

Background: Apoptosis-inducing factor (AIF) plays roles in both cell survival and death, but the significance of these activities remains unclear.
Results: The enzymatic activity of AIF promotes survival and growth of advanced cancer cells.
Conclusion: AIF supports prostate cancer cells by a mechanism involving modulation of energy metabolism.
Significance: This is the first indication that AIF supports prostate tumorigenesis.
Keywords: Cancer Biology, Cell Death, Mitochondria, Prostate Cancer, Reactive Oxygen Species (ROS)
Abstract
Apoptosis-inducing factor (AIF) promotes cell death yet also controls mitochondrial homeostasis and energy metabolism. It is unclear how these activities are coordinated, and the impact of AIF upon human disease, in particular cancer, is not well documented. In this study we have explored the contribution of AIF to the progression of prostate cancer. Analysis of archival gene expression data demonstrated that AIF transcript levels are elevated in human prostate cancer, and we found that AIF protein is increased in prostate tumors. Suppression of AIF expression in the prostate cancer cell lines LNCaP, DU145, and PC3 demonstrated that AIF does not contribute to cell toxicity via a variety of chemical death triggers, and growth under nutrient-rich conditions is largely unaffected by AIF ablation. However, under growth stress conditions, AIF depletion from DU145 and PC3 cell lines led to significant reductions in cell survival and growth that were not observed in LNCaP cells. Moreover AIF-deficient PC3 cells exhibited substantial reduction of tumorigenic growth in vivo. This reduced survival correlated with decreased expression of mitochondrial complex I protein subunits and concomitant changes in glucose metabolism. Finally, restoration of AIF-deficient PC3 cells with AIF variants demonstrated that the enzymatic activity of AIF is required for aggressive growth. Overall these studies show that AIF is an important factor for advanced prostate cancer cells and that through control of energy metabolism and redox balance, the enzymatic activity of AIF is critical for this support.
Introduction
Apoptosis-inducing factor (AIF)5 is a mitochondrial flavoprotein originally discovered through its ability to trigger cell death in various model systems (1, 2). In contrast to its pro-death capabilities AIF possesses NADH-oxidase activity that is independent of its apoptotic function (3, 4). Although an in vivo role for this activity is unclear, a number of animal studies have demonstrated the requirement of AIF for normal development. Aif-null mice die early in embryogenesis (5). Studies employing Harlequin mice, in which AIF protein levels are reduced by over 80% (6), demonstrated that AIF deficiency results in severe and progressive neuronal degeneration. This degeneration stems in part from a loss of AIF-mediated protection against cellular damage induced by oxidative stress (6). Subsequent studies have uncovered a role for AIF in regulating oxidative phosphorylation: cells lacking AIF are compromised in their respiratory chain activity (7) stemming from a loss in expression of subunits within complex I of the electron transport chain. Additional studies have provided further evidence that AIF has a vital role in maintaining mitochondrial structure and function. In mice, muscle-specific AIF loss results in severe skeletal muscle atrophy, dilated cardiomyopathy, and mitochondrial dysfunction attributed to complex I-mediated respiratory chain defects (8), whereas deletion of AIF in the brain leads to the accumulation of fragmented mitochondria with aberrant cristae (9). In humans a mutation to the AIF gene leading to defects in the respiratory chain has recently been identified in patients suffering from progressive mitochondrial encephalomyopathy (10). Overall these studies point to a central role for AIF in maintaining normal mitochondrial redox balance, distinct from its pro-death functions (11).
How the disparate activities of AIF are regulated and the overall significance of AIF to human pathologies such as cancer remains unclear. As an inducer of cell death, AIF possesses anti-neoplastic potential, yet this activity could be outweighed by the pro-survival function of AIF, and elevated AIF protein levels may benefit tumorigenesis. Indeed, it has been shown that increased AIF levels protect transformed colorectal cancer cells from stress-induced apoptosis (12). This protective effect results from a modest increase in the general cellular oxidative state, consistent with an ability of AIF to regulate cellular reactive oxygen species (ROS) levels. Increased oxidative stress, caused in part by increases in ROS (13–15), is a common feature of many developing tumors and results from tissue hypoxia caused by poor blood flow (14, 15). Although high levels of oxidative stress impair tumor development, low to moderate levels may be favorable by causing modest activation of cell stress pathways (16, 17). Furthermore, ROS are produced by many cell types as signaling molecules that stimulate proliferation (18). When coupled with metabolic changes affecting glucose consumption and macromolecule production often observed in developing cancer cells (19), AIF overexpression may reflect the ability of tumors to exploit the oxidative and metabolic capabilities of AIF in favor of tumorigenesis. In this context, the death-inducing potential of AIF must be held in check, potentially through the activity of anti-apoptotic proteins that are coordinately overexpressed.
In this study we demonstrate for the first time that AIF plays a substantial supportive role in growth and survival of aggressive prostate cancer cells. Bioinformatic investigation of publically available cancer gene expression data led to the determination that AIF transcripts are elevated in human prostate cancer tissues. Analysis of AIF protein levels shows similar increases in both human and mouse prostate cancer samples. RNAi-mediated suppression of AIF in a panel of prostate cancer cell lines demonstrate that AIF plays little role in controlling cell death in prostate cells and that cell growth and survival under nutrient-rich conditions are largely unaffected following AIF ablation. In contrast, advanced prostate cancer cells are selectively sensitive to AIF loss for survival under growth stress conditions, and tumorigenic growth of AIF-deficient cells is severely compromised in vivo. Reduced survival under growth stress correlates with decreases in protein components of the mitochondrial electron transport chain and a corresponding switch to increased glycolysis for energy production. Finally, restoration of AIF-deficient cells demonstrates that the enzymatic activity of AIF is critical to support advanced prostate cancer cell growth. These data have significant implications for control of cancer cell metabolism and identify AIF as a contributor to the aggressive phenotype of advanced prostate cancer cells.
EXPERIMENTAL PROCEDURES
Materials
Reagents were obtained as follows: GlutaMAX, RPMI 1640, DMEM, and PBS were from Invitrogen; FBS was from Hyclone; MatrigelTM was from BD Biosciences; the site-directed mutagenesis kit was from Stratagene; the QuantiChromTM glucose assay kit was from BioAssay Systems; protease inhibitor tablets were from Roche Applied Science; all other chemicals were from Sigma. Antibodies were obtained as follows: anti-AIF (Santa Cruz, sc-13116), anti-complex I 39 kDa (Invitrogen, 459100), anti-complex I 20 kDa (Invitrogen, 459210), anti-CoxIV (Invitrogen, A-21347), anti-β-actin (Sigma, A5316), and peroxidase-conjugated anti-mouse and anti-rabbit (Amersham Biosciences, NA931V and NA934V).
Oncomine Data Analysis
AIF mRNA expression in prostate cancers from 14 studies (20–33) was analyzed using Oncomine (34). Within each data set, only those samples specifically examining prostate carcinoma versus normal prostate tissue were assessed for gene expression changes. The Oncomine website details standardized normalization techniques and statistical calculations.
Tissue Microarray Analysis
A prostate cancer progression tissue microarray analysis (TMA) composed of benign prostate tissue, clinically localized prostate cancer, and hormone refractory metastatic prostate cancer has been described (35). These cases came from well classified radical prostatectomy specimens as described previously (36). Replicate tissue samples were placed in geographically distinct areas of the TMA to evaluate reproducibility within the same TMA based on location. A total of 415 tissue samples were collected from 51 patients. The TMA was soaked in xylene overnight to remove adhesive tape used for its construction. Primary antibodies were incubated before washing. Secondary anti-rabbit antibodies avidin-conjugated were applied before washing. Enzymatic reaction was completed using a streptavidin biotin detection kit (DakoCytomation, Carpinteria, CA). Protein expression was determined using a validated scoring method (33, 36, 37) where staining was evaluated for intensity and the percentage of cells staining positive. Statistical significance was determined using the Kruskal-Wallis test.
Cell Culture
PC3, DU145, and LNCaP cells were grown in RPMI 1640 containing 10% FBS supplemented with 2 mm GlutaMAX, and HEK293T cells were grown in DMEM containing 10% FBS supplemented with 2 mm GlutaMAX. All cells were maintained at 37 °C in an atmosphere of 95% air, 5% CO2 except during lentivirus infections as described below.
Plasmids
For AIF targeting by RNA interference, a 64-bp oligonucleotide containing the AIF targeting sequence GATCCTGAGCTGCCGTACA (38) was subcloned in the pRNAi plasmid (39). A fragment from this plasmid including the H1 RNA promoter and the AIF targeting cassette was then subcloned into the lentiviral plasmid FG12-GFP (40) to generate the final FG12-shAIF-GFP construct. The negative control plasmid FG12-shLacZ-GFP was constructed in the same manner except the 64-bp oligonucleotide subcloned into pRNAi contained the LacZ targeting sequence GTGACCAGCGAATACCTGT. Full oligonucleotide sequences are available upon request. For AIF RNAi-resistant restoration plasmids, silent mutations were introduced into the wild type AIF cDNA contained within pEBB-AIF (41) to generate pEBB-siMut-WT-AIF using the Stratagene site-directed mutagenesis kit as per the manufacturer's instructions. Additional rounds of mutagenesis were then performed to generate the plasmid pEBB-siMut-TVA-AIF containing the substitutions T263A and V300A. AIF cDNA was then subcloned from pEBB-siMut-WT-AIF and pEBB-siMut-TVA-AIF into the lentiviral plasmid pSL4-puro (42). The lentiviral packaging plasmids pHCMV-G, pRRE, and pRSV-rev are as described (40).
Lentivirus Production and Infection
FG12 or pSL4 derived plasmids were combined with equal amounts of lentiviral packaging plasmids and transfected into HEK293T cells by calcium phosphate precipitation as described (43). 48 h after transfection, supernatants were harvested, filtered using 0.45-μm-pore size Millex HV PVDF filter units (Millipore), and concentrated by centrifugation at 60,000 × g for 90 min. The supernatants were aspirated, and virus-containing pellets were resuspended in PBS overnight at 4 °C. One day before infection, target cells were seeded at 60,000 cells/well in 12-well plates. At the time of infection, polybrene was added to a final concentration of 25 mm, resuspended virus was added, and cells were incubated for 4 h at 37 °C in an atmosphere of 93% air, 7% CO2. Virus containing supernatants were then removed, fresh medium was added to cells, and cells were incubated for an additional 2–3 days in an atmosphere of 95% air, 5% CO2. Stable incorporation of FG12-based lentiviral DNA was determined by flow cytometry assessing GFP fluorescence (described below), whereas pSL4-based selection was performed by incubating cells in the presence of 1 μg/ml puromycin.
Cell/Tissue Lysis, SDA-PAGE, and Immunoblotting
Cell and tissue extracts were prepared in radioimmune precipitation assay lysis buffer (PBS containing 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mm DTT, 1 mm PMSF, and 1 protease inhibitor mixture tablet per 10 ml). Extracts were normalized for protein content and then separated by SDS-PAGE using 4–12% gradient SDS-PAGE gels (Invitrogen). SDS-PAGE was followed by transfer to nitrocellulose membranes (Invitrogen), which were then blocked with 5% milk in TBS containing 0.02–0.2% Tween, followed by incubation with the indicated antibodies for 1 h at room temperature. Following washing, the membranes were incubated with peroxidase-conjugated anti-mouse IgG or anti-rabbit IgG secondary antibodies for 45 min at room temperature and visualized by enhanced chemiluminescence.
Flow Cytometry and Cell Sorting
Following lentiviral infection, PC3-, LNCaP-, and DU145-derived cells were harvested by trypsinization, washed, and resuspended in PBS. GFP fluorescence was then assessed by flow cytometry using a Becton Dickinson FACSCalibur flow cytometer for analysis. For enrichment of PC3-derived cells prior to xenograft implantation experiments, the cells were harvested by trypsinization, washed with PBS, and resuspended in fresh medium. The cells were then sorted based on GFP-fluorescence using a Becton Dickinson FACSAria flow cytometer. The criterion for collection of cells was GFP fluorescence falling within the center 20% of positive cells. Following enrichment cells were expanded for further study.
Cell Growth Rate Measurements
Exponentially growing cells were harvested by trypsinization, and the total cells were determined by Coulter counting. The cells were then washed in PBS and resuspended in fresh medium at 50,000 cells/well, and replicate wells of 6-well plates were prepared. Three wells from each cell type were harvested by trypsinization at 24-h intervals for a total of 96 h, and the total number of cells in each replicate was determined by Coulter counting.
Cell Viability
The cells were seeded at 400,000 cells/well in 6-well plates and allowed to attach overnight. The cells were then left untreated or treated as follows: for 24, 48, and 72 h with arsenic trioxide (10 μm), or cisplatin (166 μm); for 24 h with hydrogen peroxide (100 and 250 μm); for 48 h with etoposide (42.5 and 85 μm). For MNNG treatments, drug was added (10, 50, 100, 250, and 500 μm); cells were incubated for 15 min at 37 °C and washed with PBS; and then fresh medium was added before incubation for an additional 48 h. The cells were harvested, washed, and resuspended in PBS containing 2 μg/ml propidium iodide. Cell viability was then determined by flow cytometry using a Becton Dickinson FACSCalibur flow cytometer.
In Vivo Tumor Growth
Exponentially growing PC3-Parental, PC3-shLacZ, and PC3-shAIF cells were harvested by trypsinization, washed, and resuspended in sterile PBS. The cells were then injected subcutaneously into the dorsal right flank (3 × 106 cells, 150 μl/injection) of 5-week-old male athymic nude mice (NCr-Foxn1nu/nu, Taconic) using 1-ml tuberculin syringes. A total of 12 animals were injected per cell type. Tumor growth was then assessed by direct measurement with calipers of two perpendicular diameters of each tumor. Tumor mass in grams was calculated by the formula 1/2a × b2, where a is the long diameter, and b is the short diameter (both in cm; 1 cm3 equals 1 g). Once tumor volumes exceeded 1500 mm3, the animals were euthanized, and the tumors were excised. A portion of each tumor was then used for protein extraction and immunoblot analysis as described above. All of the animal procedures were approved by the Institutional Animal Care and Use Committee of the Wake Forest University School of Medicine.
MatrigelTM Growth/Survival Experiments
24-well plates containing MatrigelTM were prepared by adding 100 μl of cold MatrigelTM to each well and then solidifying at 37 °C for 1 h. The cells were harvested, washed, and resuspended in fresh medium. A total of 10,000 cells were then added to coated plates in a total volume of 500 μl and allowed to grow for up to 7 days. Assessment of cellular growth and survival was performed using phase contrast microscopy.
Invasion Assay
Cell invasion was determined using BD BioCoatTM MatrigelTM invasion chambers as per the manufacturer's instructions. Briefly, RPMI 1640 containing 10% FBS supplemented with 2 mm GlutaMAX was added to the bottom chamber of either 12-well BD BioCoatTM MatrigelTM or control invasion chambers. PC3-Parental, PC3-shLacZ, and PC3-shAIF cells were harvested, washed with PBS, and resuspended in RPMI 1640 without serum. 25,000 cells in a total volume of 500 μl were then added to the upper chamber of both MatrigelTM and control coated chambers and incubated at 37 °C. At 16-, 40-, and 65-h intervals after seeding, noninvading cells were removed, and invading cells were fixed with 4% paraformaldehyde. The membranes were then excised, and invading cells were counted. The percentage of invasion was determined by the formula: ((mean number of cells invading through MatrigelTM insert membrane)/(mean number of cell migrating through control insert membrane)) × 100.
Glucose Consumption
The cells were harvested, washed, resuspended in fresh medium, and seeded in replicate 6-well plates. The cells were then incubated for 48 h at 37 °C. The medium was collected from each well, and total glucose was determined using the QuantiChromTM glucose assay kit as per the manufacturer's instructions. The glucose present in medium incubated without cells was determined and used to calculate glucose consumption of each sample. The cells from each well were harvested, and the total cell number was determined by Coulter counting. Total glucose consumption/sample was then divided by the number of cells to generate final glucose consumption/cell values for each cell line.
Lactate Secretion
The cells were harvested, washed, resuspended in fresh medium, and seeded in replicate 6-well plates. The cells were then incubated for 48 h at 37 °C. The medium was collected from each well, and the total lactate was determined using the Sigma lactate assay kit as per the manufacturer's instructions. The cells from each well were harvested, and the total cell number was determined by Coulter counting. Total lactate secreted/sample was then divided by the number of cells to generate final lactate secretion/cell values for each cell line.
Oxygen Consumption
The cells were harvested, washed and resuspended in fresh medium, and replicate wells were seeded in a 96-well plate. The oxygen-sensitive probe MitoXpress® (Luxcel Biosciences) was added to each well, and all wells were overlaid with mineral oil. Fluorescence of each well was then measured at 1.5-min intervals for a total of 200 min using a fluorescence plate reader (BMG Labtech FLUOstar Omega). The rate of oxygen consumption was determined from the slope of fluorescence versus time for each sample using arbitrary units.
Glucose Deprivation
LNCaP-, DU145-, and PC3-derived cells were harvested, washed, resuspended in either normal or glucose-free medium, and plated at 300,000 cells/well in 6-well plates. Cell viability at 0, 48, and 72 h post-seeding was determined by propidium iodide staining and flow cytometry as described above. For 2-deoxyglucose treatments, PC3-derived cells were harvested, washed, and then plated in fresh medium at 300,000 cells/well in 6-well plates. The cells were allowed to attach overnight and then left untreated or treated with 50 mm 2-deoxyglucose for 48 h. Cell viability was then determined by propidium iodide staining and flow cytometry as above.
Scratch Assay
PC3-derived cells were harvested, washed, resuspended in fresh medium, and seeded in 35-mm dishes at a total of 900,000/dish. The cells were allowed to attach overnight, and then the cells were displaced by using a p200 pipette tip to make a single scratch through the middle of each dish (44). The cells were immediately washed, fresh medium was added, and the size of each scratch was measured. The cells were then incubated for an additional 27 h, at which time the size of each scratch was measured again. For each cell line, replicate dishes were prepared, and a total of six equally spaced measurements were made on each plate. Replicate measurements were then averaged, and the distance migrated for each cell line was calculated by subtracting the distance at 27 h from the initially measured distance.
RESULTS
AIF Expression Is Elevated in Both Human Prostate Cancer and a Mouse Prostate Cancer Model
To determine whether AIF expression is altered in prostate cancer, we first employed a bioinformatics approach. Using data archived within the publically available gene expression database Oncomine (34), AIF expression data were explored specifically focusing on studies comparing prostate carcinoma to normal prostate tissue. A total of 14 studies archived within Oncomine matched this criterion, and among these 13 reported elevated AIF mRNA in prostate cancer when compared with normal prostate tissue (20–33). The extent of increased AIF expression varied from a low of 1.04-fold (essentially unchanged) to a high of 1.80-fold, with an increase in AIF expression of 25% or more demonstrated by half of the studies archived. Representative data from four studies indicating AIF transcript levels elevated by 30% or more are presented in Fig. 1A. These data indicate that AIF transcripts are often present in greater amounts in prostate cancer compared with normal prostate tissues, suggesting a possible involvement of AIF in disease progression.
FIGURE 1.

AIF is elevated in prostate cancer. A, AIF mRNA levels as assessed in four representative studies archived in the Oncomine cancer gene expression analysis database (28–30, 32). Light gray bars, AIF expression in normal prostate tissue; dark gray bars, AIF expression in prostate cancer. B, immunohistochemistry was performed on tissue microarrays derived from samples of benign (left panel), a mixture of benign and prostate carcinoma (center panels), and frank carcinoma (right panels). AIF staining is shown in brown, and hematoxylin staining is shown in blue. C, microarray samples classified as benign, prostatic intraepithelial neoplasia (PIN), or frank carcinoma (PCA) were scored for AIF staining intensity on a scale of 1–4. Note the higher proportion of samples with intense AIF staining (score 3 or 4) present in PIN and PCA samples (p < 0.0001 by Kruskal-Wallis test). D, prostate tissue was harvested from six wild type (Pten+/+) and six prostate-specific Pten-deficient (Pten−/−) mice. Protein was extracted, and AIF levels were determined by immunoblot analysis. As a loading control, extracts were immunoblotted for β-actin levels.
To correlate these changes in mRNA levels to protein content, immunohistochemical analysis was used to determine the extent of AIF overexpression in a microarray of tissues from patients that underwent radical prostatectomy (415 specimens from 51 cases). When compared with normal prostatic tissue, expression of AIF increased in both clinically localized prostate cancer and metastatic prostate tissues, and this increased expression localized to prostate epithelial cells. Representative data are shown in Fig. 1B. Median staining intensity was quantified (Fig. 1C) and showed that intensity was significantly higher in samples of prostatic intraepithelial neoplasia and frank carcinoma when compared with pathologically benign tissue (Kruskal-Wallis test p < 0.0001). To explore the potential cross-species effects of AIF expression upon prostate tumorigenesis, AIF protein levels were examined in tumors derived from a mouse model of prostate cancer. In these animals prostate-specific depletion of the tumor suppressor protein PTEN (phosphatase and tensin homolog) results in development of prostate cancer that faithfully reproduces molecular features of the human disease (45). Immunoblot analysis of PTEN−/− prostate tissues demonstrated that PTEN deletion substantially increased AIF protein levels compared with wild type prostate tissues (Fig. 1D). These data agree with those from human cancer patients and suggest that elevated AIF is a signature of prostate tumorigenesis in both humans and mice.
AIF Ablation Has Little Effect on Cell Death or in Vitro Growth of Prostate Cancer Cells
To further test the role of AIF in prostate cancer development and progression, AIF levels were suppressed in a panel of prostate cancer cell lines using an RNAi approach. Lentiviruses were used to stably infect LNCaP, DU145, and PC3 cells with shRNA sequences targeting either AIF or LacZ as a negative control (40). Cells stably incorporating hairpin DNA were detected by GFP fluorescence, and greater than 90% of cells were infected through this approach (Fig. 2A). Immunoblot analysis confirmed the near complete ablation of AIF in all targeted cells, whereas no loss of AIF expression was observed in controls (Fig. 2B). Because AIF was first identified as a molecule capable of triggering cell death, the impact of AIF ablation on the sensitivity of PC3 cells to a range of death-inducing agents was tested. Interestingly, when treated with agents to which PC3 cells are normally sensitive (MNNG, Fig. 2C; etoposide/peroxide, Fig. 2D) or resistant (arsenic trioxide/cisplatin, Fig. 2E), AIF-ablated cells demonstrated no change in sensitivity to any drug used in this panel. Similar results were obtained when treating LNCaP and DU145 cells with MNNG (data not shown). These data demonstrate that AIF does not play a significant role in controlling death in prostate cancer cells, suggesting that the activity of AIF within these cells lies outside the realm of cell death control.
FIGURE 2.

AIF ablation does not affect cell death induction in prostate cancer cells. A, lentiviruses harboring shRNA hairpins targeting LacZ or AIF and expressing GFP as a selectable marker were used to stably infect LNCaP, DU145, and PC3 cells. GFP-positive cells within each infected culture were assessed by flow cytometry. B, immunoblot analysis of stably infected LNCaP, DU145, and PC3 cells was performed to assess suppression of AIF protein expression; equal loading was confirmed by immunoblot for β-actin. C, PC3-derived cells were left untreated or treated with increasing concentrations of MNNG. Following overnight incubation, floating and attached cells were harvested, and cell viability was determined by propidium iodide staining and flow cytometry. The averages ± standard deviation of triplicate samples are shown for each data point. D, PC3-derived cells were left untreated or treated with etoposide (42.5 and 85 μm, 48 h) or hydrogen peroxide (100 and 250 μm, 24 h) before analysis as described for C. E, PC3-derived cells were treated with arsenic trioxide (ATO, 10 μm) or cisplatin (Cis, 166 μm) for the indicated periods of time before analysis as described for C.
Next, the ability of AIF-ablated cells to grow under nutrient-rich conditions was tested. Neither LNCaP (Fig. 3A) nor PC3 (Fig. 3C) cell lines demonstrated changes in growth rates following AIF ablation, indicating that these cell lines do not require AIF for growth under normal conditions in vitro. Interestingly, although DU145-shAIF was capable of cell growth, these cells grew more slowly than control DU145 cell lines (Fig. 3B), suggesting that AIF supports rapid DU145 cell growth in vitro.
FIGURE 3.
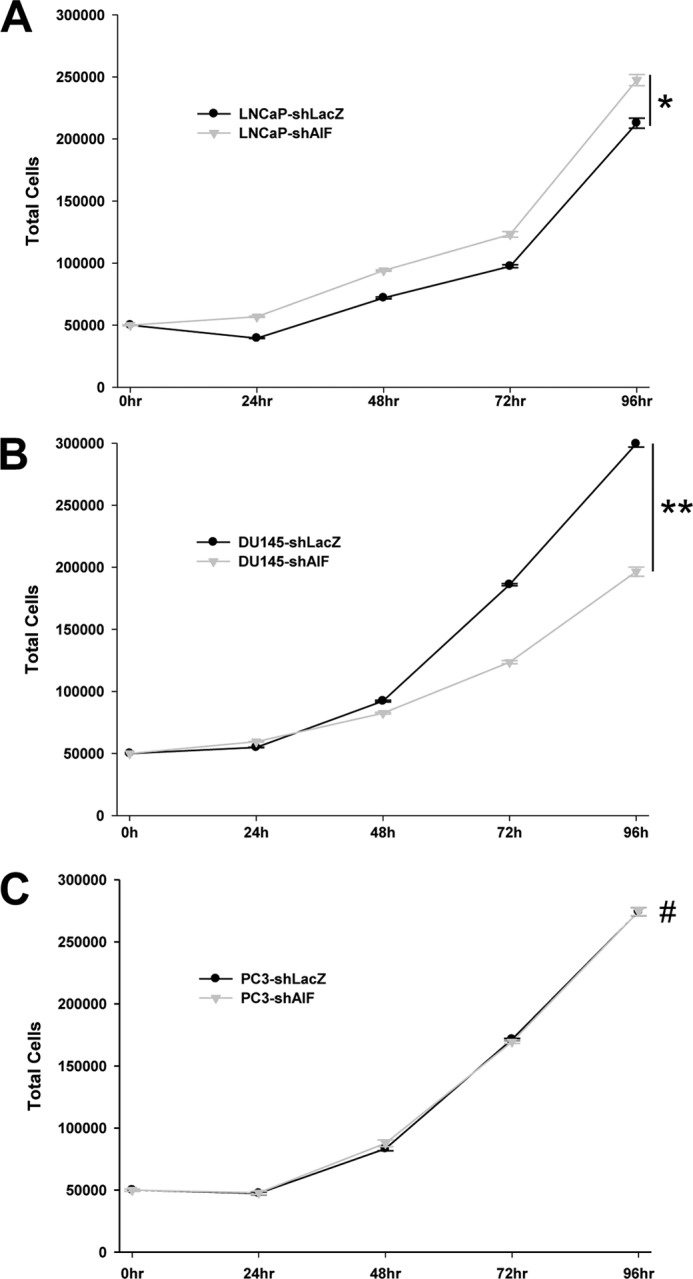
Growth of prostate cancer cells in nutrient-rich medium is not substantially affected by AIF ablation. LNCaP-derived (A), DU145-derived (B), and PC3-derived (C) cell lines were plated at 50,000 cells/well in replicate plates. The cells were then harvested every 24 h (for a total of 96 h), and the total number of cells in each well was assessed by Coulter counting. The averages ± standard deviation of triplicate samples are shown for each data point. The statistical significance of end point values was determined by Student's t test and indicates that only DU145 cells grew more slowly following AIF ablation. *, p > 0.01; **, p < 0.002; #, p > 0.5.
Advanced Prostate Cancer Cells Require AIF for Survival under Growth Stress, Invasion in Vitro, and Tumorigenesis in Vivo
Although AIF-deficient DU145 cells grew more slowly, these cells remained healthy, and no spontaneous cell death was observed. Combined with the observation that AIF ablation did not alter growth of LNCaP and PC3 cells, these data suggested that under nutrient-rich conditions, AIF is dispensable for the survival and growth of prostate cancer cells. To test the role of AIF in support of survival and proliferation under conditions of growth stress, an in vitro growth assay was performed using the three-dimensional matrix MatrigelTM. Cell growth under these conditions requires cellular metabolic changes to adapt to the complex extracellular environment, and only those cells capable of such adaptation survive and grow. Under these conditions LNCaP cells displayed normal growth and proliferation following AIF ablation (Fig. 4A, top row). In contrast, both DU145-shAIF cells (Fig. 4A, middle row) and PC3-shAIF cells (Fig. 4A, bottom row) failed to proliferate under conditions of growth stress, and a substantial portion of cells from both lineages exhibited features of cell death, including cell rounding and detachment. Thus AIF is not required for the less aggressive LNCaP cells to survive and grow, whereas the more advanced prostate cancer lines DU145 and PC3 absolutely require AIF for survival and proliferation under growth stress conditions.
FIGURE 4.

Advanced prostate cancer cells require AIF for survival under growth stress, invasion, and tumorigenesis in vivo. A, LNCaP, DU145, and PC3-derived cells were harvested, washed, suspended in fresh medium, and plated on MatrigelTM layers at a density of 10,000 cells/well. The cells were then allowed to grow for 4 days before imaging. B, PC3-derived cells were harvested, washed, and suspended in serum-free medium. Cell suspensions were added to BD-BioCoat MatrigelTM invasion chamber or control chambers and then placed into 12-well plates containing medium with serum. The cells were allowed to invade MatrigelTM layers for the indicated periods of time at which point chamber membranes were collected, the attached cells were fixed, and the total cells per membrane were counted. The percentage of invasion was determined by comparing cells on MatrigelTM-coated membranes versus uncoated control membranes. C, PC3-derived cells were injected subcutaneously into the dorsal hind flanks of male athymic nude mice. The mice were palpated daily until tumor growth was established, starting day 0 once tumors reached a size of 50 mm3.
To extend these findings and assess the ability of AIF to support an aggressive growth phenotype, PC3-derived cells were used for an in vitro invasion assay. The PC3 lines were chosen because these cells were completely insensitive to AIF ablation under nutrient-rich growth (Fig. 3C) but required AIF for survival under growth stress (Fig. 4A). As shown in Fig. 4B, PC3-shLacZ cells displayed substantial invasiveness at 40 h after seeding, yet PC3-shAIF cells were severely impaired in their invasive characteristics. We next tested whether suppression of AIF expression led to changes in growth in vivo. PC3-Parental, PC3-shLacZ, and PC3-shAIF cells were injected subcutaneously into athymic nude mice, tumors were allowed to establish, and tumor growth was followed over time (Fig. 4C). PC3-Parental and PC3-shLacZ cells displayed similar rates of tumor establishment (12 of 12 and 11 of 12 animals, respectively) and growth. In contrast PC3-shAIF cells displayed both a lower rate of tumor establishment (9 of 12 animals), as well as substantially slower growth in vivo. These data demonstrate that despite similar growth characteristics in vitro, AIF-deficient PC3 cells are substantially less able to form aggressive tumors in vivo and confirm that AIF directly contributes to growth and survival of advanced prostate cancer cells.
AIF-dependent Survival of Advanced Prostate Cancer Cells Correlates with Complex I Protein Levels and Altered Glucose Metabolism
AIF depletion often results in a loss of expression of protein components of the electron transport chain within the mitochondria, especially subunits of complex I (7). Immunoblot analysis was used to determine the status of complex I protein levels in our panel of prostate cancer cell lines (Fig. 5A). In LNCaP cells, neither the 39-kDa nor the 20-kDa complex I subunits exhibited decreased expression following AIF ablation. In contrast, both complex I subunits were reduced in DU145-shAIF and PC3-shAIF cell lines (Fig. 5A). Immunoblot analysis of tumors derived from PC3 xenografts showed that both AIF and complex I protein levels remained suppressed following growth in vivo (Fig. 5B). It is also notable that these tumors exhibited normal levels of the respiratory chain protein CoxIV, suggesting that AIF ablation did not lead to global changes in the mitochondrial proteome.
FIGURE 5.

Advanced prostate cancer cells exhibit reduced complex I expression and increased glucose consumption following AIF ablation. A, protein levels of the 39- and 20-kDa subunits of complex I were assessed in LNCaP-, DU145-, and PC3-derived cell lines by immunoblot analysis; equal loading was confirmed by immunoblot for β-actin. B, upon completion of studies shown in Fig. 4C, tumors were removed, total protein was extracted, and immunoblot analysis was performed to assess protein levels of AIF, complex I (39 kDa), and complex I (20 kDa); equal loading was confirmed by immunoblot for CoxIV. C–E, the indicated cell lines were harvested, washed, and seeded at equal densities in fresh medium. 48 h after plating, medium was collected, and the total glucose was assessed using the BioAssay systems QuantiChromTM glucose assay kit. The total number of cells in each well was assessed by Coulter counting and used to determine glucose consumption per cell.
These observations correlate with the differences observed between these three lines in sensitivity to growth stress following AIF ablation, suggesting that AIF supports metabolic and/or energy production activities necessary for survival of more advanced prostate cancer cells. A potential consequence of complex I subunit loss is a switch from energy production via oxidative phosphorylation through the mitochondrial electron transport chain to glycolysis. To test whether this switch occurs in AIF-ablated prostate cancer cells, glucose consumption was measured. As shown in Fig. 5C, glucose consumption levels in LNCaP-shAIF cells were similar to those observed in controls. In contrast, both DU145-shAIF and PC3-shAIF cells consume 2–3-fold more glucose when compared with controls (Fig. 5, D and E), suggesting a metabolic switch to glucose consumption in these two cell lines following AIF ablation.
To further define the metabolic changes induced in our cell line panel following AIF ablation lactate secretion and oxygen consumption (both indicators of oxidative phosphorylation) were measured. Similar to observations of glucose consumption, LNCaP cells displayed no change in either lactate secretion or oxygen consumption following AIF ablation (Fig. 6). DU145 cells displayed higher basal levels of lactate secretion, which were elevated further following AIF ablation, and oxygen consumption in these cells was reduced in the absence of AIF. Ablation of AIF from PC3 cells resulted in increased lactate secretion and decreased oxygen consumption, similar to the changes observed in DU145 cells. These metabolic studies correlate well with the glucose consumption experiments and confirm that a metabolic switch occurs in DU145 and PC3 cells following AIF ablation.
FIGURE 6.
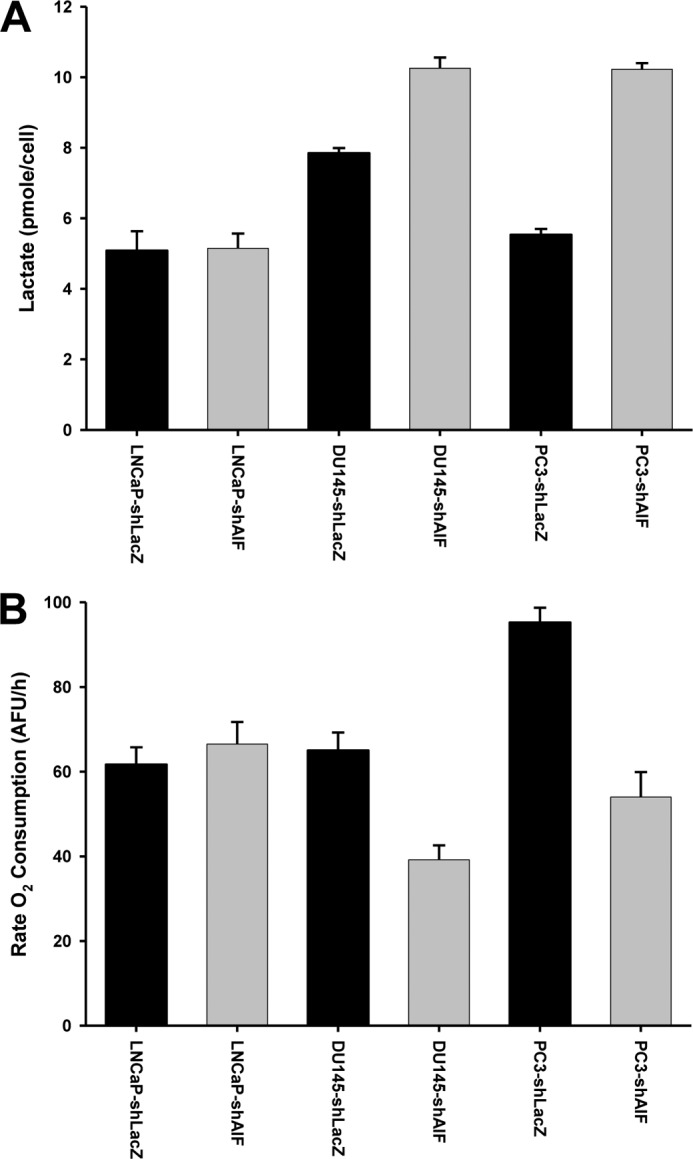
Altered lactate secretion and oxygen consumption following ablation of AIF from advanced prostate cancer cells. A, the indicated cell lines were harvested, washed, and seeded at equal densities in fresh medium. 48 h after plating, medium was collected, and secreted lactate was measured using the Sigma lactate assay kit. The total number of cells in each well was assessed by Coulter counting and used to determine lactate secretion per cell. B, the indicated cell lines were harvested, washed, and seeded at equal densities in fresh medium. MitoXpress® probe was added, and then each well was overlaid with mineral oil to avoid interference from atmospheric oxygen. Fluorescence of each well was then measured every 1.5 min for a total of 200 min.
To determine the significance of altered glucose metabolism, the viability of our panel of cells was determined following glucose withdrawal. As expected AIF ablation from LNCaP cells had little impact upon survival in glucose-free medium (Fig. 7A). In contrast both DU145 cells (Fig. 7B) and PC3 cells (Fig. 7C) displayed reduced viability in glucose-free medium following AIF ablation. To confirm the specificity of these results, PC3 cells were further subjected to survival following glycolysis inhibition with 2-deoxyglucose. As shown in Fig. 7D, PC3-shAIF cells exhibited reduced viability following 2-deoxyglucose treatment when compared with PC3-shLacZ cells. These data demonstrate that the glycolytic switch observed in advanced prostate cancer cell lines is necessary for cell survival, even under nutrient-rich conditions in vitro.
FIGURE 7.

AIF-deficient advanced prostate cancer cells are sensitive to glucose deprivation. A–C, LNCaP- (A), DU145- (B), and PC3-derived (C) cells were incubated in normal growth medium or glucose-free growth medium for 0, 48, and 72 h. Cell viability was determined by propidium iodide staining and flow cytometry as described in Fig. 2 above. The averages ± standard deviation of triplicate samples are shown for each data point. D, PC3-derived cells were incubated in normal growth medium in the absence and presence of the glycolysis inhibitor 2-deoxyglucose (50 mm) for 48 h. Cell viability was determined by propidium iodide staining and flow cytometry as described above. The averages ± standard deviation of triplicate samples are shown for each data point.
The Enzymatic Activity of AIF Is Necessary for Growth of Advanced Prostate Cancer Cells
Overall these data suggested that AIF is critical for the ability of advanced prostate cancer cell lines to survive under conditions of growth stress both in vitro and in vivo. However, the nature of how AIF supports such growth remained unclear. The NADH-oxidase activity of AIF plays a potential role in cellular energy and redox balance, raising the possibility that this activity is necessary for the support of prostate cancer cell growth. To test this concept, PC3-shAIF cells were subjected to a second round of lentiviral infection in which AIF levels were restored by the introduction of either wild type AIF or the AIF variant T263A/V300A (TVA), which harbors mutations at two key residues necessary for catalysis and is therefore enzymatically deficient (12). AIF cDNA employed within the restoration constructs contained silent mutations that render the shAIF targeting sequence of our shRNA viruses ineffective against the restored sequences. As controls, both PC3-shLacZ and PC3-shAIF cells were additionally infected with lentivirus lacking AIF cDNA (PC3-shLacZ + empty, PC3-shAIF + empty). As shown in Fig. 8A, restoration of PC3-shAIF cells with either wild type or TVA AIF viruses resulted in AIF expression above endogenous levels when compared with PC3-shLacZ or PC3-shLacZ + empty control cells. Notably, both wild type and TVA-AIF restored cells exhibited normal levels of complex I subunits, suggesting that the enzymatic activity of AIF is not necessary for maintenance of complex I expression. Glucose consumption was next assessed (Fig. 8B). Whereas WT-AIF restored glucose consumption to levels observed in control cells (shLacZ, shLacZ + empty), TVA-AIF cells continued to exhibit glucose consumption similar to the elevated levels found in shAIF and shAIF + empty cell lines. Thus, although the TVA AIF variant restored complex I levels back to wild type levels, reversion of glucose consumption did not correlate with restored expression, suggesting that the nature of energy metabolism switching following AIF ablation is not limited to the effects upon complex I expression alone.
FIGURE 8.

The enzymatic activity of AIF supports normal glucose consumption, aggressive growth, and survival under growth stress. A, immunoblot analysis of restored PC3-derived cell lines for AIF, 39-kDa complex I subunit, 20-kDa complex I subunit, and β-actin as loading control. B, glucose consumption of restored PC3 cell lines was performed as described in Figs. 3 and 6. C, MatrigelTM growth and survival of restored PC3-derived cell lines was performed as described above. D, migration assay of restored PC3-derived cell lines. The cells were seeded at 900,000 cells/35-mm dish and allowed to attach for 16 h. A p200 pipette tip was used to scratch a region from the center of each plate, the plates were immediately washed, and fresh medium was added. Gap size was then measured as time 0 distance. The cells were incubated for an additional 27 h, and gap size measurements were repeated. Distance migrated for each cell line was then determined. The averages ± standard deviation are shown for each data point, n = 15 for each cell line.
No differences in growth rates under normal conditions were observed following AIF restoration (data not shown). However, when restored cells were grown upon MatrigelTM, substantial differences became apparent (Fig. 8C): as expected PC3-shLacZ and PC3-shLacZ + empty cells grew normally, whereas both PC3-shAIF and PC3-shAIF + empty cell lines were significantly impaired in their ability to grown in MatrigelTM. Restoration of PC3-shAIF cells with wild type AIF restored normal growth in MatrigelTM. Critically, the TVA AIF variant was unable to restore normal growth despite substantial expression, demonstrating the importance of AIF enzymatic activity for support of prostate cancer cell survival. To correlate MatrigelTM growth with invasiveness, the migratory properties of restored cells was tested by a scratch assay. The cells were seeded in normal dishes at high density and allowed to attach, and a pipette tip was used to displace a single line of cells from the center of each dish. The distance of this path was immediately measured, and the rate of gap filling by each cell line was measured over time. As shown in Fig. 8D, control PC3-shLacZ and WT-AIF restored shAIF cells exhibited normal migration activity, whereas AIF-deficient and TVA-AIF restored cells migrated significantly shorter distances over the identical time period, further suggesting that AIF enzymatic activity is required for aggressive growth.
DISCUSSION
As a promoter of both cell death and cell survival, AIF is poised to regulate disparate aspects of cellular biology, yet how these dueling activities are coordinated is unclear, and a direct role for AIF as a driver of disease pathogenesis remains poorly defined. This is especially true for cancer. Decreased AIF expression has not been reported as a component in the development of any human cancer, and mutations affecting AIF activity in cancer have yet to be identified. The ability of AIF to kill tumor cells has been investigated (46, 47), yet a critical role for AIF in the execution of tumor cells has not been definitively shown. Thus, although the death activity of AIF provides the opportunity to function in a tumor-suppressive capacity, the current data do not support such a role. In contrast a small number of clinically based studies have evaluated the expression of AIF in diverse cancer cell types, and it appears that elevated AIF expression may often occur in cancer development (48–51). Unfortunately the majority of these studies do not define a direct role for AIF in controlling cancer progression. An exception is the situation of AIF in colorectal cancer (52). In this setting, elevation of AIF expression has been clearly demonstrated, and the elegant work of Urbano et al. (12) define a contributory role for AIF in both the resistance of colorectal cancer cells to chemical stress and maintenance of the tumorigenic state.
Here we demonstrate for the first time that AIF plays a supportive role in prostate cancer. We show that AIF transcript levels are elevated by up to 2-fold in prostate cancer tissues, and increased protein levels are also observed in a large cohort of clinical samples. Ablation of AIF expression in a panel of prostate cancer cell lines demonstrated that AIF does not contribute significantly to drug resistance or survival and growth under nutrient-rich conditions. Interestingly advanced prostate cancer cells demonstrate substantial reductions in survival, growth, and invasion under growth stress in vitro, and in vivo tumor growth is severely compromised following AIF ablation. These changes in growth and survival under stress conditions correlated with changes in respiratory chain protein expression and concomitant alterations in glucose metabolism that were also selective for advanced prostate cancer cells. We show that in cells sensitive to AIF ablation, the enzymatic activity of AIF is critical for the support of cancer cell survival and growth, directly linking AIF activity to cell progression. These findings suggest that AIF may represent a new molecular target for development of treatments for advanced disease.
Several questions and implications arise from these observations. First, given that early studies of AIF-mediated cell killing demonstrated that elevated AIF levels induce cell death, how do prostate cancer cells tolerate increased AIF expression? We envision three possibilities. First, the extent of elevated AIF expression in prostate cancer cells is not sufficient to trigger cell death. When compared with increased expression of oncogenes in other cancer types (53–55), elevation of AIF protein levels in prostate cancer is modest. This may suggest that the range of overexpression over which the increased AIF is of benefit is narrow and that greater levels of AIF may ultimately become toxic. A second possibility is that the death-inducing activity of AIF is blocked by up-regulation of inhibitory molecules. We have recently shown that through a ubiquitination-dependent mechanism, the pro-survival protein X-linked inhibitor of apoptosis (XIAP) is capable of preventing AIF-mediated DNA degradation (56). XIAP is also elevated in prostate cancer (35, 57), raising the possibility that XIAP blocks AIF death induction and allows the mitochondrial functions of AIF to support cancer cell survival. Finally, and most likely, AIF does not play a significant death-inducing role in prostate cells. The data presented here strongly support this model: AIF ablation in PC3 cells had no impact on sensitivity to a panel of chemical death triggers (Fig. 2, C–E), and an overexpression of wild type AIF in restored PC3-shAIF cells did not result in cellular toxicity (Fig. 8A), strongly suggesting that AIF is not a significant killer of prostate cells.
What then is the benefit of AIF expression to prostate cancer cells, and why does this benefit appear selective to more advanced prostate cancer cell types? Although the precise answers to these questions are unclear, our data directly implicate AIF as an important regulator of glucose metabolism. Tumor cells frequently utilize glycolysis as the predominant mechanism for energy production, despite sufficient oxygen and an intact mitochondrial respiratory chain (19). This switch to aerobic glycolysis, known as the Warburg effect (58), balances decreased efficiency of energy production with the shuttling of glycolytic metabolites toward biosynthetic pathways. When coupled with increased glutamine uptake as an additional source of substrates for macromolecule production (59), the Warburg effect results in a metabolic state permissive to aggressive tumor growth. Despite this metabolic switch, cancer cells often maintain a functional mitochondrial respiratory chain, and metabolites produced through activity of the TCA cycle support macromolecule biosynthesis (60). Thus, whereas tumor cells favor glycolysis, mitochondria remain capable of energy production and serve as a necessary source of raw material for macromolecular synthesis.
In this scenario AIF-mediated support of mitochondrial function is dispensable for growth under nonstress conditions such as cell culture in vitro or at well vascularized tumor sites in vivo. However, when growth stress conditions are encountered either during the early stages of primary tumor development or following metastasis, the ability to produce energy through an intact respiratory chain remains critical for tumor cell survival, and cancer cells lacking functional AIF suffer a metabolic disadvantage. Our experiments evaluating ablation of AIF from DU145 and PC3 cells are consistent with this model. A loss of AIF compromises mitochondrial energy production, forcing cells to consume more glucose to meet energy demands. Under nutrient-rich conditions, this does not prevent cell growth; however, under conditions of growth stress, the lack of mitochondrial energy production cripples cell proliferation and survival. As a less advanced prostate cancer cell line than DU145 or PC3, LNCaP cells remain wild type for a number of oncogenes involved in triggering Warburg metabolism, most notably p53, c-Myc, and HIF-1 (61–63). Thus these cells are less dependent on aerobic glycolysis for energy production and macromolecule synthesis, less addicted to glucose consumption, and capable of survival under growth stress following ablation of AIF. Defining where within the continuum of prostate cancer development the role of AIF becomes critical is an area in need of further study.
Our data show not only that AIF ablation leads to loss of complex I subunit expression but also that this loss is selective, because expression of other respiratory chain proteins remains unchanged. How this selectivity is achieved is unclear. Moreover, how AIF affects the abundance of mitochondrial proteins encoded by nuclear genes remains a significant question. A possible explanation for this phenomenon may lie in AIF control of ROS signaling. ROS has been linked to the activation of several gene expression programs, including the antioxidant response and androgen signaling pathways, both of which are of direct significance to prostate tissue (64, 65). We and others have shown that increased AIF protein levels increase cellular ROS under conditions that do not lead to cellular toxicity (12, 41), and we have found that AIF ablation in PC3 cells leads to a decrease in cellular ROS.6 These observations are consistent with the hypothesis that AIF is a direct elevator of cellular ROS. In the progression of prostate cancer from early to advanced status, many signaling pathways undergo alteration. Some of these pathways are ROS-responsive, notably the antioxidant response programs controlled by the B-loop/helix transcription factors Nrf1 and Nrf2 (66, 67) and the canonical survival pathways controlled by NF-κB (18). In advanced cancers, the ability of AIF to elevate oxidant levels may favor these co-signaling regimes. Our observations show that LNCaP cells do not exhibit loss of complex I proteins or a switch to increased glucose consumption upon AIF ablation. This may suggest an intersection between oncogene-induced changes in metabolism and the ROS modulating activities of AIF in advanced prostate cells.
In summary, herein we show that the cellular relevance of AIF activity extends beyond the realm of cell death induction and into the area of support for advanced prostate cancer cell growth and survival. Through the enzymatic activity of AIF, cellular levels of electron transport chain proteins are maintained, and levels of glucose consumption are modulated in favor of tumor cell growth. The activity of AIF in support of prostate cancer cells is restricted to cells that have achieved a more advanced status, suggesting that AIF acts at a later state in tumor development. These studies highlight the significance of AIF as a potential target for the development of new therapeutic approaches for the treatment of advanced prostate cancer.
Acknowledgments
We thank Drs. D. Lyles, S. Kridel, G. Kulik, and J. Ewald for cell lines and critical reading of the manuscript and Dr. G. Sui for extracts from mouse prostate samples and pSL4-puro. We also thank E. Rolfe, A. Hoyt, and Dr. M. Ahmed for technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grant P50 CA69568 (to A. M. C.) from the SPORE in Prostate Cancer. This work was also supported by Grant U01 CA111275 (to A. M. C.) from the Early Detection Research Network, Grant W81XWH-08-1-0045 (to J. C. W.) from the Department of Defense Prostate Cancer Research Program, and Grant RSG-09-166-01-CCG (to J. C. W.) from the American Cancer Society.
J. C. Wilkinson, unpublished observations.
- AIF
- apoptosis-inducing factor
- MNNG
- N-methyl-N′-nitro-n-nitrosoguanidine
- ROS
- reactive oxygen species
- TMA
- tissue microarray analysis
- XIAP
- X-linked inhibitor of apoptosis
- TVA
- T263A/V300A.
REFERENCES
- 1. Susin S. A., Lorenzo H. K., Zamzami N., Marzo I., Snow B. E., Brothers G. M., Mangion J., Jacotot E., Costantini P., Loeffler M., Larochette N., Goodlett D. R., Aebersold R., Siderovski D. P., Penninger J. M., Kroemer G. (1999) Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 397, 441–446 [DOI] [PubMed] [Google Scholar]
- 2. Wang X., Yang C., Chai J., Shi Y., Xue D. (2002) Mechanisms of AIF-mediated apoptotic DNA degradation in Caenorhabditis elegans. Science 298, 1587–1592 [DOI] [PubMed] [Google Scholar]
- 3. Loeffler M., Daugas E., Susin S. A., Zamzami N., Metivier D., Nieminen A. L., Brothers G., Penninger J. M., Kroemer G. (2001) Dominant cell death induction by extramitochondrially targeted apoptosis-inducing factor. FASEB J. 15, 758–767 [DOI] [PubMed] [Google Scholar]
- 4. Miramar M. D., Costantini P., Ravagnan L., Saraiva L. M., Haouzi D., Brothers G., Penninger J. M., Peleato M. L., Kroemer G., Susin S. A. (2001) NADH oxidase activity of mitochondrial apoptosis-inducing factor. J. Biol. Chem. 276, 16391–16398 [DOI] [PubMed] [Google Scholar]
- 5. Brown D., Yu B. D., Joza N., Bénit P., Meneses J., Firpo M., Rustin P., Penninger J. M., Martin G. R. (2006) Loss of AIF function causes cell death in the mouse embryo, but the temporal progression of patterning is normal. Proc. Natl. Acad. Sci. U.S.A. 103, 9918–9923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Klein J. A., Longo-Guess C. M., Rossmann M. P., Seburn K. L., Hurd R. E., Frankel W. N., Bronson R. T., Ackerman S. L. (2002) The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature 419, 367–374 [DOI] [PubMed] [Google Scholar]
- 7. Vahsen N., Candé C., Brière J. J., Bénit P., Joza N., Larochette N., Mastroberardino P. G., Pequignot M. O., Casares N., Lazar V., Feraud O., Debili N., Wissing S., Engelhardt S., Madeo F., Piacentini M., Penninger J. M., Schägger H., Rustin P., Kroemer G. (2004) AIF deficiency compromises oxidative phosphorylation. EMBO J. 23, 4679–4689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Joza N., Oudit G. Y., Brown D., Bénit P., Kassiri Z., Vahsen N., Benoit L., Patel M. M., Nowikovsky K., Vassault A., Backx P. H., Wada T., Kroemer G., Rustin P., Penninger J. M. (2005) Muscle-specific loss of apoptosis-inducing factor leads to mitochondrial dysfunction, skeletal muscle atrophy, and dilated cardiomyopathy. Mol. Cell. Biol. 25, 10261–10272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cheung E. C., Joza N., Steenaart N. A., McClellan K. A., Neuspiel M., McNamara S., MacLaurin J. G., Rippstein P., Park D. S., Shore G. C., McBride H. M., Penninger J. M., Slack R. S. (2006) Dissociating the dual roles of apoptosis-inducing factor in maintaining mitochondrial structure and apoptosis. EMBO J. 25, 4061–4073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ghezzi D., Sevrioukova I., Invernizzi F., Lamperti C., Mora M., D'Adamo P., Novara F., Zuffardi O., Uziel G., Zeviani M. (2010) Severe X-linked mitochondrial encephalomyopathy associated with a mutation in apoptosis-inducing factor. Am. J. Hum. Genet. 86, 639–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Porter A. G., Urbano A. G. (2006) Does apoptosis-inducing factor (AIF) have both life and death functions in cells? Bioessays 28, 834–843 [DOI] [PubMed] [Google Scholar]
- 12. Urbano A., Lakshmanan U., Choo P. H., Kwan J. C., Ng P. Y., Guo K., Dhakshinamoorthy S., Porter A. (2005) AIF suppresses chemical stress-induced apoptosis and maintains the transformed state of tumor cells. EMBO J. 24, 2815–2826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Movsas B., Chapman J. D., Horwitz E. M., Pinover W. H., Greenberg R. E., Hanlon A. L., Iyer R., Hanks G. E. (1999) Hypoxic regions exist in human prostate carcinoma. Urology 53, 11–18 [DOI] [PubMed] [Google Scholar]
- 14. Williams K. J., Cowen R. L., Stratford I. J. (2001) Hypoxia and oxidative stress. Tumour hypoxia. Therapeutic considerations. Breast Cancer Res. 3, 328–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ghafar M. A., Anastasiadis A. G., Chen M. W., Burchardt M., Olsson L. E., Xie H., Benson M. C., Buttyan R. (2003) Acute hypoxia increases the aggressive characteristics and survival properties of prostate cancer cells. Prostate 54, 58–67 [DOI] [PubMed] [Google Scholar]
- 16. Pinthus J. H., Bryskin I., Trachtenberg J., Lu J. P., Singh G., Fridman E., Wilson B. C. (2007) Androgen induces adaptation to oxidative stress in prostate cancer. Implications for treatment with radiation therapy. Neoplasia 9, 68–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bhat H. K., Calaf G., Hei T. K., Loya T., Vadgama J. V. (2003) Critical role of oxidative stress in estrogen-induced carcinogenesis. Proc. Natl. Acad. Sci. U.S.A. 100, 3913–3918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Morgan M. J., Liu Z. G. (2011) Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 21, 103–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dang C. V. (2012) Links between metabolism and cancer. Genes Dev. 26, 877–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yu Y. P., Landsittel D., Jing L., Nelson J., Ren B., Liu L., McDonald C., Thomas R., Dhir R., Finkelstein S., Michalopoulos G., Becich M., Luo J. H. (2004) Gene expression alterations in prostate cancer predicting tumor aggression and preceding development of malignancy. J. Clin. Oncol. 22, 2790–2799 [DOI] [PubMed] [Google Scholar]
- 21. Taylor B. S., Schultz N., Hieronymus H., Gopalan A., Xiao Y., Carver B. S., Arora V. K., Kaushik P., Cerami E., Reva B., Antipin Y., Mitsiades N., Landers T., Dolgalev I., Major J. E., Wilson M., Socci N. D., Lash A. E., Heguy A., Eastham J. A., Scher H. I., Reuter V. E., Scardino P. T., Sander C., Sawyers C. L., Gerald W. L. (2010) Integrative genomic profiling of human prostate cancer. Cancer Cell 18, 11–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu P., Ramachandran S., Ali Seyed M., Scharer C. D., Laycock N., Dalton W. B., Williams H., Karanam S., Datta M. W., Jaye D. L., Moreno C. S. (2006) Sex-determining region Y box 4 is a transforming oncogene in human prostate cancer cells. Cancer Res. 66, 4011–4019 [DOI] [PubMed] [Google Scholar]
- 23. Holzbeierlein J., Lal P., LaTulippe E., Smith A., Satagopan J., Zhang L., Ryan C., Smith S., Scher H., Scardino P., Reuter V., Gerald W. L. (2004) Gene expression analysis of human prostate carcinoma during hormonal therapy identifies androgen-responsive genes and mechanisms of therapy resistance. Am. J. Pathol. 164, 217–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wallace T. A., Prueitt R. L., Yi M., Howe T. M., Gillespie J. W., Yfantis H. G., Stephens R. M., Caporaso N. E., Loffredo C. A., Ambs S. (2008) Tumor immunobiological differences in prostate cancer between African-American and European-American men. Cancer Res. 68, 927–936 [DOI] [PubMed] [Google Scholar]
- 25. Lapointe J., Li C., Higgins J. P., van de Rijn M., Bair E., Montgomery K., Ferrari M., Egevad L., Rayford W., Bergerheim U., Ekman P., DeMarzo A. M., Tibshirani R., Botstein D., Brown P. O., Brooks J. D., Pollack J. R. (2004) Gene expression profiling identifies clinically relevant subtypes of prostate cancer. Proc. Natl. Acad. Sci. U.S.A. 101, 811–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Arredouani M. S., Lu B., Bhasin M., Eljanne M., Yue W., Mosquera J. M., Bubley G. J., Li V., Rubin M. A., Libermann T. A., Sanda M. G. (2009) Identification of the transcription factor single-minded homologue 2 as a potential biomarker and immunotherapy target in prostate cancer. Clin. Cancer Res. 15, 5794–5802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Luo J. H., Yu Y. P., Cieply K., Lin F., Deflavia P., Dhir R., Finkelstein S., Michalopoulos G., Becich M. (2002) Gene expression analysis of prostate cancers. Mol. Carcinog. 33, 25–35 [DOI] [PubMed] [Google Scholar]
- 28. Vanaja D. K., Cheville J. C., Iturria S. J., Young C. Y. (2003) Transcriptional silencing of zinc finger protein 185 identified by expression profiling is associated with prostate cancer progression. Cancer Res. 63, 3877–3882 [PubMed] [Google Scholar]
- 29. LaTulippe E., Satagopan J., Smith A., Scher H., Scardino P., Reuter V., Gerald W. L. (2002) Comprehensive gene expression analysis of prostate cancer reveals distinct transcriptional programs associated with metastatic disease. Cancer Res. 62, 4499–4506 [PubMed] [Google Scholar]
- 30. Welsh J. B., Sapinoso L. M., Su A. I., Kern S. G., Wang-Rodriguez J., Moskaluk C. A., Frierson H. F., Jr., Hampton G. M. (2001) Analysis of gene expression identifies candidate markers and pharmacological targets in prostate cancer. Cancer Res. 61, 5974–5978 [PubMed] [Google Scholar]
- 31. Singh D., Febbo P. G., Ross K., Jackson D. G., Manola J., Ladd C., Tamayo P., Renshaw A. A., D'Amico A. V., Richie J. P., Lander E. S., Loda M., Kantoff P. W., Golub T. R., Sellers W. R. (2002) Gene expression correlates of clinical prostate cancer behavior. Cancer Cell 1, 203–209 [DOI] [PubMed] [Google Scholar]
- 32. Tomlins S. A., Mehra R., Rhodes D. R., Cao X., Wang L., Dhanasekaran S. M., Kalyana-Sundaram S., Wei J. T., Rubin M. A., Pienta K. J., Shah R. B., Chinnaiyan A. M. (2007) Integrative molecular concept modeling of prostate cancer progression. Nat. Genet. 39, 41–51 [DOI] [PubMed] [Google Scholar]
- 33. Varambally S., Dhanasekaran S. M., Zhou M., Barrette T. R., Kumar-Sinha C., Sanda M. G., Ghosh D., Pienta K. J., Sewalt R. G., Otte A. P., Rubin M. A., Chinnaiyan A. M. (2002) The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 419, 624–629 [DOI] [PubMed] [Google Scholar]
- 34. Rhodes D. R., Kalyana-Sundaram S., Mahavisno V., Varambally R., Yu J., Briggs B. B., Barrette T. R., Anstet M. J., Kincead-Beal C., Kulkarni P., Varambally S., Ghosh D., Chinnaiyan A. M. (2007) Oncomine 3.0. Genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia 9, 166–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Varambally S., Yu J., Laxman B., Rhodes D. R., Mehra R., Tomlins S. A., Shah R. B., Chandran U., Monzon F. A., Becich M. J., Wei J. T., Pienta K. J., Ghosh D., Rubin M. A., Chinnaiyan A. M. (2005) Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell 8, 393–406 [DOI] [PubMed] [Google Scholar]
- 36. Rubin M. A., Zhou M., Dhanasekaran S. M., Varambally S., Barrette T. R., Sanda M. G., Pienta K. J., Ghosh D., Chinnaiyan A. M. (2002) α-Methylacyl coenzyme A racemase as a tissue biomarker for prostate cancer. JAMA 287, 1662–1670 [DOI] [PubMed] [Google Scholar]
- 37. Dhanasekaran S. M., Barrette T. R., Ghosh D., Shah R., Varambally S., Kurachi K., Pienta K. J., Rubin M. A., Chinnaiyan A. M. (2001) Delineation of prognostic biomarkers in prostate cancer. Nature 412, 822–826 [DOI] [PubMed] [Google Scholar]
- 38. Yu J., Wang P., Ming L., Wood M. A., Zhang L. (2007) SMAC/Diablo mediates the proapoptotic function of PUMA by regulating PUMA-induced mitochondrial events. Oncogene 26, 4189–4198 [DOI] [PubMed] [Google Scholar]
- 39. Kamradt M. C., Lu M., Werner M. E., Kwan T., Chen F., Strohecker A., Oshita S., Wilkinson J. C., Yu C., Oliver P. G., Duckett C. S., Buchsbaum D. J., LoBuglio A. F., Jordan V. C., Cryns V. L. (2005) The small heat shock protein αB-crystallin is a novel inhibitor of TRAIL-induced apoptosis that suppresses the activation of caspase-3. J. Biol. Chem. 280, 11059–11066 [DOI] [PubMed] [Google Scholar]
- 40. Qin X. F., An D. S., Chen I. S., Baltimore D. (2003) Inhibiting HIV-1 infection in human T cells by lentiviral-mediated delivery of small interfering RNA against CCR5. Proc. Natl. Acad. Sci. U.S.A. 100, 183–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wilkinson J. C., Wilkinson A. S., Galbán S., Csomos R. A., Duckett C. S. (2008) Apoptosis-inducing factor is a target for ubiquitination through interaction with XIAP. Mol. Cell. Biol. 28, 237–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cao P., Deng Z., Wan M., Huang W., Cramer S. D., Xu J., Lei M., Sui G. (2010) MicroRNA-101 negatively regulates Ezh2 and its expression is modulated by androgen receptor and HIF-1α/HIF-1β. Mol. Cancer 9, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Galbán S., Hwang C., Rumble J. M., Oetjen K. A., Wright C. W., Boudreault A., Durkin J., Gillard J. W., Jaquith J. B., Morris S. J., Duckett C. S. (2009) Cytoprotective effects of IAPs revealed by a small molecule antagonist. Biochem. J. 417, 765–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liang C. C., Park A. Y., Guan J. L. (2007) In vitro scratch assay. A convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2, 329–333 [DOI] [PubMed] [Google Scholar]
- 45. Wang S., Gao J., Lei Q., Rozengurt N., Pritchard C., Jiao J., Thomas G. V., Li G., Roy-Burman P., Nelson P. S., Liu X., Wu H. (2003) Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell 4, 209–221 [DOI] [PubMed] [Google Scholar]
- 46. Zhang W., Zhang C., Narayani N., Du C., Balaji K. C. (2007) Nuclear translocation of apoptosis inducing factor is associated with cisplatin induced apoptosis in LNCaP prostate cancer cells. Cancer Lett. 255, 127–134 [DOI] [PubMed] [Google Scholar]
- 47. Troutaud D., Petit B., Bellanger C., Marin B., Gourin-Chaury M. P., Petit D., Olivrie A., Feuillard J., Jauberteau M. O., Bordessoule D. (2010) Prognostic significance of BAD and AIF apoptotic pathways in diffuse large B-cell lymphoma. Clin. Lymphoma Myeloma Leuk. 10, 118–124 [DOI] [PubMed] [Google Scholar]
- 48. Skyrlas A., Hantschke M., Passa V., Gaitanis G., Malamou-Mitsi V., Bassukas I. D. (2011) Expression of apoptosis-inducing factor (AIF) in keratoacanthomas and squamous cell carcinomas of the skin. Exp. Dermatol. 20, 674–676 [DOI] [PubMed] [Google Scholar]
- 49. Li S., Wan M., Cao X., Ren Y. (2011) Expression of AIF and HtrA2/Omi in small lymphocytic lymphoma and diffuse large B-cell lymphoma. Arch. Pathol Lab. Med. 135, 903–908 [DOI] [PubMed] [Google Scholar]
- 50. Huerta S., Heinzerling J. H., Anguiano-Hernandez Y. M., Huerta-Yepez S., Lin J., Chen D., Bonavida B., Livingston E. H. (2007) Modification of gene products involved in resistance to apoptosis in metastatic colon cancer cells. Roles of Fas, Apaf-1, NFκB, IAPs, Smac/DIABLO, and AIF. J. Surg. Res. 142, 184–194 [DOI] [PubMed] [Google Scholar]
- 51. Jeong E. G., Lee J. W., Soung Y. H., Nam S. W., Kim S. H., Lee J. Y., Yoo N. J., Lee S. H. (2006) Immunohistochemical and mutational analysis of apoptosis-inducing factor (AIF) in colorectal carcinomas. APMIS 114, 867–873 [DOI] [PubMed] [Google Scholar]
- 52. Millan A., Huerta S. (2009) Apoptosis-inducing factor and colon cancer. J. Surg. Res. 151, 163–170 [DOI] [PubMed] [Google Scholar]
- 53. Norris J. L., Baldwin A. S., Jr. (1999) Oncogenic Ras enhances NF-κB transcriptional activity through Raf-dependent and Raf-independent mitogen-activated protein kinase signaling pathways. J. Biol. Chem. 274, 13841–13846 [DOI] [PubMed] [Google Scholar]
- 54. Duesbery N. S., Resau J., Webb C. P., Koochekpour S., Koo H. M., Leppla S. H., Vande Woude G. F. (2001) Suppression of ras-mediated transformation and inhibition of tumor growth and angiogenesis by anthrax lethal factor, a proteolytic inhibitor of multiple MEK pathways. Proc. Natl. Acad. Sci. U.S.A. 98, 4089–4094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yu Q., Ciemerych M. A., Sicinski P. (2005) Ras and Myc can drive oncogenic cell proliferation through individual D-cyclins. Oncogene 24, 7114–7119 [DOI] [PubMed] [Google Scholar]
- 56. Lewis E. M., Wilkinson A. S., Davis N. Y., Horita D. A., Wilkinson J. C. (2011) Nondegradative ubiquitination of apoptosis inducing factor (AIF) by X-linked inhibitor of apoptosis at a residue critical for AIF-mediated chromatin degradation. Biochemistry 50, 11084–11096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Devi G. R. (2004) XIAP as target for therapeutic apoptosis in prostate cancer. Drug News Perspect 17, 127–134 [DOI] [PubMed] [Google Scholar]
- 58. Warburg O. (1956) On the origin of cancer cells. Science 123, 309–314 [DOI] [PubMed] [Google Scholar]
- 59. Shanware N. P., Mullen A. R., DeBerardinis R. J., Abraham R. T. (2011) Glutamine. Pleiotropic roles in tumor growth and stress resistance. J. Mol. Med. 89, 229–236 [DOI] [PubMed] [Google Scholar]
- 60. DeBerardinis R. J., Thompson C. B. (2012) Cellular metabolism and disease. What do metabolic outliers teach us? Cell 148, 1132–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bernard D., Pourtier-Manzanedo A., Gil J., Beach D. H. (2003) Myc confers androgen-independent prostate cancer cell growth. J. Clin. Invest. 112, 1724–1731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ravi R. K., McMahon M., Yangang Z., Williams J. R., Dillehay L. E., Nelkin B. D., Mabry M. (1999) Raf-1-induced cell cycle arrest in LNCaP human prostate cancer cells. J. Cell. Biochem. 72, 458–469 [DOI] [PubMed] [Google Scholar]
- 63. Mabjeesh N. J., Willard M. T., Frederickson C. E., Zhong H., Simons J. W. (2003) Androgens stimulate hypoxia-inducible factor 1 activation via autocrine loop of tyrosine kinase receptor/phosphatidylinositol 3′-kinase/protein kinase B in prostate cancer cells. Clin. Cancer Res. 9, 2416–2425 [PubMed] [Google Scholar]
- 64. Mehraein-Ghomi F., Basu H. S., Church D. R., Hoffmann F. M., Wilding G. (2010) Androgen receptor requires JunD as a coactivator to switch on an oxidative stress generation pathway in prostate cancer cells. Cancer Res. 70, 4560–4568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Shiota M., Yokomizo A., Tada Y., Inokuchi J., Kashiwagi E., Masubuchi D., Eto M., Uchiumi T., Naito S. (2010) Castration resistance of prostate cancer cells caused by castration-induced oxidative stress through Twist1 and androgen receptor overexpression. Oncogene 29, 237–250 [DOI] [PubMed] [Google Scholar]
- 66. Biswas M., Chan J. Y. (2010) Role of Nrf1 in antioxidant response element-mediated gene expression and beyond. Toxicol. Appl. Pharmacol. 244, 16–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kaspar J. W., Niture S. K., Jaiswal A. K. (2009) Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic. Biol. Med. 47, 1304–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]