

Abstract
Activating point mutations in codons 12, 13, and 61 of the KRAS proto-oncogene are common in colorectal, non–small cell lung, pancreatic, and thyroid cancers. Constitutively activated KRAS mutations are strongly associated with a resistance to anti–epidermal growth factor receptor (EGFR) therapies, such as panitumumab and cetuximab used for treating metastatic colorectal carcinoma and EGFR tyrosine inhibitors used for advanced non–small cell lung cancers. Since anti-EGFR therapies are costly and may exert deleterious effects on individuals without activating mutations, KRAS mutation testing is recommended prior to the initiation of anti-EGFR therapy for these malignancies. The goal of this review is to summarize the KRAS mutation testing methods. Testing is now routinely requested in the clinical practice to provide data to assign the most appropriate anticancer chemotherapy for each given patient. Review of the most relevant literature was performed. Several areas were considered: ordering of the test, selection of the sample to be tested, and review of the testing methodologies. We found that several different methods are used for clinical KRAS mutation testing. Each of the methodologies is described, and information is provided about their performance, cost, turnaround times, detection limits, sensitivities, and specificities. We also provided “tips” for the appropriate selection and preparation of the sample to be tested. This is an important aspect of KRAS testing for clinical use, as the results of the test will affect clinical decisions with consequences for the patient.
Keywords: KRAS mutation, KRAS testing methods, personalized medicine
Introduction
Constitutively activated KRAS mutations occur in multiple human malignancies, including approximately 90% of pancreatic, approximately 30% of lung, approximately 60% of thyroid, and approximately 43% of colorectal carcinomas (CRC).1-3 Currently, within the United States, clinical KRAS mutation testing is performed on CRC, thyroid, endometrial, pancreatic, and non–small cell lung cancers (http://www.amptestdirectory.org/index.cfm). Research on KRAS began in 1964 when it was first identified as the cause of leukemia virus–induced rat sarcoma.4 Later, the Kirsten rat sarcoma retroviral oncogene sequence was cloned and used to identify the human homolog gene, now known as KRAS or KRAS2 (Kirsten rat sarcoma virus 2 homolog).5 Constitutively activating KRAS mutations were first seen when abnormal KRAS gene sequences from human malignancies could transform NIH3T3 cells.6 Since these early studies, an enormous amount of data on KRAS gene function in normal cells and malignancies has accumulated, and KRAS sequence analysis is now a routine part of patient care. Here, we will briefly review KRAS gene function and the current molecular methods employed in clinical KRAS mutation testing.4-11
KRAS is located at 12p12.1, spans approximately 38 kb, and encodes a 188–amino acid residue with a molecular weight of 21.6 kDa. KRAS normally functions in signal transduction cascades initiated by the binding of epidermal growth factor receptor (EGFR), hepatocyte growth factor, and insulin-like growth factor to their receptors.8-11 When activated wild-type KRAS binds GTP, this results in a conformational change that allows the protein to bind and activate over 20 known downstream effectors, including Raf, Braf, mTOR, MEK1 and 2, ERK, AKT, and PIK3CA. These downstream effectors exert many different effects, including apoptosis suppression, promotion of cell growth, cell transformation, angiogenesis, migration, and differentiation.8-13 KRAS functions as a molecular binary switch that alternates between a GTP-bound “active” state and a GDP-bound “off” state, with each state having a specific molecular conformation. While KRAS has intrinsic GTPase activity, the hydrolysis rate constant is too low to be physiologically relevant, but specific “GTPase activating proteins” can increase hydrolysis roughly 100,000-fold. In turn, guanine nucleotide exchange factor proteins promote GTP binding by lowering the affinity of KRAS for bound GDP and catalyzing its replacement with GTP.12,13 Activating KRAS mutations are point mutations mostly affecting KRAS amino acid residues 12, 13, and 61, all of which decrease the intrinsic KRAS and GTPase activating protein–promoted GTP hydrolysis, resulting in constitutive KRAS activation.12,13 Interestingly, KRAS mutations occur early in the development of CRC and late in non–small cell lung cancers.14-16
Following the failure of one or more prior chemotherapeutic regimens, anti-EGFR therapies are often used for locally advanced or metastatic CRC (mCRC) and non–small cell lung cancers. The anti-EGFR antibodies, panitumumab and cetuximab, are employed in treating mCRC, while EGFR tyrosine inhibitors are used to treat lung cancers unresponsive to previous treatments. Numerous studies have demonstrated that activating KRAS mutations are strongly associated with a resistance to anti-EGFR therapies, although one recent study may have found an exception for the G13D KRAS mutation in mCRC.17-21 Consequently, the National Comprehensive Cancer Network recommends all patients with CRC or non–small cell lung cancer being considered for anti-EGFR therapy to be tested for KRAS mutations.22
Request for KRAS Testing
The clinician typically initiates KRAS testing requests for a patient. Usually, the testing is performed on tumor tissue removed from the patient during a previous surgery or biopsy procedure. Typically, patients undergoing KRAS testing are those with high-stage tumors that require adjuvant therapy. If metastatic disease is present, it is important to clarify that the sample needed for testing is not the primary tumor but rather a representative tissue sample from the metastatic lesion. DNA is usually extracted from FFPE tissue blocks. It is the pathologist’s responsibility to identify the best tumor section to be subjected to testing. This includes evaluation of the slide with cut tissue or the tissue block, followed by microdissection and macrodissection for tumor enrichment, to eliminate portions of necrotic tumor and nonneoplastic tissue. Typically, tumor-enriched areas will have relatively easily identified histology and can be dissected away from benign tissue. This process is often aided by having the fixed tissue cut and placed on an unstained slide and having a standard H&E-stained slide of the same tissue cut available for comparison. Alternatively, the tissue block may be cut, as is depicted in Figure 1A and 1B. This allows the pathologist to identify exactly where the tumor-rich areas are versus tumor-poor areas.
Figure 1.
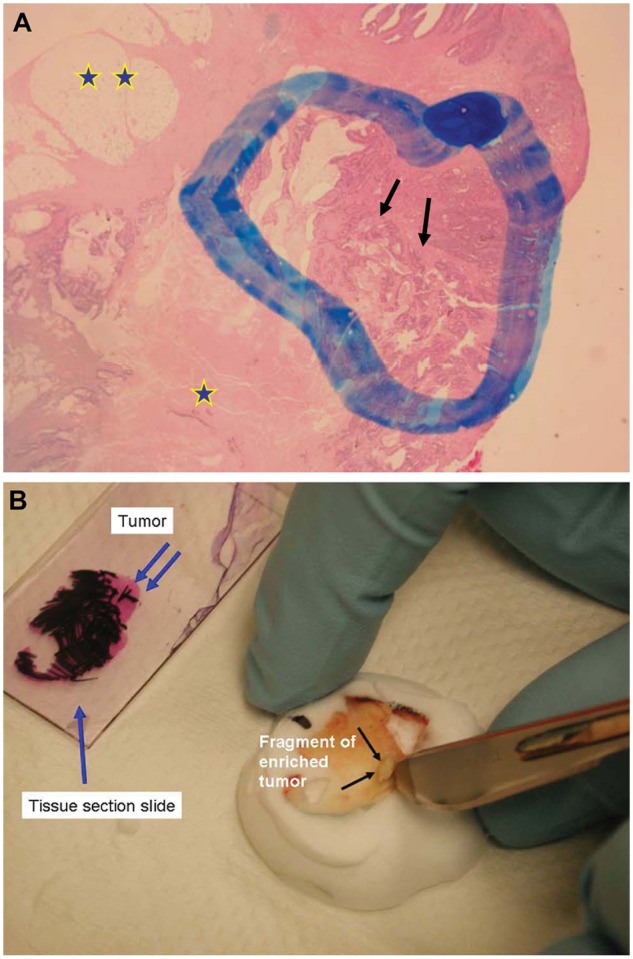
(A) The pathologist selects the tissue block most representative of the tumor. First, evaluation of the H&E slide (arrow) will allow selection of the areas most rich with the tumor (double arrow), eliminating areas of tumor necrosis (star) and of nonneoplastic tissue (double star). The tumor areas are highlighted with a marker. (B) The tissue section slide previously marked (single blue arrow) is superimposed to the corresponding paraffin block, and the tumor area is identified. The selected tumor-enriched areas may be easily cut out from the tissue block (black arrows) and sent for KRAS analysis. Alternatively, tumor-rich tissue areas (blue arrows) may be scraped off an unstained slide with a razor blade and the DNA extracted for testing.
KRAS Mutation Testing Methods
More than 60 methods have been employed in KRAS testing, most of which fall under the categories of sequencing, high-resolution melting analysis (HRM), single-strand conformation polymorphism (SSCP), denaturing gradient gel electrophoresis (DGGE), denaturing high-performance liquid chromatography (DHPLC), array/strip analysis, and allele-specific PCR. All of these techniques have been successfully applied to clinical KRAS testing, and each has its unique feature. Here, we will briefly discuss each of these techniques and compare their sensitivities, specificities, turnaround times, and costs.
Sample preparation
Clinical testing for CRC and non–small cell lung cancers is commonly performed on snap-frozen, formalin-fixed, or FFPE tissue blocks. DNA is usually isolated with commercial kits. Often, a pathologist will examine the specific areas of the slide so that only areas with viable tumor are selected for testing (Fig. 1A and 1B). Clinical testing for KRAS mutations on solid tumors can be challenging for several reasons. First, DNA from such sources requires special preparation, as 1) it can be fragmented and cross-linked to other biomolecules, making amplification difficult, especially for longer DNA sequences; 2) it may contain PCR inhibitors that must be removed before amplification; and 3) formalin fixation can introduce DNA sequence alterations.23,24 Additionally, the analysis of tumors for mutations can be complicated by a high ratio of benign stromal to tumor cells, diluting out the possibly mutant DNA with wild-type KRAS sequences. This can be especially difficult with postchemotherapy tumors and desmoplastic pancreatic tumors, where the number of tumor cells can be very low. Lastly, biopsy sizes may be small, limiting the amount of DNA available, and many tumors are KRAS heterozygous, having both mutant and wild-type KRAS, further diluting mutant DNA. For these reasons, and because false-positive or -negative results can result in grossly inappropriate patient care, KRAS solid tumor testing requires the highest sensitivity and specificity as possible. In addition, tissue samples subjected to the stringent conditions of bone decalcification do not yield intact DNA for molecular analysis.
Sequencing Methods in KRAS Testing
Sanger sequencing and pyrosequencing of PCR-amplified DNA are commonly employed in KRAS mutation analysis. Sanger sequencing of PCR-amplified genomic DNA is considered the gold-standard technique by many researchers, as it can identify all mutated base pairs and identify small insertions and deletions, and it is often used to confirm results obtained from techniques such as HRM, SSCP, and DGGE, methods that do not identify specific base changes.25,26 Both methods are DNA polymerase based, with analysis based on analyzing the termination of DNA synthesis.25-27
Sanger sequencing
Sanger sequencing is based on the synthesis of complementary DNA by DNA polymerase with 2 different sets of nucleotide bases being present: the natural 2′-deoxy form (dNTP) and 2′,3′-dideoxynucleotide (ddNTP). All 4 bases are present (A, T, G, C) in each nucleotide set. Incorporation of a ddNTP results in the termination of DNA synthesis; with the optimal ratio of dNTPs to ddNTPs, a set of nested fragments is produced, each with different nucleoside monophosphate units. By labeling each of the ddNTPs with 4 different fluorophores, the DNA sequence can be read by analyzing the fluorescent emissions as the fragments pass through a laser detection system. With automated Sanger sequencing, “raw” fluorescent signals are typically analyzed by both visual inspection and with software that removes cross-talk, corrects for variations in fragment migration, and normalizes emission intensities. Typically, calling a specific base is considered valid when the probability of error is 1% or less. The Sanger sequencing method can sequence up to 800 bases at one time, which is more than sufficient for KRAS analyses.27
Sanger sequencing is often used to analyze KRAS mutation status and has the advantage of interrogating essentially all possible base substitutions. Most commonly, PCR primers are used to amplify the genomic DNA immediately flanking known KRAS point mutations. Although this method works well, its limit of detection is relatively modest compared to other techniques used in KRAS mutation analyses. Multiple studies have found Sanger sequencing to require at least 15% to 50% of the sample DNA to be a KRAS mutant for reliable detection, which is a detection limit insufficient for clinical samples low in tumor cells or DNA. In most studies, this detection limit was calculated by increasing the percentage of wild-type KRAS DNA in a wild-type/mutant KRAS DNA mix.27-32 Specifically, samples with less than 10% tumor cells showed significantly fewer KRAS mutations than those with more than 10% tumor cells.28 Interestingly, whole-genome amplification followed by standard PCR and Sanger sequencing increased the KRAS mutation detection limit in one study but lowered it in another.32,33 The reason for this discrepancy is unknown but likely due to initial DNA quality and differences in amplication methods. Finally, one study found Sanger sequencing to have lower sensitivity in detecting base substitutions at the first position of codon 12 compared to other substitutions. It also had an 11.1% false-positive rate and 6.1% false-negative rate with an automated interpretation algorithm with a 10% threshold, indicating that manual review of all Sanger sequencing data is needed.26
Pyrosequencing
Pyrosequencing is a DNA polymerase–based sequencing method that measures the release of inorganic pyrophosphates in several reactions, the last of which creates photons from luciferase activity. Pyrosequencing analyzes the addition of a specific dNTP in limiting amounts to a reaction in DNA polymerase–mediated DNA synthesis on a complementary DNA strand immobilized on a solid support. When a complementary dNTP is added to the reaction, the DNA polymerase extends the primer and pauses when it hits a noncomplementary base. After completion of a washing step, the reaction continues following the addition of a new complementary base. The pyrophosphate released with incorporation of a dNTP is converted to ATP by sulfurylase, which is subsequently degraded by luciferin to a photon. The light is usually measured by a photomultiplier tube, avalanche photodiode, or charge-coupled device camera. After completion of a washing step, a new dNTP and other required reagents are added to the reaction. For reactions analyzing an adenine addition, dATP is added to the reaction (Fig. 2). The limit of pyrosequencing is due to the fact that it can only sequence approximately 40 to 50 bases, and it cannot accurately measure homopolymer repeats greater than 5 nucleotides.27 However, for the analysis of PCR KRAS amplicons, these constraints are usually insignificant.
Figure 2.
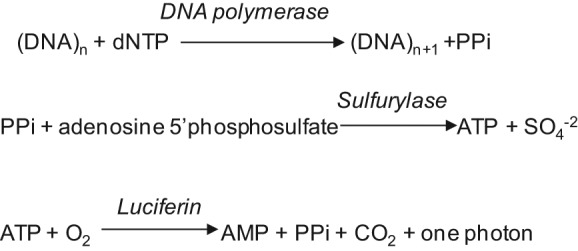
The sequencing primer binds a complementary ssDNA molecule to be sequenced. DNA polymerase is added, and each dNTP is added and removed from the reaction. When a base is added to the DNA being synthesized, PPi is released and subsequently converted to ATP by ATP sulfurylase in the presence of adenosine 5′ phosphosulfate. The ATP is used in the conversion of luciferin to oxyluciferin, generating visible light proportional to the ATP concentration. The light is usually measured by a photomultiplier tube, avalanche photodiode, or charge-coupled device camera.
Like Sanger sequencing, pyrosequencing is commonly used to analyze KRAS mutation status and is usually performed on immobilized PCR-amplified amplicons derived from genomic DNA. Most studies have demonstrated that the detection limit of pyrosequencing is approximately 1.25% to 6% mutant DNA.26,30,32,34-36 The increased sensitivity of pyrosequencing is especially useful where KRAS mutant DNA is likely to be heavily diluted in wild-type DNA, such as CRC with microsatellite instability and significant inflammatory infiltrates or desmoplastic pancreatic tumors with few malignant cells and abundant stromal cells.35 Additionally, because the detection of each pyrosequencing light emission is more uniform than the detection of different fluorophores in Sanger sequencing, interpreting low-level signals is less subjective in pyrosequencing than in Sanger sequencing. In contrast to most molecular diagnostic techniques, Sanger sequencing and pyrosequencing can both detect double KRAS mutants, although the clinical significance of double mutants is presently unknown.26 Finally, Sanger sequencing and pyrosequencing assay sensitivities can be increased by slide macrodissection for tumor enrichment (Fig. 1A).
Sequencing variations
Because sequencing technologies may lack sufficient sensitivity for some clinical samples, efforts have been made to increase assay sensitivity. For example, Arcila et al.37 employed locked nucleic acid primers (containing 2′-O, 4′-C methylene bridges, which increase nucleic acid hybridization specificity) in the initial PCR amplification of the KRAS genomic DNA. In comparison to standard Sanger sequencing, locked nucleic acid primers detected 14% more KRAS exon 2 mutations (36% v. 45%). This 14% increase in KRAS mutations was detected by analyzing 308 CRC samples previously subjected to standard oligonucleotide amplification and Sanger sequencing to the same protocol employing locked nucleic acid primers. In 308 samples, 121 were KRAS mutation positive with standard amplification, and 140 were positive with locked nucleic acid primer amplification, which is a 14% increase in detection sensitivity.37 Similarly, the same group employed matrix-assisted laser desorption/ionization time-of-flight mass spectrometry to investigate KRAS amplicon mutation status and achieved a modest 5% increase in mutation detection. Presently, Sanger sequencing is considered the “gold standard” for KRAS mutation testing, although other techniques, such as array assays and allele-specific PCR analyses, work well and have greater sensitivities than sequencing.
High-Resolution Melting Analysis (HRM)
HRM is also commonly used to analyze KRAS mutation status. Typically, regions flanking the KRAS point mutations to be investigated are PCR amplified. The resulting amplicons are heat denatured and subsequently renatured at 40°C to 50°C in the presence of appropriate buffers and of a fluorescent dye that emits more strongly when bound to dsDNA than ssDNA. The mix is heated, and the fluorescent intensity is measured and compared to wild-type and mutant controls. Multiple data acquisitions (~25) are taken per degree of temperature increase, and sample denaturation points are compared. dsDNA denaturation and the concomitant lowering of fluorescent emission intensities, occurring at different temperatures, differentiate mutant and wild-type sequences. Temperature changes can range from a few fractions of a degree to nearly 10°C, depending on the size of the amplicon and on the base differences between the sample and the control.25,26 The detection limit of HRM is in the range of 3% to 10% mutant DNA intermixed with wild-type DNA.26,30,33 Most researchers who have compared HRM to other KRAS mutation testing methods have found it to be suitable and cost-effective for clinical testing. Therefore, HRM is often the recommended KRAS testing method for high-throughput testing.25,30,33,38-41 For paraffin-embedded samples, the sensitivity and specificity of HRM have been reported as 88% and 80%, respectively, where sequence analysis was used to determine accuracy and the calculations were performed employing Microsoft Excel (Redmond, WA) and the R statistical package.39 However, Franklin et al.25 compared HRM, allele-specific PCR, and Sanger sequencing and found that HRM had a specificity of 61% when mutations were confirmed by Sanger sequencing, reporting a significant overestimation of the actual number of mutations. For this reason, it is recommended that positive HRM results be confirmed by sequencing or allele-specific PCR.24,26,30,41
Single-Strand Conformation Polymorphism (SSCP) and Denaturing Gradient Gel Electrophoresis (DGGE) for KRAS Testing
SSCP and DGGE are less often employed for KRAS testing than other methods. For both techniques, DNA surrounding the KRAS point mutations is PCR amplified before analysis. In SSCP, the amplicon is heat denatured, rapidly renatured, and subjected to electrophoresis with amplified wild-type KRAS co-run as a control. An ssDNA segment with a point mutation will assume different secondary and tertiary structures, resulting in a different migration pattern compared to the wild-type control.
In DGGE, control duplex wild-type dsDNA is compared to the sample dsDNA that may carry a point mutation. The 2 DNA duplexes are subjected to electrophoresis under mild denaturing conditions. Under such conditions, duplexes carrying mutations will often adopt a more complex, or different, 3-dimensional conformation than duplexes not carrying mutations. As a result, they will migrate at a different rate during gel electrophoresis. In general, SSCP and DGGE are less efficient and accurate than DNA sequencing; hence, sequencing has largely supplanted this testing method. However, these techniques may still have value as screening methods for mutations if they are verified by DNA sequencing or allele-specific PCR.38,42,43 In addition, like HRM, SSCP and DGGE do not identify mutated bases; thus, for this reason, samples testing positive for mutations using these methods should be confirmed by sequencing or allele-specific PCR. Lastly, the detection efficiency of both methods falls with increasing amplicon size, becoming difficult with amplicons over 200 base pairs and impossible with amplicons of 300 base pairs.
The accuracy of duplex analysis has been increased by employing DHPLC to analyze amplicon mixtures. Wild-type amplified KRAS DNA is mixed with similarly amplified tumor KRAS DNA, denatured, renatured, and subjected to DHPLC. Tumor samples carrying base changes will form heteroduplexes with wild-type amplified DNA, which will typically have a more complex tertiary structure, and elute more rapidly than wild-type homoduplexes.44 Although the detection sensitivity for this technique is as low as 1% mutant DNA, it also does not identify the specific mutation, and further mutation confirmation testing may be necessary.
Array or Strip Assay for KRAS Mutation Detection
Many different variations of array/strip assays have been successfully employed in KRAS mutation analysis. Since such platforms need only to identify a low number of mutations, KRAS arrays are far less complex than most array systems.45 Most assays begin with isolating genomic DNA, with possible KRAS mutations, from sources such as a frozen or formalin-fixed tumor or feces from individuals suspected of having CRC.45,46 Like many other protocols, the strip assay for KRAS mutation detection starts with PCR amplification of the DNA immediately surrounding the KRAS point mutations. The DNA is then denatured and hybridized to a solid matrix with specific DNA sequences carrying different KRAS mutations. Usually, the hybridized amplified sequences are detected by their attachment to a specific label, such as a terminal biotinylated moiety, that can bind a streptavidin-conjugated fluorophore following several washing steps. The detection limit of most array/strip assays is high, and as little as 0.1% to 1% mutant DNA diluted in wild-type DNA can be detected. Several studies have demonstrated high sensitivities and specificities of up to 100%.45-48 Interestingly, like sequencing, this technique can detect rare cases where a given sample has more than one type of KRAS mutation, although it only detects those mutations present on the analysis strip. Due to the high sensitivity of the assays, this technique has been successfully applied to KRAS mutation detection in samples that are likely to contain large amounts of nonmutant DNA, including blood and feces.46,49
Some of these assays have increased the detection of low-abundance KRAS mutations by employing peptide nucleic acid polymers (PNA). PNA is a synthetic polymer composed of N-(2-aminoethyl)-glycine units linked by peptide bonds with bases attached by methylene carbonyl bonds. PNA/DNA hybrids are stronger than DNA/DNA hybrids. PNA sequences overlap and bind strongly to wild-type KRAS sequences and can block 3′ sequence–overlapping DNA-based primers attenuating PCR initiation. The same PNA sequences do not bind to mutant KRAS DNA, allowing normal PCR round initiation and increasing the fraction of mutant KRAS DNA amplified.46
Allele-Specific PCR
Taq DNA polymerase initiates the PCR reaction from mismatched template-primer 3′-termini at 10−3 to 10−6 lower amplification efficiency, depending on the mismatch type.50 Allele-specific PCR employs the poor initiation of the Taq polymerase from mismatched primers by using primers with 3′ ends that match the KRAS mutations, allowing the detection of minute concentrations of mutant KRAS. In some systems, allele-specific PCR technology can detect one mutant allele in a background of 104 wild-type alleles.51 Since the Taq polymerase does initiate at a low level from mismatched primers, KRAS primers with mutant sequences will sometimes initiate from wild-type sequences, giving a false-positive result at high Cp values.50 For this reason, allele-specific PCR results are typically considered positive at Cp values of 35 and lower.50,51 Since most tumors have unmutated KRAS, many allele-specific PCR protocols also amplify wild-type KRAS sequences as internal controls. Amplification of wild-type KRAS sequences is useful. These sequences represent internal PCR controls and are used for comparison to amplified mutant sequences, which, if successfully amplified, should have different Cp values as compared to wild-type DNA.50,51
BRAF and NRAS Testing
CRC may also exhibit BRAF and NRAS mutations in about 4.7% and 2.6% of cases, respectively.52 Activating mutation of these genes is also associated with a resistance to panitumumab or cetuximab therapies.52,53 For this reason, the National Comprehensive Cancer Network recommends that all patients with CRC being considered for anti-EGFR therapy be tested for BRAF mutations.22 The testing methods reviewed here are also commonly used for BRAF testing. For example, Arcila et al.37 applied the same analysis comparing Sanger sequencing with standard versus locked nucleic acid primer amplification and found that the latter amplification method also gave better BRAF mutation detection. NRAS mutation testing is not presently recommended by the National Comprehensive Cancer Network, although the test is offered by some molecular diagnostic companies, such as Quest Diagnostics (Madison, NJ).
Conclusion
The National Comprehensive Cancer Network currently recommends KRAS testing for all patients with CRC or non–small cell lung cancer being considered for anti-EGFR therapy.22 Here, we reviewed several of the more commonly employed techniques used to analyze KRAS mutation status. While all techniques have efficacy, they differ in sensitivity and accuracy. Presently, Sanger sequencing, pyrosequencing, and allele-specific PCR are the most commonly used analysis methods, although various array/strip methods are likely to become more common in the future.46,49
Comparisons of different techniques have shown different sensitivities. For example, Sanger sequencing requires the presence of 15% to 50% mutant DNA for the test result to be positive. On the other hand, pyrosequencing requires about 5% and allele-specific PCR requires about 1% of the same material to generate a positive test result.26,30,32,34-36,50,51 Array/strip assays appear to reach the highest sensitivity, with some assays detecting 0.1% mutant DNA.45-49 Conversely, HRM, SSCP, and DGGE often show relatively high false-positive rates compared to other techniques and often require additional testing with different methodologies to confirm the mutation detected.24,26,30,38,41-44 These techniques hold value for large-volume screening protocols, such as research applications, but provide little value in most molecular diagnostic laboratory applications. For most of the techniques listed above, the turnaround times are similar, although mutation confirmation testing for HRM, SSCP, and DGGE prove more time consuming. The advantages and disadvantages of each of these techniques are summarized in Table 1. In the future, as new anti-EGFR therapies become available, the need for KRAS testing will probably increase. It is likely that newer, more sensitive, and cheaper KRAS mutation diagnostic techniques will become available, ultimately fostering the development of personalized medicine.
Table 1.
Summary of the Relative Strengths and Weaknesses of Each of the KRAS Mutation Analysis Methods
| Testing method | Method base | Detection limita | Advantages | Disadvantages | References |
|---|---|---|---|---|---|
| Sanger sequencing | Dideoxynucleotide chain termination with fluorophore detection | 15%-50% | Provides the entire amplified sequence | Detection sensitivity lower than other methods | 28-33 |
| Pyrosequencing | Light detection following nucleotide incorporation | 1.25%-6% | Provides the entire amplified sequence | Cannot read homopolymeric runs over ~5 nucleotides | 27, 31, 33, 35-37 |
| High-resolution melting analysis | Differential melting curves between mutant and wild-type DNA duplexes | 3%-10% | Quick and useful for large-scale initial screening | No DNA sequence, low specificity | 26, 27, 39-42 |
| Single-strand conformation polymorphism and denaturing gradient gel electrophoresis | PCR amplification followed by analysis of heat denaturation curves | 1% | Quick and useful for large-scale initial screening | No DNA sequence, low specificity, testing can be complex | 39, 43-45 |
| Array/strip assay | KRAS DNA amplification followed by binding to a solid matrix | 0.1%-1% | High sensitivity and specificity | Does not test for KRAS mutations not on test strip | 46, 47 |
| Allele-specific PCR | Detection based on excluding mismatched primer-target sequences | 0.1%-1% | High sensitivity and specificity | No DNA sequence, not valid at high Cp values (>35) | 51, 52 |
The detection limit is defined as the minimum percentage of mutant KRAS DNA that can be detected when diluted into wild-type DNA.
Footnotes
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
- 1. Bos JL. RAS oncogenes in human cancer: a review. Cancer Res. 1989;49:4682-9 [PubMed] [Google Scholar]
- 2. Andreyev HJ, Norman AR, Cunningham D, et al. Kirsten ras mutations in patients with colorectal cancer: the ‘RASCAL II’ study. Br J Cancer. 2001;85:692-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bos JL, Fearon ER, Hamilton SR, et al. Prevalence of ras gene mutations in human colorectal cancers. Nature. 1987;327:293-7 [DOI] [PubMed] [Google Scholar]
- 4. Harvey JJ. An unidentified virus which causes the rapid production of tumors in mice. Nature. 1964;204:1104-5 [DOI] [PubMed] [Google Scholar]
- 5. Chang EH, Gonda MA, Ellis RW, Scolnick EM, Lowy DR. Human genome contains four genes homologous to transforming genes of Harvey and Kirsten murine sarcoma viruses. Proc Natl Acad Sci U S A. 1982;79:4848-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shih C, Padhy LC, Murray M, Weinberg RA. Transforming genes of carcinomas and neuroblastomas introduced into mouse fibroblasts. Nature. 1981;290:261-4 [DOI] [PubMed] [Google Scholar]
- 7. Zuber J, Tchernitsa OI, Hinzmann B, et al. A genome-wide survey of RAS transformation targets. Nat Genet. 2000;24(2):144-52 [DOI] [PubMed] [Google Scholar]
- 8. Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer. 2003;3:459-65 [DOI] [PubMed] [Google Scholar]
- 9. Downward J. Targeting RAS signaling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11-22 [DOI] [PubMed] [Google Scholar]
- 10. Graziani A, Gramaglia D, dalla Zonca P, Comoglio PH. Hepatocyte growth factor/scatter factor stimulates the Ras-guanine nucleotide exchanger. J Biol Chem. 1993;268:9165-8 [PubMed] [Google Scholar]
- 11. Hu YP, Patil SB, Panasiewicz M, et al. Heterogeneity of receptor function in colon carcinoma cells determined by cross-talk between type I insulin-like growth factor receptor and epidermal growth factor receptor. Cancer Res. 2008;68:8004-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rajalingam K, Schreck R, Rapp UR, Albert S. Ras oncogenes and their downstream targets. Biochim Biophys Acta. 2007;1773:1177-95 [DOI] [PubMed] [Google Scholar]
- 13. Jancík S, Drábek J, Radzioch D, Hajdúch M. Clinical relevance of KRAS in human cancers. J Biomed Biotechnol. 2010;2010:150960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pretlow TP, Pretlow TG. Mutant KRAS in aberrant crypt foci (ACF): initiation of colorectal cancer? Biochim Biophys Acta. 2005;1756:83-96 [DOI] [PubMed] [Google Scholar]
- 15. McLellan EA, Owen RA, Stepniewska KA, Sheffield JP, Lemoine NR. High frequency of K-ras mutations in sporadic colorectal adenomas. Gut. 1993;34:392-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sugio K, Kishimoto Y, Virmani AK, Hung JY, Gazdar AF. K-ras mutations are a relatively late event in the pathogenesis of lung carcinomas. Cancer Res. 1994;54:5811-5 [PubMed] [Google Scholar]
- 17. Amado RG, Wolf M, Peeters M, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626-34 [DOI] [PubMed] [Google Scholar]
- 18. Karapetis CS, Khambata-Ford S, Jonker DJ, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359:1757-65 [DOI] [PubMed] [Google Scholar]
- 19. Linardou H, Dahabreh IJ, Kanaloupiti D, et al. Assessment of somatic k-RAS mutations as a mechanism associated with resistance to EGFR-targeted agents: a systematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. Lancet Oncol. 2008;9:962-72 [DOI] [PubMed] [Google Scholar]
- 20. Massarelli E, Varella-Garcia M, Tang X, et al. KRAS mutation is an important predictor of resistance to therapy with epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. Clin Cancer Res. 2007;13:2890-6 [DOI] [PubMed] [Google Scholar]
- 21. De Roock W, Jonker DJ, Di Nicolantonio F, et al. Association of KRAS p.G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. JAMA. 2010;304:1812-20 [DOI] [PubMed] [Google Scholar]
- 22. National Comprehensive Cancer Network NCCI Clinical Practice Guidelines in Oncology: non small cell lung cancer (version 2.2009) and colon cancer (version 3.2009). Available from: http://www.nccn.org/professionals/physician_gls/PDF/colon.ncl.pdf and http://www.nccn.org/professionals/physician_gls/PDF/colon.pdf
- 23. Gilbert MT, Haselkorn T, Bunce M, et al. The isolation of nucleic acids from fixed, paraffin-embedded tissues: which methods are useful when? PLoS One. 2007;2:e537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Williams C, Pontén F, Moberg C, et al. A high frequency of sequence alterations is due to formalin fixation of archival specimens. Am J Pathol. 1999;155:1467-71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Franklin WA, Haney J, Sugita M, Bemis L, Jimeno A, Messersmith WA. KRAS mutation: comparison of testing methods and tissue sampling techniques in colon cancer. J Mol Diagn. 2010;12:43-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tsiatis AC, Norris-Kirby A, Rich RG, et al. Comparison of Sanger sequencing, pyrosequencing, and melting curve analysis for the detection of KRAS mutations: diagnostic and clinical implications. J Mol Diagn. 2010;12:425-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Metzker ML. Emerging technologies in DNA sequencing. Genome Res. 2005;15:1767-6 [DOI] [PubMed] [Google Scholar]
- 28. Weichert W, Schewe C, Lehmann A, et al. KRAS genotyping of paraffin-embedded colorectal cancer tissue in routine diagnostics: comparison of methods and impact of histology. J Mol Diagn. 2010;12:35-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pang NK, Nga ME, Chin SY, et al. KRAS and BRAF mutation analysis can be reliably performed on aspirated cytological specimens of metastatic colorectal carcinoma. Cytopathology. 2011;22:358-64 [DOI] [PubMed] [Google Scholar]
- 30. Ibrahem S, Seth R, O’Sullivan B, Fadhil W, Taniere P, Ilyas M. Comparative analysis of pyrosequencing and QMC-PCR in conjunction with high resolution melting for KRAS/BRAF mutation detection. Int J Exp Pathol. 2010;91:500-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gao J, Li YY, Sun PN, Shen L. Comparative analysis of dideoxy sequencing, the KRAS StripAssay and pyrosequencing for detection of KRAS mutation. World J Gastroenterol. 2010;16:4858-64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ogino S, Kawasaki T, Brahmandam M, et al. Sensitive sequencing method for KRAS mutation detection by pyrosequencing. J Mol Diagn. 2005;7:413-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. van Eijk R, van Puijenbroek M, Chhatta AR, et al. Sensitive and specific KRAS somatic mutation analysis on whole-genome amplified DNA from archival tissues. J Mol Diagn. 2010;12:27-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dufort S, Richard MJ, de Fraipont F. Pyrosequencing method to detect KRAS mutation in formalin-fixed and paraffin-embedded tumor tissues. Anal Biochem. 2009;391:166-8 [DOI] [PubMed] [Google Scholar]
- 35. Packham D, Ward RL, Ap Lin V, Hawkins NJ, Hitchins MP. Implementation of novel pyrosequencing assays to screen for common mutations of BRAF and KRAS in a cohort of sporadic colorectal cancers. Diagn Mol Pathol. 2009;18:62-71 [DOI] [PubMed] [Google Scholar]
- 36. Sundström M, Edlund K, Lindell M, et al. KRAS analysis in colorectal carcinoma: analytical aspects of pyrosequencing and allele-specific PCR in clinical practice. BMC Cancer. 2010;10:660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Arcila M, Lau C, Nafa K, Ladanyi M. Detection of KRAS and BRAF mutations in colorectal carcinoma roles for high-sensitivity locked nucleic acid-PCR sequencing and broad-spectrum mass spectrometry genotyping. J Mol Diagn. 2011;13:64-73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Whitehall V, Tran K, Umapathy A, et al. A multicenter blinded study to evaluate KRAS mutation testing methodologies in the clinical setting. J Mol Diagn. 2009;11:543-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gonzalez-Bosquet J, Calcei J, Wei JS, et al. Detection of somatic mutations by high-resolution DNA melting (HRM) analysis in multiple cancers. PLoS One. 2011;6:e14522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Deschoolmeester V, Boeckx C, Baay M, et al. KRAS mutation detection and prognostic potential in sporadic colorectal cancer using high-resolution melting analysis. Br J Cancer. 2010;103:1627-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Er TK, Chang YS, Yeh KT, Chang TJ, Chang JG. Comparison of two different screening methods for the KRAS mutation in colorectal cancer. Clin Lab. 2010;56:175-86 [PubMed] [Google Scholar]
- 42. Half E, Bercovich D, Rozen P. Familial adenomatous polyposis. Orphanet J Rare Dis. 2009;4:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hayes VM, Westra JL, Verlind E, et al. New comprehensive denaturing-gradient-gel-electrophoresis assay for KRAS mutation detection applied to paraffin-embedded tumours. Genes Chromosomes Cancer. 2000;29:309-14 [PubMed] [Google Scholar]
- 44. Lilleberg SL, Durocher J, Sanders C, Walters K, Culver K. High sensitivity scanning of colorectal tumors and matched plasma DNA for mutations in APC, TP53, K-RAS, and BRAF genes with a novel DHPLC fluorescence detection platform. Ann N Y Acad Sci. 2004;1022:250-6 [DOI] [PubMed] [Google Scholar]
- 45. Kriegshäuser G, Auner V, Schuster E, et al. KRAS mutation analysis in genomic DNA isolated from formalin-fixed paraffin-embedded ovarian tissue: evaluation of a strip-based reverse-hybridisation assay. J Clin Pathol. 2011;64:252-6 [DOI] [PubMed] [Google Scholar]
- 46. Prix L, Uciechowski P, Böckmann B, Giesing M, Schuetz AJ. Diagnostic biochip array for fast and sensitive detection of K-ras mutations in stool. Clin Chem. 2002;48:428-35 [PubMed] [Google Scholar]
- 47. Ausch C, Buxhofer-Ausch V, Oberkanins C, et al. Sensitive detection of KRAS mutations in archived formalin-fixed paraffin-embedded tissue using mutant-enriched PCR and reverse-hybridization. J Mol Diagn. 2009;11:508-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fariña Sarasqueta A, Moerland E, de Bruyne H, et al. SNaPshot and StripAssay as valuable alternatives to direct sequencing for KRAS mutation detection in colon cancer routine diagnostics. J Mol Diagn. 2011;13:199-205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yen LC, Yeh YS, Chen CW, et al. Detection of KRAS oncogene in peripheral blood as a predictor of the response to cetuximab plus chemotherapy in patients with metastatic colorectal cancer. Clin Cancer Res. 2009;15:4508-13 [DOI] [PubMed] [Google Scholar]
- 50. Huang MM, Arnheim N, Goodman MF. Extension of base mispairs by Taq DNA polymerase: implications for single nucleotide discrimination in PCR. Nucleic Acids Res. 1992;20:4567-73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kimura H, Kasahara K, Kawaishi M, et al. Detection of epidermal growth factor receptor mutations in serum as a predictor of the response to gefitinib in patients with non-small-cell lung cancer. Clin Cancer Res. 2006;12:3915-21 [DOI] [PubMed] [Google Scholar]
- 52. De Roock W, Claes B, Bernasconi D, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11:753-62 [DOI] [PubMed] [Google Scholar]
- 53. Di Nicolantonio F, Martini M, Molinari F, et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol. 2008;26:5705-12 [DOI] [PubMed] [Google Scholar]