

Background: Sortilin and p75NTR induce neuronal apoptosis by binding pro-neurotrophins during development and following neuronal injury.
Results: Sortilin interacts with an extracellular juxtamembrane 23-amino acid sequence of p75NTR.
Conclusion: Despite binding being mediated through extracellular interactions, the intracellular domain of sortilin regulates p75NTR shedding and apoptosis.
Significance: Mapping may allow design of compounds inhibiting neuronal cell death by blocking the interaction between sortilin and p75NTR.
Keywords: Apoptosis, Neurotrophins, Receptors, RIP, Shedding, p75, Sortilin
Abstract
Neurotrophins comprise a group of neuronal growth factors that are essential for the development and maintenance of the nervous system. However, the immature pro-neurotrophins promote apoptosis by engaging in a complex with sortilin and the p75 neurotrophin receptor (p75NTR). To identify the interaction site between sortilin and p75NTR, we analyzed binding between chimeric receptor constructs and truncated p75NTR variants by co-immunoprecipitation experiments, surface plasmon resonance analysis, and FRET. We found that complex formation between sortilin and p75NTR relies on contact points in the extracellular domains of the receptors. We also determined that the interaction critically depends on an extracellular juxtamembrane 23-amino acid sequence of p75NTR. Functional studies further revealed an important regulatory function of the sortilin intracellular domain in p75NTR-regulated intramembrane proteolysis and apoptosis. Thus, although the intracellular domain of sortilin does not contribute to p75NTR binding, it does regulate the rates of p75NTR cleavage, which is required to mediate pro-neurotrophin-stimulated cell death.
Introduction
Neurotrophins including NGF, BDNF, and neurotrophin-3 (NT3)3 constitute a group of neuronal growth factors that are important for the development, maintenance, and differentiation of the nervous system (1). However, neurotrophins can also be released in an immature form known as pro-neurotrophins (proNTs), which display distinct and often opposing biological activities to those of their mature counterparts (2–4). Hence, by forming a heterotrimeric complex with sortilin and the p75 neurotrophin receptor (p75NTR), proNGF and proBDNF induce apoptosis during development and cell senescence, as well as following neuronal injury (5–7). In addition, proNT3 has recently been found to promote apoptosis in superior and spiral ganglion neurons by binding sortilin and p75NTR (8, 9). Binding of proNTs is achieved by binding of the pro-domain by sortilin, whereas p75NTR interacts with the mature part of the neurotrophin (5). However, sortilin and p75NTR also exist as a preformed receptor complex, interacting in the absence of ligand (5).
Sortilin is a neuronal type I receptor belonging to the Vps10p domain receptor family (10, 11). Extracellularly, sortilin contains a ligand-binding 10-bladed β-propeller followed by a 10CC module (12). A short cytoplasmic tail contains several internalization and sorting motifs, which facilitate the trafficking and sorting functions of sortilin (13). p75NTR is a member of the TNF receptor superfamily and is best known for its role in neuronal apoptosis and neurodegeneration (14). Its extracellular domain consists of four tandemly arranged ligand-binding domains followed by a ∼60-amino acid juxtamembrane stalk region. As for other members of the TNF receptor superfamily, a death domain is found in the cytoplasmic domain linked to the transmembrane sequence via a 60-amino acid p75NTR-specific “chopper” module (15). The p75NTR intracellular domain (ICD) has been reported to promote apoptosis, as well as cell survival in a cell type-specific manner if released into the cytosol (16–19). This is achieved by regulated intramembrane proteolysis (RIP) whereby the stalk region of p75NTR is first cleaved by the α-secretase TACE/ADAM17, generating the C-terminal fragment. The remaining membrane-tethered stub is subsequently cleaved by presenilin-dependent γ-secretase, releasing the p75NTR ICD to the cytosol for signaling (20).
We have previously demonstrated that sortilin increases p75NTR affinity for proNTs. For example, cells expressing p75NTR alone bind proNGF with an approximate Kd value of ∼15 nm, whereas co-expression of sortilin increases affinity for proNGF ∼100-fold to Kd = ∼160 pm (5). This dramatically decreases the effective concentration of proNTs required for apoptotic signaling by p75NTR. Here we identify the extracellular domains in sortilin and p75NTR that are responsible for receptor heterodimerization. Although the intracellular domain of sortilin is not involved in receptor interactions, we found that it regulates RIP of p75NTR and proNT-stimulated apoptotic signaling, suggesting additional roles for sortilin in p75NTR-mediated apoptosis.
EXPERIMENTAL PROCEDURES
DNA Work
The pcDNA3.1/Zeo(−) containing cDNA encoding human wild type sortilin, sortilinmut (Y792A, L795A, and Leu829 to Leu830 deleted), IL2Recd,tm-sortilinmut,icd, and sortilintailless (truncated at position Cys783) have previously been described (13, 21, 22). For construction of sortilinecd-IL2Rtm,icd, a fragment was amplified by standard PCR techniques using the α-subunit of the human interleukin-2 receptor (IL2R)/pcDNA3.1/Zeo(−) as the template and an upstream primer encoding part of the transmembrane domain of IL2R and part of the luminal domain of sortilin, and a downstream primer containing a cytosolic sequence of IL2R. Using the native luminal BspEI site of sortilin and a 3′ primer-generated AflII site, the fragment was ligated into predigested sortilinmut/pcDNA3.1/Zeo(−). PCR-mediated overlap extension was used to fuse the extracellular and transmembrane domains of sortilin and HA-tagged p75NTR with the β-galactosidase Δα and Δω, respectively, generating sortilin-β-galΔα and HA-p75NTR-β-galΔω. An upstream fragment encoding part of the extracellular and transmembrane domains of sortilin in combination with part of β-galactosidase Δα was amplified using sortilinmut/pcDNA 3.1/Zeo(−) as the template. A downstream fragment encoding β-galactosidaseΔα was amplified using the template Δα/pwzl/Neo (23). The upstream fragment containing HA-p75NTR extracellular and transmembrane domains and part of β-galactosidase Δω was generated using HA-p75NTR/pcDNA3.1/G418 as the template. A downstream fragment encoding β-galactosidase Δω was amplified using Δω/pwzl/Hygro (23) as the template. Following amplification of overlapping PCR products, sortilin fusion protein was inserted into sortilinmut/pcDNA3.1/Zeo(−) using the native luminal BspEI site and the 3′ primer-generated AflII site, whereas HA-p75NTR fusion protein was ligated into predigested pcDNA 3.1/G418(−) using a primer-generated 5′-NotI and the 3′ AflII sites.
To make deletion and truncation expression constructs of p75NTR (p75NTRΔC1 (1–29, 66–425), p75NTRΔC1,2 (1–29, 109–425), p75NTRΔC1,2,3,4 (1–29, 190–425), p75NTRΔstalk (1–227, 251–425), p75NTRstalk,tm,icd, (228–425), and p75NTRICD (274–425)) and YFP- or CFP-tagged versions of these (34), a modified pCDNA3 (Invitrogen) backbone was used. The rat p75NTR signal peptide including a Kozak sequence (nucleotides −29 to +87) was inserted between the KpnI and EcoRV restriction sites, generating the vector pCDNA3-SP. p75NTR coding sequences were amplified under standard PCR conditions and cloned into pCDNA3-SP using the primer-generated EcoRV and NheI sites. In cases where p75NTR variants were fused to a fluorophore, YFP and CFP were amplified by PCR from peYFP-N1 and peCFP-N1 (Clontech), using primers incorporating 5′ EcoRV and NheI restriction sites and a 3′ stop codon and a HindIII site. Enhanced YFP and CFP were cloned in frame between the EcoRV and HindIII restriction sites of pCDNA3-SP, generating the vectors pCDNA3-YFP/CFP. p75NTR coding sequences were amplified by PCR with a 5′ EcoRV and a 3′ NheI restriction site. p75NTR coding sequences were then cloned between the EcoRV and NheI restriction sites of pCDNA3-YFP/CFP to generate in-frame fusion proteins. p75NTRtm,icd (251–425) was constructed as previously described (24) and fused to YFP as described above. To generate sortilin-YFP, full-length human sortilin, including the sequences encoding the signal and pro-peptides, and the Kozak sequence were amplified by PCR, thereby generating a 3′-Kpn site and a 5′-Nhe site. The fragment was inserted into pCDNA3-YFP. The p75NTR-Gal4 reporter construct for the p75NTR cleavage reporter assay was cloned by inserting rat p75NTR cDNA into the pcDNA3-GVP vector containing Gal4 DNA binding and VP16 transcription activating domains and a neomycin selection cassette. The plasmid was digested with MunI, and a Gal4-responsive 9×UAS Luc2P cassette from the pGL4.35 reporter vector (Promega) was inserted by blunt end cloning after filling in the recessed 3′ ends with Klenow fragment.
Cell Lines and Culturing
HEK293 cells and rat Schwannoma cells (RN22 cells) were cultured in DMEM (Lonza) supplemented with 10% FCS, 100 units of penicillin, and 100 μg/ml streptomycin (Invitrogen) in 5% CO2 at 37 °C. Transfections were carried out with FuGENE 6 transfection reagent (Roche Applied Science), and stably transfected clones were selected in medium containing 150 (HEK293 cells) or 300 (RN22 cells) μg/ml Zeocin (Invitrogen) and/or 400 μg/ml Geneticin (Invitrogen).
Antibodies and Proteins
Anti-neurotensin receptor-3, recognizing the extracellular domain of sortilin, was purchased from BD Transduction Laboratories (612100) and anti-p75NTR antibody (ab10494) from Abcam. Anti-p75NTR ICD 9992, for detecting p75NTR ICD, was a generous gift from Professor Moses V. Chao (Skirball Institute of Biomolecular Medicine, New York University School of Medicine). Anti-IL2Rα (I6152) and anti-HA tag (H6908) antibodies were from Sigma. Anti-GFP (11814460001) was purchased from Roche Applied Science. Recombinant His6-tagged soluble human sortilin (sol-sortilin) was produced and purified as previously described (22). The luminal domain of human p75NTR (Met1–Asn250) and human RET receptor tyrosine kinase (Met1–Arg635), all fused to the Fc region of IgG1, were purchased from R & D Systems. Recombinant human furin cleavage-resistant proNGF, proNT3, and wild type proBDNF were a gift from Professor Elisabeth Schwarz (Institute for Biotechnology, Martin-Luther-Universität, Halle-Wittenberg) (25). Recombinant mouse proNGF used for RN22 cell death assays was purchased from Chemicon. Mouse NGF was from Austral Biologicals (GF-022) and neurotensin from Sigma (N6383).
Co-immunoprecipitation
Stably or transiently transfected HEK293 cells were incubated with or without ligand in PBS (with 1 mm CaCl2 and MgCl2) for 90 min at room temperature and then treated with the reducible protein cross-linker dithiobis succinimidylpropionate (Pierce) according to the manufacturer's instructions. After washing, the cells were lysed on ice for 10 min in TNE buffer (20 mm Tris, pH 8, 10 mm EDTA) supplemented with 1% Nonidet P-40 and complete protease inhibitor mixture (Roche Applied Science). Samples were immunoprecipitated overnight at 4 °C by use of Gammabind G-Sepharose beads (Amersham Biosciences) coupled with anti-p75NTR (10494) or anti-GFP antibodies. Nonspecific binding was removed by washing beads five times in TBS supplemented with 0.05% Tween 20, and the proteins were eluted by boiling samples in reducing sample buffer (20 mm dithioerythritol, 2.5% SDS). Protein samples were subjected to reducing SDS-PAGE, Western blotting, and visualization using the Fuji film LAS1000 imaging system. The blots were stripped in 62.5 mm Tris-HCl, pH 6.7, 100 mm 2-mercaptoethanol, and 2% SDS for 30 min at 50 °C under rotation. Co-immunoprecipitation performed with p75NTR variants was visualized with the Odyssey infrared imaging system.
β-Galactosidase Complementation Assay
HEK293 cells were seeded into white 96-well tissue culture plates with clear bottoms (Sigma) at a density of 30,000 cells/well. The cells were then transfected with sortilin-β-galΔα and/or HA-p75NTR-β-galΔω and after 48 h incubated with or without proNTs for 30 min at 37 °C. β-galactosidase activity was assessed by adding 100 μl of Gal-Screen substrate (50 μl of both solution A and solution B) (Applied Biosystems) to each well. The plates were incubated at room temperature for 1 h, and luminescence was measured with a Victor3 1420 multilabel counter.
Surface Plasmon Resonance Analysis
Analyses were performed on a Biacore 3000 instrument equipped with CM5 sensor chips and activated as previously described (22). sol-sortilin was immobilized at a density of ∼72 fmol/mm2 in 10 mm sodium acetate, pH 4.0, and the remaining sites were blocked with 1 m ethanolamine, pH 8.5. Injection of proteins was done in running buffer at 25 °C (10 mm HEPES, pH 7.4, containing 150 mm NaCl, 1.5 mm CaCl2, 1 mm EGTA, and 0.005% P20). Binding was expressed in relative response units as the response obtained from the flow cell containing immobilized receptor minus the response obtained when using an activated and blocked but uncoupled flow cell.
Cell Population FRET Assay
The FRET of p75NTR molecules within a population of HEK293 cells was determined 48 h post-transfection of triplicate wells of transiently transfected cells and untransfected cells (to determine background fluorescence). The cells were washed once in PBS, pH 7.4, harvested in PBS, and centrifuged for 30 s at 10,000 × g. Cell pellets were resuspended in 200 μl of PBS and loaded into 96-well black microtiter plates (Greiner) for immediate analysis. Fluorescence was recorded simultaneously for the donor fluorophore at 430 ± 10 nm/480 ± 10 nm and the acceptor fluorophore at 485 nm/530 ± 10 nm, and FRET was measured at 430 ± 10 nm/530 ± 10 nm on a POLARstar OPTIMA multidetection microplate reader (BMG Labtech). FRET was calculated on cells as a FRET ratio, which is the fractional increase in YFP emission caused by FRET (26) according to the formula described in Ref. 34.
Receptor Processing Experiments
HEK293 cells or stably transfected polyclonal RN22 cells were seeded into poly-l-lysine-coated 24-well culture tissue plates at a density of 40,000 cells/well. HEK293 cells were transfected after 24 h and used for shedding experiments after an additional 48 h. Prior to treatment with 200 nm phorbol 12-myristate 13-acetate (PMA) (Calbiochem) cells were incubated for 1 h with 200 nm compound E (Alexis Biochemicals) or 1 μm epoxomicin (Sigma) in DMEM supplemented with 10% FCS. The cells were then incubated for 3 h (HEK293 cells) or 6 h (RN22 cells), after which they were lysed in TNE buffer supplemented with 1% Nonidet P-40 and complete protease inhibitor mixture and subjected to Western blotting.
Primary cultures of superior cervical ganglion neurons were established from postnatal day 3–5 sortilin knock-out mice (7) as described in Ref. 27. Dissociated neurons were seeded into poly-l-ornithine (Sigma) and 3 μg/ml laminin (Invitrogen) coated wells in neurobasal A medium supplemented with 1% nonessential amino acids, 1% GlutaMAX (all from Gibco), 0.2% B27 (Invitrogen), and 10 ng/ml NGF. 24 h post-seeding, the cells were starved for 4 h and subsequently added 1 μm epoxomicin, 10 ng/ml NGF and 10% FCS for 1 h followed by the addition of 200 nm PMA for 3 h. The cells were harvested in TNE buffer supplemented with 1% Nonidet P-40 and complete protease inhibitor mixture and subjected to Western blotting using HRP-conjugated secondary antibodies and Supersignal West Femto Sensitivity Substrate from Pierce.
RN22 Cell Death Assay
Polyclonal stable RN22 cell lines were seeded into poly-l-lysine-coated black 96-well tissue culture plates (PerkinElmer) at a density of 7500 cells/well. After 24 h, the cells were washed three times in DMEM without phenol red, supplemented with 100 units of penicillin and 100 μg/ml streptomycin, 1% GlutaMAX (Invitrogen), 1.5 μg/ml insulin, 50 μg/ml transferrin, 30 nm selenium, and 30 nm triiodothyronine (all from Sigma) and incubated with or without proNGF in a final volume of 100 μl of the same medium. Following 72 h of incubation, the number of dead and live cells was scored with the MultiTox-Fluor multiplex cytotoxicity assay (Promega) measuring fluorescence with a Victor3 1420 multilabel counter. To correct for differences in cell numbers among wells, the death signal was related to the life signal from the same well.
Luciferase Assay
HEK293 cells stably co-expressing p75NTR-Gal4 and luciferase under the UAS promoter were seeded in 24-well tissue culture plates at a density of 30,000 cells/well. After 24 h, some cells were transiently transfected with variants of sortilin. 72 h post-seeding, the cells were analyzed for luciferase activity using the luciferase assay from Promega. Prior to analysis, the cultures were treated 1 h with epoxomicin (1 μm) and in some cases with the α-secretase inhibitor TAPI-2 (20 μm) followed by the addition of PMA (200 nm) for 3 h. Luminescence was measured with a POLARstar OPTIMA plate reader.
Statistics
The results were analyzed by Student's t test, with p < 0.05 being considered a significant level of difference. In Figs. 5 and 6, the effect of wild type sortilin was compared with control and with the effect of sortilin mutants.
FIGURE 5.

The intracellular domain of sortilin regulates RIP of p75NTR. A, representative (n = 6) Western blot (WB) of HEK293 cell lysates, which were transiently co-transfected with p75NTR and sortilin constructs as indicated and treated for 3 h with 200 nm PMA, 200 nm γ-secretase inhibitor compound E, or 1 μm proteasome inhibitor epoxomicin as indicated. B, densitometric quantification of the amount of p75NTR ICD in each condition in A relative to the amount of full-length p75NTR. The data represent the means ± S.E. of six experiments. C, schematic representation of the Gal4/UAS assay. D, luminescence resulting from luciferase activity present in lysates of HEK293 cells stably co-expressing cDNAs encoding p75NTR-Gal4 and luciferase under the UAS promoter. The results were obtained following 3 h of treatment with 200 nm PMA and/or 20 μm α-secretase inhibitor TAPI-2 as indicated. PMA induced a ∼60% increase in the amount of p75NTR-ICD-Gal4-mediated luciferase activity (the data represent the means ± S.E. of three experiments). E, luciferase luminescence mediated by p75NTR ICD-Gal4 generated in HEK293 cells stably expressing p75NTR-Gal4 and luciferase under the UAS promotor and transiently transfected with sortilin variants as indicated. 48 h post-transfection, the cells were treated with 200 nm PMA for 3 h and analyzed for luciferase activity (the data represent the means ± S.E. of three experiments). F, luciferase activity in untreated HEK293 cells stably expressing p75NTR-Gal4 and luciferase and transiently transfected with wild type sortilin or control DNA. Cont. transf., control transfected; CTF, C-terminal fragment.
FIGURE 6.

Sortilin intracellular domain promotes RIP of p75NTR and proNGF-dependent cell death in RN22 cells. A, representative (n = 6) Western blots (WB) of lysates from RN22 cells that were stably transfected with sortilin constructs as shown and treated for 6 h with 200 nm PMA, 200 nm compound E, or 1 μm epoxomicin as indicated. B, densitometric quantification of the amount of p75NTR ICD in each condition in A relative to the amount of full-length p75NTR. The data represent the means ± S.E. of six experiments. C, quantification of proNGF-induced cell death relative to no proNT addition in polyclonal RN22 cell lines stably expressing various sortilin constructs. The cells were incubated with proNGF as indicated for 72 h, whereupon cell death was quantified using the MultiTox-Fluor multiplex cytotoxicity assay. *, p < 0.05; **, p < 0.01. D, Western blot (n = 3) of lysates from SCG neurons isolated from wild type and sortilin knock-out mice cultured in the presence of 1 μm epoxomicin for 1 h followed by the addition of 200 nm PMA for 3 h. CTF, C-terminal fragment; Untransf., untransfected.
RESULTS
ProNGF Increases Affinity between Sortilin and p75NTR
To map binding between sortilin and p75NTR, we established a co-immunoprecipitation protocol using stably or transiently transfected HEK293 cells. These cells were then treated with the cell-permeable, reducible protein cross-linker dithiobis succinimidylpropionate and lysates subjected to co-immunoprecipitation, reducing SDS-PAGE, and Western blotting. For our studies, we used the well characterized sortilin variant sortilinmut, which contains two disrupted internalization motifs, one being the tyrosine-based YXXØ sequence (792YSVL) and the other being a deletion of the C-terminal dileucine motif (829LL) (13). Whereas wild type sortilin is mainly localized in perinuclear compartments (10, 28), sortilinmut is predominantly expressed on the cell surface (13). Results obtained using this construct are therefore directly comparable with those obtained with chimeric receptor constructs lacking intracellular domain sequences, which are also mainly expressed on the cell surface. First, we established that sortilinmut and p75NTR bind and evaluated the effect of both proNGF and NGF on this interaction. Following cross-linking and immunoprecipitation of p75NTR, sortilinmut was clearly co-immunoprecipitated, showing that sortilinmut and p75NTR interact in the absence of ligand (Fig. 1) consistent with previous reports (5, 7). Treatment with 25 nm NGF, prior to cross-linking, had no effect on sortilinmut-p75NTR complex formation. In contrast, 25 nm proNGF increased the interaction 2.3-fold ± 0.4 (n = 4, p < 0.04), consistent with the notion that proNGF is a shared ligand between sortilin and p75NTR, whereby the pro-domain of proNGF binds sortilin and the mature domain binds p75NTR (5).
FIGURE 1.
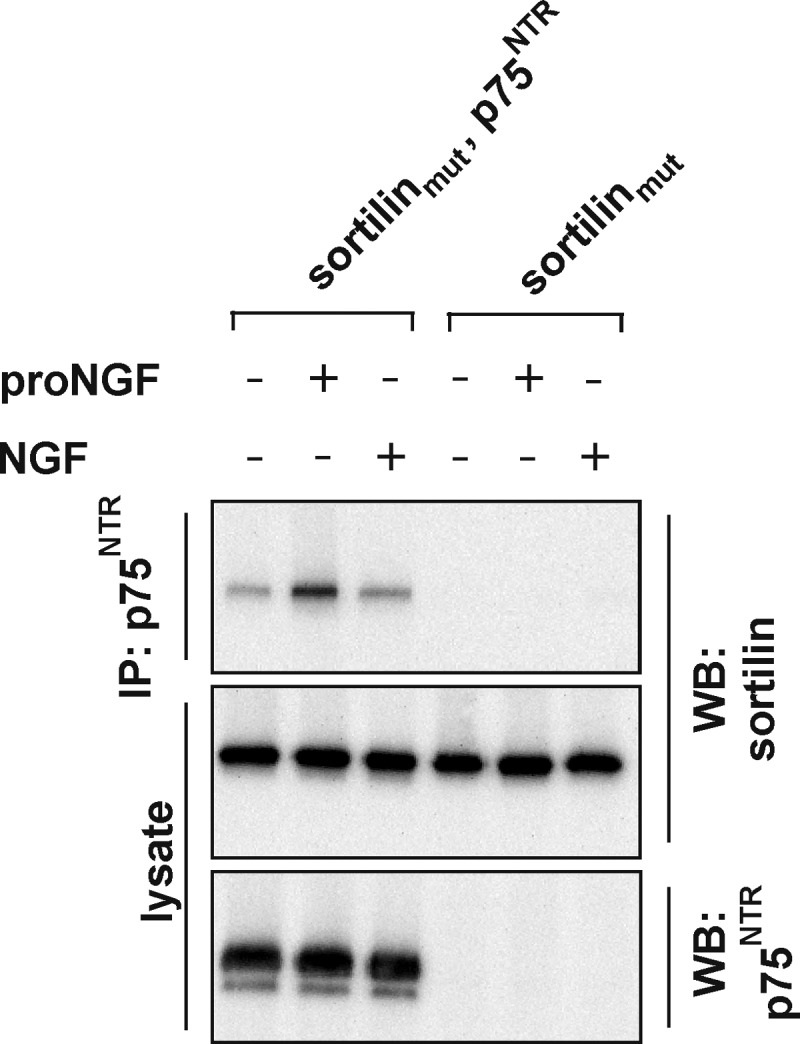
Effect of pro- and mature NGF on the sortilin-p75NTR interaction. Representative (n = 4) Western blot (WB) of lysates of HEK293 cells stably expressing either sortilinmut or sortilinmut and p75NTR incubated with 25 nm proNGF or NGF as indicated. The cells were cross-linked with dithiobis succinimidylpropionate, and the lysates were immunoprecipitated (IP) with anti-p75NTR antibodies prior to SDS-PAGE and Western blotting. Sortilin is co-immunoprecipitated by p75NTR, and the interaction is stabilized by proNGF but not NGF.
p75NTR Binding Properties of the Extracellular and Transmembrane Domain of Sortilin
We next evaluated whether the extracellular and transmembrane domains of sortilin are required for it to bind to p75NTR. To do this, co-immunoprecipitations were carried out using a truncated sortilin variant lacking the entire cytoplasmic domain (sortilintailless). HEK293 cells co-expressing sortilintailless and p75NTR were cross-linked in the absence or presence of 25 nm proNGF, and lysates were immunoprecipitated with anti-p75NTR antibodies. Sortilintailless clearly bound p75NTR, demonstrating that residues within the extracellular domain and/or the transmembrane region account for complex formation in the absence of ligand (Fig. 2A). As expected, the interaction between sortilintailless and p75NTR was again further increased in the presence of proNGF (2.1-fold ± 0.3, n = 4, p < 0.02).
FIGURE 2.

p75NTR binding epitopes within the extracellular and transmembrane domains of sortilin. A, representative (n = 4) Western blot (WB) of immunoprecipitates (IP) and lysates of p75NTR and sortilintailless, lacking its intracellular domain, compared with sortilinmut. Stable cultures of HEK293 cells expressing either sortilintailless or sortilintailless and p75NTR were incubated with or without proNGF and cross-linked with dithiobis succinimidylpropionate. Precipitates and crude lysates were then resolved by SDS-PAGE and Western blotted as indicated. B, schematic representation of expression constructs of sortilin and HA-p75NTR lacking their cytoplasmic domains and fused to one of two weakly interacting subunits of β-galactosidase that were used in the β-galactosidase complementation assay. Upon dimerization of receptor chimeras, the β-galactosidase active site and thus activity is reconstituted. C, measurement of the luminescence of β-galactosidase in HEK293 cells transfected with HA-p75NTR- and sortilin-β-galactosidase fusion expression constructs. Co-transfected cells produced significantly higher luminescence than single- and nontransfected cells (p < 0.03). The data represent the means ± S.E. of three experiments. D, representative Western blot (n = 4) of crude lysates from transfectants used in C. E, luminescence of the β-galactosidase substrate added to cells transfected with HA-p75NTR- and sortilin-β-galactosidase expression constructs and incubated for 30 min with increasing concentrations of proNGF, proBDNF, or proNT3. In contrast to BSA, at 40 pm all three proNTs increased complex formation (means ± S.E. of three to six experiments).
To confirm these findings in another cellular context, we developed a sensitive protocol for monitoring sortilin and p75NTR complex formation using the β-galactosidase complementation assay (23). This assay is based on two weakly interacting subunits of β-galactosidase that upon dimerization reconstitute the active site measured by luminescence (23). We substituted the cytoplasmic domains of sortilin and HA-tagged p75NTR with each of the two β-galactosidase subunits (Fig. 2B). The constructs were then transiently transfected into HEK293 cells and β-galactosidase activity measured using a chemiluminescent substrate. Whereas expression of each construct alone only resulted in a weak signal, co-expression increased enzyme activity ∼13-fold, suggesting that the extracellular and/or the transmembrane domain of sortilin and p75NTR also interact in this paradigm (Fig. 2, C and D). The addition of increasing amounts of proNGF, proBDNF, or proNT3 from as low as 40 pm proNT increased the interaction of sortilin with p75NTR (Fig. 2E). These results further show that proBDNF and proNT3 bind with very high affinity to sortilin and p75NTR, similar to what we previously have reported for proNGF (5).
Sortilin and p75NTR Interact through Their Extracellular Domains
To investigate whether p75NTR interacts with the extracellular domain of sortilin, we constructed a sortilin-interleukin-2 receptor (IL2R) chimeric receptor designated sortilinecd-IL2Rtm,icd that combined the extracellular domain of sortilin and the transmembrane region and intracellular domain of IL2R (Fig. 3A). The binding properties of sortilinecd-IL2Rtm,icd with p75NTR were then tested in transfected HEK293 cells by co-immunoprecipitation experiments. We found that sortilinecd-IL2Rtm,icd readily interacted with p75NTR and that proNGF increased this interaction 1.9-fold ± 0.1, n = 5, p < 0.002 (Fig. 3B). These findings were further substantiated by surface plasmon resonance analysis using the commercially available fusion protein of the extracellular domain of p75NTR linked to the constant region of human IgG (p75NTRecd-Fc). When p75NTRecd-Fc was injected onto a Biacore sensor chip containing immobilized soluble sortilin (sol-sortilin), binding was clearly evident (Fig. 3C). To ensure that binding did not stem from the Fc region of the fusion protein, binding to a RET-Fc tyrosine-kinase fusion protein was probed under similar conditions. RET-Fc did not bind sol-sortilin (Fig. 3C), confirming that the binding between sortilin and p75NTR relies on residues within these extracellular domains.
FIGURE 3.

Residues within the extracellular domain of sortilin account for binding to p75NTR. A, schematic representation of chimeric sortilin variants used to map the domain required to interact with p75NTR. B, representative (n = 5) Western blots (WB) of lysates and immunoprecipitates (IP) of p75NTR and sortilinecd-IL2Rtm,icd and sortilinmut, with and without proNGF treatment. Cross-linked proteins were immunoprecipitated with anti-p75NTR antibodies and precipitates were Western blotted with anti-sortilin antibodies. C, representative sensorgram (n = 3) obtained from surface plasmon resonance analysis of 20 nm p75NTRecd-Fc (solid line) and 20 nm RET-Fc (broken line) injected onto a Biacore sensor chip containing immobilized sol-sortilin. D, representative sensorgram (n = 3) from surface plasmon resonance analysis of 20 nm p75NTRecd-Fc poured onto a Biacore sensor chip with immobilized sol-sortilin in the absence (solid line) or presence of 20 μm neurotensin (broken line). The response for neurotensin alone has been subtracted. E, representative (n = 4) Western blots of lysates and immunoprecipitates of p75NTR and sortilin variants with or without treatment with proNGF.
We next addressed whether p75NTR engages the ligand-binding site in the 10-bladed β-propeller domain of sortilin. To do this, we carried out surface plasmon resonance competition studies with excess amounts of the tridecapeptide neurotensin. Neurotensin binds the tunnel of the β-propeller of sortilin and thereby sterically blocks access of all known ligands to sortilin (11, 12). Because neurotensin has a low molecular weight, binding to immobilized sortilin elicits only a minor signal, in terms of response units. However, compared with the signal obtained from binding of higher molecular weight proteins, competitive binding of neurotensin results in a greatly reduced signal overall. However, we were only able to block binding between sortilin and p75NTRecd-Fc to a minor extent, suggesting that p75NTR predominantly binds sortilin outside the neurotensin binding area (Fig. 3D).
We next determined whether p75NTR also interacts with the sortilin intracellular domain. To address this, we made an additional sortilin-IL2R-based chimeric receptor by substituting the intracellular domain of IL2R with that from sortilinmut, thereby generating IL2Recd,tm-sortilinmut,icd, (Fig. 3A). The chimera-encoding plasmid was then transfected into HEK293 cells together with p75NTR, and binding of the sortilin intracellular domain was assessed by co-immunoprecipitation experiments. Binding of full-length IL2R to p75NTR acted as the control. We were not able to co-immunoprecipitate IL2Recd,tm-sortilinmut icd or IL2R, demonstrating that the intracellular domain of sortilin does not interact with p75NTR (Fig. 3E).
Defining a Sortilin-binding Region of the p75NTR Extracellular Domain
To map the interaction site(s) of the extracellular domain at which p75NTR binds sortilin, co-immunoprecipitation experiments using various truncated p75NTR variants and sortilin were carried out. p75NTR variants include N-terminally truncated forms lacking one or more ligand binding repeats and a p75NTR variant lacking 23 amino acids (Thr228–Asp250) of the extracellular juxtamembrane stalk region (p75NTRΔstalk) (Fig. 4A). Following co-immunoprecipitation, we found that all truncated p75NTR variants bound equally well to sortilinecd-IL2Rtm,icd (Fig. 4B) except for p75NTRΔstalk, which had greatly reduced binding to sortilinecd-IL2Rtm,icd. However, in the presence of 25 nm proNGF, p75NTRΔstalk was co-immunoprecipitated with sortilinecd-IL2Rtm,icd (Fig. 4C). Because p75NTRΔstalk retained the ability to associate with sortilin via the shared ligand proNGF, reduced binding in the absence of proNGF cannot be ascribed to incorrect folding or improper cell surface expression of p75NTRΔstalk. Rather, these data suggest that the stalk region of p75NTR is responsible for its constitutive interaction with sortilin.
FIGURE 4.

The stalk region of p75NTR binds sortilin. A, schematic representation of truncated and deletion p75NTR receptor variants used to map the domain of p75NTR required to interact with sortilin. B, representative (n = 4) Western blots (WB) of immunoprecipitates (IP) and lysates of HEK293 cells that were transiently co-transfected with sortilinecd-Il2Rtm,icd and p75NTR receptor variants. The most C-terminal 23 amino acids of the p75NTR stalk region interact with sortilin. C, representative (n = 3) Western blots of immunoprecipitates and lysates from HEK293 transiently expressing sortilinecd-Il2Rtm,icd and p75NTR receptor variants as indicated. p75NTRΔstalk binds exogenous proNGF (25 nm) and thereby interacts with sortilinecd-Il2Rtm,icd. D, FRET analysis of sortilin and p75NTR interactions. HEK293 cells were transiently co-transfected with the indicated constructs and analyzed 48 h post-transfection. Full-length p75NTR and sortilin induced FRET above background, whereas p75NTRΔstalk did not, indicating that the stalk region of p75NTR is required for the interaction with sortilin. The data represent the means ± S.E. of three experiments. E, representative (n = 3) Western blot of lysates and immunoprecipitates of sortilinecd-IL2Rtm,icd and YFP-tagged p75NTR variants demonstrating that p75NTRΔstalk does not significantly interact with sortilinecd-IL2Rtm,icd. Constructs were transiently transfected into HEK293 cells, and p75NTR-YFP receptor constructs were immunoprecipitated with anti-GFP antibodies. F, representative (n = 3) Western blots of lysates and immunoprecipitates of sortilinecd-Il2Rtm,icd and YFP-tagged p75NTR variants demonstrating that the 23 amino acids of the p75NTR stalk region are sufficient to rescue extracellular binding of two otherwise nonoverlapping sortilin and p75NTR constructs.
To substantiate these results, we subjected p75NTRΔstalk and sortilin to FRET analysis. To do this, full-length p75NTR and p75NTRΔstalk were C-terminally fused with CFP to generate p75NTR-CFP and p75NTRΔstalk-CFP, respectively. Constructs were transiently transfected into HEK293 cells together with sortilin fused to YFP (i.e., sortilin-YFP). Because constitutive ligand-independent homodimerization of p75NTR is not mediated through the intracellular domain (29), FRET originating from cells co-transfected with the ICD of p75NTR fused to either CFP or YFP (i.e., p75NTRICD-CFP and p75NTRICD-YFP) served as a negative control. The extent of FRET between p75NTR-CFP and sortilin-YFP was increased 3-fold relative to background; however, the extent of FRET between p75NTRΔstalk-CFP and sortilin-YFP was significantly lower, being equivalent to that of p75NTRICD-CFP and p75NTRICD-YFP (Fig. 4D). We also assessed the binding properties of YFP-tagged p75NTR receptor constructs by co-immunoprecipitation using sortilinecd-IL2Rtm,icd as a binding partner. Similar to FRET analysis, p75NTRΔstalk-YFP had a reduced binding to sortilinecd-IL2Rtm,icd compared with p75NTR-YFP (Fig. 4E). However, it is noteworthy that a deletion of the stalk region of p75NTR abrogates binding to sortilin as measured by FRET, indicating that the extracellular domains alone mediate the binding between sortilin and p75NTR.
Next, we sought to determine whether the 23 amino acids in the stalk region of p75NTR alone are sufficient for binding to the sortilin extracellular domain. Co-immunoprecipitation experiments were therefore performed using a truncated variant of p75NTR-YFP designated p75NTRstalk,tm,icd-YFP containing only the stalk, transmembrane, and intracellular domains of p75NTR. Because sortilinecd-IL2Rtm,icd was used as a binding partner, only the 23 amino acids of the p75NTR stalk region overlap with sortilin (Fig. 4A). We found that the presence of the stalk region fully rescued binding of the p75NTR extracellular domain with sortilinecd-IL2Rtm,icd (Fig. 4F). This strongly suggests that the residues Thr228–Asp250 within the C-terminal stalk region of p75NTR constitute a core-binding surface of the p75NTR extracellular domain for sortilin.
Sortilin Intracellular Domain Regulates RIP of p75NTR and proNGF-dependent Apoptosis
Given that the interaction between sortilin and p75NTR maps to the extracellular domain, we sought to determine whether this domain is sufficient to affect p75NTR function. Depending on the molecular context, one function of p75NTR relates to its ability to induce apoptosis, which requires RIP of p75NTR (16, 17). Because sortilin is essential for proNT-dependent apoptosis through p75NTR (5–9), we measured the accumulation of p75NTR ICD by Western blotting within HEK293 cells transfected with wild type sortilin, sortilinmut, and sortilintailless in the presence of proteasome inhibitors. To induce p75NTR RIP, experiments were carried out in the presence of phorbol ester (PMA), which increases α-secretase activity via the protein kinase C pathway (30). Expression of any sortilin construct caused a nonsignificant increase in the amount of p75NTR C-terminal fragment. By contrast, the sortilin constructs had significant but differential effects on p75NTR ICD production. p75NTR control-transfected cells produced a low but obvious amount of p75NTR ICD. Co-transfection with wild type sortilin significantly increased the amount of p75NTR ICD after 3 h (Fig. 5, A and B). Sortilinmut increased formation of p75NTR ICD to an intermediate nonsignificant level, and formation of p75NTR ICD in the presence of sortilintailless was even more reduced.
To confirm these findings in another cellular context, we assessed the effect of sortilin on p75NTR ICD release using the Gal4/UAS system (31). This assay is based on binding of the nuclear protein Gal4 to the UAS element, which controls the nuclear transcription of luciferase. Because Gal4 is fused to the C-terminal end of p75NTR, only shedding and release of p75NTR ICD-Gal4 and subsequent translocation to the nucleus induces transcription of luciferase, which can be measured by a chemiluminescent substrate (Fig. 5C). To validate our assay, HEK293 cells were stably co-transfected with p75NTR-Gal4 and UAS-luciferase cDNA and treated with PMA. PMA significantly increased luciferase activity compared with that observed in untreated cells, consistent with the notion that PMA increases RIP of p75NTR (Fig. 5D). Furthermore, the effect of PMA was fully inhibited in the presence of an α-secretase inhibitor (TAPI-2), showing that RIP of p75NTR is required for up-regulation of luciferase transcription and activity (Fig. 5D). To test the effect of sortilin variants, p75NTR-Gal4 and UAS-luciferase stably transfected HEK293 cells were transiently transfected with wild type sortilin, sortilinmut, or sortilintailless and treated with PMA. Wild type sortilin significantly increased luciferase activity compared with that in control transfected cells, an effect observable even without PMA stimulation (Fig. 5, E and F). Again, sortilinmut and sortilintailless did not significantly induce p75NTR ICD-Gal4-mediated transcription above that of the control plasmid.
We next assessed the accumulation of endogenous p75NTR ICD in rat RN22 Schwannoma cells, which naturally express high levels of p75NTR and have previously been demonstrated to undergo RIP (16). Because RN22 cells only express very low amounts of endogenous sortilin, we generated polyclonal stable RN22 cell lines for wild type sortilin, sortilinmut, and sortilintailless and measured the accumulation of p75NTR ICD by Western blotting following addition of PMA and proteasome inhibitors. As for HEK293 cells, we found that wild type sortilin significantly increased p75NTR ICD formation, whereas sortilinmut induced a less pronounced nonsignificant accumulation of p75NTR ICD (Fig. 6, A and B). Sortilintailless did not affect p75NTR ICD accumulation, which was again comparable with that observed in untransfected RN22 cells.
Because sortilin apparently regulates RIP of p75NTR and given that RIP has been associated with neuronal apoptosis (16), we next determined the ability of wild type sortilin, sortilinmut, and sortilintailless to regulate proNT-dependent apoptosis. We established a proNGF-induced cell death assay using polyclonal stable RN22 sortilin-expressing cell lines. Cells were then incubated with increasing amounts of proNGF, and death was assessed after 72 h using the MultiTox-Fluor multiplex cytotoxicity assay, which measures activity of proteases released from necrotic cells. Untransfected RN22 cells did not die upon proNGF stimulation. However, proNGF significantly stimulated cell death in cells transfected with wild type sortilin in a dose-dependent manner (Fig. 6C). Cells expressing sortilinmut exhibited an intermediate rate of death, whereas cells transfected with sortilintailless were largely unaffected. This strongly suggests that the intracellular domain of sortilin regulates proNGF-dependent cell death through p75NTR in RN22 cells.
To test the effect of sortilin on p75NTR RIP under physiological conditions, we measured accumulation of p75NTR ICD within primary cultures of SCG neurons isolated from sortilin knock-out mice. SCG neurons endogenously express high amounts of sortilin and p75NTR, which has previously been shown to undergo RIP upon exposure to PMA (16). In the presence of PMA, neurons devoid of sortilin showed reduced rates of p75NTR RIP compared with SCG neurons isolated from wild type littermates (Fig. 6D). These findings, together with the results obtained from HEK293 and RN22 cells, collectively suggest that sortilin promotes RIP of p75NTR, resulting in increased release of p75NTR ICD and ultimately in apoptosis.
DISCUSSION
Here we have mapped the binding site between sortilin and p75NTR and found that the interaction relies on residues within their extracellular domains. Although we found no evidence that the sortilin intracellular domain interacts with p75NTR, we found that it does regulate RIP of p75NTR and p75NTR-dependent cell death signaling.
The present results confirm previous findings that co-expression of sortilin and p75NTR generates a high affinity receptor complex for proNTs, which increases p75NTR affinity for proNTs by more than 2 orders of magnitude (5). Furthermore, the current study is the first demonstration that picomolar concentrations of proBDNF and proNT3 (∼40 pm) increase complex formation between sortilin and p75NTR, similar to what has been reported for proNGF (5). Mapping of the interaction site between sortilin and p75NTR revealed that the extracellular domain of sortilin alone is sufficient to mediate an interaction with full-length p75NTR. Unfortunately, because of the structure of the sortilin extracellular domain (12), it is not possible to truncate the receptor further and still maintain the structural integrity of either the 10-bladed β-propeller or the 10CC domain. Thus, we were unable to further map the region of the extracellular domain of sortilin required for its constitutive interaction with p75NTR. However, based on the finding that addition of a non-neurotrophin sortilin ligand, neurotensin, had little effect on the direct interaction between the extracellular domain of sortilin and p75NTR, we conclude that the ligand-binding tunnel of the 10-bladed β-propeller domain of sortilin is unlikely to contain the p75NTR interaction site and that the 10CC domain is more likely to mediate this interaction.
Using a variety of truncated p75NTR receptor constructs, we determined that the extracellular domain of p75NTR is alone sufficient to mediate the co-association with sortilin in the absence of ligand. Deletion of the cysteine-rich ligand-binding domains of p75NTR had no effect on sortilin binding; however, deletion of 23 amino acids within the membrane-proximal region of the p75NTR stalk domain significantly reduced the ability of p75NTR to interact with sortilin. Remarkably, a construct containing only the most membrane-proximal 23 amino acids of the extracellular domain was sufficient to restore binding between p75NTR and sortilin, indicating that this is the major sortilin-binding site of p75NTR. These findings further support our notion that the 10CC domain of sortilin is likely to contain the p75NTR-binding site because the 10CC domain is expected to be located relatively close to the plasma membrane surface (12).
By use of Western blotting and an assay that measures p75NTR ICD-dependent transcription of luciferase, we analyzed the effect of sortilin on p75NTR RIP. We found that sortilin increases RIP of p75NTR, leading to significantly increased levels of p75NTR ICD. Moreover, the increased RIP of p75NTR was accompanied by an increase in cell death signaling mediated by p75NTR. Our finding that sortilin expression facilitates the generation of the p75NTR ICD is consistent with our data and published reports that proNTs facilitate both an interaction between p75NTR and sortilin and the generation of a p75NTR ICD-dependent cell death signal (5, 7, 8, 16). Importantly, the YXXØ and dileucine trafficking motifs of sortilin were required for sortilin to increase the rate of p75NTR RIP and to promote cell death in the presence of proNGF in RN22 cells. Interestingly, the dileucine motif together with the YXXØ motif are both required for sortilin endocytosis (13), and the sortilin mutant used herein has both of these motifs specifically mutated. The tailless sortilin mutant also lacks these sequences. Our findings that sortilinmut and sortilintailless engage in a complex with p75NTR but fail to stimulate RIP of p75NTR and RN22 cell death indicate that sortilin-dependent apoptosis is the result of two events, which require high affinity binding of proNTs, as well as endocytosis of sortilin. Because generation of p75NTR ICD occurs predominately in endosomes where γ-secretase activity is concentrated (32, 33), it is tempting to speculate that sortilin facilitates internalization of p75NTR or fragments thereof destined for endosomes. However, additional studies would be required to elucidate the mechanism of sortilin trafficking in relation to RIP of p75NTR and neuronal cell death.
In summary, we have mapped the binding site between sortilin and p75NTR to the extracellular juxtamembrane stalk region of p75NTR. The interaction between sortilin and p75NTR is further strengthened by the pro-apoptotic proNT ligands, which promote generation of the p75NTR ICD (16). Furthermore, sortilin-dependent release of the p75NTR ICD correlates with proNT-dependent apoptosis facilitated by sortilin. We therefore conclude that sortilin mediates cell death not only by interacting with p75NTR as a co-receptor for proNTs but also through generation of p75NTR cleavage fragments and release of p75NTR ICD.
Acknowledgments
We thank Prof. Helen M. Blau (Institute for Regenerative Medicine and Stem Cell Biology, Stanford University School of Medicine) for providing us with constructs encoding mutated subunits of β-galactosidase and Prof. Moses V. Chao (Skirball Institute of Biomolecular Medicine, New York University School of Medicine) for the anti-p75NTR ICD antibody. Prof. Elisabeth Schwarz (Institute for Biotechnology, Martin-Luther-Universität, Halle-Wittenberg) generously supplied us with proNTs. Anne Marie Bundsgaard, Anja Aagaard, and Benedicte Vestergaard (Department of Biomedicine, University of Aarhus) are thanked for excellent technical assistance. Rowan Tweedale (Queensland Brain Institute, The University of Queensland) is acknowledged for critical reading of the manuscript.
This work was supported by the by the Lundbeck Foundation, the National Health and Medical Research Council of Australia, the Finnish Graduate School of Neuroscience, the Sigrid Juselius Foundation, and Academy of Finland Center of Excellence program.
- NT3
- neurotrophin-3
- ecd
- extracellular domain
- ICD
- intracellular domain
- IL2R
- interleukin-2 receptor
- p75NTR
- p75 neurotrophin receptor
- proNT
- pro-neurotrophin
- RIP
- regulated intramembrane proteolysis
- SCG
- superior cervical ganglion
- sol-sortilin
- soluble sortilin
- tm
- transmembrane domain
- UAS
- upstream activating sequences
- PMA
- phorbol 12-myristate 13-acetate.
REFERENCES
- 1. Frade J. M., Barde Y. A. (1998) Nerve growth factor. Two receptors, multiple functions. Bioessays 20, 137–145 [DOI] [PubMed] [Google Scholar]
- 2. Lee R., Kermani P., Teng K. K., Hempstead B. L. (2001) Regulation of cell survival by secreted proneurotrophins. Science 294, 1945–1948 [DOI] [PubMed] [Google Scholar]
- 3. Beattie M. S., Harrington A. W., Lee R., Kim J. Y., Boyce S. L., Longo F. M., Bresnahan J. C., Hempstead B. L., Yoon S. O. (2002) ProNGF induces p75-mediated death of oligodendrocytes following spinal cord injury. Neuron 36, 375–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Harrington A. W., Leiner B., Blechschmitt C., Arevalo J. C., Lee R., Mörl K., Meyer M., Hempstead B. L., Yoon S. O., Giehl K. M. (2004) Secreted proNGF is a pathophysiological death-inducing ligand after adult CNS injury. Proc. Natl. Acad. Sci. U.S.A. 101, 6226–6230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nykjaer A., Lee R., Teng K. K., Jansen P., Madsen P., Nielsen M. S., Jacobsen C., Kliemannel M., Schwarz E., Willnow T. E., Hempstead B. L., Petersen C. M. (2004) Sortilin is essential for proNGF-induced neuronal cell death. Nature 427, 843–848 [DOI] [PubMed] [Google Scholar]
- 6. Teng H. K., Teng K. K., Lee R., Wright S., Tevar S., Almeida R. D., Kermani P., Torkin R., Chen Z. Y., Lee F. S., Kraemer R. T., Nykjaer A., Hempstead B. L. (2005) ProBDNF induces neuronal apoptosis via activation of a receptor complex of p75NTR and sortilin. J. Neurosci. 25, 5455–5463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jansen P., Giehl K., Nyengaard J. R., Teng K., Lioubinski O., Sjoegaard S. S., Breiderhoff T., Gotthardt M., Lin F., Eilers A., Petersen C. M., Lewin G. R., Hempstead B. L., Willnow T. E., Nykjaer A. (2007) Roles for the pro-neurotrophin receptor sortilin in neuronal development, aging and brain injury. Nat. Neurosci. 10, 1449–1457 [DOI] [PubMed] [Google Scholar]
- 8. Tauris J., Gustafsen C., Christensen E. I., Jansen P., Nykjaer A., Nyengaard J. R., Teng K. K., Schwarz E., Ovesen T., Madsen P., Petersen C. M. (2011) Proneurotrophin-3 may induce sortilin-dependent death in inner ear neurons. Eur. J. Neurosci. 33, 622–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yano H., Torkin R., Martin L. A., Chao M. V., Teng K. K. (2009) Proneurotrophin-3 is a neuronal apoptotic ligand. Evidence for retrograde-directed cell killing. J. Neurosci. 29, 14790–14802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Petersen C. M., Nielsen M. S., Nykjaer A., Jacobsen L., Tommerup N., Rasmussen H. H., Roigaard H., Gliemann J., Madsen P., Moestrup S. K. (1997) Molecular identification of a novel candidate sorting receptor purified from human brain by receptor-associated protein affinity chromatography. J. Biol. Chem. 272, 3599–3605 [DOI] [PubMed] [Google Scholar]
- 11. Nykjaer A., Willnow T. E. (2012) Sortilin. A receptor to regulate neuronal viability and function. Trends Neurosci. 35, 261–270 [DOI] [PubMed] [Google Scholar]
- 12. Quistgaard E. M., Madsen P., Grøftehauge M. K., Nissen P., Petersen C. M., Thirup S. S. (2009) Ligands bind to sortilin in the tunnel of a ten-bladed β-propeller domain. Nat. Struct. Mol. Biol. 16, 96–98 [DOI] [PubMed] [Google Scholar]
- 13. Nielsen M. S., Madsen P., Christensen E. I., Nykjaer A., Gliemann J., Kasper D., Pohlmann R., Petersen C. M. (2001) The sortilin cytoplasmic tail conveys Golgi-endosome transport and binds the VHS domain of the GGA2 sorting protein. EMBO J. 20, 2180–2190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dechant G., Barde Y. A. (2002) The neurotrophin receptor p75NTR. Novel functions and implications for diseases of the nervous system. Nat. Neurosci. 5, 1131–1136 [DOI] [PubMed] [Google Scholar]
- 15. Coulson E. J., Reid K., Baca M., Shipham K. A., Hulett S. M., Kilpatrick T. J., Bartlett P. F. (2000) Chopper, a new death domain of the p75 neurotrophin receptor that mediates rapid neuronal cell death. J. Biol. Chem. 275, 30537–30545 [DOI] [PubMed] [Google Scholar]
- 16. Kenchappa R. S., Zampieri N., Chao M. V., Barker P. A., Teng H. K., Hempstead B. L., Carter B. D. (2006) Ligand-dependent cleavage of the P75 neurotrophin receptor is necessary for NRIF nuclear translocation and apoptosis in sympathetic neurons. Neuron 50, 219–232 [DOI] [PubMed] [Google Scholar]
- 17. Kenchappa R. S., Tep C., Korade Z., Urra S., Bronfman F. C., Yoon S. O., Carter B. D. (2010) p75 neurotrophin receptor-mediated apoptosis in sympathetic neurons involves a biphasic activation of JNK and up-regulation of tumor necrosis factor-α-converting enzyme/ADAM17. J. Biol. Chem. 285, 20358–20368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ceni C., Kommaddi R. P., Thomas R., Vereker E., Liu X., McPherson P. S., Ritter B., Barker P. A. (2010) The p75NTR intracellular domain generated by neurotrophin-induced receptor cleavage potentiates Trk signaling. J. Cell Sci. 123, 2299–2307 [DOI] [PubMed] [Google Scholar]
- 19. Kommaddi R. P., Thomas R., Ceni C., Daigneault K., Barker P. A. (2011) Trk-dependent ADAM17 activation facilitates neurotrophin survival signaling. FASEB J. 25, 2061–2070 [DOI] [PubMed] [Google Scholar]
- 20. Skeldal S., Matusica D., Nykjaer A., Coulson E. J. (2011) Proteolytic processing of the p75 neurotrophin receptor. A prerequisite for signalling? Neuronal life, growth and death signalling are crucially regulated by intra-membrane proteolysis and trafficking of p75NTR. Bioessays 33, 614–625 [DOI] [PubMed] [Google Scholar]
- 21. Hermey G., Sjøgaard S. S., Petersen C. M., Nykjaer A., Gliemann J. (2006) Tumour necrosis factor α-converting enzyme mediates ectodomain shedding of Vps10p-domain receptor family members. Biochem. J. 395, 285–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Munck Petersen C., Nielsen M. S., Jacobsen C., Tauris J., Jacobsen L., Gliemann J., Moestrup S. K., Madsen P. (1999) Propeptide cleavage conditions sortilin/neurotensin receptor-3 for ligand binding. EMBO J. 18, 595–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mohler W. A., Blau H. M. (1996) Gene expression and cell fusion analyzed by lacZ complementation in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 93, 12423–12427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jung K. M., Tan S., Landman N., Petrova K., Murray S., Lewis R., Kim P. K., Kim D. S., Ryu S. H., Chao M. V., Kim T. W. (2003) Regulated intramembrane proteolysis of the p75 neurotrophin receptor modulates its association with the TrkA receptor. J. Biol. Chem. 278, 42161–42169 [DOI] [PubMed] [Google Scholar]
- 25. Hauburger A., Kliemannel M., Madsen P., Rudolph R., Schwarz E. (2007) Oxidative folding of nerve growth factor can be mediated by the pro-peptide of neurotrophin-3. FEBS Lett. 581, 4159–4164 [DOI] [PubMed] [Google Scholar]
- 26. Meyer B. H., Segura J. M., Martinez K. L., Hovius R., George N., Johnsson K., Vogel H. (2006) FRET imaging reveals that functional neurokinin-1 receptors are monomeric and reside in membrane microdomains of live cells. Proc. Natl. Acad. Sci. U.S.A. 103, 2138–2143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zareen N., Greene L. A. (2009) Protocol for culturing sympathetic neurons from rat superior cervical ganglia (SCG). J. Vis. Exp. 23, e988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jacobsen L., Madsen P., Jacobsen C., Nielsen M. S., Gliemann J., Petersen C. M. (2001) Activation and functional characterization of the mosaic receptor SorLA/LR11. J. Biol. Chem. 276, 22788–22796 [DOI] [PubMed] [Google Scholar]
- 29. Vilar M., Charalampopoulos I., Kenchappa R. S., Simi A., Karaca E., Reversi A., Choi S., Bothwell M., Mingarro I., Friedman W. J., Schiavo G., Bastiaens P. I., Verveer P. J., Carter B. D., Ibáñez C. F. (2009) Activation of the p75 neurotrophin receptor through conformational rearrangement of disulphide-linked receptor dimers. Neuron 62, 72–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schlöndorff J., Blobel C. P. (1999) Metalloprotease-disintegrins. Modular proteins capable of promoting cell-cell interactions and triggering signals by protein-ectodomain shedding. J. Cell Sci. 112, 3603–3617 [DOI] [PubMed] [Google Scholar]
- 31. Sotthibundhu A., Sykes A. M., Fox B., Underwood C. K., Thangnipon W., Coulson E. J. (2008) β-Amyloid(1–42) induces neuronal death through the p75 neurotrophin receptor. J. Neurosci. 28, 3941–3946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bronfman F. C., Escudero C. A., Weis J., Kruttgen A. (2007) Endosomal transport of neurotrophins. Roles in signaling and neurodegenerative diseases. Dev. Neurobiol. 67, 1183–1203 [DOI] [PubMed] [Google Scholar]
- 33. Urra S., Escudero C. A., Ramos P., Lisbona F., Allende E., Covarrubias P., Parraguez J. I., Zampieri N., Chao M. V., Annaert W., Bronfman F. C. (2007) TrkA receptor activation by nerve growth factor induces shedding of the p75 neurotrophin receptor followed by endosomal γ-secretase-mediated release of the p75 intracellular domain. J. Biol. Chem. 282, 7606–7615 [DOI] [PubMed] [Google Scholar]
- 34. Sykes A. M., Palstra N., Abankwa D., Hill J. M., Skeldal S., Matusica D., Venkatraman P., Hancock J. F., Coulson E. J. (October 26, 2012) J. Biol. Chem. 10.1074/jbc.M112.382903 [DOI] [PMC free article] [PubMed] [Google Scholar]