poor prognosis for hepatocellular carcinoma. Gastroenterology 139, 2146–2157 [DOI] [PubMed] [Google Scholar]
- 13. Fu W. J., Li J. C., Wu X. Y., Yang Z. B., Mo Z. N., Huang J. W., Xia G. W., Ding Q., Liu K. D., Zhu H. G. (2010) Small interference RNA targeting Krüppel-like factor 8 inhibits the renal carcinoma 786–0 cells growth in vitro and in vivo. J. Cancer Res. Clin. Oncol. 136, 1255–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu L., Liu N., Xu M., Liu Y., Min J., Pang H., Zhang N., Zhang H. (2012) Lentivirus-delivered Krüppel-like factor 8 small interfering RNA inhibits gastric cancer cell growth in vitro and in vivo. Tumour Biol 33, 53–61 [DOI] [PubMed] [Google Scholar]
- 15. Cox B. D., Natarajan M., Stettner M. R., Gladson C. L. (2006) New concepts regarding focal adhesion kinase promotion of cell migration and proliferation. J. Cell. Biochem. 99, 35–52 [DOI] [PubMed] [Google Scholar]
- 16. Schnell O., Romagna A., Jaehnert I., Albrecht V., Eigenbrod S., Juerchott K., Kretzschmar H., Tonn J. C., Schichor C. (2012) Krüppel-like factor 8 (KLF8) is expressed in gliomas of different WHO grades and is essential for tumor cell proliferation. PLoS ONE 7, e30429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wan W., Zhu J., Sun X., Tang W. (2012) Small interfering RNA targeting Krüppel-like factor 8 inhibits U251 glioblastoma cell growth by inducing apoptosis. Mol. Med. Report 5, 347–350 [DOI] [PubMed] [Google Scholar]
- 18. Wei H., Wang X., Gan B., Urvalek A. M., Melkoumian Z. K., Guan J. L., Zhao J. (2006) Sumoylation delimits KLF8 transcriptional activity associated with the cell cycle regulation. J. Biol. Chem. 281, 16664–16671 [DOI] [PubMed] [Google Scholar]
- 19. Urvalek A. M., Lu H., Wang X., Li T., Yu L., Zhu J., Lin Q., Zhao J. (2011) Regulation of the oncoprotein KLF8 by a switch between acetylation and sumoylation. Am. J. Transl. Res. 3, 121–132 [PMC free article] [PubMed] [Google Scholar]
- 20. Lu H., Wang X., Li T., Urvalek A. M., Yu L., Li J., Zhu J., Lin Q., Peng X., Zhao J. (2011) Identification of poly (ADP-ribose) polymerase-1 (PARP-1) as a novel Krüppel-like factor 8-interacting and -regulating protein. J. Biol. Chem. 286, 20335–20344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mehta T. S., Lu H., Wang X., Urvalek A. M., Nguyen K. H., Monzur F., Hammond J. D., Ma J. Q., Zhao J. (2009) A unique sequence in the N-terminal regulatory region controls the nuclear localization of KLF8 by cooperating with the C-terminal zinc-fingers. Cell Res. 19, 1098–1109 [DOI] [PubMed] [Google Scholar]
- 22. Urvalek A. M., Wang X., Lu H., Zhao J. (2010) KLF8 recruits the p300 and PCAF co-activators to its amino terminal activation domain to activate transcription. Cell Cycle 9, 601–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhao J., Bian Z. C., Yee K., Chen B. P., Chien S., Guan J. L. (2003) Identification of transcription factor KLF8 as a downstream target of focal adhesion kinase in its regulation of cyclin D1 and cell cycle progression. Mol. Cell. 11, 1503–1515 [DOI] [PubMed] [Google Scholar]
- 24. Zerfaoui M., Errami Y., Naura A. S., Suzuki Y., Kim H., Ju J., Liu T., Hans C. P., Kim J. G., Abd Elmageed Z. Y., Koochekpour S., Catling A., Boulares A. H. (2010) Poly(ADP-ribose) polymerase-1 is a determining factor in Crm1-mediated nuclear export and retention of p65 NF-κB upon TLR4 stimulation. J. Immunol. 185, 1894–1902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang Z. Q., Stingl L., Morrison C., Jantsch M., Los M., Schulze-Osthoff K., Wagner E. F. (1997) PARP is important for genomic stability but dispensable in apoptosis. Genes Dev. 11, 2347–2358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang X., Lu H., Urvalek A. M., Li T., Yu L., Lamar J., DiPersio C. M., Feustel P. J., Zhao J. (2011) KLF8 promotes human breast cancer cell invasion and metastasis by transcriptional activation of MMP9. Oncogene 30, 1901–1911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhao J., Pestell R., Guan J. L. (2001) Transcriptional activation of cyclin D1 promoter by FAK contributes to cell cycle progression. Mol. Biol. Cell 12, 4066–4077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pierce A. J., Johnson R. D., Thompson L. H., Jasin M. (1999) XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 13, 2633–2638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jirawatnotai S., Hu Y., Michowski W., Elias J. E., Becks L., Bienvenu F., Zagozdzon A., Goswami T., Wang Y. E., Clark A. B., Kunkel T. A., van Harn T., Xia B., Correll M., Quackenbush J., Livingston D. M., Gygi S. P., Sicinski P. (2011) A function for cyclin D1 in DNA repair uncovered by protein interactome analyses in human cancers. Nature 474, 230–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Franken N. A., Rodermond H. M., Stap J., Haveman J., van Bree C. (2006) Clonogenic assay of cells in vitro. Nat. Protoc. 1, 2315–2319 [DOI] [PubMed] [Google Scholar]
- 31. Zhao J., Guan J. L. (2009) Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev. 28, 35–49 [DOI] [PubMed] [Google Scholar]
- 32. Brenner J. C., Ateeq B., Li Y., Yocum A. K., Cao Q., Asangani I. A., Patel S., Wang X., Liang H., Yu J., Palanisamy N., Siddiqui J., Yan W., Cao X., Mehra R., Sabolch A., Basrur V., Lonigro R. J., Yang J., Tomlins S. A., Maher C. A., Elenitoba-Johnson K. S., Hussain M., Navone N. M., Pienta K. J., Varambally S., Feng F. Y., Chinnaiyan A. M. (2011) Mechanistic rationale for inhibition of poly(ADP-ribose) polymerase in ETS gene fusion-positive prostate cancer. Cancer Cell 19, 664–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sajish M., Zhou Q., Kishi S., Valdez D. M., Jr., Kapoor M., Guo M., Lee S., Kim S., Yang X. L., Schimmel P. (2012) Trp-tRNA synthetase bridges DNA-PKcs to PARP-1 to link IFN-γ and p53 signaling. Nat. Chem. Biol. 15, 547–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yoon J. H., Ahn S. G., Lee B. H., Jung S. H., Oh S. H. (2012) Role of autophagy in chemoresistance. Regulation of the ATM-mediated DNA-damage signaling pathway through activation of DNA-PKcs and PARP-1. Biochem. Pharmacol. 83, 747–757 [DOI] [PubMed] [Google Scholar]
- 35. Yang X. J., Grégoire S. (2006) A recurrent phospho-sumoyl switch in transcriptional repression and beyond. Mol. Cell 23, 779–786 [DOI] [PubMed] [Google Scholar]
- 36. Hietakangas V., Anckar J., Blomster H. A., Fujimoto M., Palvimo J. J., Nakai A., Sistonen L. (2006) PDSM, a motif for phosphorylation-dependent SUMO modification. Proc. Natl. Acad. Sci. U.S.A. 103, 45–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yang S. H., Galanis A., Witty J., Sharrocks A. D. (2006) An extended consensus motif enhances the specificity of substrate modification by SUMO. EMBO J. 25, 5083–5093 [DOI] [PMC free article] [PubMed] [Google Scholar]