

Background: LIP5 helps regulate the membrane fission and recycling activities of the ESCRT pathway.
Results: Biochemical and structural studies reveal how LIP5 binds different ESCRT-III proteins.
Conclusion: ESCRT-III proteins bind independently to different LIP5 sites, and the LIP5-CHMP5 structure reveals a novel interaction mode.
Significance: The results help explain how LIP5 can activate VPS4 ATPases to act on ESCRT-III filaments.
Keywords: NMR, Protein Chemistry, Protein Complexes, Protein Structure, Protein-Protein Interactions, CHMP5, ESCRT-III, Endosomal Sorting Complexes Required for Transport, LIP5, MIT
Abstract
The endosomal sorting complex required for transport (ESCRT) pathway remodels membranes during multivesicular body biogenesis, the abscission stage of cytokinesis, and enveloped virus budding. The ESCRT-III and VPS4 ATPase complexes catalyze the membrane fission events associated with these processes, and the LIP5 protein helps regulate their interactions by binding directly to a subset of ESCRT-III proteins and to VPS4. We have investigated the biochemical and structural basis for different LIP5-ligand interactions and show that the first microtubule-interacting and trafficking (MIT) module of the tandem LIP5 MIT domain binds CHMP1B (and other ESCRT-III proteins) through canonical type 1 MIT-interacting motif (MIM1) interactions. In contrast, the second LIP5 MIT module binds with unusually high affinity to a novel MIM element within the ESCRT-III protein CHMP5. A solution structure of the relevant LIP5-CHMP5 complex reveals that CHMP5 helices 5 and 6 and adjacent linkers form an amphipathic “leucine collar” that wraps almost completely around the second LIP5 MIT module but makes only limited contacts with the first MIT module. LIP5 binds MIM1-containing ESCRT-III proteins and CHMP5 and VPS4 ligands independently in vitro, but these interactions are coupled within cells because formation of stable VPS4 complexes with both LIP5 and CHMP5 requires LIP5 to bind both a MIM1-containing ESCRT-III protein and CHMP5. Our studies thus reveal how the tandem MIT domain of LIP5 binds different types of ESCRT-III proteins, promoting assembly of active VPS4 enzymes on the polymeric ESCRT-III substrate.
Introduction
The endosomal sorting complex required for transport (ESCRT)3 pathway mediates membrane remodeling and fission events required for intraluminal vesicle formation at multivesicular bodies, the abscission stage of cytokinesis, and the budding of many enveloped viruses, including HIV-1 (reviewed in Refs. 1–9). ESCRT factors are recruited to their different sites of action by sequential protein-protein and protein-membrane interactions that are initiated by membrane-specific adaptors such as the HRS-STAM complex (multivesicular body vesicle formation), CEP55 (cytokinesis), and retroviral Gag proteins (virus budding). These adaptors then bind and recruit early-acting ESCRT factors such as ESCRT-I, ESCRT-II, and ALIX, which in turn recruit downstream ESCRT-III and VPS4 ATPase complexes.
The actual membrane fission events appear to be catalyzed by the concerted activities of the ESCRT-III and VPS4 complexes. Humans express 12 related ESCRT-III-like proteins, which can be subdivided into eight different subfamilies, termed CHMP1–7 (charged multivesicular body proteins 1–7) and IST1. The CHMP1, CHMP2, and CHMP4 subfamilies each have multiple related members, which carry letter designations (CHMP1A and CHMP1B, etc.). The different ESCRT-III subunits share a common architecture, comprising a conserved core domain that mediates membrane binding and filament formation and a C-terminal tail that can either fold back on the core to autoinhibit polymerization or open to expose protein-binding sites (reviewed in Ref. 10). The core domain is a four-helix bundle consisting of a long helical hairpin and two smaller helices that pack against the open end of the hairpin (10–13). The C-terminal tails can vary considerably in sequence and length but commonly contain sequence elements termed microtubule-interacting and trafficking (MIT)-interacting motifs (MIMs) that can bind the MIT domains found in VPS4 and many other ESCRT-III-associated proteins (reviewed in Ref. 14). Although the mechanistic details remain to be determined, current models generally posit that ESCRT-III proteins form membrane-bound filaments that recruit other cofactors and draw opposing membranes together to promote fission (15–17).
The energy for ESCRT-mediated vesicle formation is ultimately provided by the VPS4 ATPase, which is expressed as a single protein in yeast (Vps4p) and two related VPS4 proteins in humans and other mammals (VPS4A and VPS4B/SKD1) (reviewed in Ref. 18). VPS4 ATPases function late in the ESCRT pathway to release assembled ESCRT-III subunits from the membrane (19–22). VPS4 enzymes arrive immediately prior to the membrane fission step of virus budding and abscission (8, 23–26) and may play an active role in the fission process, possibly by remodeling or reorganizing the membrane-bound ESCRT-III filaments. Like other AAA (ATPases associated with diverse cellular activites) ATPases, VPS4 proteins can form hexameric rings, and the active enzyme appears to be a single-ring hexamer,4 although double-ring dodecamers and other higher order oligomeric assemblies can also form in vitro (19, 27–31). Each VPS4 subunit comprises an N-terminal MIT domain, an ATPase cassette that contains canonical large and small AAA-ATPase domains, and a β-domain that is inserted within the small ATPase domain. The MIT domain binds MIM elements within different ESCRT-III substrates, the AAA-ATPase cassette mediates nucleotide hydrolysis and ring formation, and the three-stranded sheet of the β-domain interacts with the activator protein LIP5 (yeast Vta1p) (18).
ESCRT assembly and activities are regulated at multiple different stages, but VPS4 recruitment, assembly, and activation appear to be a particularly important regulatory node. VPS4 enzymes are recruited to their sites of action, at least in part, by binding to a variety of different and often partially redundant MIM elements that are exposed upon ESCRT-III filament formation (19, 32–38). In addition to these general ESCRT-III interactions, two binary complexes, CHMP1-IST1 and LIP5-CHMP5, play particularly important roles in VPS4 recruitment and activation. CHMP1 (yeast Vps46p/Did2p) and IST1 (Ist1p) bind one another and help recruit VPS4 to sites of action (12, 13, 33, 39–43). LIP5/Vta1p stimulates VPS4 oligomerization and ATPase activity in vitro and in vivo (28, 34, 40, 43–49) and makes a particularly high affinity interaction with CHMP5 (yeast Vps60p) (44, 50). These two complexes appear to act synergistically because they exhibit synthetic multivesicular body sorting phenotypes in Saccharomyces cerevisiae (51, 52). Moreover, the tandem MIT domains within LIP5 can bind MIM elements within both CHMP1 and IST1, implying that the complexes probably interact directly in cells (12, 33, 34, 41, 42).
Comprehensive genetic and biochemical studies in yeast have provided a model for Vps4p cofactor recruitment in which the protein is initially present in the cytoplasm as a lower order multimer (either a monomer or dimer) that is recruited to core ESCRT-III polymers with the help of the Did2p-Ist1p complex. This assembly then recruits Vta1p, which helps to promote assembly of the active Vps4p enzyme (33). It is not yet clear precisely how Vps60p/CHMP5 fits into this scheme, but a leading model is that Vps60p acts through Vta1p very late in the process to help stimulate Vps4p to recycle ESCRT components back into the cytoplasm (34). Consistent with this idea, recombinant Vps60p can stimulate Vps4p ATPase activity in a Vta1p-dependent fashion. Vta1p is required for Vps60p endosomal localization, but the converse is not true, and indeed, Vps60p antagonizes Vta1p endosomal localization (34). In mammalian cells, however, CHMP5 and LIP5 form stable complexes in the cytoplasm, and the two proteins are therefore likely to be recruited together in this case (53). CHMP5 activities appear to be more important for some ESCRT-dependent processes than others because siRNA depletion of CHMP5 inhibits multivesicular body sorting, but does not inhibit HIV-1 budding (44).
A remarkably complex network of different MIM-MIT interactions plays a critical role in recruiting different effector proteins to the surface of polymerized ESCRT-III filaments. The 12 different human ESCRT-III subunits contain distinct classes of MIMs that can be displayed singly (as in CHMP5) (34, 50) or in tandem (as in IST1) (42) and can even bind differently to different MIT domains (as in CHMP1B) (36, 54). Moreover, humans express more than a dozen different MIT domain-containing proteins (14, 55–57), whose MIT domains can be arrayed either singly (as in VPS4 enzymes) or in tandem (as in LIP5) (58). Although MIT domains are simple three-helix bundles (59, 60), they can engage their MIM ligands in a variety of different ways. To date, four different classes of MIM elements have been characterized structurally: MIM1 motifs form short amphipathic helices that bind in the groove between MIT helices 2 and 3 (35, 36), MIM2 motifs are extended strands that bind in the groove between MIT helices 1 and 3 (37, 61), MIM3 motifs are longer amphipathic helices that bind in the same groove (54), and MIM4 motifs are related to MIM1 motifs but form a more extended helix and make more polar interactions (62). In all of these cases, the MIM ligand binds along a MIT groove in an orientation that is parallel to the first MIT helix. Despite these advances in our understanding of the molecular basis for specific MIM-MIT interactions, we do not yet have a complete inventory of all possible MIM-MIT interactions or a full understanding of the “code” that dictates how different MIMs select their specific MIT domain-binding partners.
LIP5 performs its central regulatory function within the ESCRT pathway by binding ESCRT-III proteins through its N-terminal tandem MIT domain and by binding VPS4 ATPases through its C-terminal VSL (Vta1/SBP1/LIP5) domain (see Fig. 1A) (45, 48). These interactions allow LIP5 to coordinate ESCRT-III and VPS4 activities both during normal cellular homeostasis and also apparently in response to interferon signaling during innate immune responses. In the latter case, Leis and co-workers (63, 64) have reported that covalent modification of CHMP5 and other ESCRT-III proteins with the ubiquitin-like protein ISG15 blocks the LIP5-CHMP5 and LIP5-VPS4 interactions and thereby inhibits enveloped viruses from using the ESCRT pathway to bud from cells. To define better how LIP5 can mediate its different regulatory functions, we have characterized how LIP5 interacts with CHMP5 and other ESCRT-III proteins and determined the interdependence of different LIP5-ligand interactions both in vitro and in cells.
FIGURE 1.

Domain organization of human LIP5 and structure of the LIP5(MIT)2 domain. A, domain organization and residue numbering for human LIP5. B, ribbon diagram of LIP5(MIT)2, with MIT1 in green, MIT2 in blue, and helix 4 in aqua. C, superposition of the MIT helices from LIP5(MIT)2 and Vta1p(MIT)2 (gray) (Protein Data Bank code 2RKK (58)).
EXPERIMENTAL PROCEDURES
Expression Constructs and Plasmids
Bacterial and mammalian expression constructs and antibodies are provided in supplemental Table 1.
Peptides Used in LIP5(MIT)2 Binding Studies
Peptides corresponding to CHMP1B(169–199), CHMP1B(181–199), CHMP2A(206–222), CHMP3(201–222), CHMP4B(187–202), CHMP6(166–181), IST1(321–339), and IST1(340–366) were synthesized and purified by reverse-phase C18 HPLC at the University of Utah Peptide Synthesis Core. Expected masses were confirmed by MALDI-TOF-MS: CHMP1B(169–199), calculated = 3285 Da and experimental = 3283 Da; CHMP1B(181–199), calculated = 2185 Da and experimental = 2184 Da; CHMP2A(206–222), calculated = 2000 Da and experimental = 1999 Da; CHMP3(201–222), calculated = 2566 Da and experimental = 2565 Da; CHMP4B(187–202), calculated = 1662 Da and experimental = 1661 Da; CHMP6(166–181), calculated = 1834 Da and experimental = 1834 Da; acetylated and amidated IST1(321–339), calculated = 2053 Da and experimental = 2053 Da; and IST1(340–366), calculated = 3017 Da and experimental = 3016 Da. Peptide concentrations were determined by amino acid analysis or absorbance at 280 nm.
LIP5 and CHMP5 Expression
LIP5 and CHMP5 proteins with N-terminal GST or His10 (hereafter referred to as His) affinity tags (supplemental Table 1) were expressed in BL21-CodonPlus(DE3)- RIPL Escherichia coli (Agilent). Cells were grown in ZYP-5052 autoinduction medium (65) at 37 °C for 5 h, transferred to 19 °C for 16–24 h, harvested by centrifugation at 4000 × g for 10 min, and stored frozen at −80 °C. Immobilized GST fusion proteins used in biosensor binding studies were captured from crude supernatants onto the biosensor chips using immobilized anti-GST antibodies, whereas all other GST and all His fusion proteins were purified to homogeneity as described below.
LIP5 and CHMP5 Purification
LIP5 Proteins
GST fusions of LIP5(1–173), LIP5(1–170), LIP5(1–168), LIP5(1–168)(W147D), and LIP5(1–166) and His fusions of LIP5 (full-length, residues 1–307), LIP5(1–183), LIP5(1–159), and LIP5(1–158) were purified as described below, with all procedures performed at 4 °C unless noted otherwise. In each case, affinity chromatography was used as the first purification step.
For GST fusion proteins, 10 g of cell paste was thawed, resuspended in 50 ml of GST lysis buffer (50 mm Tris-HCl (pH 8.0), 150 mm NaCl, 1 mg/ml lysozyme, 0.125% deoxycholate, 1 mm DTT, and protease inhibitors (aprotinin, leupeptin, pepstatin, and PMSF)), lysed by sonication, and clarified by centrifugation at 15,000 rpm (Beckman JA-20 rotor) for 45 min. Supernatants were filtered through 0.45-μm syringe filters (Corning) and loaded onto 5-ml glutathione-Sepharose columns (GE Healthcare Life Sciences). The matrices were washed with 20 column volumes of GST wash buffer (50 mm Tris-HCl (pH 8.0), 150 mm NaCl, and 1 mm DTT), 20 column volumes of high salt GST wash buffer (GST wash buffer plus 500 mm NaCl), and 20 column volumes of GST wash buffer. Bound proteins were eluted with 60 ml of GST wash buffer plus 20 mm reduced l-glutathione (Sigma). For His fusion proteins, cell pellets were thawed, resuspended, and lysed as described above but in His lysis buffer (50 mm Tris-HCl (pH 8.0), 150 mm NaCl, 1 mg/ml lysozyme, 0.125% deoxycholate, 25 mm imidazole, and protease inhibitors). Clarified lysates were filtered and loaded onto a 5-ml nickel-nitrilotriacetic acid-agarose affinity column (Qiagen). Matrices were washed with 20 column volumes of His buffer (50 mm Tris-HCl (pH 8.0), 150 mm NaCl, and 25 mm imidazole), 20 column volumes of high salt His buffer (His buffer plus 500 mm NaCl), and 20 column volumes of His buffer. Bound proteins were eluted with 60 ml of His buffer plus 750 mm imidazole.
Eluted GST and His fusion proteins were processed to remove the affinity tags. Fusion proteins were dialyzed against 2 liters of proteolysis buffer I (50 mm Tris-HCl (pH 8.0), 100 mm NaCl, and 5 mm EDTA) for 4 h, tobacco etch virus or PreScission protease was added at a 1:150 enzyme:substrate ratio, and the reactions were dialyzed for an additional 12–15 h against 2 liters of proteolysis buffer II (50 mm Tris-HCl (pH 8), 100 mm NaCl, 0.5 mm EDTA, and 1 mm DTT) at 4 °C (PreScission protease) or room temperature (tobacco etch virus protease). Proteolytic processing was generally quantitative and left either two or three non-native residues at the N terminus (Gly-His (referred to hereafter as GH), tobacco etch virus; or Gly-Pro-His (referred to hereafter as GPH), PreScission protease), which are not included in our numbering schemes.
Processed LIP5 proteins were purified by ion exchange chromatography (LIP5(1–183) using Q-Sepharose and all other LIP5 proteins using SP-Sepharose; GE Healthcare Life Sciences) with elution gradients from 50 to 500 mm NaCl in 50 mm Tris-HCl (pH 8) and 1 mm DTT. Fractions containing LIP5 proteins were concentrated and purified to homogeneity by gel filtration using Superdex 75 columns (GE Healthcare Life Sciences). The buffer used for gel filtration was either 25 mm Tris-HCl (pH 8), 150 mm NaCl, and 1 mm DTT (proteins used for binding studies) or 20 mm sodium phosphate (pH 6.3), 100 mm NaCl, 0.1 mm EDTA, and 0.5 mm DTT (proteins used for NMR spectroscopy). These procedures yielded ∼1 μmol/liter of culture. Expected protein masses were confirmed by electrospray ionization mass spectrometry: GPH-LIP5(1–307), calculated = 34,170 Da and experimental = 34,167 Da; GPH-LIP5(1–183), calculated = 21,121 Da and experimental = 21,119 Da; GH-LIP5(1–173), calculated = 19,980 Da and experimental = 19,978 Da; GH-LIP5(1–170), calculated = 19,622 Da and experimental = 19,621 Da; GH-LIP5(1–168), calculated = 19,379 Da and experimental = 19,378 Da; GH-LIP5(1–168)(W147D), calculated = 19,308 Da and experimental = 19,308 Da; GH-LIP5(1–166), calculated = 19,223 Da and experimental = 19,222 Da; GPH-LIP5(1–159), calculated = 18,640 Da and experimental = 18,638 Da; and GPH-LIP5(1–158), calculated = 18,582 Da and experimental = 18,581 Da.
CHMP5 Proteins
Cells expressing His-CHMP5(139–195) or His-CHMP5(139–191)(A191C) were thawed and resuspended (10 g of cell paste/50 ml of lysis buffer) in denaturing lysis buffer (50 mm Tris-HCl (pH 8), 500 mm NaCl, 8 m urea, and 20 mm imidazole) and lysed by sonication, and the lysate was clarified by centrifugation at 15,000 rpm (Beckman JA-20 rotor) for 45 min. Supernatants were filtered through 0.45-μm syringe filters and loaded onto a 5-ml equilibrated nickel-nitrilotriacetic acid-agarose affinity column. The matrix was washed with 20 column volumes of denaturing lysis buffer, followed by 20 column volumes of denaturing lysis buffer without urea. His fusion proteins were eluted with 60 ml of 50 mm Tris-HCl (pH 8), 500 mm NaCl, and 750 mm imidazole and processed using tobacco etch virus protease as described for LIP5. Peptides were concentrated, dried, resuspended in 2 ml of aqueous solution containing 5% acetic acid and 30% acetonitrile, and purified by reverse-phase C18 HPLC in aqueous solution containing 0.1% TFA using a linear gradient of 30–70% acetonitrile. Peak fractions were pooled and lyophilized. Masses were confirmed by MALDI-TOF-MS: GH-CHMP5(139–195), calculated = 6294 Da and experimental = 6294 Da; and GH-CHMP5(139–191)(A191C), calculated = 5930 Da and experimental = 5931 Da. This protocol yielded ∼0.25 μmol of peptide/liter of culture.
Fluorescent Labeling of CHMP5 for Binding Studies
CHMP5(139–191)(A191C) was fluorescently labeled in a 93-μl reaction that contained 0.8 mm CHMP5(139–191)(A191C) peptide, 4.0 mm Oregon Green® 488 maleimide (Invitrogen), and 1 m triethylamine in dimethylacetamide for 4 h at 20 °C. The reaction was quenched by the addition of acetic acid to 5%, and the conjugated peptide was purified by reverse-phase C18 HPLC. The labeled peptide was dissolved in water to a final concentration of 27 μm (ϵ496 = 70,000 cm−1 m−1 in 20 mm HEPES (pH 7.5) and 100 mm NaCl) and stored at −20 °C. The mass of the dye-labeled peptide was confirmed by MALDI-TOF-MS: dye-labeled GH-CHMP5(139–191)(A191C), calculated = 6394 Da and experimental = 6396 Da.
Biosensor Binding Assays
Biosensor binding experiments were performed as described previously (36). Briefly, GST-LIP5 and GST-CHMP5 proteins were expressed as described above. Cells from 10-ml cultures were resuspended in 4 ml of lysis buffer (50 mm Tris-HCl (pH 7.0), 300 mm NaCl, 5% (w/v) glycerol, 0.125% sodium deoxycholate, 5 mm 2-mercaptoethanol, 1 mg/ml lysozyme, and protease inhibitors) and sonicated, and the lysates were clarified by centrifugation for 30 min at 13,000 rpm in a microcentrifuge. GST-fused proteins were captured directly onto anti-GST antibody-derivatized CM5 sensor chips. Pure LIP5 proteins or CHMP1B, CHMP2A, CHMP3, CHMP4B, CHMP5, and CHMP6 or IST1 peptides were diluted to the designated concentrations in binding buffer (25 mm Tris-HCl (pH 7.2), 150 mm NaCl, 1 mm DTT, 0.1 mg/ml BSA, and 0.01% Tween 20) and injected in triplicate (50 μl/min, 25 °C), and binding data were collected. Binding constants were obtained by fitting the equilibrium responses to 1:1 binding models (see Fig. 6, B–E). Each binding isotherm was measured one to five times, and mean KD values ± S.D. are reported as described in the legend to Fig. 6.
FIGURE 6.

Biosensor analyses of LIP5(MIT)2 binding to CHMP5. A, summaries of LIP5 and CHMP5 domain structures, secondary structures, interaction sites (red), and estimated equilibrium dissociation constants (KD, pm). KD values derived from global fits of a single binding experiment are denoted, with the fitting errors shown in parentheses. All other KD values are averages of multiple independent measurements, and the reported errors are either the range (n = 2) or S.D. (n ≥ 3). B–E, representative sensorgrams and fits showing the interactions between wild-type LIP5(MIT)2 and full-length CHMP5 (residues 1–219) (B), LIP5(MIT)2 and CHMP5(139–195) (C), LIP5(MIT)2(W147D) and CHMP5(139–195) (D), and LIP5(MIT)2 and CHMP5(139–195)(L163D,L167D,L170D,L174D) (E). Duplicates of the binding experiments are shown in blue and black, global experimental fits are shown in orange, protein concentrations are shown for a subset of the curves, and fitted dissociation constants for these specific experiments are shown with estimated errors in parentheses. All proteins designated LIP5(MIT)2 correspond to the LIP5(1–168) construct.
Fluorescence Binding Assays
Fluorescence anisotropy binding measurements were performed in Corning 384-well black plates on a Tecan Infinite 200 plate reader with excitation at 485 nm and detection at 535 nm. 60-μl binding reactions (30 min, 25 °C) contained 2-fold dilutions of LIP5 proteins and 250 pm dye-labeled CHMP5(139–191)(A191C) peptide in binding buffer. KD values were obtained by fitting the increase in fluorescence anisotropy (ΔFA) with the 1:1 binding equation ΔFA = (0.5/[CHMP5]ΔFAmax)([CHMP5] + [LIP5] + KD)(([CHMP5] + [LIP5] + KD)2 − 4[CHMP5][LIP5])1/2 using KaleidaGraph (Synergy Software). Each binding isotherm was measured at least four times, and mean KD values ± S.D. are reported.
Analytical Ultracentrifugation
Analytical ultracentrifugation data were collected at 4 °C using a Beckman Optima XL-A centrifuge at a rotor speed of 11,000 rpm for 5.6, 11.2, and 22.4 μm LIP5 in 25 mm Tris-HCl (pH 8.0), 150 mm NaCl, and 1 mm tris(2-carboxyethyl)phosphine. Data were globally fit to ideal single-species models with fixed or floating molecular masses (data not shown) using the nonlinear least squares algorithm in the HETEROANALYSIS software. Protein partial specific volumes and solvent densities were calculated with the program SEDNTERP (version 1.09).
NMR Sample Preparation
Isotope-labeled GST-LIP5(1–168), His-LIP5(1–183), and His-CHMP5(139–195) were expressed in minimal autoinduction medium (15N-labeled proteins) (65) or M9 minimal medium (15N/13C-labeled proteins) and purified as described above. NMR samples were prepared in 25 mm sodium phosphate (pH 6.3), 50 mm NaCl, 0.1 mm EDTA, 0.5 mm DTT, 40 μm sodium azide, 90% H2O, and 10% D2O. The following samples were prepared: 1) 0.5 mm 15N/13C-labeled LIP5(1–183) (resonance assignments and structure determination for unbound LIP5(MIT)2), 2) 0.5 mm 15N/13C-labeled LIP5(1–168) + 0.5 mm unlabeled CHMP5(139–195) (resonance assignments for bound LIP5(MIT)2 and collection of a half-filtered three-dimensional NOESY) (see Fig. 8), 3) 0.5 mm unlabeled LIP5(1–168) + 0.5 mm 15N/13C-labeled CHMP5(139–195) (resonance assignments for bound CHMP5(139–195)), 4) 0.8 mm 15N/13C-labeled LIP5(1–168) + 0.8 mm 15N/13C-labeled CHMP5(139–195) (NOE distance constraints for complex structure determination), 5) 0.5 mm 15N-labeled LIP5(1–168) + 0.5 mm unlabeled CHMP5(139–195) (JNH for bound LIP5(MIT)2), 6) 0.5 mm 15N-labeled LIP5(1–168) + 0.5 mm unlabeled CHMP5(139–195) + 3% (w/v) C12E5/n-hexanol (JNH + DNH for bound LIP5(MIT)2) (66), 7) 0.5 mm unlabeled LIP5(1-168) + 0.5 mm 15N-labeled CHMP5(139–195) (JNH for bound CHMP5(139–195)), 8) 0.5 mm unlabeled LIP5(1–168) + 0.5 mm 15N-labeled CHMP5(139–195) + 3% (w/v) C12E5/n-hexanol (JNH + DNH for bound CHMP5(139–195)), 9) 0.150 mm 15N-labeled LIP5(1–168) + 0.150 mm unlabeled CHMP5(139–195), and 10) 0.150 mm 15N-labeled LIP5(1-183) + 0.150 mm unlabeled CHMP5(139–195) (chemical shift comparisons for LIP(MIT)2 in the two complexes; data not shown).
FIGURE 8.

NMR data and structure calculations for LIP5(MIT)2-CHMP5(139–195). A, two orthogonal views of the LIP5(MIT)2-CHMP5(139–195) complex shown with overlays of the 10 lowest energy structures. The left view shows that CHMP5 residues 155–189 (red) are well ordered, and the right view shows that LIP5 residues 1–163 are well ordered. Disordered residues at the protein termini (gray) were omitted from the structures shown in Fig. 7 and from the statistics reported in Table 1. We concluded that CHMP5 helix 4 is not involved in LIP5(MIT)2 binding because 1) there were no intermolecular NOEs between CHMP5 helix 4 and LIP5(MIT)2; 2) residual dipolar coupling values for CHMP5 helix 4 were small, indicating that this helix might not have a unique orientation (unlike helices 5 and 6); 3) resonances within this helix were sharper and relaxed more slowly than those in the rest of the protein; and 4) deleting this helix did not affect LIP5 binding affinity (Fig. 6A, compare the equivalent LIP5(MIT)2 binding affinities of the CHMP5(139–195) and CHMP5(150–195) constructs). B, backbone chemical shift assignments showing Cα and Cβ connectivities for LIP5(122–131), a portion of helix H6 at the MIT1-MIT2 interface. Amide strips from three-dimensional CBCA(CO)NH (A) and HNCACB (B) spectra are shown, with blue and red horizontal lines showing sequential connections of Cα and Cβ resonances, respectively. C, representative three-dimensional 13C-edited NOESY (black) used in determining the LIP5-CHMP5 structure and three-dimensional 13C-half-filtered NOESY (red) used to confirm intermolecular NOEs. Intermolecular NOEs are labeled for LIP5 Met-97 QE and CHMP5 Leu-183 QD2 (a), Leu-183 QD1 (b), Ala-186 QB (c), Ala-187 QB (d), Leu-183 HA (e), Leu-183 HN (f), and Ala-187 HN (g); LIP5 Trp-147 HD1 and CHMP5 Leu-158 QD1 (h), Leu-158 QD2 (i), and Leu-163 QD1/2 (j); and LIP5 Thr-150 QG2 and CHMP5 Leu-170 QD1 (k), Leu-170 QD2 and Leu-167 QD2 (l), Leu-167 QD1 (m), Leu-167 HG (n), Leu-167 HB2 (o), Glu-166 HB2 (p), Glu-166 HB3 (q), Leu-167 HA (r), Glu-166 HA (s), and Leu-167 HN (t). Asterisks denote incomplete filtering of the Trp-147 HD1 diagonal signal.
NMR Data Collection and Resonance Assignment
NMR data for resonance assignments, residual dipolar couplings, and titrations were recorded on a Varian INOVA 600-MHz spectrometer, and data for NOE constraints (three-dimensional NOESY) were recorded on a DirectDrive 900-MHz spectrometer, both equipped with cryogenic probes. Data were processed with FELIX 2007 (Felix NMR, Inc.) using in-house processing macros. Chemical shift assignments were obtained from analysis of standard triple-resonance three-dimensional NMR experiments (67) using SPARKY 3 (79) and AutoAssign (68). Assignments were obtained for 93% (LIP5(1–183)) and 92% (LIP5(1–168)-CHMP5(139–195) complex) of the available 1H and 13C/15N resonances, and complete assignments were obtained for the main chain atoms (HN, N, Cα, and Cβ).
Structure Calculations
NMR structures were determined from NMR-derived distance constraints, φ and ψ torsion angle constraints, and residual dipolar couplings. Interproton NOE intensities and coordinates were obtained from three-dimensional 1H-(13C,15N)-1H NOESY (60-ms mixing time) using SPARKY 3. A half-filtered three-dimensional 1H-(13C,15N)-1H NOESY (60-ms mixing time) was recorded on sample 3 and used to confirm intermolecular NOEs at the complex interface (see Fig. 8). Main chain phi and psi torsion angle ranges were estimated from 13Cα chemical shifts using TALOS+ (69, 70). Chemical shift analyses indicated that all prolines were in trans-conformations. Amide NH residual dipolar couplings (1DNH) were obtained from two-dimensional 1H-15N HSQC, TROSY-HSQC, and in-phase anti-phase (IPAP) HSQC spectra in isotropic (samples 5 and 7) and aligned (samples 6 and 8) solutions. Dipolar couplings for residues in regular α-helical regions were included in calculations. NOEs were assigned using the automated NOE assignment module implemented in CYANA 3.0 (71, 72). Table 1 summarizes the NMR-derived constraints and structure statistics. Structures were initially calculated using CYANA and refined using XPLOR-NIH 2.29. Distance, φ/ψ torsion angle, and dipolar coupling constraint files were converted to XPLOR format using the software PDBSTAT (73). Stereochemical assignments from CYANA were retained, and the upper bounds of NOE distance constraints were increased by 10%. An improved simulated annealing protocol in XPLOR was implemented, with updated features (74), including a backbone hydrogen bond potential term (HBDB) and a torsion angle potential term (RAMA). Models were visualized and figures were prepared in PyMOL (DeLano Scientific). NMR constraints were deposited in the Biological Magnetic Resonance Bank (LIP5(MIT)2:18681 and LIP5-CHMP5:18682).
TABLE 1.
NMR structure statistics
Statistics are for the well ordered regions LIP5(1–163) and LIP(1–163)-CHMP5(155–189), deposited in the Protein Data Bank. r.m.s.d., root mean square deviation.
| LIP5(1–183) | LIP5(1–168) + CHMP5(139–195)a | |
|---|---|---|
| Distance constraints (total)a | 3107 | 3286 |
| Intraresidue (i = j) | 692 | 798 |
| Sequential (|i − j| = 1) | 851 | 894 |
| Medium-range (|i − j| = 2–4) | 740 | 782 |
| Long-range (|i − j| ≥ 5) | 824 | 668 |
| Intermolecular | 1443 | |
| Stereospecific assignmentsa | 44 | 60 |
| Dihedral φ/ϕ angle constraintsa | 318 | 359 |
| Dipolar coupling constraintsa | ||
| DNH for LIP5 | 82 | |
| DNH for CHMP5 | 34 | |
| Backbone hydrogen bondsa,b | 110 | 132 |
| Structure statisticsa | ||
| Violations (mean, S.D.) | ||
| Distance constraints (Å) | 0.024 (0.002) | 0.030 (0.002) |
| Dihedral angle constraints | 0.454° (0.131°) | 0.353° (0.011°) |
| DNH dipolar coupling constraints (Hz) | 0.582 (0.032) | |
| Maximum distance constraint violation (Å) | 0.50 | 0.52 |
| Maximum dihedral angle violation | 5.0° | 5.5° |
| Maximum dipolar coupling violation (Hz) | 1.2 | |
| Deviation from ideal geometry (r.m.s.d.)a | ||
| Bond lengths (Å) | 0.004 (0.000) | 0.003 (0.000) |
| Bond angles | 0.472° (0.016°) | 0.459° (0.025°) |
| Impropers | 0.325° (0.025°) | 0.414° (0.046°) |
| Mean r.m.s.d. (Å) | ||
| Aligned residuesc | LIP5 (3–49,52–162) | LIP5 (3–49,54–74,78–131,135–163) + CHMP5 (155–176,181–188) |
| Backbone atoms (Cα, C, N, O)d | 0.55 ± 0.13 | 0.70 ± 0.10 |
| Ramachandran (φ/ψ) (%)a | ||
| Favored | 96 | 94 |
| Additionally allowed | 4 | 5 |
| Generously allowed | 0 | 1 |
a Statistics are for the well-ordered regions, LIP51–163 and LIP1–163-CHMP5155–189, deposited at the PDB.
b Hydrogen bonds were identified and their distance/angle properties optimized using the Hydrogen Bond Database (HBDB) potential in XPLOR-NIH.
c Residues selected for alignment have φ/ψ order parameter values >0.9. This parameter was measured using PDBSTAT.
d Mean r.m.s.d. values and S.D. were determined relative to the average reference structure.
VPS4B(SAB)-LIP5 Binding Titrations
15N-Labeled His-VPS4B(SAB) (small ATPase and beta; residues 300–423) was expressed in M9 medium and affinity-purified, and the His affinity tag was removed using PreScission protease as described above for His-LIP5 proteins. The processed protein was dialyzed against 20 mm sodium phosphate (pH 6.3), 100 mm NaCl, 0.5 mm DTT, and 0.1 mm EDTA and further purified in the same buffer by gel filtration on a Superdex 75 column. Unlabeled His-LIP5(VSL) (residues 260–307) was expressed in autoinduction medium and purified as described for VPS4B(SAB). Expected masses were confirmed by electrospray ionization mass spectrometry: 15N (99%), GPH-VPS4B(SAB), calculated = 14,075 Da and experimental = 14,073 Da; and GPH-LIP5(VSL), calculated = 5770 Da and experimental = 5770 Da. Typical yields were 0.5 and 1.0 μmol/liter of culture for 15N-labeled VPS4B(SAB) and LIP5(VSL), respectively. Purified 15N-labeled VPS4B(SAB), LIP5(VSL), and LIP5 proteins were buffer-exchanged, using a Vivaspin 20 centrifugal concentrator (5-kDa molecular mass cutoff; Sartorius), into NMR buffer (20 mm sodium phosphate (pH 6.3), 100 mm NaCl, 0.5 mm DTT, 0.1 mm EDTA, and 8% D2O) through multiple rounds of concentration and dilution. Separate NMR samples were prepared for each titration point and contained 75 μm 15N-labeled VPS4B(SAB) and the indicated amounts of either LIP5(VSL) or LIP5 (see Fig. 9, B–D). 1H-15N HSQC and TROSY-HSQC spectra were recorded for 15N-labeled VPS4B(SAB)-LIP5(VSL) and 15N-labeled VPS4B(SAB)-LIP5, respectively, at 28 °C on a Varian INOVA 600 spectrometer. NMR spectra were of poor quality for LIP5 concentrations above 550 μm, limiting saturation of VPS4B(SAB) to ∼70% (see dashed lines in Fig. 9B) and resulting in larger errors for the fitted KD values.
FIGURE 9.

Equilibrium dissociation constants for the LIP5(MIT)2-CHMP1B and LIP5(VSL)-VPS4B complexes are not affected by LIP5 sequence elements outside the minimal binding domains. A, biosensor binding isotherms for CHMP1B(181–199) binding to full-length and C-terminally truncated LIP5 constructs. Data points for each curve were measured in triplicate, and the reported errors in the KD measurements are the estimated errors associated with fitting to simple 1:1 binding models. B, binding isotherms derived from NMR chemical shift perturbations of three different 15N-labeled VPS4B(SAB) backbone amide residues induced by binding of LIP5 (closed symbols, dashed lines) or LIP5(VSL) (open symbols, solid lines). KD values and S.D. reported for LIP5 and LIP5(VSL) binding were from averages of the KD values derived from the individual fits of the 15N-labeled VPS4B(SAB) A, B, and C amide isotherms (see C and D). C, overlaid 1H-15N HSQC spectra of 75 μm 15N-labeled VPS4B(SAB) alone (black) or in the presence of 90 μm LIP5(VSL) (red). Resonances used to derive the binding isotherms shown in B are labeled. D, overlaid 1H-15N TROSY-HSQC spectra of 75 μm 15N-labeled VPS4B(SAB) alone (black) or in the presence of 90 μm full-length LIP5(1–307) (red). Shifted resonances are labeled as described for C. Note that the chemical shifts induced by binding of full-length LIP5 and LIP5(VSL) alone are very similar. Our estimated dissociation constant for the human LIP5(VSL)-VPS4B(SAB) interaction (∼200 μm) is considerably weaker than a previous estimate of the KD for full-length LIP5 binding to immobilized VPS4B/SKD1 (53 nm), but that measurement was made under conditions in which avidity effects could have contributed to the observed KD because the dimeric LIP5 protein was binding immobilized VPS4B/SKD1 (46). Our dissociation constant is ∼3-fold weaker than that reported previously for the yeast Vta1p(VSL)-Vps4p(SAB) interaction (48), and this agreement seems reasonable given that the proteins are from different species and that the previous assay was also configured so that avidity effects could have contributed to the measured binding affinity.
Amide chemical shifts were analyzed using NMRViewJ (One Moon Scientific, Inc.), and shift changes were calculated using ΔHN = (ΔH2 + (ΔN/6.5)2)1/2. Equilibrium dissociation constants (KD) were obtained by fitting to the 1:1 binding equation ΔHN = (0.5/[VPS4B(SAB)]ΔHNmax)([VPS4B(SAB)] + [LIP5] + KD)(([VPS4B(SAB)] + [LIP5] + KD)2 − 4[VPS4B][LIP5])1/2 using KaleidaGraph.
Co-immunoprecipitations
HEK293T cells were seeded at 6 × 105 cells/well in a 6-well plate, transfected with the designated expression vectors (1.5 μg of pEGFP-VPS4A and pcDNA3.1-VPS4A; 3 μg of pCAG-Myc-LIP5, pCAG-Myc-LIP5(Y278A), pCAG-Myc-LIP5(M64A), pCAG-Myc-LIP5(W147D), pCAG-CHMP5-Myc, and pCAG-CHMP5-Myc(L4D)) using the manufacturer's calcium phosphate transfection protocol (Clontech CalPhos mammalian transfection kit), and incubated for 48 h at 37 °C and 5% CO2. Cells were lysed with 300 μl of Nonidet P-40 buffer (0.5% Nonidet P-40, 50 mm Tris-HCl (pH 8.8), 5% glycerol, and 150 mm NaCl) supplemented with protease inhibitor mixture (Sigma) for 30 min on ice. Cell lysates were clarified by centrifugation at 16,000 × g, incubated with anti-VPS4A (1:250 dilution; our rabbit polyclonal antibody designated UT829) or anti-Myc (1:1000 dilution; Covance) antibody for 30 min on ice, incubated with protein A-agarose beads (8-ml slurry; Millipore) for 2 h on ice, and washed three times with Nonidet P-40 buffer. Bound proteins were detected by Western blotting.
siRNA Depletion of CHMP5
HEK293T cells were seeded at 2.0 × 105 cells/well in a 6-well plate and incubated for 24 h at 37 °C and 5% CO2. siRNAs specific for CHMP5 (sense, 5′-AGAAUAUGGUCAAGCAGAAtt, where “t” represents non-complementary overhanging deoxynucleotides) or luciferase (sense, 5′-CGUACGCGGAAUACUUCGAtt) were transfected using Lipofectamine RNAiMAX (Invitrogen) at a final concentration of 40 nm for each oligoribonucleotide according to the manufacturer's instructions. Transfected cells were incubated for 24 h, and fresh medium was added. Cells were cotransfected a second time with the same siRNAs (40 nm each), pEGFP-VPS4A (1.5 μg), and pCAG-Myc-LIP5 (3 μg) using Lipofectamine 2000 (Invitrogen) and incubated for 48 h. Cell lysates were co-immunoprecipitated as described above.
RESULTS
LIP5 Residues 1–163 Comprise a Tandem MIT Domain
We used NMR spectroscopy to determine the solution structure of the N-terminal half of human LIP5 (residues 1–183) (Fig. 1 and Table 1). The structure revealed that residues 1–163 comprise a globular domain that contains two MIT modules, as well as an ordered strand at the N terminus and a type I β-turn at the C terminus (Fig. 1B). This entire domain is hereafter generically referred to as LIP5(MIT)2, except in cases in which it is important to denote the precise position of the C terminus (see below). LIP5 residues 164–170 form a poorly ordered mobile strand that extends beyond the final turn, and residues beyond 170 lack persistent structure.
Each MIT module within LIP5(MIT)2 is a three-helix bundle, and the bundles are connected by a nine-residue linker that includes a short helix (H4, residues 77–80). The two MIT modules are oriented at approximately right angles and interact through an extensive interdomain interface formed by the first two helices of each module. As expected, LIP5(MIT)2 resembles the analogous tandem MIT domain of the S. cerevisiae homolog Vta1p, and the six equivalent MIT helices overlay with a backbone atom (N, Cα, and C) root mean square deviation of 2.2 Å (Fig. 1C) (58). However, Vta1p(MIT)2 lacks the LIP5(MIT)2 N-terminal strand (residues 1–13) and the C-terminal turn (residues 156–159). The N-terminal strand of LIP5(MIT)2 folds back and packs into the groove between MIT1 helices 1 and 3, with key interactions made by Leu-4, Leu-7, Pro-8, Leu-10, and Pro-11. These interactions are reminiscent of an intermolecular crystal packing interaction seen in the crystal structure of the MIT domain from yeast Vps4p binding nonspecifically to a proline-rich sequence within Vps2p (35). Sequence alignments indicate that the N-terminal strand of human LIP5(MIT)2 is well conserved in mammalian LIP5 proteins but can vary in sequence and length in other vertebrate and metazoan species (Fig. 2A).
FIGURE 2.

Alignments of LIP5(MIT)2 domains and CHMP5 leucine collar motifs from different metazoan species. A, sequence alignment of the LIP5(MIT)2 domains from nine different metazoan species. Positions of secondary structure elements for human LIP5(MIT)2 are shown above, and the conserved Trp-147 residue within the CHMP5-binding site is highlighted in red. Aligned sequences are Homo sapiens (Q9NP79), Pongo abelii (Q5R5W5), Bos taurus (Q32L63), Mus musculus (Q9CR26), Gallus gallus (E1BRP3), Xenopus. laevis (Q6IP85), Danio rerio (Q5RJ50), Drosophila melanogaster (Q9W0B3), and Caenorhabditis elegans (O45812). UniProt accession numbers are shown in parentheses. The alignment was performed using only the LIP5(MIT)2 domains. B, sequence alignment of the LIP5(MIT)2-binding regions from CHMP5 proteins of nine different metazoan species. The positions of helices H4–H6 in human CHMP5 are shown above, but with H4 shown in an open box because this helix does not contact LIP5. The six conserved leucine residues of the leucine collar motif are highlighted in red. Aligned sequences are H. sapiens (Q9NZZ3), P. abelii (Q5RBR3), B. taurus (F1MZV2), M. musculus (Q9D7S9), G. gallus (E1BSI4), X. laevis (Q6DD52), D. rerio (E9QG19), D. melanogaster (Q9VVI9), and C. elegans (O16458). The alignment was performed using intact CHMP5 proteins, and the LIP5(MIT)2-binding regions were extracted from that alignment.
MIM1 Elements from CHMP1B and Other ESCRT-III Proteins Bind LIP5 MIT1
Mutational, spectroscopic, and binding analyses were performed to determine how different ESCRT-III proteins can bind LIP5(MIT)2. Biosensor analyses were initially used to survey the binding of immobilized GST-LIP5(MIT)2 to constructs that spanned MIMs from six different ESCRT-III proteins (Fig. 3A and Table 2). The tightest binder was a construct that spanned the C-terminal region of CHMP1B (positions 169–199; KD = 2.9 ± 0.4 μm) (Fig. 3A, closed squares). This longer CHMP1B construct was initially tested because the C terminus of CHMP1B can bind different MIT domains either by forming a MIM1 motif, in which the C-terminal half of the final CHMP1B helix (residues ∼180–196) binds in the helix 2/3 groove (36), or by forming a MIM3 motif, in which a more extended helix (residues ∼174–196) binds in the helix 1/3 groove (54). As shown in Fig. 3B, a minimal CHMP1B construct that spanned just the MIM1 helix (CHMP1B(181–199)) bound LIP5(MIT)2 with full affinity (KD = 3.3 ± 0.1 μm) (Fig. 3B, compare open and closed diamonds). This dissociation constant is 10-fold tighter than the MIM1-type interaction made by CHMP1B with the VPS4A MIT domain (36) and ∼4-fold tighter than the MIM3-type interaction made with the spastin MIT domain (54). Hence, the C-terminal CHMP1B MIM1 helix is an unusually good binding partner for LIP5(MIT)2.
FIGURE 3.

ESCRT-III MIM1 elements bind in the helix 2/3 groove of LIP5 MIT1. A, survey of the interactions between LIP5(MIT)2 and human ESCRT-III MIM elements. Shown are biosensor-derived isotherms for peptides from the designated ESCRT-III proteins interacting with immobilized GST-LIP5(MIT)2 (see inset for key). Note that MIM1 elements from CHMP1B, CHMP2A, IST1, and CHMP3 all bind, whereas MIM2 elements from CHMP4B, CHMP6, and IST1 do not. B, CHMP1B MIM1 binds the helix 2/3 groove of LIP5 MIT1. Shown are binding isotherms for CHMP1B(181–199) (closed diamonds) or CHMP1B(169–199) (all other curves) binding to immobilized wild-type or mutant LIP5(MIT)2 proteins (see inset for key). WT + C5139–195 denotes CHMP1B(169–199) binding to wild-type LIP5(MIT)2 in the presence of saturating concentrations of CHMP5(139–195). Note that the LIP5(MIT)2 mutation M64A (MIT1 helix 2/3 groove) reduced CHMP1B(181–199) binding significantly, whereas mutations in each of the other available grooves did not. These data imply that the CHMP1B MIM1 element binds in the helix 2/3 groove of the first LIP5 MIT module of LIP5(MIT)2 and that CHMP1B MIM1 binding is not affected by CHMP5(139–195) binding to MIT2.
TABLE 2.
ESCRT-III MIM binding to wild-type and mutant LIP5(MIT)2
| LIP5(MIT)2 | LIP5(MIT)2 (L4D,L7D) (MIT1-H13) | LIP5(MIT)2 (M64A) (MIT1-H23) | LIP5(MIT)2 (L95A) (MIT2-H13) | LIP5(MIT)2 (Y119A) (MIT2-H23) | |
|---|---|---|---|---|---|
| μm | μm | μm | μm | μm | |
| CHMP1B(169–199) (MIM1) | 2.9 ± 0.4a (n = 5)b | 5.8 (0.1) (n = 1) | 160 ± 52 (n = 2) | 3.0 ± 0.9 (n = 2) | 3.0 ± 0.7 (n = 2) |
| CHMP1B(169–199) (MIM1) + CHMP5(139–195) | 2.5 ± 0.3 (n = 2) | 5.8 (0.1) (n = 1) | 150 (3) (n = 1) | 2.2 (0.1) (n = 1) | 2.8 (0.1) (n = 1) |
| CHMP1B(181–199) (MIM1) | 3.3 (0.1) (n = 1) | NDc | 240 (10) (n = 1) | 3.1 (0.1) (n = 1) | ND |
| CHMP2A(206–222) (MIM1) | 32 (1) (n = 1) | 67 (1) (n = 1) | 900 (10) (n = 1) | 29 (1) (n = 1) | 32 (1) (n = 1) |
| CHMP3(201–222) (MIM1) | 330 (10) (n = 1) | 520 (10) (n = 1) | >400 (n = 1) | 350 (10) (n = 1) | 280 (10) (n = 1) |
| CHMP4B(187–202) (MIM2) | >800 (n = 1) | >800 (n = 1) | >800 (n = 1) | >800 (n = 1) | >800 (n = 1) |
| CHMP6(166–181) (MIM2) | >400 (n = 1) | >400 (n = 1) | >400 (n = 1) | >400 (n = 1) | >400 (n = 1) |
| IST1(340–366) (MIM1) | 24 ± 6 (n = 3) | 40 ± 14 (n = 2) | 220 (60) (n = 1) | 24 ± 3 (n = 3) | 22 ± 5 (n = 2) |
| IST1(321–339) (MIM2) | >400 (n = 3) | >400 (n = 3) | >400 (n = 1) | >400 (n = 2) | >400 (n = 2) |
a Dissociation constants are reported in units of μm.
b Fitting errors from single binding isotherms are shown in parentheses; ± errors are either differences between two measurements (n = 2) or S.D. (n ≥ 3). ND, measurement not done. H13 and H23 denote the mutation places on the MIT domain, a H13 mutation is located in the groove between helices 1 and 3, and a H23 mutation is in the groove between helices 2 and 3.
c ND, measurement not done.
Terminal MIM1 elements from CHMP2A(206–222) and IST(340–366) bound with intermediate affinities (KD = 32 ± 1 and 24 ± 6 μm, respectively) (Fig. 3A and Table 2), and the MIM1 element from CHMP3(201–222) bound weakly (KD = 300 ± 10 μm). Binding was not detectable for any of the MIM2 constructs tested: CHMP4B(187–202), CHMP6(166–181), and IST1(321–339) (Fig. 3A and Table 2). Thus, LIP5(MIT)2 can bind MIM1 elements from multiple ESCRT-III proteins but apparently cannot bind MIM2 elements. Our data generally agree well with a previous study (50), except that the equilibrium dissociation constant we measured for the CHMP1B constructs (∼3 μm) was higher than that in the previous report (EC50 = 25 nm). In that case, however, the tighter binding measurement may have been enhanced by avidity effects because the experiment was configured so that dimeric LIP5 was binding to immobilized CHMP1B.
Mutational and spectroscopic analyses were performed to map the CHMP1B MIM1-binding site on LIP5(MIT)2. As noted above, MIMs can bind MIT domains in the grooves between helices 2 and 3 (the binding site for MIM1 and MIM4 motifs) or between helices 1 and 3 (the binding site for MIM2 and MIM3 motifs). LIP5(MIT)2 therefore presents four candidate MIM-binding grooves (although binding in the helix 1/3 groove of the first LIP5 MIT module would require displacement of the N-terminal strand). The CHMP1B MIM1-binding site was mapped by testing binding to four different LIP5(MIT)2 constructs, each of which carried point mutations in a different MIT binding groove. As shown in Fig. 3B, CHMP1B(169–199) bound with similar affinities to wild-type LIP5(MIT)2 and three mutant constructs. However, the LIP5(MIT)2 mutation M64A within the helix 2/3 groove of MIT1 weakened CHMP1B(169–199) binding by >50-fold. This mutation had a similar inhibitory effect on the binding of CHMP1B(181–199) (Table 2). Thus, the CHMP1B MIM1 motif appears to bind in the helix 2/3 groove of LIP5 MIT1. The LIP5(MIT)2 groove mutant M64A also uniquely reduced binding of all of the other ESCRT-III MIM1 elements tested, indicating that all of these MIM1 elements also bind in the helix 2/3 groove of LIP5 MIT1 (Table 2).
In a complementary mapping approach, we used 1H-15N HSQC-NMR spectroscopy to identify which 15N-labeled LIP5(MIT)2 backbone amide resonances were perturbed by CHMP1B(181–199) binding (Fig. 4, A–C). LIP5(MIT)2 backbone amide resonances that shifted upon addition of CHMP1B(181–199) (Fig. 4D, red) mapped almost exclusively to the surface presented by helices 2 and 3 of the first LIP5(MIT)2 MIT domain (green). Thus, the two mapping approaches agreed well and are consistent with a structure-based model for MIM1-like binding of CHMP1B(181–199) (Fig. 4D, left, gray helix). We therefore conclude that CHMP1B binds the helix 2/3 surface of the first MIT module of LIP5, likely via a MIM1-MIT interaction.
FIGURE 4.

Chemical shift mapping of the CHMP1B-binding site on LIP5(MIT)2. A and B show two different regions of the overlaid 1H-15N HSQC spectra of 15N-labeled LIP5(MIT)2 either free (red) or in complex with CHMP1B(181–199) (blue; 1:2 ratio of LIP5(MIT)2 to CHMP1B(181–199), >99.8% bound). Amide resonances are labeled with their amino acid assignments. The magnitude of chemical shift changes upon CHMP1B(181–199) binding was calculated using the expression ΔHN = (ΔH2 + (ΔN/6.5)2)1/2, and residues with ΔHN > 0.05 ppm are indicated with arrows. The average ΔHN for resonances not shifted by peptide binding was ∼0.01 ppm. C, ΔHN chemical shift changes induced by CHMP1B(181–199) binding for all backbone amides within LIP5(MIT)2. D, positions of LIP5(MIT)2 backbone amide chemical shifts induced by CHMP1B(181–199) binding. Both sides of LIP5(MIT)2 are shown (space-filling models), with shifted residues shown in red (ΔHN ≥ 0.05 ppm) and unshifted residues in MIT1 and MIT2 shown in green and blue, respectively. The position of a (hypothetical) bound MIM1 helix was modeled by overlaying the LIP5 MIT1 module onto the structure of the VPS4A MIT-CHMP1A(180–191) complex (Protein Data Bank code 2JQ9) (36). The chemical shift mapping data indicate that CHMP1B(181–199) binds on the helix 2/3 face of the first LIP5 MIT module, in good agreement with the mutational analysis shown in Fig. 3B. Very similar mapping data were also obtained for the CHMP2A(206–222) construct (data not shown).
Novel CHMP5 Element Binds the Second LIP5 MIT Module
A previous study showed that LIP5 binds CHMP5 with unusually high affinity (50), suggesting that the interaction with this ESCRT-III family member may be unique. The minimal LIP5-binding site was mapped to CHMP5 residues 149–173 (50), and we therefore initially mapped and quantified LIP5 binding using a CHMP5 fragment that spanned this region (CHMP5(139–195)). These studies employed a fluorescence polarization assay in which CHMP5(139–191) was covalently linked to an Oregon Green® 488 dye, and LIP5 binding was quantified by monitoring increases in fluorescence polarization. Normalized binding isotherms for the full-length LIP5 protein (residues 1–307) and a series of C-terminal truncation mutants are shown in Fig. 5. In excellent agreement with a previous report (50), full-length LIP5 bound CHMP5(139–195) with a dissociation constant of 2.0 ± 0.2 nm, whereas a control LIP5(260–307) construct corresponding to the C-terminal VSL domain alone bound only very weakly, if at all (KD > 10 μm). Analytical ultracentrifugation analyses confirmed that the LIP5 protein formed a tight dimer, as expected based upon structural studies of the dimeric C-terminal VSL domain (48, 58) (data not shown). However, monomeric LIP5(MIT)2-containing constructs that lacked the VSL domain also bound CHMP5(139–195) with similar affinities, implying that neither dimerization nor the VSL domain contributed to CHMP5 binding (e.g. LIP5(1–183) KD = 1.8 ± 0.2 nm) (Fig. 5 and data not shown). Tighter binding was observed for several intermediate-length LIP5(MIT)2 constructs such as LIP5(1–168) that terminated near residue 170 (KD = 80 ± 40 pm) (Fig. 5), but even shorter LIP(MIT)2 constructs such as LIP5(1–159) bound CHMP5(139–195) with dissociation constants that were similar to those of the full-length LIP5 protein (KD = 1.3 ± 0.3 nm) (see Fig. 5). These data demonstrate that CHMP5(139–195) binds the tandem MIT domain of LIP5. The tight-binding LIP5(1–168) construct was used for all subsequent binding analyses, but we believe that all of the different LIP5(MIT)2-containing constructs make similar interactions with CHMP5(139–195) (see below).
FIGURE 5.

CHMP5(139–195) binds tightly to the LIP5(MIT)2 domain. Fluorescence anisotropy binding isotherms show the binding of fluorescently labeled CHMP5(139–191)(A191C) to a series of different LIP5 constructs (see inset for key). Plotted points are averages of at least four independent anisotropy measurements for each LIP5 concentration, and error bars show S.D. in the measurements. Curves show fits of these isotherms to simple 1:1 binding models. Reported KD values are means ± S.D. from the fits to at least four independent binding isotherms.
Biosensor binding analyses were used to quantify LIP5(MIT)2 binding to a series of different immobilized GST-CHMP5 constructs. In good agreement with the fluorescence polarization data, LIP5(MIT)2 bound tightly to full-length GST-CHMP5, with an estimated dissociation constant of 100 ± 2 pm in this assay (Fig. 6, A and B). All other CHMP5 constructs that contained residues 150–195 bound with similar affinities (e.g. Fig. 6C and summarized in Fig. 6A), whereas the control N-terminal CHMP5(1–120) fragment did not bind. These data indicate that the LIP5(MIT)2-binding site spans predicted CHMP5 helices H5 and H6 and that LIP5 binding is not autoinhibited by the upstream core domain of CHMP5, which again agrees well with a previous study (50).
Structure of the LIP5(MIT)2-CHMP5(139–195) Complex
To define the structural basis for the unusual LIP5-CHMP5 interaction, we determined the solution structure of the LIP5(MIT)2-CHMP5(139–195) complex (Fig. 7 and Table 1). As expected from the tight-binding affinity, the complex was in slow exchange on the NMR time scale, and the spectra were of high quality, particularly for a complex of 26 kDa (Fig. 8 and Table 1). As illustrated in Fig. 7A, the LIP5(MIT)2 backbone structure did not change significantly upon CHMP5 binding. The LIP5(MIT)2-binding site comprises CHMP5 helices 5 and 6 and the linkers that flank either side of helix 5 (residues 155–189), which bind perpendicular to the helical bundle and wrap around three sides of LIP5 MIT2 (Fig. 7, B and C). These elements all make extensive contacts with LIP5 MIT2 and are well defined in the structure (Fig. 8). The N-terminal end of CHMP5 helix 5 crosses the MIT2 helix 6/7 groove, which would occlude binding of other ligands in this groove (e.g. MIM1 and MIM4 elements). The C-terminal end of CHMP5 helix 5 and the helix 5/6 linker sit in the groove between the two LIP5 MIT domains and make limited contacts with MIT1 helix 1 (Fig. 7C). CHMP5 helix 4 was also present and adopted a helical conformation but did not contact the MIT domain and appears to adopt many different orientations in solution (Fig. 8 and legend).
FIGURE 7.

Structure of the LIP5(MIT)2-CHMP5(139–195) complex. A, overlay of the LIP5(MIT)2 domains from the apo (gray) and LIP5(MIT)2-CHMP5(139–195) (MIT1, green; MIT2, blue) structures. LIP5(MIT)2 domains overlay with a backbone atom root mean square deviation of 1.2 Å. B and C, orthogonal views of the LIP5(MIT)2-CHMP5(139–195) complex, with labels on selected CHMP5 leucine collar residues and on the LIP5(MIT)2 Trp-147 residue. For clarity, only well ordered residues (positions 155–189) from the CHMP5(139–195) construct are shown (see Fig. 8). Candidate intermolecular salt bridges and hydrogen bonds are LIP5 Lys-15–CHMP5 Asp-168, LIP5 Arg-22–CHMP5 Glu-178, LIP5 Arg-30–CHMP5 Asp-184, LIP5 Arg-146–CHMP5 Glu-164, LIP5 His-153–CHMP5 Glu-173, LIP5 Lys-157–CHMP5 Glu-173, LIP5 Lys-112–CHMP5 Tyr-182, LIP5 Lys-116–CHMP5 Ser-180, and LIP5 Asn-154–CHMP5 Glu-166.
The amphipathic CHMP5 helices 5 and 6 and linkers create a hydrophobic binding collar that includes a series of six leucine residues, and we therefore term this motif the “leucine collar” (Fig. 7B). All six leucine residues are invariant across mammals, and conservative substitutions occur only in the first leucine in other metazoan species (Fig. 2B). The leucine collar makes complementary hydrophobic interactions with LIP5 MIT2 residues in helix 5 (Tyr-93, Met-97, and Tyr-100), helix 6 (Ile-115 and Tyr-119), and helix 7 (Trp-147 and Thr-150). The complex is also stabilized by strong charge complementarity because the CHMP5 binding surface includes 13 Asp and Glu residues, whereas the LIP5(MIT)2 surface includes 11 basic residues (MIT1 helix 1: Lys-15, His-19, Arg-22, and Arg-29; and MIT2: Lys-96, Lys-112, Lys-116, Lys-143, Arg-146, His-153, and Lys-157) (see the legend to Fig. 7 for a list of intermolecular salt bridges and hydrogen bonds). Thus, the unusually high affinity of the LIP5-CHMP5 interaction appears to result from a combination of extensive hydrophobic interactions made by the extended leucine collar, together with charge complementarity between the basic LIP5 MIT2 module and the highly acidic CHMP5 binding site. The interaction buries a total surface area of 2540 Å2, which is substantially greater than any previously characterized MIM-MIT interaction and which is consistent with the unusually high binding affinity.
As noted above, LIP5 constructs that terminated in the vicinity of residue 170 exhibited somewhat tighter CHMP5 binding, and we therefore considered the possibility that the LIP5(1–168)-CHMP5(139–195) complex might have an unusual or aberrant structure. To test this possibility, we compared the 1H-15N HSQC-NMR spectrum of the higher affinity 15N-labeled LIP5(1–168)-CHMP5(139–195) complex with that of the lower affinity 15N-labeled LIP5(1–183)-CHMP5(139–195) complex. The two spectra were superimposable, with no significant backbone amide chemical shift changes except at the extreme termini, where the two LIP5(MIT)2 constructs differ in length and sequence (data not shown). We therefore concluded that all of the different LIP5(MIT)2-CHMP5 complexes under study likely have very similar structures. We do not have a definitive explanation for the underlying cause of the higher binding affinities seen for LIP5 constructs that terminate near residue 170, but we speculate that it may reflect a favorable charge interaction between the (non-native) LIP5 C termini and the neighboring CHMP5 Arg-151 residue.
Mutational Analyses of the LIP5-CHMP5 Interface
The LIP5(MIT)2-CHMP5(139–195) structure was used to identify mutations on either side of the interface that were predicted to destabilize the complex. On the CHMP5 side, we tested the effects of two different sets of mutations within the leucine collar motif, one in which the central two leucine residues of helix 5 were mutated to aspartates (L167D,L170D) and a second in which all four helix 5 leucines were mutated (L163D,L167D,L170D,L174D). Consistent with the presumed importance of the leucine collar, the double mutation reduced CHMP5(139–195) binding affinity by >8000-fold, and the quadruple mutation reduced CHMP5(139–195) binding to undetectable levels (Fig. 6, A and E). The LIP5(MIT)2-CHMP5(139–195) structure also suggested that Trp-147 was likely to play a key role on the LIP5 side of the interface. We found that a W147D mutation in LIP5(MIT)2 reduced CHMP5(139–195) binding by 3000-fold in the biosensor binding assay (Fig. 6, A and D), and the detrimental effect was even greater in the fluorescence polarization binding assay (data not shown). Thus, our mutational analyses supported the validity of the LIP5(MIT)2-CHMP5(139–195) structure and identified non-binding mutants that could be used in cell-based studies.
Lack of Cooperativity or Autoinhibition in LIP5-Ligand Interactions
Although many components of the ESCRT pathway engage in autoinhibitory and/or cooperative binding interactions, the structure of the LIP5(MIT)2-CHMP5 complex did not reveal any conformational changes that appeared likely to influence LIP5 MIT1 binding to ESCRT-III MIM1 motifs. This lack of binding cooperativity was confirmed in biosensor binding experiments that showed that saturation of the MIT2 binding site with CHMP5(133–195) did not alter the binding affinity of CHMP1B(169–199) for LIP5(MIT)2 (Fig. 3B, compare closed diamonds and open squares; and Table 2). Thus, the two different types of ESCRT-III ligands bind the LIP5 MIT modules without positive or negative cooperativity.
It has been suggested that the terminal domains of Vta1p may interact and mutually autoinhibit their different ligand interactions (58), so we also tested for autoinhibition within LIP5 by comparing the binding affinities of different ligands for full-length LIP5 versus the isolated LIP5(MIT)2 and LIP5(VSL) domains. Our binding assays were configured to measure differences in intrinsic ligand binding affinities rather than avidity effects. As discussed above, CHMP5(139–195) bound equally tightly to full-length LIP5 and to a minimal LIP5(MIT)2 construct (Figs. 5 and 6), implying that downstream LIP5 sequences did not autoinhibit CHMP5 binding to the LIP5 MIT2 module. Similarly, biosensor binding analyses showed that CHMP1B(181–199) bound with the same affinity to LIP5 constructs that spanned the full-length protein (residues 1–307), the MIT2 domain, and the downstream linker (residues 1–259) or the MIT2 domain alone (residues 1–168) (Fig. 9A). These experiments indicate that MIM1 binding to the LIP5 MIT1 module is not autoinhibited by downstream LIP5 sequences.
Finally, NMR chemical shift titration analyses were used to quantify binding of the LIP5 constructs to VPS4B (Fig. 9, B–D). These binding studies utilized a construct that comprised the small ATPase and inserted β-domains of VPS4B (termed SAB for small ATPase and beta, residues 300–423). As shown in Fig. 9 (C and D), binding of either full-length LIP5 or the LIP5(VSL) domain alone (residues 260–307) induced very similar backbone amide chemical shift changes in the same subset of residues within 15N-labeled VPS4B(SAB), indicating that both LIP5 constructs bound in a very similar fashion. Chemical shift titration data for three shifted residues (designated A, B, and C) were independently fit to single-site binding models in each complex, yielding similar equilibrium dissociation constants for VPS4B(SAB) binding to either full-length LIP5 (KD = 170 ± 30 μm) or the VSL domain alone (KD = 233 ± 1 μm) (Fig. 9B). Thus, our data indicate that external LIP5 elements do not autoinhibit ligand binding to any of the three known protein interaction sites within the LIP5(MIT)2 and LIP5(VSL) domains.
Interdependence of LIP5-Ligand Binding in Vivo
Co-immunoprecipitation experiments were used to test the relevance of our in vitro binding analyses and to determine the interdependence of different LIP5-ligand interactions in a cellular context. As shown in Fig. 10A, Myc-LIP5 protein co-immunoprecipitated efficiently with the wild-type VPS4A-EGFP protein (where EGFP is enhanced GFP; first lane), but this interaction was inhibited by LIP5 point mutations that blocked binding to the VPS4(SAB) domain (Y278A mutation in the LIP5(VSL) domain, positive control based upon the Vta1p(VSL)-Vps4a(SAB) structure (48)) (second lane), CHMP1B and other ESCRT-III MIM1 interactions (M64A mutation in the helix 2/3 groove of LIP5 MIT1) (third lane), or CHMP5 binding (W147D mutation in LIP5 MIT2) (fourth lane). None of these mutations altered LIP5 expression levels, implying that they acted by disrupting protein-protein interactions rather than by destabilizing the protein. The interdependence of the different LIP5-ligand interactions was confirmed and extended by experiments that demonstrated the following. 1) Endogenous CHMP5 and VPS4A proteins co-immunoprecipitated with Myc-LIP5 (Fig. 10B, first lane), and both of these interactions were inhibited by a LIP5 point mutation that inhibited CHMP5 binding (W147D mutation in LIP5 MIT2) (second lane). 2) VPS4A and endogenous LIP5 proteins both co-immunoprecipitated with CHMP5-Myc (Fig. 10C, first lane), and both of these interactions were inhibited by a CHMP5 mutation that blocked LIP5 binding (L163D,L167D,L170D,L174D mutation (denoted L4D) in the CHMP5 binding motif) (compare the second lane). 3) siRNA depletion of CHMP5 inhibited VPS4 binding to LIP5 (Fig. 10D, compare the first and second lanes). 4) Point mutations on either side of the LIP5-CHMP5 interface inhibited VPS4A-EGFP co-immunoprecipitation of both CHMP5-Myc and Myc-LIP5 (Fig. 10E, compare the second and third lanes with the first lane). Taken together, these experiments demonstrated that VPS4A forms stable precipitable complexes with both LIP5 and CHMP5 in cells and that these complexes form only if LIP5 can interact with both CHMP5 and CHMP1B (and/or other MIM1-containing ESCRT-III proteins).
FIGURE 10.
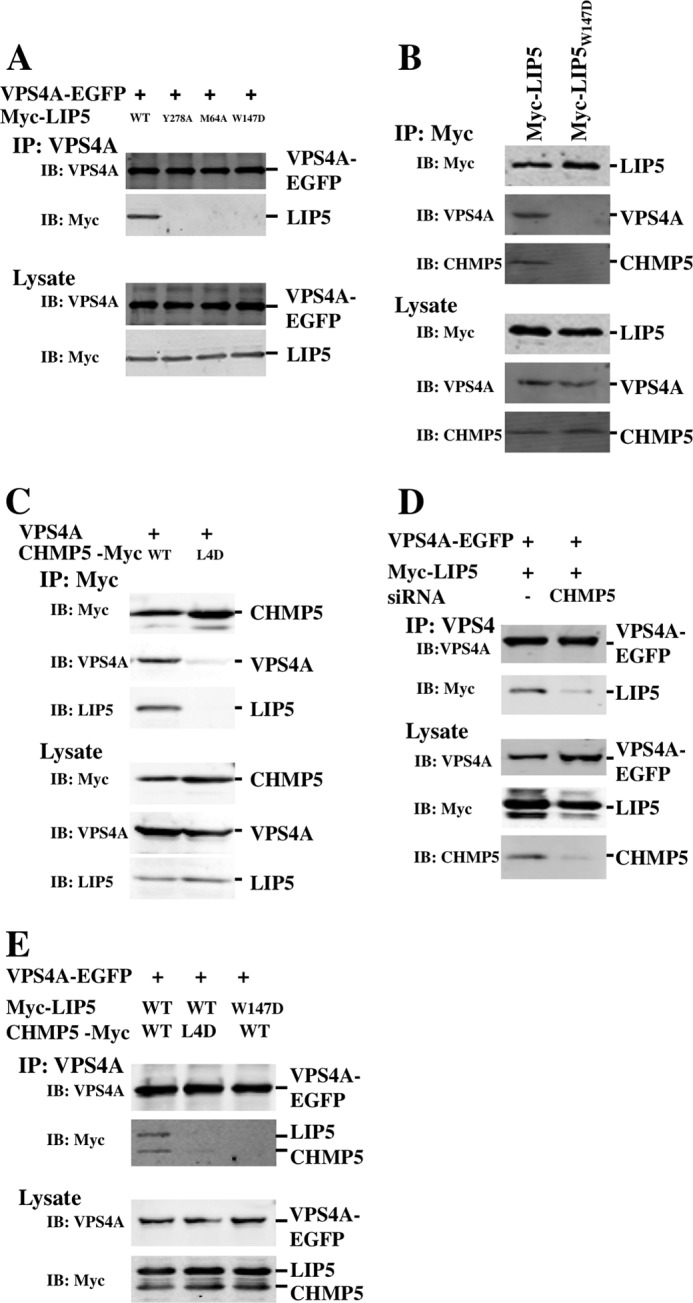
Immunoprecipitations of LIP5-ligand complexes. A, LIP5 co-immunoprecipitated with VPS4A (first lane), but the interaction was inhibited by LIP5 point mutations that blocked VPS4 (LIP5(Y278A); second lane), CHMP1B (LIP5(M64A); third lane) or CHMP5 (LIP5(W147D); fourth lane) binding. B, endogenous VPS4A and CHMP5 proteins co-immunoprecipitated with LIP5 (first lane) but not with the LIP5(W147D) mutant (second lane). C, VPS4A and endogenous LIP5 co-immunoprecipitated with CHMP5 (first lane) but not with a CHMP5 protein that contained leucine collar mutations (CHMP5(L163D,L167D,L170D,L174D (L4D); second lane). D, LIP5 co-immunoprecipitated with VPS4A (first lane), but the interaction was inhibited by siRNA depletion of the CHMP5 protein (second lane). E, LIP5 and CHMP5 co-immunoprecipitated with VPS4A (first lane), but the interactions were inhibited by a CHMP5 mutation (L163D,L167D,L170D,L174D) that inhibited LIP5 binding (second lane) or by a LIP5 mutation (W147D) that inhibited CHMP5 binding (third lane). IP, immunoprecipitation; IB, immunoblotting.
DISCUSSION
Like many other cellular machines that must function transiently, the ESCRT pathway is tightly regulated to ensure that the machinery localizes to correct sites of action, forms properly, and assembles and disassembles with “all or none” behavior. ESCRT regulation occurs at a number of different steps and via a number of different mechanisms, including the use of membrane-specific adaptor proteins, ordered protein-protein and protein-membrane interactions, post-translational modifications such as ubiquitylation and phosphorylation, autoinhibition, allostery, and avidity (1–9). VPS4 activation is one of the highly regulated steps in the ESCRT pathway and is governed primarily by the actions of the activator protein LIP5. LIP5 comprises two well ordered domains: a tandem MIT domain at the N terminus and a VSL dimerization domain at the C terminus, separated by a disordered linker region of ∼100 residues (Fig. 1) (58). Our analyses have helped to reveal how LIP5 connects VPS4 to the ESCRT-III lattice by demonstrating that the tandem LIP5 MIT domain must make two different types of interactions with CHMP5 and CHMP1B (and/or other ESCRT-III proteins) in order for LIP5 and CHMP5 to form precipitable cellular complexes with VPS4. We did not find any evidence that the VSL and MIT domains of LIP5 communicate directly (e.g. Figs. 5 and 9). Rather, our data support a model in which multiple weak ligand interactions, together with avidity effects resulting from LIP5 dimerization, cooperate to recruit LIP5 to oligomeric ESCRT-III assemblies and activate VPS4 at the proper time and place.
CHMP1B Interactions with LIP5(MIT)2
Our analyses agree well with previous studies that mapped LIP5/Vta1p binding to the C terminus of CHMP1B/Did2p (Fig. 3) (34, 50). However, a previous study mapped the interaction of yeast Did2p to the second MIT module of Vta1p (58), whereas we found that CHMP1B binds the first MIT module of human LIP5. Our studies therefore provide functions for both LIP5 MIT modules because CHMP1B and other ESCRT-III MIM1 elements bind the first MIT module and CHMP5 binds the second MIT module. Our mutational and NMR spectroscopic data are consistent with a model in which the terminal helix of CHMP1B makes a MIM1-like interaction with the helix 2/3 groove of the first LIP5 MIT module. CHMP1B/Did2p is an unusual ESCRT-III protein in that it binds well to the MIT domains from both VPS4 and LIP5 (33, 34, 39, 40, 50, 58). This is apparently possible because the helix 2/3 grooves of the MIT domains from VPS4 and LIP5 are similar in character. In particular, LIP5 MIT1 residues that comprise the predicted MIM1-binding groove (Leu-40, Met-43, Met-47, Leu-60, Met-64, and Leu-67) retain the hydrophobic character and are generally similar to the corresponding residues within the analogous VPS4A MIT groove (Gln-32, Val-36, Leu-40, Cys-60, Leu-64, and Arg-67). Our structure-based alignments also predict that an important intermolecular salt bridge formed in the VPS4A-CHMP1A complex (VPS4A MIT Asp-65/Glu-68–CHMP1A Arg-121) (35, 36) is conserved in the LIP5-CHMP1B complex (as LIP5 Asp-65/Glu-68–CHMP1A Arg-121). Conversely, the chemical character of the MIM1-binding site on the second LIP5 MIT module (the helix 6/7 groove) is significantly more polar in character, with charged residues at Asp-126, Arg-142, and Arg-146, and this likely explains why CHMP1B MIM1 does not bind in this groove.
LIP5(MIT)2 failed to bind MIM2 elements from CHMP4B, CHMP6, or IST1 or to bind the MIM3 element from CHMP1B, all of which should bind in MIT helix 1/3 grooves. The helix 1/3 groove of the first MIT domain of LIP5(MIT)2 is filled by the N-terminal strand of LIP5, which will occlude MIM2 and MIM3 binding unless this strand is displaced. It is less clear why MIM2 or MIM3 elements did not bind the equivalent helix 5/7 groove of the second LIP5 MIT domain, but there are several substitutions versus the VPS4A MIT helix 1/3 groove that could reduce MIM2 binding affinities. Alternatively, the terminal type I turn of LIP5(MIT)2 orients the mobile LIP5 residues 164–170 along this groove, and this may sterically inhibit ligand binding. In summary, although the tandem MIT domain of LIP5 has four different grooves that could, in principle, engage up to four different MIM elements, we found evidence only for MIM1 element binding in the helix 2/3 groove of the first MIT module.
CHMP5 Binding to LIP5(MIT)2
Our biochemical and structural studies demonstrated that CHMP5 helices 5 and 6 and adjacent linkers form an amphipathic leucine collar that wraps almost completely around the second MIT module of LIP5(MIT)2. This is the first example in which an ESCRT-III MIM element binds perpendicular to and outside of the MIT grooves (designated a MIM5-MIT interaction) (Fig. 11). The interaction is also the most extensive MIM-MIT interaction seen to date in terms of both buried surface area and binding affinity (Figs. 6 and 7). Interactions with the first LIP5 MIT module are minimal, however, and CHMP5 binding does not alter the affinity of CHMP1B binding to MIT1 (Fig. 3B). The binding of CHMP5 would clearly occlude the binding of other MIM elements in the helix 6/7 groove but does not obviously alter the accessibility of the helix 5/7 groove, which could therefore, in principle, bind an additional unidentified ligand(s).
FIGURE 11.

Comparison of the different MIM-MIT complexes. Shown are structures of the following complexes: human VPS4A MIT-CHMP1A(181–199) (Protein Data Bank code 2JQ9), human VPS4A MIT-CHMP6(166–181) (code 2K3W), human spastin MIT-CHMP1B(148–194) (code 3EAB), human AMSH MIT-CHMP3(200–222) (code 2XZE), LIP5 MIT2-CHMP5(155–189) (code 2LXM). In each case, MIT helices 1–3 are shown in red, orange, and yellow, respectively.
A previous study mapped the yeast Vps60p/CHMP5 interaction to the second MIT module of Vta1p/LIP5 (58). Nevertheless, there again appear to be significant differences between the analogous metazoan and yeast interactions because the primary Vta1p interaction site was mapped to Vps60p helix 4 (34), whereas the LIP5 interaction site spans CHMP5 helices 5 and 6, and CHMP5 helix 4 does not contact LIP5(MIT)2 (Figs. 6–8) (53). In most ESCRT-III proteins, the fifth helix and surrounding regions can fold back onto the core domain and make autoinhibitory interactions (12, 53, 75, 76). We found, however, that LIP5(MIT)2 bound with the same affinity to full-length CHMP5 as to the minimal CHMP5 binding site (Fig. 6A), implying either that CHMP5 does not exhibit autoinhibition or that the LIP5-binding site on CHMP5 is exposed in the “closed” CHMP5 conformation (53).
LIP5-Ligand Interactions in Cells
We also performed a series of co-immunoprecipitation experiments to examine the interdependence of different LIP5-ligand interactions within cells. Our data support the relevance of our LIP5-CHMP5 structural studies because structure-based mutations on either side of the LIP5-CHMP5 interface inhibited complex formation both in vitro (Fig. 6) and in cells (Fig. 10). Furthermore, our data indicate that both of the LIP5(MIT)2 interactions that we characterized are required to make stable precipitable cellular VPS4 complexes with both LIP5 and CHMP5. Specifically, mutations or siRNA depletions that disrupted the LIP5-CHMP5 interaction blocked VPS4A co-immunoprecipitation with both proteins. Similarly, a mutation in the first MIT domain of LIP5 that inhibited the binding of CHMP1B (and other MIM1-containing ESCRT-III proteins) also inhibited co-precipitation of VPS4A with LIP5 (Fig. 10A). In the absence of detailed localization and functional studies, we can only speculate about the nature of these VPS4 complexes and where in the cell they form. Nevertheless, our data are consistent with published models in which LIP5 and CHMP5 are initially tightly associated in the cytoplasm (53). Formation of stable ESCRT-III-associated LIP5-VPS4 complexes might then require both a direct LIP5(VSL)-VPS4(SAB) interaction and a supporting interaction between the CHMP1B MIM1 element and the first LIP5 MIT domain (33, 45). It is not yet clear why CHMP5 is also required to form this complex, but it was recently shown that CHMP5 can bind the ALIX paralog BROX at a site located just downstream of the LIP5-binding site (77). BROX and ALIX both contain Bro1 domains, and the two proteins can function interchangeably in initiating some ESCRT assembly reactions (78). We therefore speculate that the BROX-CHMP5 interaction might also contribute to targeting the LIP5-CHMP5 complex to membrane-associated ESCRT-III assemblies (although it is alternatively possible that CHMP5 helps target BROX to these sites). Once recruited, LIP5 presumably promotes VPS4 assembly and activates the enzyme to remodel the ESCRT-III polymer and release ESCRT factors back into the cytoplasm.
Acknowledgments
We thank Dr. Debra Eckert for help with analytical ultracentrifugation experiments and Dr. J. Nicholas Francis for help with HPLC purification of CHMP5 peptides.
This work was supported, in whole or in part, by National Institutes of Health Grants AI051174 and GM082545 (to W. I. S.).
This article was selected as a Paper of the Week.

This article contains supplemental Table 1.
The atomic coordinates and structure factors (codes 2LXL and 2LXM) have been deposited in the Protein Data Bank (http://wwpdb.org/).
All constructs used in this study have been deposited in the DNASU database (http://dnasu.asu.edu/DNASU/).
N. Monroe, H. Han, D. G. Malgorzata, D. Eckert, M. A. Karren, F. Whitby, W. I. Sundquist, and C. P. Hill, unpublished data.
- ESCRT
- endosomal sorting complex required for transport
- MIT
- microtubule-interacting and trafficking
- MIM
- MIT-interacting motif
- HSQC
- heteronuclear single-quantum coherence
- TROSY
- transverse relaxation optimized spectroscopy.
REFERENCES
- 1. Hanson P. I., Cashikar A. (2012) Multivesicular body morphogenesis. Annu. Rev. Cell Dev. Biol. 28, 337–362 [DOI] [PubMed] [Google Scholar]
- 2. Rusten T. E., Vaccari T., Stenmark H. (2012) Shaping development with ESCRTs. Nat. Cell Biol. 14, 38–45 [DOI] [PubMed] [Google Scholar]
- 3. Weiss E. R., Göttlinger H. (2011) The role of cellular factors in promoting HIV budding. J. Mol. Biol. 410, 525–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Caballe A., Martin-Serrano J. (2011) ESCRT machinery and cytokinesis: the road to daughter cell separation. Traffic 12, 1318–1326 [DOI] [PubMed] [Google Scholar]
- 5. Henne W. M., Buchkovich N. J., Emr S. D. (2011) The ESCRT pathway. Dev. Cell 21, 77–91 [DOI] [PubMed] [Google Scholar]
- 6. Hurley J. H., Hanson P. I. (2010) Membrane budding and scission by the ESCRT machinery: it's all in the neck. Nat. Rev. Mol. Cell Biol. 11, 556–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hurley J. H. (2010) The ESCRT complexes. Crit. Rev. Biochem. Mol. Biol. 45, 463–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Guizetti J., Gerlich D. W. (2012) ESCRT-III polymers in membrane neck constriction. Trends Cell Biol. 22, 133–140 [DOI] [PubMed] [Google Scholar]
- 9. McCullough J., Colf L. A., Sundquist W. I. (2012) Membrane fission reactions of the mammalian ESCRT pathway. Annu. Rev. Biochem., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lata S., Schoehn G., Solomons J., Pires R., Göttlinger H. G., Weissenhorn W. (2009) Structure and function of ESCRT-III. Biochem. Soc. Trans. 37, 156–160 [DOI] [PubMed] [Google Scholar]
- 11. Muzioł T., Pineda-Molina E., Ravelli R. B., Zamborlini A., Usami Y., Göttlinger H., Weissenhorn W. (2006) Structural basis for budding by the ESCRT-III factor CHMP3. Dev. Cell 10, 821–830 [DOI] [PubMed] [Google Scholar]
- 12. Bajorek M., Schubert H. L., McCullough J., Langelier C., Eckert D. M., Stubblefield W. M., Uter N. T., Myszka D. G., Hill C. P., Sundquist W. I. (2009) Structural basis for ESCRT-III protein autoinhibition. Nat. Struct. Mol. Biol. 16, 754–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xiao J., Chen X. W., Davies B. A., Saltiel A. R., Katzmann D. J., Xu Z. (2009) Structural basis of Ist1 function and Ist1-Did2 interaction in the multivesicular body pathway and cytokinesis. Mol. Biol. Cell 20, 3514–3524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hurley J. H., Yang D. (2008) MIT domainia. Dev. Cell 14, 6–8 [DOI] [PubMed] [Google Scholar]
- 15. Fabrikant G., Lata S., Riches J. D., Briggs J. A., Weissenhorn W., Kozlov M. M. (2009) Computational model of membrane fission catalyzed by ESCRT-III. PLoS Comput. Biol. 5, e1000575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Elia N., Fabrikant G., Kozlov M. M., Lippincott-Schwartz J. (2012) Computational model of cytokinetic abscission driven by ESCRT-III polymerization and remodeling. Biophys. J. 102, 2309–2320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Boura E., Różycki B., Chung H. S., Herrick D. Z., Canagarajah B., Cafiso D. S., Eaton W. A., Hummer G., Hurley J. H. (2012) Solution structure of the ESCRT-I and -II supercomplex: implications for membrane budding and scission. Structure 20, 874–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hill C. P., Babst M. (2012) Structure and function of the membrane deformation AAA ATPase Vps4. Biochim. Biophys. Acta 1823, 172–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Babst M., Wendland B., Estepa E. J., Emr S. D. (1998) The Vps4p AAA ATPase regulates membrane association of a Vps protein complex required for normal endosome function. EMBO J. 17, 2982–2993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ghazi-Tabatabai S., Saksena S., Short J. M., Pobbati A. V., Veprintsev D. B., Crowther R. A., Emr S. D., Egelman E. H., Williams R. L. (2008) Structure and disassembly of filaments formed by the ESCRT-III subunit Vps24. Structure 16, 1345–1356 [DOI] [PubMed] [Google Scholar]
- 21. Saksena S., Wahlman J., Teis D., Johnson A. E., Emr S. D. (2009) Functional reconstitution of ESCRT-III assembly and disassembly. Cell 136, 97–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wollert T., Wunder C., Lippincott-Schwartz J., Hurley J. H. (2009) Membrane scission by the ESCRT-III complex. Nature 458, 172–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Elia N., Sougrat R., Spurlin T. A., Hurley J. H., Lippincott-Schwartz J. (2011) Dynamics of endosomal sorting complex required for transport (ESCRT) machinery during cytokinesis and its role in abscission. Proc. Natl. Acad. Sci. U.S.A. 108, 4846–4851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Baumgärtel V., Ivanchenko S., Dupont A., Sergeev M., Wiseman P. W., Kräusslich H. G., Bräuchle C., Müller B., Lamb D. C. (2011) Live-cell visualization of dynamics of HIV budding site interactions with an ESCRT component. Nat. Cell Biol. 13, 469–474 [DOI] [PubMed] [Google Scholar]
- 25. Jouvenet N., Simon S. M., Bieniasz P. D. (2011) Visualizing HIV-1 assembly. J. Mol. Biol. 410, 501–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jouvenet N., Zhadina M., Bieniasz P. D., Simon S. M. (2011) Dynamics of ESCRT protein recruitment during retroviral assembly. Nat. Cell Biol. 13, 394–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gonciarz M. D., Whitby F. G., Eckert D. M., Kieffer C., Heroux A., Sundquist W. I., Hill C. P. (2008) Biochemical and structural studies of yeast Vps4 oligomerization. J. Mol. Biol. 384, 878–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Scott A., Chung H. Y., Gonciarz-Swiatek M., Hill G. C., Whitby F. G., Gaspar J., Holton J. M., Viswanathan R., Ghaffarian S., Hill C. P., Sundquist W. I. (2005) Structural and mechanistic studies of VPS4 proteins. EMBO J. 24, 3658–3669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Landsberg M. J., Vajjhala P. R., Rothnagel R., Munn A. L., Hankamer B. (2009) Three-dimensional structure of AAA ATPase Vps4: advancing structural insights into the mechanisms of endosomal sorting and enveloped virus budding. Structure 17, 427–437 [DOI] [PubMed] [Google Scholar]
- 30. Xiao J., Xia H., Yoshino-Koh K., Zhou J., Xu Z. (2007) Structural characterization of the ATPase reaction cycle of endosomal AAA protein Vps4. J. Mol. Biol. 374, 655–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hartmann C., Chami M., Zachariae U., de Groot B. L., Engel A., Grütter M. G. (2008) Vacuolar protein sorting: two different functional states of the AAA-ATPase Vps4p. J. Mol. Biol. 377, 352–363 [DOI] [PubMed] [Google Scholar]
- 32. Davies B. A., Azmi I. F., Payne J., Shestakova A., Horazdovsky B. F., Babst M., Katzmann D. J. (2010) Coordination of substrate binding and ATP hydrolysis in Vps4-mediated ESCRT-III disassembly. Mol. Biol. Cell 21, 3396–3408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shestakova A., Hanono A., Drosner S., Curtiss M., Davies B. A., Katzmann D. J., Babst M. (2010) Assembly of the AAA ATPase Vps4 on ESCRT-III. Mol. Biol. Cell 21, 1059–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Azmi I. F., Davies B. A., Xiao J., Babst M., Xu Z., Katzmann D. J. (2008) ESCRT-III family members stimulate Vps4 ATPase activity directly or via Vta1. Dev. Cell 14, 50–61 [DOI] [PubMed] [Google Scholar]
- 35. Obita T., Saksena S., Ghazi-Tabatabai S., Gill D. J., Perisic O., Emr S. D., Williams R. L. (2007) Structural basis for selective recognition of ESCRT-III by the AAA ATPase Vps4. Nature 449, 735–739 [DOI] [PubMed] [Google Scholar]
- 36. Stuchell-Brereton M. D., Skalicky J. J., Kieffer C., Karren M. A., Ghaffarian S., Sundquist W. I. (2007) ESCRT-III recognition by VPS4 ATPases. Nature 449, 740–744 [DOI] [PubMed] [Google Scholar]
- 37. Kieffer C., Skalicky J. J., Morita E., De Domenico I., Ward D. M., Kaplan J., Sundquist W. I. (2008) Two distinct modes of ESCRT-III recognition are required for VPS4 functions in lysosomal protein targeting and HIV-1 budding. Dev. Cell 15, 62–73 [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 38. Merrill S. A., Hanson P. I. (2010) Activation of human VPS4A by ESCRT-III proteins reveals ability of substrates to relieve enzyme autoinhibition. J. Biol. Chem. 285, 35428–35438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nickerson D. P., West M., Odorizzi G. (2006) Did2 coordinates Vps4-mediated dissociation of ESCRT-III from endosomes. J. Cell Biol. 175, 715–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lottridge J. M., Flannery A. R., Vincelli J. L., Stevens T. H. (2006) Vta1p and Vps46p regulate the membrane association and ATPase activity of Vps4p at the yeast multivesicular body. Proc. Natl. Acad. Sci. U.S.A. 103, 6202–6207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Agromayor M., Carlton J. G., Phelan J. P., Matthews D. R., Carlin L. M., Ameer-Beg S., Bowers K., Martin-Serrano J. (2009) Essential role of hIST1 in cytokinesis. Mol. Biol. Cell 20, 1374–1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bajorek M., Morita E., Skalicky J. J., Morham S. G., Babst M., Sundquist W. I. (2009) Biochemical analyses of human IST1 and its function in cytokinesis. Mol. Biol. Cell 20, 1360–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nickerson D. P., West M., Henry R., Odorizzi G. (2010) Regulators of Vps4 ATPase activity at endosomes differentially influence the size and rate of formation of intralumenal vesicles. Mol. Biol. Cell 21, 1023–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ward D. M., Vaughn M. B., Shiflett S. L., White P. L., Pollock A. L., Hill J., Schnegelberger R., Sundquist W. I., Kaplan J. (2005) The role of LIP5 and CHMP5 in multivesicular body formation and HIV-1 budding in mammalian cells. J. Biol. Chem. 280, 10548–10555 [DOI] [PubMed] [Google Scholar]
- 45. Azmi I., Davies B., Dimaano C., Payne J., Eckert D., Babst M., Katzmann D. J. (2006) Recycling of ESCRTs by the AAA-ATPase Vps4 is regulated by a conserved VSL region in Vta1. J. Cell Biol. 172, 705–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fujita H., Umezuki Y., Imamura K., Ishikawa D., Uchimura S., Nara A., Yoshimori T., Hayashizaki Y., Kawai J., Ishidoh K., Tanaka Y., Himeno M. (2004) Mammalian class E Vps proteins, SBP1 and mVps2/CHMP2A, interact with and regulate the function of an AAA-ATPase SKD1/Vps4B. J. Cell Sci. 117, 2997–3009 [DOI] [PubMed] [Google Scholar]
- 47. Shiflett S. L., Ward D. M., Huynh D., Vaughn M. B., Simmons J. C., Kaplan J. (2004) Characterization of Vta1p, a class E Vps protein in Saccharomyces cerevisiae. J. Biol. Chem. 279, 10982–10990 [DOI] [PubMed] [Google Scholar]
- 48. Yang D., Hurley J. H. (2010) Structural role of the Vps4-Vta1 interface in ESCRT-III recycling. Structure 18, 976–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Haas T. J., Sliwinski M. K., Martínez D. E., Preuss M., Ebine K., Ueda T., Nielsen E., Odorizzi G., Otegui M. S. (2007) The Arabidopsis AAA ATPase SKD1 is involved in multivesicular endosome function and interacts with its positive regulator LYST-INTERACTING PROTEIN5. Plant Cell 19, 1295–1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shim S., Merrill S. A., Hanson P. I. (2008) Novel interactions of ESCRT-III with LIP5 and VPS4 and their implications for ESCRT-III disassembly. Mol. Biol. Cell 19, 2661–2672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rue S. M., Mattei S., Saksena S., Emr S. D. (2008) Novel Ist1-Did2 complex functions at a late step in multivesicular body sorting. Mol. Biol. Cell 19, 475–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dimaano C., Jones C. B., Hanono A., Curtiss M., Babst M. (2008) Ist1 regulates Vps4 localization and assembly. Mol. Biol. Cell 19, 465–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Shim S., Kimpler L. A., Hanson P. I. (2007) Structure/function analysis of four core ESCRT-III proteins reveals common regulatory role for extreme C-terminal domain. Traffic 8, 1068–1079 [DOI] [PubMed] [Google Scholar]
- 54. Yang D., Rismanchi N., Renvoisé B., Lippincott-Schwartz J., Blackstone C., Hurley J. H. (2008) Structural basis for midbody targeting of spastin by the ESCRT-III protein CHMP1B. Nat. Struct. Mol. Biol. 15, 1278–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tsang H. T., Connell J. W., Brown S. E., Thompson A., Reid E., Sanderson C. M. (2006) A systematic analysis of human CHMP protein interactions: additional MIT domain-containing proteins bind to multiple components of the human ESCRT-III complex. Genomics 88, 333–346 [DOI] [PubMed] [Google Scholar]
- 56. Ciccarelli F. D., Proukakis C., Patel H., Cross H., Azam S., Patton M. A., Bork P., Crosby A. H. (2003) The identification of a conserved domain in both spartin and spastin, mutated in hereditary spastic paraplegia. Genomics 81, 437–441 [DOI] [PubMed] [Google Scholar]
- 57. Rigden D. J., Liu H., Hayes S. D., Urbé S., Clague M. J. (2009) Ab initio protein modelling reveals novel human MIT domains. FEBS Lett. 583, 872–878 [DOI] [PubMed] [Google Scholar]
- 58. Xiao J., Xia H., Zhou J., Azmi I. F., Davies B. A., Katzmann D. J., Xu Z. (2008) Structural basis of Vta1 function in the multivesicular body sorting pathway. Dev. Cell 14, 37–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Scott A., Gaspar J., Stuchell-Brereton M. D., Alam S. L., Skalicky J. J., Sundquist W. I. (2005) Structure and ESCRT-III protein interactions of the MIT domain of human VPS4A. Proc. Natl. Acad. Sci. U.S.A. 102, 13813–13818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Takasu H., Jee J. G., Ohno A., Goda N., Fujiwara K., Tochio H., Shirakawa M., Hiroaki H. (2005) Structural characterization of the MIT domain from human Vps4b. Biochem. Biophys. Res. Commun. 334, 460–465 [DOI] [PubMed] [Google Scholar]
- 61. Samson R. Y., Obita T., Freund S. M., Williams R. L., Bell S. D. (2008) A role for the ESCRT system in cell division in archaea. Science 322, 1710–1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Solomons J., Sabin C., Poudevigne E., Usami Y., Hulsik D. L., Macheboeuf P., Hartlieb B., Göttlinger H., Weissenhorn W. (2011) Structural basis for ESCRT-III CHMP3 recruitment of AMSH. Structure 19, 1149–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Pincetic A., Kuang Z., Seo E. J., Leis J. (2010) The interferon-induced gene ISG15 blocks retrovirus release from cells late in the budding process. J. Virol. 84, 4725–4736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kuang Z., Seo E. J., Leis J. (2011) The mechanism of inhibition of retrovirus release from cells by interferon-induced gene ISG15. J. Virol. 85, 7153–7161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Studier F. W. (2005) Protein production by autoinduction in high density shaking cultures. Protein Expr. Purif. 41, 207–234 [DOI] [PubMed] [Google Scholar]
- 66. Rückert M., Otting G. (2000) Alignment of biological macromolecules in novel nonionic liquid crystalline media for NMR experiments. J. Am. Chem. Soc. 122, 7793–7797 [Google Scholar]
- 67. Cavanagh J., Fairbrother W. J., Palmer A. G., 3rd, Rance M., Skelton N. J. (2007) Protein NMR Spectroscopy: Principles and Practice, Elsevier Academic Press, San Diego [Google Scholar]
- 68. Zimmerman D. E., Kulikowski C. A., Huang Y., Feng W., Tashiro M., Shimotakahara S., Chien C., Powers R., Montelione G. T. (1997) Automated analysis of protein NMR assignments using methods from artificial intelligence. J. Mol. Biol. 269, 592–610 [DOI] [PubMed] [Google Scholar]
- 69. Cornilescu G., Delaglio F., Bax A. (1999) Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR 13, 289–302 [DOI] [PubMed] [Google Scholar]
- 70. Shen Y., Delaglio F., Cornilescu G., Bax A. (2009) TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 44, 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Guntert P. (2004) Automated NMR structure calculation with CYANA. Methods Mol. Biol. 278, 353–378 [DOI] [PubMed] [Google Scholar]
- 72. Güntert P. (2009) Automated structure determination from NMR spectra. Eur. Biophys. J. 38, 129–143 [DOI] [PubMed] [Google Scholar]
- 73. Bhattacharya A., Tejero R., Montelione G. T. (2007) Evaluating protein structures determined by structural genomics consortia. Proteins 66, 778–795 [DOI] [PubMed] [Google Scholar]
- 74. Schwieters C. D., Kuszewski J. J., Clore G. M. (2006) Using Xplor-NIH for NMR molecular structure determination. Prog. NMR Spectrosc. 48, 47–62 [Google Scholar]
- 75. Lin Y., Kimpler L. A., Naismith T. V., Lauer J. M., Hanson P. I. (2005) Interaction of the mammalian endosomal sorting complex required for transport (ESCRT) III protein hSnf7-1 with itself, membranes, and the AAA+ ATPase SKD1. J. Biol. Chem. 280, 12799–12809 [DOI] [PubMed] [Google Scholar]
- 76. Lata S., Roessle M., Solomons J., Jamin M., Göttlinger H. G., Svergun D. I., Weissenhorn W. (2008) Structural basis for autoinhibition of ESCRT-III CHMP3. J. Mol. Biol. 378, 818–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Mu R., Dussupt V., Jiang J., Sette P., Rudd V., Chuenchor W., Bello N. F., Bouamr F., Xiao T. S. (2012) Two distinct binding modes define the interaction of Brox with the C-terminal tails of CHMP5 and CHMP4B. Structure 20, 887–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Popov S., Popova E., Inoue M., Göttlinger H. G. (2009) Divergent Bro1 domains share the capacity to bind human immunodeficiency virus type 1 nucleocapsid and to enhance virus-like particle production. J. Virol. 83, 7185–7193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Goddard T. D., Kneller D. G. (2001) SPARKY 3, University of California, San Francisco [Google Scholar]