

Background: The Mre11-Rad50-Nbs1 (MRN) complex and the ubiquitin E3 ligase RNF8 play important roles in DNA DSB repair.
Results: RNF8 interacts with and ubiquitinates Nbs1 to promote binding of Nbs1 to DSBs and HR-mediated DSB repair.
Conclusion: Nbs1 ubiquitination by RNF8 is important for Nbs1 recruitment to DSBs and HR-mediated repair of DSBs.
Significance: These studies help to understand how ubiquitination modifications contribute to DSB repair and genome stability maintenance in mammalian cells.
Keywords: DNA Damage Response, DNA Repair, Homologous Recombination, Post-translational Modification, Ubiquitination, Double Strand Break (DSB) Repair, Nbs1, RNF8
Abstract
Ubiquitination plays an important role in the DNA damage response. We identified a novel interaction of the E3 ubiquitin ligase RNF8 with Nbs1, a key regulator of DNA double-strand break (DSB) repair. We found that Nbs1 is ubiquitinated both before and after DNA damage and is a direct ubiquitination substrate of RNF8. We also identified key residues on Nbs1 that are ubiquitinated by RNF8. By using laser microirradiation and live-cell imaging, we observed that RNF8 and its ubiquitination activity are important for promoting optimal binding of Nbs1 to DSB-containing chromatin. We also demonstrated that RNF8-mediated ubiquitination of Nbs1 contributes to the efficient and stable binding of Nbs1 to DSBs and is important for HR-mediated DSB repair. Taken together, these studies suggest that Nbs1 is one important target of RNF8 to regulate DNA DSB repair.
Introduction
Homologous recombination (HR)3 and non-homologous end joining (NHEJ) are two major pathways to repair DSBs (1, 2). HR is error-free, involving extended end resection of the DSB ends to generate single-stranded DNA (ssDNA) and utilizing the homologous sister chromatid as a template (3). Classical NHEJ is through the Ku-dependent pathway to religate DSB ends, which can be error-prone when DSB ends are not compatible (4). Microhomology-mediated end joining (MMEJ) is a major type of alternative NHEJ when the Ku-dependent classical NHEJ is deficient. MMEJ involves short processing of the ends and religation of DNA ends at microhomology sequences (5, 6), and this pathway requires the functions of the Mre11-Rad50-Nbs1 (MRN) complex and CtIP (7–11).
Upon formation of DSBs, the MRN complex recognizes the lesions and recruits ATM (ataxia telangiectasia mutated) to DSBs (12–14). Activation of ATM leads to phosphorylation of H2AX (γ-H2AX) (15). MDC1 is recruited to the damage sites by binding to γ-H2AX through its C-terminal BRCT domain (16). Subsequently, the E3 ubiquitin ligase RNF8 is recruited to DSBs through interaction of the N-terminal FHA domain of RNF8 with MDC1 at the ATM phosphorylation sites of MDC1 (17). Meanwhile, Nbs1 is recruited to DSB-flanking regions by MDC1 through binding of the FHA/BRCT domains of Nbs1 with the CK2 phosphorylation sites of MDC1 (18–23). Thus, MDC1 serves as a scaffold protein to recruit RNF8 and MRN to DSB-flanking chromatin regions.
Upon DNA damage, ubiquitination occurs at DSB sites, and a number of proteins involved in the ubiquitination process are accumulated at DSBs and play critical roles in mediating DNA damage responses (24, 25). For instance, BRCA1, a key player of HR-mediated DSB repair, contains an N-terminal RING domain that interacts with BARD1 to form a heterodimeric E3 ubiquitin ligase (26, 27). The BRCA1-BARD1 complex then interacts with the E2 conjugating enzyme UbcH5C and promotes Lys-6-linked ubiquitin chain formation (28, 29).
More recent studies demonstrate that RNF8 and its ubiquitination activity play crucial roles in the DNA damage response. RNF8 interacts with Ubc13 and can catalyze Lys-63-linked ubiquitination of H2A and H2AX at DSB sites (30). RNF8 can also interact with other E2s, including UbcH8 and UbcH5C, for mediating the DNA damage response (31–35). Importantly, RNF8 was found to be critical for localizing BRCA1 to DSBs (17, 36–38). The ubiquitination activity of RNF8 is required for recruiting RAP80, which contains two ubiquitin interacting motifs, to DSBs (38). Through formation of the RAP80-ABRA1-BRCA1 complex, BRCA1 is recruited to DSBs (39–42). RNF8 is also important for recruiting another ubiquitin E3 ligase, RNF168, and 53BP1 to DSBs (17, 36, 37, 43, 44). Consistent with its critical roles in mediating the ubiquitination-dependent recruitment of repair factors, RNF8-deficient cells are defective in G2/M checkpoint and exhibit sensitivity to ionizing radiation (IR) (17, 36, 37).
In this study, we demonstrate a novel interaction of RNF8 with MRN. We found that RNF8 directly binds to Nbs1 and ubiquitinates Nbs1. By mass spectrometry analysis, we identified Nbs1 Lys-435 as the key residue ubiquitinated by RNF8. We also showed that RNF8 and RNF8 ubiquitination activity are needed for efficient localization of Nbs1 to DSBs. More specifically, we demonstrated that RNF8-mediated ubiquitination of Nbs1 contributes significantly to Nbs1 localization to DSBs. By using EGFP-based DSB repair substrates, we demonstrated that RNF8 is important for promoting HR while being dispensable for MMEJ. Further analysis revealed that RNF8-mediated ubiquitination of Nbs1 is important for promoting DSB repair by HR. These studies reveal a new mechanism underlying the role of RNF8 in mediating DNA damage responses through ubiquitinating Nbs1 and promoting optimal and stable recruitment of MRN to DSBs for DSB repair by HR.
EXPERIMENTAL PROCEDURES
Cell Culture, Transfection, Retroviral Infection, and RNA Interference
U2OS and 293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum in the presence of antibiotics. Sf21 insect cells were cultured in Grace's insect medium (Invitrogen) containing 10% fetal bovine serum. Sf9 insect cells were cultured in Sf-900 II SFM medium (Invitrogen). 293T and U2OS cells were transfected by a calcium chloride method. U2OS cells stably expressing FLAG-tagged RNF8 (wild-type and mutants) and Myc-tagged Nbs1 (wild-type and mutants), with shRNA-resistant silent mutations, were generated by retroviral infection using pBabepuro vector, followed by puromycin selection.
Silencing of endogenous RNF8, Nbs1, Mre11, MDC1, and H2AX by shRNAs was performed by retroviral infection using the vector pMKO-puro expressing corresponding shRNAs. The shRNA target sequences for Mre11 and Nbs1 were previously described (45), and the following shRNAs were designed by Dharmacon: for RNF8sh GGACAAUUAUGGACAACAAGA; for MDC1sh GUCUCCCAGAAGACAGUGAUU; and for H2AXsh GGGACGAAGCACUUGGUAA. Enhanced blue fluorescence protein (EBFP)-marked shRNAs were generated by replacing the puromycin marker of pMKO-puro with EBFP-puromycin fusion protein (EBFP obtained from Addgene plasmid 14983 (46)), followed by retroviral infection and puromycin selection.
Plasmid Construction
FLAG, Myc, and/or HA tag epitopes were fused with RNF8 (RNF8 cDNA was kindly provided by J. Lukas), Nbs1, Mre11, Rad50, or ubiquitin in pcDNA3 vector. GST-tagged Nbs1 and RNF8 were made in pGEX-6P-1 (GE Healthcare), and EGFP-tagged Nbs1, RNF8, and Mre11 were generated in EGFP-C1 (CLONTECH) or EGFP-pBabepuro. mRFP-tagged Nbs1 (wild-type and mutants) were generated in mRFP-N1. The indicated point mutations for RNF8, Nbs1, and ubiquitin (HA-ubiquitin wild type was kindly provided by P. R. Yew) were generated by site-directed mutagenesis (QuikChange, Stratagene). The indicated RNF8 deletion mutants were generated by PCR amplification and ligation of fragments.
Whole Cell Lysis, Co-immunoprecipitation, Immunofluorescence, and Antibodies
Cells were lysed in NETN (150 mm NaCl, 1 mm EDTA, 20 mm Tris-Cl, pH 8.0, 0.5% Nonidet P-40 (v/v)) containing protease and phosphatase inhibitors (50 mm sodium fluoride, NaF, and 0.1 mm sodium orthovanadate, Na3VO4). Cleared cell lysates were then collected for immunoprecipitation and/or subjected to SDS-PAGE as described (47). Immunostaining and immunofluorescence microscopy analysis were performed as described (47, 48).
RNF8 polyclonal antibody was generated by immunizing rabbits with GST-fused RNF8 fragment (amino acids 1–324) and affinity-purified. Polyclonal antibodies against Mre11 (D27) and Nbs1 (D29) were described previously (49, 50). Other antibodies include anti-Rad50 and anti-Myc-9E10 (Novus Biologicals), anti-Ku70 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), anti-FLAG-M2 (Sigma), anti-HA and anti-MDC1 (Sigma), anti-H2AX (Bethyl), and anti-GST and anti-RPA2 (Oncogene).
Protein Purification
Nbs1 and Mre11-Rad50 were expressed in Sf9 insect cell by baculovirus infection and purified as described (51). Full-length RNF8 was expressed in Sf9 cells and harvested at 48 h after baculoviral infection. The cell pellet was resuspended in NTA Buffer A (20 mm Tris-HCl, pH 7.5, 500 mm KCl, 10% glycerol, 0.1% Triton X-100, 2 mm 2-mercaptoethanol) containing 5 mm imidazole and protease inhibitors. Cleared lysates were prepared by sonication followed by ultracentrifugation at 3,500 × g for 50 min at 4 °C. The lysates were loaded onto a nickel-NTA-Sepharose (Qiagen) column and washed with 5 column volumes of NTA buffer A containing 5 mm imidazole, followed by 10 column volumes of NTA buffer A containing 60 mm imidazole. Proteins were sequentially eluted with NTA elution buffer (20 mm Tris-HCl, pH 7.5, 150 mm KCl, 10% glycerol, 0.1% Triton X-100, 2 mm 2-mercaptoethanol, 2 mm dithiothreitol (DTT), 250 mm imidazole, and protease inhibitors).
GST Pull-down Assay
GST-RNF8 proteins were expressed in Sf21 insect cells. The insect cell pellets were lysed with sonication buffer (50 mm Tris-Cl, pH 7.9, 150 mm NaCl, 1 mm EDTA, 0.5% Nonidet P-40, protein inhibitors). Supernatants from lysates, cleared by centrifugation, were incubated with glutathione-Sepharose beads (Amersham Biosciences) at 4 °C, followed by three washes with sonication buffer. For binding experiments, glutathione-Sepharose beads bound to GST or GST-RNF8 were incubated with purified Mre11, Rad50, and Nbs1 proteins for 2 h at 4 °C. The beads were washed three times with sonication buffer and then subjected to SDS-PAGE.
In Vivo and in Vitro Ubiquitination Assay
For the in vivo ubiquitination assay, U2OS cells were transfected with HA-tagged ubiquitin (Ub) and Myc-tagged Nbs1. 40 h post-transfection, cells were harvested and lysed in 1% SDS lysis buffer (50 mm Tris-HCl, pH 7.5, 1% SDS) and boiled for 10 min. Lysates were cleared by centrifugation at 14,000 × g for 10 min. Supernatant was diluted 1:10 with NETN buffer (150 mm NaCl, 5 mm EDTA, 50 mm Tris-HCl, pH 7.5, 0.1% Nonidet P-40) and then immunoprecipitated with mouse anti-Myc antibody (9E10) at 4 °C overnight. The beads were washed three times with ice-cold NETN buffer and then subjected to SDS-PAGE.
For the in vitro ubiquitination assay, ubiquitination reactions were conducted at 37 °C for 1 h in a total volume of 20 μl containing 2 μg of HA-tagged ubiquitin, 25 ng of rabbit E1 (Boston Biochem), 0.25 μg of UbcH5c (Boston Biochem), 50 ng of RNF8, 1 μg of Myc-tagged Nbs1, and ubiquitination buffer (50 mm Tris-HCl, pH7.5, 5 mm MgCl2, 2 mm NaF, 5 mm ATP, 0.6 mm DTT, 50 mm KCl). The reactions were stopped by 1× protein loading buffer and then subjected to SDS-PAGE.
Laser Microirradiation and Live-cell Imaging
DSBs were generated in live-cell nuclei by laser-induced microirradiation using a picosecond short-pulsed green laser (a diode-pumped second harmonic 532-nm Nd:YAG laser microbeam with 76-MHz repetition rate, 12-ps pulse duration) coupled to a Zeiss Axiovert microscope for live cell, time lapse image capture (Laboratory of Dr. Michael W. Berns, University of California at San Diego, La Jolla, CA (52)). The average laser power used for DNA cutting was 16 milliwatts (post-objective), and the total energy delivered per focused laser spot was 480 mJ. Fluorescence intensities of the laser microirradiated areas were calculated using ImageJ software (National Institutes of Health), with the cellular background fluorescence intensity subtracted from the laser-induced damage site intensity. Each data point is the average of 10 independent measurements.
DSB Repair Assays and Fluorescence-activated Cell Sorting (FACS) Analysis
EGFP-based DSB repair substrates for HR and MMEJ were previously described (11). I-SceI was expressed by retroviral infection of HA-I-SceI, and 7 days later, cells were collected for FACS analysis of EGFP-positive events. FACS analysis was performed using a BD Accuri C6 flow cytometer and accompanying data analysis software (BD Biosciences).
Mass Spectrometry Analysis
Nbs1 was first ubiquitinated by the in vitro ubiquitination reactions described above. The ubiquitinated Nbs1 sample was denatured in urea and then reduced and alkylated with tris(2-carboxyethyl)phosphine hydrochloride (Roche Applied Science) and chloroacetamide (Sigma-Aldrich), respectively. The sample was then digested overnight with trypsin (Promega) according to the manufacturer's specifications.
The protein digest was pressure-loaded onto a 250-μm inner diameter fused silica capillary (Polymicro Technologies) column with a Kasil frit packed with 3 cm of 5-μm C18 resin (Phenomenex). After desalting, the loading column was connected to a 100-μm inner diameter fused silica capillary (Polymicro Technologies) analytical column with a 5-μm pulled tip, packed with 10 cm of 5-μm C18 resin (Phenomenex).
The column was placed in line with an 1100 quaternary HPLC pump (Agilent Technologies), and the eluted peptides were electrosprayed directly into an LTQ Orbitrap Velos mass spectrometer (Thermo Scientific). The buffer solutions used were 5% acetonitrile, 0.1% formic acid (buffer A), 80% acetonitrile, 0.1% formic acid (buffer B); and 500 mm ammonium acetate, 5% acetonitrile, 0.1% formic acid (buffer C). A six-step MudPIT was run with salt pulses of 0, 20, 40, 70, and 100% buffer C and 90% buffer C, 10% buffer B. The 120-min elution gradient had the following profile: 10% buffer B beginning at 10 min to 45% buffer B at 90 min to 100% buffer B from 100 min to 110 min. A cycle consisted of one full scan mass spectrum (300–1600 m/z) followed by 20 data-dependent collision-induced dissociation MS/MS spectra. Application of mass spectrometer scan functions and HPLC solvent gradients was controlled by the Xcalibur data system (Thermo Scientific).
MS/MS spectra were extracted using RawXtract (version 1.9.9) (53). MS/MS spectra were searched with the Sequest algorithm (54) against a human IPI database concatenated to a decoy database in which the sequence for each entry in the original database was reversed (55). The Sequest search was performed using full enzyme specificity, static modification of cysteine due to carboxyamidomethylation (57.02146), and differential modification of lysine due to ubiquitination (114.04296). Sequest search results were assembled and filtered using the DTASelect (version 2.0) algorithm (56). The peptide identification false positive rate was kept below 1%, and all peptide spectrum matches had less than 10 ppm mass error.
RESULTS
The MRN Complex Interacts with RNF8
Because both Nbs1 and RNF8 are recruited by MDC1 to DSBs through a direct interaction with MDC1 (17–23, 36–38), we asked whether RNF8 may possibly associate with MRN. Co-immunoprecipitation (co-IP) showed that FLAG-RNF8 interacts with Myc-tagged Nbs1, Mre11, or Rad50 when they were expressed in 293T cells (Fig. 1A), suggesting that RNF8 interacts with the MRN complex. However, these results cannot distinguish which component of MRN interacts with RNF8, because overexpressed Myc-Nbs1, Myc-Mre11, or Myc-Rad50 can form a complex with endogenous counterparts. When Nbs1 and RNF8 were co-expressed in insect cells, their interaction was also observed (supplemental Fig. 1). We then purified Nbs1 and the Mre11-Rad50 complex from insect cells and showed that purified Nbs1 but not Mre11-Rad50 interacts with RNF8 (Fig. 1B). These data suggest that RNF8 binds to MRN through a direct interaction with Nbs1.
FIGURE 1.

The MRN complex interacts with RNF8 through Nbs1. A, 293T cells were transfected with FLAG-RNF8 and Myc-Nbs1 or Myc-Mre11 (left) or FLAG-RNF8 and Myc-Rad50 (right). 48 h after transfection, whole cell lysates were collected and immunoprecipitated with anti-Myc antibody, followed by immunoblotting. B, purified Nbs1 (left) or purified Mre11-Rad50 complex (right) proteins were incubated with GST-RNF8 coupled to glutathione-agarose beads and probed with antibodies as indicated. C, 293T cells were co-transfected with FLAG-RNF8 and Myc-tagged Mre11, Rad50, and Nbs1 (MRN). Transfected cells were treated with 10 Gy of IR. 1 h later, whole cell lysates were collected for imunoprecipitation with anti-Myc antibody and probed with antibodies as indicated. D, 293T cells were treated with 10 Gy of IR, and 1 h later, whole cell lysates were collected for immunoprecipitation with anti-RNF8 antibody or control rabbit anti-mouse IgG (RαM). Immunoblotting was performed using anti-Nbs1, anti-Mre11, and anti-RNF8 antibodies. E, 293T cells were transfected with Myc-Nbs1 and FLAG-RNF8 WT, FLAG-RNF8-R42A FHA mutant, or mock (−). Whole cell lysates were immunoprecipitated with anti-Myc antibody, and immunoblotting was performed.
To examine whether the Nbs1 and RNF8 interaction is regulated upon DNA damage, we performed co-IP of RNF8 and MRN before and after IR. Neither overexpressed MRN nor endogenous MRN showed a difference in association with RNF8 before and after IR in 293T cells and U2OS cells (Fig. 1, C and D) (data not shown). These data suggest that MRN interacts with RNF8 in the absence of DNA damage and that IR-induced damage does not significantly regulate their interactions. These data also suggest that MRN and RNF8 can interact before they are recruited by MDC1 to DSBs. This is further supported by the observation that the FHA mutant of RNF8, RNF8-R42A, defective in binding to MDC1 at ATM phosphorylation sites (17, 36–38), does not show a difference in its interaction with Nbs1 (Fig. 1E).
Nbs1 Is Ubiquitinated by RNF8
The direct interaction of Nbs1 with RNF8 prompted us to investigate whether Nbs1 is ubiquitinated by RNF8. Ubiquitination of Nbs1 was readily detected before and after DNA damage (Fig. 2A), which is different from the previous report that Nbs1 ubiquitination is triggered by IR treatment (57). The Nbs1-K686A/K690A (KK) mutant, which was defective in binding with Mre11 (48), is as effectively ubiquitinated as wild-type Nbs1 (Fig. 2B), suggesting that formation of the MRN complex is not necessarily required for RNF8 to ubiquitinate Nbs1.
FIGURE 2.

Nbs1 is ubiquitinated by RNF8. A, in vivo ubiquitination assay of 293T cells transfected with HA-Ub and Myc-Nbs1 and treated with or without 10 Gy of IR. After a 1-h recovery, cells were lysed and immunoprecipitated with anti-Myc antibody. Ubiquitinated Nbs1 was probed with anti-HA antibody. B, in vivo ubiquitination assay of 293T cells transfected with HA-Ub and Myc-Nbs1 WT, K686A/K690A mutant (KK), or mock (−). Cells were lysed and subjected to immunoprecipitation with anti-Myc antibody. Ubiquitinated Nbs1 was probed with anti-HA antibody. C, left, in vivo ubiquitination assay. U2OS cells were transfected with Myc-Nbs1 and HA-ubiquitin, followed by retroviral infection of RNF8 shRNA. 40 h later, cells were treated with or without 10 Gy of IR. After a 1-h recovery, cells were lysed, immunoprecipitated with anti-Myc antibody, and then probed with anti-HA antibody for ubiquitinated Nbs1. Right, Western blot shows silencing of endogenous RNF8 by shRNAs (*, nonspecific band), with Ku70 used as a loading control. D, in vitro ubiquitination assay. Recombinant Nbs1 protein was purified from Sf9 cells and added in combination with Ub machinery components (HA-Ub, E1, UbcH5C, Ubc13, and RNF8 as indicated). After a 1-h incubation, samples were boiled in SDS sample buffer and subjected to immunoblotting with anti-HA antibody to show ubiquitinated Nbs1. E, in vitro ubiquitination assay. Recombinant Nbs1 protein was added in combination with Ub machinery components (HA-ubiquitin, E1, UbcH5C, and RNF8-WT or RNF8-C403S as indicated). After a 1-h incubation, samples were boiled in SDS sample buffer and subjected to immunoblotting with anti-HA antibody for revealing Nbs1 ubiquitination. F, in vivo ubiquitination assay of 293T cells transfected with Myc-Nbs1 and HA-ubiquitin (WT, K6R, K48R, or K63R mutant) plasmids. Cells were lysed and immunoprecipitated with anti-Myc antibody. Ubiquitinated Nbs1 was shown by anti-HA immunoblotting. IB, immunoblot.
To show whether RNF8 contributes to Nbs1 ubiquitination, we examined Nbs1 ubiquitination with or without suppression of RNF8 by shRNA. Nbs1 ubiquitination was significantly reduced before and after IR when RNF8 was depleted (Fig. 2C), suggesting that RNF8 is involved in ubiquitination of Nbs1. In vitro ubiquitination using purified proteins further confirmed that Nbs1 is a substrate of RNF8 (Fig. 2D). Consistent with this, the RNF8 RING domain mutant RNF8-C403S was defective in Nbs1 ubiquitination (Fig. 2E). Interestingly, we observed that Nbs1 ubiquitination was much stronger in the presence of E2 UbcH5C than with Ubc13 (Fig. 2D). To identify the ubiquitin linkage of Nbs1 ubiquitination, we examined different types of ubiquitin mutants (K6R, K48R, and K63R). Expression of HA-Ub K6R but not K48R or K63R reduced Nbs1 ubiquitination (Fig. 2F), suggesting that Lys-6-linked ubiquitin chain formation is one of the major types of Nbs1 ubiquitination. UbcH5C has been shown to support formation of Lys-6 ubiquitin linkages catalyzed by BRCA1-BARD1 (29). Collectively, these data suggest that RNF8 promotes ubiquitination of Nbs1, likely with E2 UbcH5C.
RNF8-mediated Ubiquitination of MRN Is Independent of MDC1
We showed that the RNF8 FHA mutant RNF8-R42A defective in MDC1 binding interacts normally with Nbs1 (Fig. 1E), suggesting that the interaction of RNF8 with MDC1 is not required for the Nbs1 and RNF8 interaction, which can occur before Nbs1 is recruited to DSB-surrounding areas by MDC1. We further showed that the RNF8-R42A mutant effectively ubiquitinated Nbs1 as wild-type RNF8 (Fig. 3A). In addition, ubiquitination of Nbs1 was not changed when the expression of MDC1 or H2AX was suppressed (Fig. 3B). These studies suggest that ubiquitination of Nbs1 by RNF8 can occur without the recruitment of Nbs1 and RNF8 by MDC1 to DSB-flanking regions upon H2AX phosphorylation.
FIGURE 3.

RNF8-mediated ubiquitination of Nbs1 is independent of MDC1. A, in vivo ubiquitination assay. U2OS cells stably expressing FLAG-tagged RNF8-WT or RNF8-R42A FHA mutant, with endogenous RNF8 silenced by shRNAs, were transfected with Myc-Nbs1 and HA-ubiquitin. 40 h later, cells were lysed, immunoprecipitated with anti-Myc antibody, and then probed with anti-HA antibody for revealing ubiquitinated Nbs1. B, in vivo ubiquitination assay. U2OS cells were transfected with Myc-Nbs1 and HA-ubiquitin and then retrovirally infected with or without MDC1 or H2AX shRNA viruses. 40 h later, cells were lysed, immunoprecipitated with anti-Myc antibody, and then probed with anti-HA antibody for showing ubiquitinated Nbs1. Immunoblots show silencing of H2AX and MDC1, with Ku70 used as a loading control. C, top, schematic drawing of RNF8 protein domain structure, including FHA and RING domains. Bottom, in vivo ubiquitination assay of 293T cells transfected with Myc-Nbs1 or FLAG-tagged RNF8 (full-length 1–485, N-terminal deletion 148–485, C-terminal deletion 1–400, or combined N-terminal and C-terminal deletion 148–400) plasmids. Cells were lysed and immunoprecipitated with anti-Myc antibody, followed by immunoblotting with the indicated antibodies. *, nonspecific band. IB, immunoblot.
To identify which regions of RNF8 mediate its interaction with Nbs1, we deleted either or both the N terminus and C terminus of RNF8. The N-terminal deletion mutant RNF8(148–485) and the N- and C-terminal double deletion mutant RNF8(148–400) failed to bind to Nbs1, whereas deletion of the C terminus of RNF8 (RNF8(1–400)) did not change the binding with Nbs1 (Fig. 3C). These studies suggest that the interaction of RNF8 with Nbs1 is mediated by the N terminus of RNF8.
Nbs1 Lys-435 Is a Key Residue Ubiquitinated by RNF8
To identify which residues of Nbs1 are ubiquitinated by RNF8, we performed mass spectrometry analysis on Nbs1 recovered from the RNF8-mediated in vitro ubiquitination assays. Nbs1 peptides containing ubiquitinated Lys-435 and Lys-502 were recovered (Fig. 4A). To examine whether these two sites are indeed ubiquitinated in vivo, we generated and expressed Nbs1-K435R, -K502R, and -K435R/K502R mutants in 293T cells and showed that Nbs1 ubiquitination was significantly reduced in the Nbs1-K435R mutant, with a minor reduction in the Nbs1-K502R mutant (Fig. 4B). Combining K435R and K502R mutations further reduced the level of Nbs1 ubiquitination. In vitro ubiquitination assays using purified proteins further confirmed that mutating Lys-435 on Nbs1 impaired RNF8-mediated ubiquitination of Nbs1 (Fig. 4C). These studies suggest that Nbs1 Lys-435 is a key residue ubiquitinated by RNF8.
FIGURE 4.
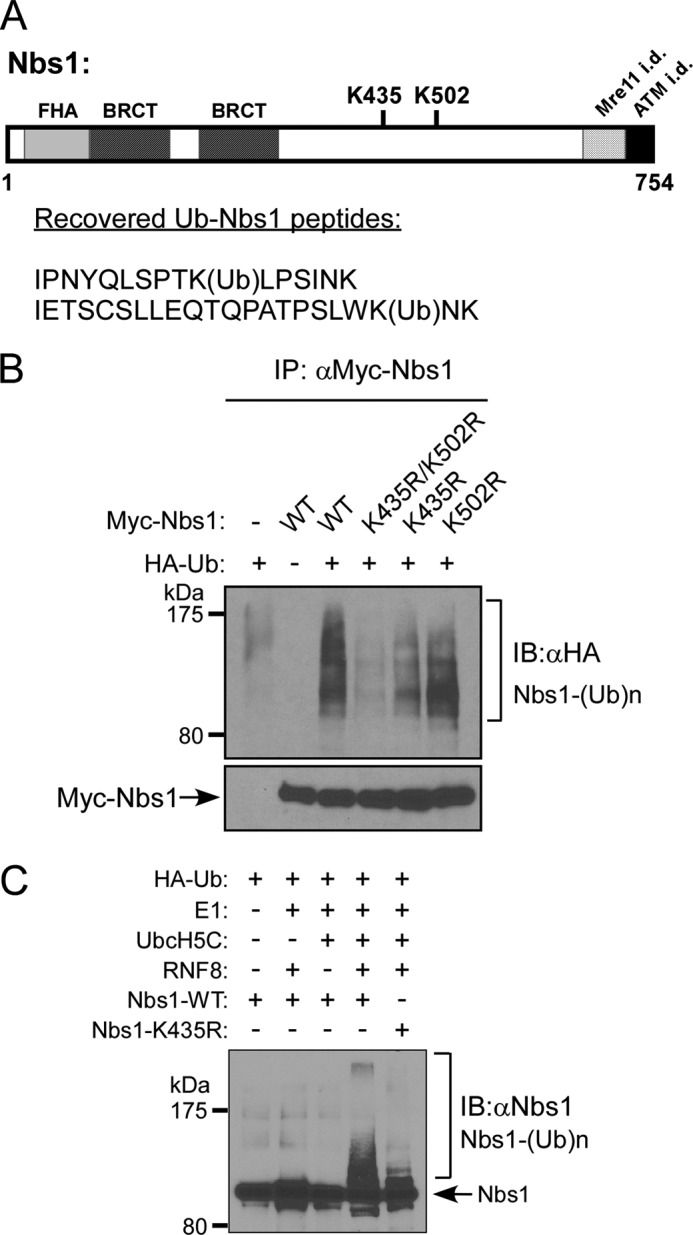
The interaction of Nbs1 with RNF8 is important for RNF8 to ubiquitinate Nbs1 at key residue Lys-435. A, ubiquitinated Nbs1 was subjected to mass spectrometry analysis, as described under “Experimental Procedures.” Recovered peptides reveal two putative RNF8 ubiquitination sites, Lys-435 (K435) and Lys-502 (K502), indicated within the schematic drawing of the Nbs1 protein domain structure, which includes the FHA domain, BRCT domains, and Mre11- and ATM-interacting domains (i.d.). B, in vivo ubiquitination assay of 293T cells transfected with HA-ubiquitin and Myc-tagged Nbs1 (WT, K435R/K502R, K435R, or K502R) plasmids. Cells were lysed and immunoprecipitated with anti-Myc antibody. Ubiquitinated Nbs1 was detected by anti-HA immunoblotting. C, in vitro ubiquitination assay. Recombinant Nbs1 (WT or K435R) proteins were purified from Sf9 cells and added in combination with Ub machinery components (HA-ubiquitin, E1, UbcH5C, and RNF8). After a 1-h incubation, samples were boiled in SDS sample buffer and subjected to immunoblotting with anti-HA antibody. Ubiquitinated Nbs1 was probed as indicated. IB, immunoblot.
RNF8 Promotes Efficient Recruitment of Nbs1 to DSB-flanking Chromatin Regions
Although the Nbs1 interaction with RNF8 and Nbs1 ubiquitination by RNF8 are not regulated by DNA damage, such interaction and ubiquitination may facilitate Nbs1 function to repair DSBs upon their recruitment to DSBs after DNA damage. We examined whether Nbs1 ubiquitination by RNF8 may modulate Nbs1 recruitment to DSBs. We utilized laser microirradiation to generate DSBs (52) and performed live-cell, time-lapse imaging to monitor EGFP-Nbs1 recruitment to the damage sites. Although it was described that Nbs1 is recruited to DSBs when RNF8 is deficient (17), our quantitative analysis revealed that Nbs1 localization to DSBs was reduced even at the early times after laser treatment when endogenous RNF8 was silenced by stably expressing RNF8 shRNAs marked by EBFP (Fig. 5A). This suggests that RNF8 may promote and/or stabilize the recruitment of Nbs1 to DSBs. Inactivation of Mre11 or Nbs1 also significantly reduced RNF8 recruitment to DSBs (supplemental Fig. 2), which is probably due to impaired ATM activation in Mre11- or Nbs1-deficient cells causing impaired H2AX phosphorylation and recruitment of RNF8 and MDC1 to chromatin-flanking DSB regions (15).
FIGURE 5.

RNF8 promotes efficient recruitment of Nbs1 to DSB-flanking chromatin regions. A, U2OS cells stably expressing EBFP-marked RNF8 shRNAs or control (MKO-EBFP) were transfected with EGFP-Nbs1 and subjected to laser-induced microirradiation to generate DSBs. Live-cell imaging was performed at the indicated time points. The absolute intensity of EGFP-Nbs1 recruitment was determined (described under “Experimental Procedures”), with error bars representing S.D. Representative cell images show recruitment of EGFP-Nbs1 to damage sites, with red lines showing the laser-cutting path in precut cells and red arrows indicating localization to damage sites (middle). The immunoblot shows silencing of endogenous RNF8 by EBFP-marked shRNAs (*, nonspecific band), with Ku70 used as a loading control. B and C, U2OS cells stably expressing FLAG-RNF8 WT, RING-domain mutant C403S (B) or FHA domain mutant R42A (C), with endogenous RNF8 silenced by EBFP-marked shRNAs, were transfected with EGFP-Nbs1. Laser microirradiation, live cell imaging, and quantitation of EGFP-Nbs1 recruitment were performed as in A. The red lines show the laser-cutting path in precut cells, and red arrows show localization to damage sites. Western blot shows expression of FLAG-RNF8 variants, with Ku70 used as a loading control.
We then examined Nbs1 recruitment in the context of RNF8 mutants. U2OS cells were stably expressed with shRNA-resistant FLAG-tagged RNF8 wild-type (WT) or the RING domain point mutant (C403S), impaired in its ubiquitination function, and endogenous RNF8 was depleted by shRNAs (RNF8sh). Upon laser-induced microirradiation, Nbs1 recruitment was decreased in cells expressing the RNF8 RING mutant C403S, compared with cells reconstituted with RNF8-WT (Fig. 5B), suggesting that the ubiquitination activity of RNF8 is important for efficient recruitment of Nbs1 to DSBs.
We showed that deletion of the C terminus of RNF8 (deletion of amino acids 401–485) did not influence the interaction of Nbs1 with RNF8 (Fig. 3C). Because this deletion still contains the RING domain of RNF8, this result suggests that the ubiquitination activity of RNF8 is not required for regulating the Nbs1 and RNF8 interaction. Thus, impaired Nbs1 recruitment to DSBs in the presence of RNF8 RING mutant is not due to modulation of Nbs1 and RNF8 interactions by RNF8-mediated ubiquitination of Nbs1.
RNF8 is recruited to DSBs by MDC1 through the binding of its FHA domain with MDC1 (17, 36–38). Because the interaction of Nbs1 with RNF8 and the ubiquitination of Nbs1 by RNF8 can occur before DNA damage, we asked whether the physical presence of RNF8 at DSB-flanking sites is needed for Nbs1 recruitment. We monitored Nbs1 recruitment to DSBs in the RNF8-R42A FHA mutant cells with endogenous RNF8 silenced by RNF8sh. Nbs1 recruitment to DSBs was reduced in the RNF8-R42A FHA mutant cells compared with wild-type RNF8 cells (Fig. 5C). These data suggest that the recruitment of RNF8 to DSB-flanking sites is important for stable binding of Nbs1 to DSBs. Therefore, although the interaction of Nbs1 with RNF8 and ubiquitination of Nbs1 by RNF8 occur at similar levels before and after DNA damage, stable binding of Nbs1 to chromosomal DSBs may require the Nbs1 and RNF8 interaction and active ubiquitination of Nbs1 by RNF8 at DSB-surrounding regions.
RNF8-mediated Ubiquitination of Nbs1 Is Important for Efficient Recruitment of Nbs1 to DSBs
Because the ubiquitination activity of RNF8 is important for efficient recruitment of Nbs1 to DSBs, we asked whether RNF8-mediated Nbs1 ubiquitination is important for Nbs1 DSB recruitment. Interestingly, the recruitment of Nbs1 ubiquitination mutants Nbs1-K435R and Nbs1-K435R/K502R was decreased, as compared to Nbs1-WT (Fig. 6A). The recruitment of Mre11 to DSBs is also defective in the Nbs1-K435R and Nbs1-K435R/K502R mutants (supplemental Fig. 3). This suggests that RNF8-mediated Nbs1 ubiquitination is important for promoting MRN recruitment at DSBs.
FIGURE 6.

RNF8-mediated ubiquitination of Nbs1 is important for efficient recruitment of Nbs1 to DSBs. A, U2OS cells were transfected with EGFP-Nbs1 WT or Nbs1 ubiquitination mutants (K435R/K502R or K435R), with endogenous Nbs1 silenced by EBFP-marked shRNAs. Laser microirradiation and live-cell imaging were performed at the indicated time points, and the absolute intensities of EGFP-Nbs1 recruitments were determined, with error bars representing S.D. B, 293T cells were transfected with HA-MDC1 and Myc-tagged Nbs1 (WT, K435R/K502R, or K435R) plasmids. Whole cell lysates were immunoprecipitated with anti-Myc antibody, and immunoblotting was performed as indicated.
To understand how Nbs1 ubiquitination may contribute to Nbs1 recruitment to DSBs, we examined whether Nbs1 ubiquitination by RNF8 changes the interaction of Nbs1 with MDC1. Myc-tagged Nbs1 and HA-tagged MDC1 were co-expressed in 293T cells, and co-immunoprecipitation revealed that the interaction of Nbs1 with MDC1 was not changed when Lys-435 or Lys-435 and Lys-502 were mutated (Fig. 6B). These data suggest that RNF8-mediated ubiquitination of Nbs1 is not involved in the regulation of the interaction of Nbs1 with MDC1. Thus, the recruitment defects of Nbs1 ubiquitination mutants are not caused by the modulation of Nbs1 with MDC1 interaction.
RNF8-mediated Ubiquitination of Nbs1 Is Important for HR
It has been shown that MRN is important for both HR and MMEJ (7–9, 58). Whereas HR requires extensive end resection, MMEJ only needs limited end resection at DSBs. We utilized EGFP-based repair assays for HR and MMEJ, as described previously (11) (supplemental Fig. 4). U2OS cells with a single integrated copy of the HR or MMEJ repair substrate were silenced for endogenous RNF8 by shRNA. I-SceI was induced by retroviral infection, followed by FACS analysis of EGFP-positive events to assay for DSB repair by HR or MMEJ (Fig. 7A). Interestingly, different from the MRN complex, when the RNF8 expression was suppressed by shRNA, HR was impaired, but MMEJ was not reduced but rather increased. These data suggest that RNF8 is important for HR but dispensable for MMEJ. Increased MMEJ is either due to the fact that MMEJ is used as an alternative repair pathway when HR is deficient or that RNF8 plays a role in suppressing MMEJ.
FIGURE 7.
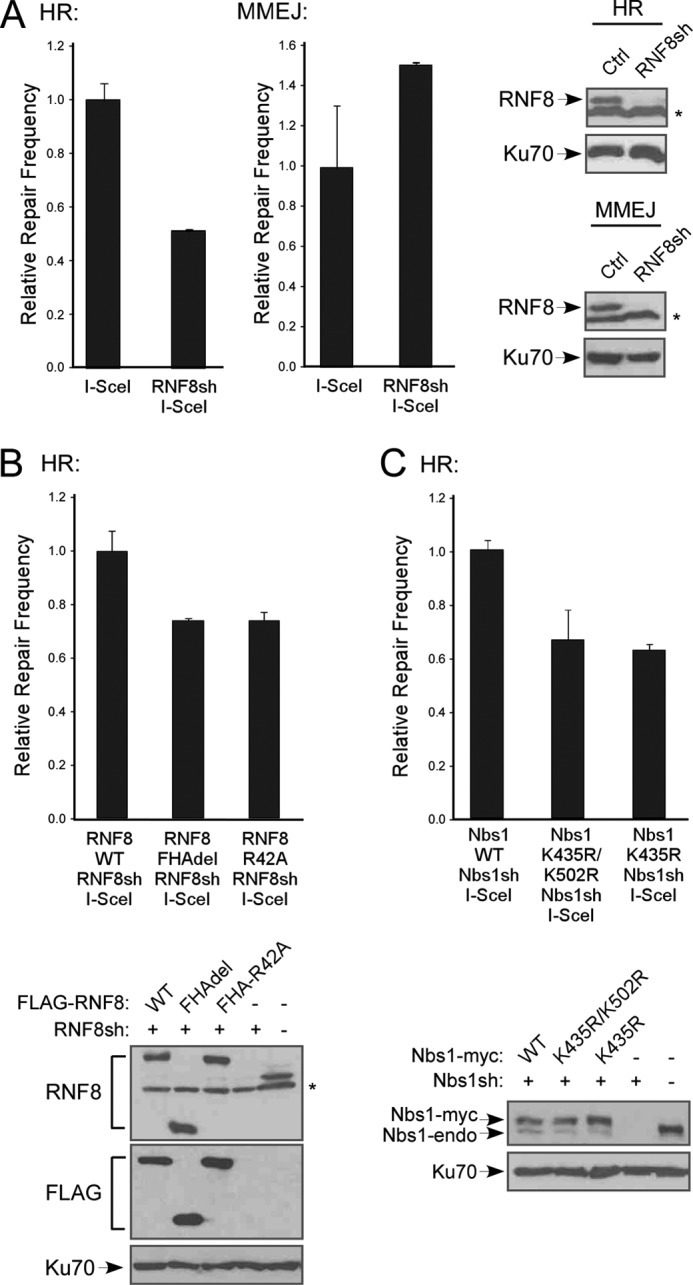
RNF8-mediated ubiquitination of Nbs1 promotes DSB repair by HR. A, U2OS cells were stably integrated with EGFP-based repair substrates for HR and MMEJ (described previously (11)) and stably expressing shRNAs against RNF8 or control. I-SceI was induced by retroviral infection, and 7 days later, cells were collected for FACS analysis of EGFP-positive cells. The immunoblot shows silencing of RNF8 (*, nonspecific band), with Ku70 used as a loading control. B, U2OS cells carrying HR repair substrate were stably expressed with FLAG-RNF8 WT or the indicated mutants, with endogenous RNF8 silenced by RNF8sh. I-SceI was induced and FACS analysis was performed. Immunoblot shows expression of RNF8 variants, with Ku70 used as a loading control. C, U2OS cells carrying HR repair substrate were stably expressed with Nbs1-Myc WT or the indicated mutants, with endogenous Nbs1 silenced by Nbs1sh. I-SceI was induced, and FACS analysis was performed. Immunoblot shows expression of Nbs1 variants, with Ku70 used as a loading control. Error bars, S.D.
We then examined the RNF8 FHA domain mutant RNF8-R42A and FHA deletion mutant (RNF8-FHAdel) in the HR repair assay. I-SceI expression was induced in U2OS cells carrying the HR repair substrate stably expressing FLAG-tagged RNF8 WT or the R42A and FHAdel mutants with endogenous RNF8 silenced by shRNAs. HR frequency was decreased in cells reconstituted with RNF8-R42A or RNF8-FHAdel mutants, as compared with RNF8-WT (Fig. 7B). These data suggest that the RNF8 FHA domain, possibly through mediating RNF8 recruitment to DSBs by MDC1, is important for HR.
To further explore how RNF8-mediated ubiquitination of Nbs1 is involved in DSB repair, we examined the Nbs1 ubiquitination mutants K435R and K435R/K502R in the HR assay. I-SceI was expressed in U2OS cells carrying the HR repair assay substrates and stably expressing Nbs1-Myc WT or ubiquitination mutants, with endogenous Nbs1 silenced by shRNA. The HR frequency was decreased in cells expressing Nbs1 ubiquitination mutants (Fig. 7C). Thus, RNF8-mediated ubiquitination of Nbs1 is important for MRN function in HR. We also found that IR-induced RPA foci formation was reduced in the Nbs1-K435R and Nbs1-K435R/K502R mutants (supplemental Fig. 5), suggesting that RNF8-mediated ubiquitination of Nbs1 is important for promoting end resection.
DISCUSSION
Accumulating evidence suggests a critical role of ubiquitination in mediating DNA damage responses (25, 59). Upon DNA damage, MDC1 is recruited to DSBs by γH2AX through a direct interaction of its BRCT domains with γH2AX (16). Both RNF8 and Nbs1 interact with MDC1, and such interactions are important for RNF8 and Nbs1 to be recruited to DSB-flanking regions (17–23, 36–38). By co-immunoprecipitation, we demonstrate that RNF8 interacts with MRN, and this interaction remains at similar levels before and after DNA damage. By using purified Nbs1, Mre11, and Rad50, we showed that RNF8 directly interacts with Nbs1 through the N terminus of RNF8. Because the interactions of RNF8 and Nbs1 remain at similar levels before and after DNA damage, the recruitment of RNF8 and Nbs1 by MDC1 to DSBs upon DNA damage is not necessarily required for establishing the Nbs1 and RNF8 interaction. Consistently, the interaction of Nbs1 and RNF8 is not changed in the RNF8 FHA mutant R42A (Fig. 1E), which is defective in binding to MDC1 and being recruited to DSBs (17, 36–38). Thus, RNF8 is associated with MRN constitutively before and after MRN is recruited to DSBs.
We further demonstrate that Nbs1 is ubiquitinated by RNF8. During the preparation of this manuscript, damage-induced Nbs1 ubiquitination by Skp2 was reported (57). However, different from the published data, we observed similar ubiquitination levels of Nbs1 before and after DNA damage in multiple cell lines. When RNF8 expression is suppressed by shRNAs, Nbs1 ubiquitination is reduced in vivo. Furthermore, in vitro ubiquitination assays showed that RNF8 but not its RING domain mutant can directly ubiquitinate Nbs1, suggesting that Nbs1 is a direct substrate of RNF8 for ubiquitination. By performing mass spectrometry analysis, we identified two new ubiquitination sites, Lys-435 and Lys-502, on Nbs1. Mutating Lys-435 significantly reduces Nbs1 ubiquitination, whereas mutating Lys-502 only has a minor effect. These data suggest that Lys-435 is a key site for Nbs1 ubiquitination. In vitro analysis showed that the Nbs1-K435R mutant is defective in RNF8-mediated ubiquitination, indicating that Lys-435 is an important site ubiquitinated by RNF8. Constitutive ubiquitination of Nbs1 detected before and after DNA damage is consistent with the observation that the interaction of RNF8 and Nbs1 is not changed in response to DNA damage.
Various E2s have been shown to interact with RNF8 to promote ubiquitination (31–35, 59). RNF8 interacts with Ubc13 to ubiquitinate H2A and H2AX at DSB sites (17, 36). By using in vitro ubiquitination assays, we found that UbcH5C exhibits much stronger activity than Ubc13 to promote RNF8-mediated ubiquitination of Nbs1, mainly through Lys-6-linked polyubiquitination. Both Lys-6- and Lys-63-based ubiquitination have been reported to be present at the DNA damage sites (40). BRCA1-BARD1 promotes Lys-6-based polyubiquitination by interacting with UbcH5C (29). Recent studies also reveal a critical function of RNF8 in promoting Lys-48-based polyubiquitination of Ku80 and Chk2 to regulate NHEJ and the checkpoint response (32). Thus, RNF8 functions with different E2s to mediate ubiquitination of multiple key players in the DNA damage response cascade to regulate DSB repair and checkpoint control.
MRN plays a critical role in DNA DSB repair and is required for both HR and MMEJ (7–9, 58, 60). Interestingly, we found that although RNF8 is needed for HR, it is dispensable for MMEJ, suggesting that MRN carries out certain DSB repair functions independent of RNF8. It also suggests an important role of RNF8 to promote error-free HR activity but not MMEJ, which is often associated with genome instability.
Although Nbs1 is still recruited to DSBs when RNF8 is defective (17), our quantitative analysis revealed that the abundance of Nbs1 recruited to DSBs is significantly reduced when RNF8 expression is suppressed or when the ubiquitination activity of RNF8 is impaired. This suggests that RNF8 and certain RNF8-mediated ubiquitination events are required for efficient recruitment of Nbs1 to DSBs.
By using Nbs1-K435R, we showed that RNF8-mediated ubiquitination of Nbs1 is important for efficient recruitment and stable association of Nbs1 with DSBs. Currently, it is not clear how RNF8-mediated ubiquitination of Nbs1 modulates Nbs1 associations with DSBs. Because deleting the C terminus of RNF8, including the RING domain, does not influence the interaction of RNF8 with Nbs1, RNF8-mediated ubiquitination of Nbs1 seems not to affect the interaction of Nbs1 with RNF8. We also showed that the K435R/K502R ubiquitination-defective Nbs1 mutant interacts with MDC1 at similar levels as wild-type RNF8. These data suggest that defective binding of Nbs1 ubiquitination mutants to DSBs is not caused by impaired interactions of Nbs1 with RNF8 or with MDC1.
We further showed that Nbs1 recruitment to DSBs is compromised in cells expressing the RNF8 FHA mutant, which is defective in localizing to DSBs due to loss of interaction with MDC1 (17, 36–38). Thus, the presence of RNF8 at DSB-flanking sites is needed for stable interaction of Nbs1 with DBSs. Two possible mechanisms may be involved. First, although Nbs1 interacts with RNF8 before and after DNA damage, such interactions at DSB sites may be important for stabilizing Nbs1 binding to DSB-flanking sites. Second, Nbs1 ubiquitination may be a dynamic process balanced by ubiquitination and deubiquitination activities. Thus, the presence of RNF8 at DSBs after DNA damage may be needed for maintaining sufficient levels of ubiquitinated Nbs1 at DSBs, although Nbs1 can be ubiquitinated by RNF8 before recruitment to DSBs. Ubiquitinated Nbs1 at DSBs is required for promoting stable Nbs1 interactions with DSBs.
How does ubiquitinated Nbs1 support more stable interactions of Nbs1 with chromosomal DSBs? One possibility is that ubiquitination modifications of Nbs1 at sites such as Lys-435 and Lys-502 may be recognized by certain damage-responsive proteins containing ubiquitin-binding domains (61, 62). When these ubiquitin-binding domain proteins are localized to DSBs upon DNA damage, they recruit more Nbs1 with them through binding to ubiquitinated Nbs1. Alternatively, ubiquitin-binding domain proteins present at DSBs may stabilize and strengthen the interaction of ubiquitinated Nbs1 with DSB-flanking chromatin regions through their binding with ubiquitinated Nbs1.
It was also described that Nbs1 is ubiquitinated by Skp2 at the C terminus of Nbs1 (sites Lys-735 and Lys-751) to promote ATM activation (57). Our studies reveal an independent pathway of Nbs1 ubiquitination by RNF8 at Lys-435 to regulate Nbs1 recruitment to DSBs. Both of these ubiquitination mechanisms are important for HR-mediated DSB repair. Thus, Nbs1 is ubiquitinated by different pathways to modulate its function in DSB repair and checkpoint activation. Multiple levels of Nbs1 ubiquitination may contribute to the regulation of different steps of HR-mediated DSB repair, leading to more efficient and precise regulation of DSB repair in response to cellular damaging signals.
In summary, our study reveals a novel interaction of Nbs1 with RNF8 and suggests a new mechanism underlying Nbs1 ubiquitination by the RNF8 pathway to promote DSB repair through HR. These studies further help our understanding of how ubiquitination is regulated to facilitate DSB repair and to maintain genome stability in mammalian cells.
Acknowledgments
We thank members of the Wu laboratory for helpful discussions. We thank Drs. Junjie Chen (University of Texas MD Anderson Cancer Center), Maria Jasin (Memorial Sloan-Kettering Cancer Center), Jiri Lukas (Institute of Cancer Biology, Denmark), Tanya Paull (University of Texas, Austin, TX), Manuel Stucki (University of Zurich), P. Renee Yew (University of Texas Health Science Center, San Antonio, TX), and Xiaochun Yu (University of Michigan) for kindly providing valuable reagents.
This work was supported, in whole or in part, by National Institutes of Health Grants CA102361, GM080677, CA140972, and CA102361-07S1 (to X. W.); CA80100 (to T. H.); and P41 RR011823 (to J. R. Y.). This work was also supported by the Beckman Laser Institute Inc. Foundation (to M. W. B.).

This article contains supplemental Figs. 1–5.
- HR
- homologous recombination
- NHEJ
- non-homologous end joining
- DSB
- double-strand break
- MMEJ
- microhomology-mediated end joining
- MRN
- Mre11-Rad50-Nbs1
- γ-H2AX
- phosphorylated H2AX
- IR
- ionizing radiation
- EGFP
- enhanced green fluorescent protein
- EBFP
- enhanced blue fluorescence protein
- NTA
- nitrilotriacetic acid
- Ub
- ubiquitin
- IP
- immunoprecipitation
- BRCT
- BRCA1 C terminus domain
- FHA
- forkhead-associated.
REFERENCES
- 1. Symington L. S., Gautier J. (2011) Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 45, 247–271 [DOI] [PubMed] [Google Scholar]
- 2. Pâques F., Haber J. E. (1999) Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 63, 349–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Moynahan M. E., Jasin M. (2010) Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell Biol. 11, 196–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lieber M. R. (2010) The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 79, 181–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McVey M., Lee S. E. (2008) MMEJ repair of double-strand breaks (director's cut). Deleted sequences and alternative endings. Trends Genet. 24, 529–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nussenzweig A., Nussenzweig M. C. (2007) A backup DNA repair pathway moves to the forefront. Cell 131, 223–225 [DOI] [PubMed] [Google Scholar]
- 7. Xie A., Kwok A., Scully R. (2009) Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nat. Struct. Mol. Biol. 16, 814–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rass E., Grabarz A., Plo I., Gautier J., Bertrand P., Lopez B. S. (2009) Role of Mre11 in chromosomal nonhomologous end joining in mammalian cells. Nat. Struct. Mol. Biol. 16, 819–824 [DOI] [PubMed] [Google Scholar]
- 9. Dinkelmann M., Spehalski E., Stoneham T., Buis J., Wu Y., Sekiguchi J. M., Ferguson D. O. (2009) Multiple functions of MRN in end-joining pathways during isotype class switching. Nat. Struct. Mol. Biol. 16, 808–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bennardo N., Cheng A., Huang N., Stark J. M. (2008) Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 4, e1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang H., Shao Z., Shi L. Z., Hwang P. Y., Truong L. N., Berns M. W., Chen D. J., Wu X. (2012) CtIP protein dimerization is critical for its recruitment to chromosomal DNA double-stranded breaks. J. Biol. Chem. 287, 21471–21480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. D'Amours D., Jackson S. P. (2002) The Mre11 complex. At the crossroads of DNA repair and checkpoint signalling. Nat. Rev. Mol. Cell Biol. 3, 317–327 [DOI] [PubMed] [Google Scholar]
- 13. Lee J. H., Paull T. T. (2007) Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene 26, 7741–7748 [DOI] [PubMed] [Google Scholar]
- 14. Uziel T., Lerenthal Y., Moyal L., Andegeko Y., Mittelman L., Shiloh Y. (2003) Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 22, 5612–5621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Polo S. E., Jackson S. P. (2011) Dynamics of DNA damage response proteins at DNA breaks. A focus on protein modifications. Genes Dev. 25, 409–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Stucki M., Clapperton J. A., Mohammad D., Yaffe M. B., Smerdon S. J., Jackson S. P. (2005) MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 123, 1213–1226 [DOI] [PubMed] [Google Scholar]
- 17. Mailand N., Bekker-Jensen S., Faustrup H., Melander F., Bartek J., Lukas C., Lukas J. (2007) RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell 131, 887–900 [DOI] [PubMed] [Google Scholar]
- 18. Chapman J. R., Jackson S. P. (2008) Phospho-dependent interactions between NBS1 and MDC1 mediate chromatin retention of the MRN complex at sites of DNA damage. EMBO Rep. 9, 795–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wu L., Luo K., Lou Z., Chen J. (2008) MDC1 regulates intra-S-phase checkpoint by targeting NBS1 to DNA double-strand breaks. Proc. Natl. Acad. Sci. U.S.A. 105, 11200–11205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Melander F., Bekker-Jensen S., Falck J., Bartek J., Mailand N., Lukas J. (2008) Phosphorylation of SDT repeats in the MDC1 N terminus triggers retention of NBS1 at the DNA damage-modified chromatin. J. Cell Biol. 181, 213–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Spycher C., Miller E. S., Townsend K., Pavic L., Morrice N. A., Janscak P., Stewart G. S., Stucki M. (2008) Constitutive phosphorylation of MDC1 physically links the MRE11-RAD50-NBS1 complex to damaged chromatin. J. Cell Biol. 181, 227–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Celeste A., Fernandez-Capetillo O., Kruhlak M. J., Pilch D. R., Staudt D. W., Lee A., Bonner R. F., Bonner W. M., Nussenzweig A. (2003) Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat. Cell Biol. 5, 675–679 [DOI] [PubMed] [Google Scholar]
- 23. Lukas C., Melander F., Stucki M., Falck J., Bekker-Jensen S., Goldberg M., Lerenthal Y., Jackson S. P., Bartek J., Lukas J. (2004) Mdc1 couples DNA double-strand break recognition by Nbs1 with its H2AX-dependent chromatin retention. EMBO J. 23, 2674–2683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Al-Hakim A., Escribano-Diaz C., Landry M. C., O'Donnell L., Panier S., Szilard R. K., Durocher D. (2010) The ubiquitous role of ubiquitin in the DNA damage response. DNA Repair 9, 1229–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Messick T. E., Greenberg R. A. (2009) The ubiquitin landscape at DNA double-strand breaks. J. Cell Biol. 187, 319–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Brzovic P. S., Rajagopal P., Hoyt D. W., King M. C., Klevit R. E. (2001) Structure of a BRCA1-BARD1 heterodimeric RING-RING complex. Nat. Struct. Biol. 8, 833–837 [DOI] [PubMed] [Google Scholar]
- 27. Hashizume R., Fukuda M., Maeda I., Nishikawa H., Oyake D., Yabuki Y., Ogata H., Ohta T. (2001) The RING heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by a breast cancer-derived mutation. J. Biol. Chem. 276, 14537–14540 [DOI] [PubMed] [Google Scholar]
- 28. Nishikawa H., Ooka S., Sato K., Arima K., Okamoto J., Klevit R. E., Fukuda M., Ohta T. (2004) Mass spectrometric and mutational analyses reveal Lys-6-linked polyubiquitin chains catalyzed by BRCA1-BARD1 ubiquitin ligase. J. Biol. Chem. 279, 3916–3924 [DOI] [PubMed] [Google Scholar]
- 29. Wu-Baer F., Lagrazon K., Yuan W., Baer R. (2003) The BRCA1/BARD1 heterodimer assembles polyubiquitin chains through an unconventional linkage involving lysine residue K6 of ubiquitin. J. Biol. Chem. 278, 34743–34746 [DOI] [PubMed] [Google Scholar]
- 30. Plans V., Scheper J., Soler M., Loukili N., Okano Y., Thomson T. M. (2006) The RING finger protein RNF8 recruits UBC13 for lysine 63-based self polyubiquitylation. J. Cell Biochem. 97, 572–582 [DOI] [PubMed] [Google Scholar]
- 31. Zhang S., Chea J., Meng X., Zhou Y., Lee E. Y., Lee M. Y. (2008) PCNA is ubiquitinated by RNF8. Cell Cycle 7, 3399–3404 [DOI] [PubMed] [Google Scholar]
- 32. Feng L., Chen J. (2012) The E3 ligase RNF8 regulates KU80 removal and NHEJ repair. Nat. Struct. Mol. Biol. 19, 201–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lok G. T., Sy S. M., Dong S. S., Ching Y. P., Tsao S. W., Thomson T. M., Huen M. S. (2012) Differential regulation of RNF8-mediated Lys48- and Lys63-based poly-ubiquitylation. Nucleic Acids Res. 40, 196–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bekker-Jensen S., Rendtlew Danielsen J., Fugger K., Gromova I., Nerstedt A., Lukas C., Bartek J., Lukas J., Mailand N. (2010) HERC2 coordinates ubiquitin-dependent assembly of DNA repair factors on damaged chromosomes. Nat. Cell Biol. 12, 80–86 [DOI] [PubMed] [Google Scholar]
- 35. Ito K., Adachi S., Iwakami R., Yasuda H., Muto Y., Seki N., Okano Y. (2001) N-terminally extended human ubiquitin-conjugating enzymes (E2s) mediate the ubiquitination of RING-finger proteins, ARA54 and RNF8. Eur. J. Biochem. 268, 2725–2732 [DOI] [PubMed] [Google Scholar]
- 36. Huen M. S., Grant R., Manke I., Minn K., Yu X., Yaffe M. B., Chen J. (2007) RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell 131, 901–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kolas N. K., Chapman J. R., Nakada S., Ylanko J., Chahwan R., Sweeney F. D., Panier S., Mendez M., Wildenhain J., Thomson T. M., Pelletier L., Jackson S. P., Durocher D. (2007) Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science 318, 1637–1640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang B., Elledge S. J. (2007) Ubc13/Rnf8 ubiquitin ligases control foci formation of the Rap80/Abraxas/Brca1/Brcc36 complex in response to DNA damage. Proc. Natl. Acad. Sci. U.S.A. 104, 20759–20763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kim H., Chen J., Yu X. (2007) Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science 316, 1202–1205 [DOI] [PubMed] [Google Scholar]
- 40. Sobhian B., Shao G., Lilli D. R., Culhane A. C., Moreau L. A., Xia B., Livingston D. M., Greenberg R. A. (2007) RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science 316, 1198–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang B., Matsuoka S., Ballif B. A., Zhang D., Smogorzewska A., Gygi S. P., Elledge S. J. (2007) Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science 316, 1194–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yan J., Kim Y. S., Yang X. P., Li L. P., Liao G., Xia F., Jetten A. M. (2007) The ubiquitin-interacting motif containing protein RAP80 interacts with BRCA1 and functions in DNA damage repair response. Cancer Res. 67, 6647–6656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stewart G. S., Panier S., Townsend K., Al-Hakim A. K., Kolas N. K., Miller E. S., Nakada S., Ylanko J., Olivarius S., Mendez M., Oldreive C., Wildenhain J., Tagliaferro A., Pelletier L., Taubenheim N., Durandy A., Byrd P. J., Stankovic T., Taylor A. M., Durocher D. (2009) The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell 136, 420–434 [DOI] [PubMed] [Google Scholar]
- 44. Doil C., Mailand N., Bekker-Jensen S., Menard P., Larsen D. H., Pepperkok R., Ellenberg J., Panier S., Durocher D., Bartek J., Lukas J., Lukas C. (2009) RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell 136, 435–446 [DOI] [PubMed] [Google Scholar]
- 45. Olson E., Nievera C. J., Lee A. Y., Chen L., Wu X. (2007) The Mre11-Rad50-Nbs1 complex acts both upstream and downstream of ataxia telangiectasia mutated and Rad3-related protein (ATR) to regulate the S-phase checkpoint following UV treatment. J. Biol. Chem. 282, 22939–22952 [DOI] [PubMed] [Google Scholar]
- 46. Ai H. W., Shaner N. C., Cheng Z., Tsien R. Y., Campbell R. E. (2007) Exploration of new chromophore structures leads to the identification of improved blue fluorescent proteins. Biochemistry 46, 5904–5910 [DOI] [PubMed] [Google Scholar]
- 47. Olson E., Nievera C. J., Liu E., Lee A. Y., Chen L., Wu X. (2007) The Mre11 complex mediates the S-phase checkpoint through an interaction with replication protein A. Mol. Cell Biol. 27, 6053–6067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chen L., Nievera C. J., Lee A. Y., Wu X. (2008) Cell cycle-dependent complex formation of BRCA1·CtIP·MRN is important for DNA double-strand break repair. J. Biol. Chem. 283, 7713–7720 [DOI] [PubMed] [Google Scholar]
- 49. Wu X., Avni D., Chiba T., Yan F., Zhao Q., Lin Y., Heng H., Livingston D. (2004) SV40 T antigen interacts with Nbs1 to disrupt DNA replication control. Genes Dev. 18, 1305–1316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wu X., Ranganathan V., Weisman D. S., Heine W. F., Ciccone D. N., O'Neill T. B., Crick K. E., Pierce K. A., Lane W. S., Rathbun G., Livingston D. M., Weaver D. T. (2000) ATM phosphorylation of Nijmegen breakage syndrome protein is required in a DNA damage response. Nature 405, 477–482 [DOI] [PubMed] [Google Scholar]
- 51. Paull T. T., Gellert M. (1998) The 3′ to 5′ exonuclease activity of Mre 11 facilitates repair of DNA double-strand breaks. Mol. Cell 1, 969–979 [DOI] [PubMed] [Google Scholar]
- 52. Botvinick E. L., Berns M. W. (2005) Internet-based robotic laser scissors and tweezers microscopy. Microsc. Res. Tech. 68, 65–74 [DOI] [PubMed] [Google Scholar]
- 53. McDonald W. H., Tabb D. L., Sadygov R. G., MacCoss M. J., Venable J., Graumann J., Johnson J. R., Cociorva D., Yates J. R., 3rd (2004) MS1, MS2, and SQT. Three unified, compact, and easily parsed file formats for the storage of shotgun proteomic spectra and identifications. Rapid Commun. Mass Spectrom. 18, 2162–2168 [DOI] [PubMed] [Google Scholar]
- 54. Eng J. K., Mccormack A. L., Yates J. R., 3rd (1994) An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5, 976–989 [DOI] [PubMed] [Google Scholar]
- 55. Peng J., Elias J. E., Thoreen C. C., Licklider L. J., Gygi S. P. (2003) Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis. The yeast proteome. J. Proteome Res. 2, 43–50 [DOI] [PubMed] [Google Scholar]
- 56. Tabb D. L., McDonald W. H., Yates J. R., 3rd (2002) DTASelect and contrast. Tools for assembling and comparing protein identifications from shotgun proteomics. J. Proteome Res. 1, 21–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wu J., Zhang X., Zhang L., Wu C. Y., Rezaeian A. H., Chan C. H., Li J. M., Wang J., Gao Y., Han F., Jeong Y. S., Yuan X., Khanna K. K., Jin J., Zeng Y. X., Lin H. K. (2012) Skp2 E3 ligase integrates ATM activation and homologous recombination repair by ubiquitinating NBS1. Mol. Cell 46, 351–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Buis J., Wu Y., Deng Y., Leddon J., Westfield G., Eckersdorff M., Sekiguchi J. M., Chang S., Ferguson D. O. (2008) Mre11 nuclease activity has essential roles in DNA repair and genomic stability distinct from ATM activation. Cell 135, 85–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Panier S., Durocher D. (2009) Regulatory ubiquitylation in response to DNA double-strand breaks. DNA Repair 8, 436–443 [DOI] [PubMed] [Google Scholar]
- 60. Jazayeri A., Falck J., Lukas C., Bartek J., Smith G. C., Lukas J., Jackson S. P. (2006) ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat. Cell Biol. 8, 37–45 [DOI] [PubMed] [Google Scholar]
- 61. Harper J. W., Schulman B. A. (2006) Structural complexity in ubiquitin recognition. Cell 124, 1133–1136 [DOI] [PubMed] [Google Scholar]
- 62. Hicke L., Schubert H. L., Hill C. P. (2005) Ubiquitin-binding domains. Nat. Rev. Mol. Cell Biol. 6, 610–621 [DOI] [PubMed] [Google Scholar]