

Abstract
Cancer is now known as a disease of genomic alterations. Mutational analysis and genomics profiling in recent years have advanced the field of lung cancer genetics/genomics significantly. It is becoming more accepted now that the identification of genomic alterations in lung cancer can impact therapeutics, especially when the alterations represent “oncogenic drivers” in the processes of tumorigenesis and progression. In this review, we will highlight the key driver oncogenic gene mutations and fusions identified in lung cancer. The review will summarize and report the available demographic and clinicopathological data as well as molecular details behind various lung cancer gene alterations in the context of race. We hope to shed some light into the disparities in the incidence of various genetic mutations among lung cancer patients of different racial backgrounds. As molecularly targeted therapy continues to advance in lung cancer, racial differences in specific genetic/genomic alterations can have an important impact in the choices of therapeutics and in our understanding of the drug sensitivity/resistance profile. The most relevant genes in lung cancer described in this review include the following: EGFR, KRAS, MET, LKB1, BRAF, PIK3CA, ALK, RET, and ROS1. Commonly identified genetic/genomic alterations such as missense or nonsense mutations, small insertions or deletions, alternative splicing, and chromosomal fusion rearrangements were discussed. Relevance in current targeted therapeutic drugs was mentioned when appropriate. We also highlighted various targeted therapeutics that are currently under clinical development, such as the MET inhibitors and antibodies. With the advent of next-generation sequencing, the landscape of genomic alterations in lung cancer is expected to be much transformed and detailed in upcoming years. These genomic landscape differences in the context of racial disparities should be emphasized both in tumorigenesis and in drug sensitivity/resistance. It is hoped that such effort will help to diminish racial disparities in lung cancer outcome in the future.
Keywords: lung cancer, racial disparities, cancer gene, targeted therapy
Introduction
Lung cancer is one of the most common human cancers and represents the leading cause of cancer mortality in the United States (US). There were an estimated 221,130 new cases and 156,940 deaths in 2011.1 The overall survival rate of lung cancer patients remains poor despite available standard treatments. Recent advances in the fields of mutational analysis and molecularly targeted therapy made it possible to develop new receptor kinase inhibitors such as erlotinib and gefitinib (against epidermal growth factor receptor [EGFR]) and most recently crizotinib (against rearranged anaplastic lymphoma kinase [ALK]) and antibodies such as cetuximab (against EGFR) and bevacizumab (against vascular endothelial growth factor [VEGF]).2 These discoveries yielded better response rates and have marked a new era of paradigm change in targeted lung cancer personalized therapy. Moreover, in recent years, lung cancer molecular profiling has been largely fueled by the tremendously fast scientific and technological advancement in cancer genome research with high-output genomic analysis platforms. Mutational cancer gene analysis has shifted from single gene analysis and later gene family analysis to more recently high-throughput next-generation global genome sequencing analysis, including sequencing of the whole cancer genome, exome, transcriptome, or epigenome. The vast amount of genomic information in a generation is expected to transform our current understanding of lung cancer and would in turn usher a new era of personalized lung cancer therapy. In fact, a number of institutions have already begun to integrate molecular profiling and even clinical next-generation sequencing (NGS) into routine lung cancer diagnosis to empower treatment decisions.3 The increasingly growing genetic and genomic database of lung cancer now sheds light into the racial differences of lung cancer genetics across different human populations. These findings may translate into an important force in the development of treatment algorithm and therapeutic choices in the new era of genomics-guided lung cancer personalized medicine. Nonetheless, the full spectra of the disparities in lung cancer genetics among different human populations are still lacking for the most part. In this review, we attempt to provide a concise summary of the current understanding and available data on the racial differences of lung cancer genes among different human populations.
Race and Lung Cancer Outcome Disparities among Different Populations
Health disparities in the US are a recognized phenomenon that has been well documented in the literature (Fig. 1). Discrepancies between African American and white lung cancer patient populations are supported by data from the 2000-2008 Surveillance, Epidemiology, and End Results (SEER) database.4 It exhibits substantial disparities: the average annual age-adjusted incidence for lung cancer between 2000 and 2008 was 75.2 per 100,000 for African Americans and 64.9 per 100,000 for whites (data from the SEER 17 Registry database for 2000-2008). Notably, African American lung cancer patients were less likely to receive surgical treatment compared to their white counterparts. Women, older patients, and patients in lower socioeconomic classes displayed significantly lower rates of surgery compared to patients in higher socioeconomic classes.4 Further evidence from SEER 17 data using age-specific analyses shows that African Americans under the age of 50 years are almost twice as likely to develop lung cancer as whites in the same age group. The mean age at diagnosis was 66 years in African Americans compared with 71 years in whites.5 In the age group between 40 and 54 years, African American men are 2 to 4 times more likely to develop lung cancer than white men after adjusting for smoking history.6 Health outcome disparities among diverse racial groups in the US are multifactorial and compound. Although socioeconomic and cultural differences across racial groups can account for some of the current disparities, recent evidence from the evolving field of lung cancer genetics research is beginning to transform the way we address these differences in health outcomes from a macroscopic intervention into an increasingly personalized molecular approach.
Figure 1.
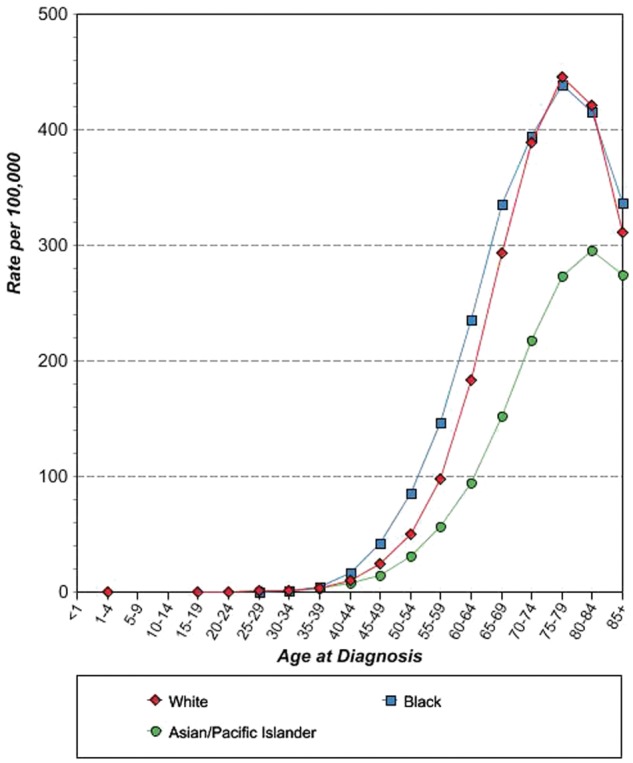
Age-specific SEER lung cancer incidence rates in the United States: 2000-2008. Reproduced with permission from the National Cancer Institute‥
Lung Cancer: Genes and Genome
Copy Number Variations
Copy number alterations in lung cancer have been studied using dense single-nucleotide polymorphism (SNP) arrays, providing us with further insight about the molecular basis of the disease. Weir et al.7 identified 57 significantly recurrent events from a cohort of 371 tumors. The most commonly identified event was chromosome 14q13.3 amplification, accounting for 12% of all the tumor samples. A novel proto-oncogene involved in a significant fraction of lung adenocarcinomas was identified as NKX2-1 (NK2 homeobox 1, also called TITF1), which resides in the 14q13.3 amplification interval and encodes a lineage-specific transcription factor. Interestingly, a recent study examining the TITF1 protein and genomic expression in non–small cell lung cancer (NSCLC) using integrative immunohistochemistry (for protein expression), FISH, and qPCR (for gene copy number) analysis revealed that the protein versus genomic patterns of TITF1 have opposing roles in NSCLC prognosis and may occur preferentially in different subsets of NSCLC patients with distinct oncogenic mutations.8 Broet et al.9 reported significantly higher rates of copy number gain on 16p13.13 and 16p13.11 in East Asian patients’ tumor samples while higher rates of genomic loss on 19p13.3 and 19p13.11 occurred in white patients. A novel oncogene FUS was found to be frequently associated with gain in copy number in the 16p region in lung adenocarcinoma in never smokers in addition to the finding of MYC gene copy number gain.10 Both EGFR and KRAS gene copy number gains have been found to occur more frequently in tumors harboring the activating mutations of the respective oncogene.11
Mutations
The unprecedented advances in lung cancer genome analysis in recent years have revolutionized our understanding of the disease at a deeper molecular scale. First, the analysis of entire gene families (e.g., protein kinome, lipid kinome, and tyrosine phosphatome) as part of DNA mutational profiling of cancer genes in lung cancer unveiled vital information about the molecular structure of the disease. Protein mutations of the RAS/RAF/MEK/MAP kinase signaling pathway were studied in the first of its kind large-scale system.12 The study showed that serine/kinase BRAF was frequently mutated in human cancer at a frequency of 66% in malignant melanoma and at a less dramatic rate in other types of cancer including lung cancer (2% in primary adenocarcinoma). The discovery of cancer-associated mutations was driven by systemic resequencing of the cancer genome. A recent study intending to discover new somatic mutations in 188 human lung adenocarcinomas13 revealed over 1,000 somatic mutations after DNA sequencing of 623 genes with known or suspected cancerous activity. It identified 26 genes with a significantly high mutagenesis rate, possibly implicating them in tumorigenesis. Other frequently mutated genes include tyrosine kinases such as EGFR homolog ERBB4 and multiple Ephrin receptor genes such as EPHA3, VEGFR2 (KDR), and NTKR. These studies provide us with insight into key signaling pathways in lung adenocarcinoma tumorigenesis, which can serve as novel molecular targets for future therapeutic development.
In the following, we will provide a review with emphasis on the molecular genetic variations in several key molecular targets that are documented in lung cancer literature (EGFR, BRAF, KRAS, MET, LKB1, and PIK3CA). The review was focused also on the racial differences of these cancer genes among human populations and the implication of such differences in the future of personalized cancer therapy.
EGFR (HER1 or ERBB1)
EGFR is the key paradigm of molecular targeted therapy in lung cancer, which is now commonly used in the clinical setting worldwide (Fig. 2). Current available drugs that target EGFR can be divided into 2 categories: small-molecule EGFR tyrosine kinase inhibitor (TKI)—gefitinib and erlotinib—and monoclonal anti-EGFR antibody—cetuximab. The genomic discoveries in EGFR and the resultant targeted treatment opened a new window of opportunity for our renewed understanding in lung cancer biology and therapy. This paradigm shift has mainly been fueled by study findings, which reported that a specific cluster of EGFR gene mutations in lung adenocarcinoma resulted in enhanced sensitivity and clinical response to EGFR kinase inhibitors gefitinib14,15 and erlotinib.16 The EGFR mutation database massively grew following research efforts covering thousands of patient tumor samples. EGFR mutations that exist in NSCLC were found to be predominantly somatic, while only a few including T790M were found to be germline in nature. Exons 18 to 21 within the tyrosine kinase domain were the most heavily sequenced region, as it is considered to harbor the mutational hot spots. There are also other EGFR mutations that reside outside these hot spot exons, some having a unique impact on TKI sensitivity, albeit occurring at a relatively lower frequency; for example, the E884K mutation in exon 22 is more sensitizing to gefitinib but confers insensitivity to erlotinib.17,18 The majority (85%) of the currently identified EGFR kinase mutations can be attributed to the L858R missense mutation in exon 21 and short in-frame deletion variants in exon 19,19,20 both being found sensitizing to EGFR TKIs. Several reports suggest that EGFR mutations may carry a prognostic value.21,22 Exon 19 deletions and L858R were found to exist in a subset of NSCLC patients with unique clinical characteristics. These patients were usually never or light female smokers with an adenocarcinoma histology.
Figure 2.

Spectrum of EGFR oncogenic driver mutations among different racial groups with NSCLC. The different color shades represent EGFR mutational rates reported by different studies. Data on the African American and Latin American cohorts are based on a limited number of studies available.46,55-58 Data on the Asian and white cohorts are abundant over recent years, and several representative studies were selected for graphical representation here.23,24,28,46,56,101,143
Interestingly, the frequency of EGFR mutations differed among different racial groups in the population. EGFR mutations were highly prevalent in the Asian patient population at 30% compared to only 7% in whites.23 Other studies frequently reported similar EGFR mutational frequencies, ranging from 19.6% to 40.1% among different ethnicities within the Asian population.24-30 The EGFR mutational frequency was found to be even higher among Asian never smokers at a rate range between 48% to 75.3%31-33 (Fig. 3). In 2009, the IRESSA Pan-Asia Study (IPASS) results, which compared gefitinib with carboplatin-paclitaxel, were reported. The trial was conducted on never or light smokers of Asian descent with adenocarcinoma (higher frequency of sensitizing EGFR mutations) as a mutation-enriching strategy for the study population. The trial demonstrated that the gefitinib-treated group had a 12-month progression-free survival rate of 24.9% compared to 6.7% in the chemotherapy-treated group.34 It was concluded that the presence of EGFR mutations was a strong predictive factor for improved outcome with gefitinib therapy as a first-line therapy. Remarkably, patients with no identifiable EGFR mutations, despite possessing the clinical parameters of higher probability of having the mutations, in fact developed a worse outcome with gefitinib therapy. Other subsequent phase 3 studies in different parts of the world using both gefitinib and erlotinib tested in the first-line therapy setting to compare with cytotoxic chemotherapy further substantiated the paradigm of first-line EGFR TKI use among patients proven to harbor the EGFR-sensitizing mutations in a genotype-informed targeted therapy paradigm.35
Figure 3.

Spectrum of oncogenic driver mutations in Asian never smokers with lung adenocarcinoma.31-33 The data were collected from 3 different studies to represent the mutational frequency range of different genes among the same population. N/A = not available.
EGFR gene copy number has also been intensely investigated in lung cancer. It was found to be increased in 30% to 50% of NSCLC patients.36 Accordingly, some studies reported that EGFR gene copy number may play a predictive role in EGFR TKI therapy response.37,38 Nevertheless, other studies reported that these earlier findings were confounded by the fact that a high EGFR copy number can significantly co-exist with EGFR mutations. Furthermore, differences in assay platforms for gene copy numbers such as FISH and genomic real-time qPCR, in addition to tissue quality variations, can further confound the results and their interpretations. With the growth of EGFR TKI prescription use in the lung cancer clinic, clinical acquired resistance became inevitable. The predominant resistant mechanism was found to be EGFR T790M mutation in exon 20, which is part of the hydrophobic “gatekeeper” residue within the kinase domain, which confers resistance to both gefitinib and erlotinib.39-41 The T790M mutation accounts for almost half the cases of acquired resistance.42 Other documented resistance cases were associated with primary resistance mutations such as exon 20 insertions (D770_N771 insNPG)43 and acquired resistance mutations such as exon 19 missense mutation D761Y.44,45 It is noteworthy that N771GY and A767-V769dup, which are novel somatic insertion mutations in exon 20, and S768N, a missense mutation, were all identified in the African American cohort.46,47 In East Asian patients, S768I, a somatic EGFR mutation, was reported with evidence, suggesting a potential role in EGFR TKI resistance.24,27,48,49 Yet, these acquired genetic resistances were not exclusive to any racial group. MET genomic amplification has also been reported to be implicated in acquired EGFR TKI resistance.50,51 More novel mechanisms of acquired resistance have emerged from larger tumor rebiopsy studies, which also include PIK3CA mutation, EGFR amplification, and small cell lung cancer (SCLC) transformation.52
Although abundant research data were collected on the frequency of EGFR mutations in the Asian and white populations, corresponding information regarding the African American population remains deficient. Health disparities exist between African Americans and the rest of the US population, resulting in inferior health outcomes.5,53 The prevalence of EGFR mutations in the African American population varies significantly in the documented literature. In one study by Yang et al.,54 EGFR mutations were found in only 2.4% of African Americans compared to 14.1% in whites. Another supporting study found that only 2% of African American patients expressed activating EGFR mutations versus 17% in whites.46 Yet, in a recent study by Cote et al.,55 it was found that EGFR mutations existed in 11.9% of NSCLC tumors of African American patients versus 15.6% in whites, that is, more comparable frequencies. This finding was supported by Reinersman et al.,56 who reported EGFR mutations in 19% of the African American NSCLC samples versus 13% in whites, arguing that all patients with advanced lung adenocarcinoma should undergo mutational analysis before the initiation of therapy. Interestingly, in 2 recent studies conducted in a Latin American cohort, the EGFR mutational frequency was significantly higher at 30.4% to 33.2%.57,58
The EGFR mutational status seems also to vary between the primary lung tumor and the corresponding metastases. Often, the EGFR mutations would be present in the primary lung tumor but appear to be absent in the metastases. According to several studies, the discordance rate of EGFR mutations ranged between 16.2% to 32.5%.59-62
Emerging clinical data in studies testing a molecularly matched targeted therapy approach particularly in mutation-enriching patient populations using clinicopathological parameters, for example, the IPASS, have now strengthened the notion that molecular tumor selection by profiling trumps clinical selection.63 The IPASS also paved the road for the arrival of the first-line use of EGFR TKI (erlotinib and gefitinib) in sensitizing EGFR mutation–positive advanced NSCLC patients. Going forward, this position is likely to be even more strengthened by emerging genomic analysis of the lung cancer genome among different populations. The declining cost of high-throughput tumor molecular profiling would also facilitate and further justify this approach of genotype-informed therapy decision in lieu of “clinical profiling” or “racial profiling” for therapy decisions.
KRAS
KRAS encodes a GTPase that plays the role of a central mediator of downstream growth factor receptor signaling and therefore is critical for cell proliferation, survival, and differentiation (Fig. 4). KRAS gene mutations are uncommon in squamous cell carcinoma but can be present in approximately 15% to 25% of lung adenocarcinomas.64 The mutations are missense mutations primarily in codons 12 and 13 of (exon)1. In the vast majority of cases, KRAS mutations were found in EGFR wild-type tumors; hence, EGFR and KRAS mutations were mutually exclusive.65-67 Although mutant KRAS has well-established poor prognostication utility, there are conflicting data in the literature regarding its use as a predictive biomarker in NSCLC.65,68 Interestingly, most studies support the notion that KRAS mutations are less common in Asians compared to their white counterparts.9,69,70 In 2 studies conducted in a population of Chinese NSCLC patients, the KRAS mutation was found to range between 3.8% and 8%. This is dramatically low compared to the NSCLC white patient population in which studies including 2 meta-analyses suggested a prevalence rate ranging between 18% to 26%.23,46,56,71,72 Worthy of note, KRAS mutational frequency among Asian never smokers was detected at an even lower range between 1.92% to 3.5%.31-33 Yet, as with EGFR, there is scarce and inconsistent evidence regarding the KRAS mutational status in the African American group, raising the issue of health disparities in research. Some studies suggest that there is no significant difference between African Americans and whites in KRAS mutation frequency.46,73 In a recent study, KRAS mutations were found to be more likely in whites, with 26% versus 17% in African Americans.56 The KRAS mutational frequency rate seems to be quite similar in the Latin American population, ranging between 14.6% to 16.6%.57,58 It is noteworthy that KRAS mutations tend to be less common in never smokers compared to former or current smokers.69
Figure 4.

Spectrum of KRAS oncogenic driver mutations among different racial groups with NSCLC. The different color shades represent KRAS mutational rates reported by different studies. Data on the African American and Latin American cohorts are based on a limited number of available studies.46,56-58,73 Data on the white cohort are based on multiple studies including 2 meta-analyses of 22 studies with 1,470 NSCLC patients.23,46,56,71-73 Data on the Asian cohort are based on studies conducted in the Chinese and Korean populations.143-145
As with EGFR, the KRAS mutational status seems to also vary between the primary tumor and the corresponding metastases. Several studies observed the absence of KRAS mutations from metastases at a discordance rate of 22.5% to 26% in KRAS-positive lung cancer patients.60-62 Currently, no effective targeted therapeutics targeting mutated KRAS exists for clinical use, although a number of agents such as MEK inhibitors are actively undergoing clinical trial investigation.
MET (c-Met proto-oncogene, HGF receptor): oncogenic mutations, amplification/overexpression, and alternative splicing
The MET proto-oncogene is located on human chromosome 7, where EGFR and hepatocyte growth factor (HGF) genes also reside (Fig. 5). MET encodes a receptor tyrosine kinase (RTK), which binds to its natural ligand, HGF, also called scatter factor (SF).74 The ligand-receptor binding induces a conformational change in the MET receptor that facilitates receptor phosphorylation and activation. In the context of malignancy, the MET-HGF/SF pathway has been strongly implicated as a mediator of pleiotropic effects such as tumor growth, survival, branching morphogenesis, motility and migration, cell scattering, invasion, tumor angiogenesis, and metastasis.75-77 According to a recent study by Rikova et al.,78 the MET receptor ranked first in the number of tyrosinated phosphopeptides in NSCLC tissue samples among members of the RTK family, thus supporting the notion that MET is a key driver RTK in the lung oncogenic process. Furthermore, cross-talk was discovered between MET and EGFR signaling pathways in lung cancer.79,80
Figure 5.

Spectrum of MET mutations among different racial groups with NSCLC.89 The frequency of MET mutations is presented in accordance of the findings with the histological subtypes of lung cancer and racial groups.
Oncogenic mutations in MET were first identified by Schmidt et al.81-83 in 1997 in which germline mutations were found to occur in 100% of hereditary papillary renal cell carcinomas and somatic mutations in MET were found in 10% to 15% of sporadic papillary renal cell carcinomas. More recently, MET gene mutational research efforts revealed various MET mutations in both SCLC and NSCLC, particularly in the nonkinase domain regions. These mutations were clustered in 2 main hot spots: the extracellular ligand-binding semaphorin (Sema) domain and the regulatory juxtamembrane (JM) domain.84-87 Interestingly, the JM mutations of MET 84 and also the alternative splicing variants that skip the JM domain87,88 have been found to be oncogenic and could result in enhanced tyrosine phosphorylation, which in turn activate multiple downstream pathways that increase cell motility and metastasis.88
NSCLC commonly overexpresses MET, often in its activated form, and there can be frequent co-expression with EGFR.85 In fact, MET overexpression has been found to inversely correlate with survival in lung cancer, among other human malignancies.85 Although the MET receptor has been extensively studied, there is insufficient literature regarding its mutational profile across different racial groups. In a recent study by Krishnaswamy et al.,89 lung cancer tissue samples were obtained from patients of different racial backgrounds for mutational sequencing. Notably, in this study, most of the MET mutations were found to be of germline nature. Interestingly, preclinical studies in germline met mutations in a murine transgenic model reveal mutation- and background-associated differences in tumor profiles.90 The most frequently detected MET mutation, N375S, occurred in 13% of East Asians compared to only 2.6% in whites and none among African Americans. MET mutations were also more likely to exist in male smokers and squamous cell carcinoma. This study shows that MET displays different mutational profiles across different racial groups, and thus, it would be very interesting to expand on these findings and investigate the MET gene alterations across larger populations. The Sema domain housed all the nonsynonymous mutations except R988C and T1010I in the JM domain. The previously mentioned N375S mutation, which existed at a higher frequency in East Asians, was a nonsynonymous mutation in the Sema domain. On the contrary, R988C was only identified in whites and African Americans. This study did not detect any nonsynonymous mutations in the tyrosine kinase domain; however, whites and East Asians were found to possess a synonymous SNP, 3912C>T (D1304), in the tyrosine kinase domain more frequently than African Americans. Another earlier study identified that the somatic exon 14–skipping splice variant of MET occurred in 1.3% of a US lung cancer cohort.85 This finding was echoed by similar results of 1.7% in another study focused on a Japanese cohort.91 The Japanese study was also able to identify an increased MET copy number in 5.6% of the patients (P = 0.041).
To this end, the potential impact of MET alterations in tumors in the therapeutic sensitivity profile is still relatively uncertain. A large number of MET and HGF targeting agents have been undergoing clinical trial studies in recent years, and 2 of them, tivantinib (ARQ197, a highly selective non–ATP-competitive TKI) and onartuzumab (MetMAb, a 1-arm monoclonal antibody), have already entered into phase 3 randomized clinical studies.92,93 The phase 3 non-squamous NSCLC study for tivantinib (ARQ197) has been completed and interim analysis showed no statistically significant difference in overall survival benefit between tivantinb plus erlotinib versus erlotinib plus placebo. Nonetheless, there is a progression-free survival benefit of the tivantinib arm over placebo arm. MET amplification94 and HGF autocrine signaling95 have been found to predict MET inhibition sensitivity in human cancers. On the other hand, MET mutations can pose varying degrees of impact on MET TKI sensitivity/resistance profiles in preclinical studies.18,96 Clearly, further investigations into the role of MET alterations in predicting therapeutic sensitivities are urgently warranted.97 Nonetheless, this task could prove to be more daunting than expected due to at least the versatility of the receptor oncogenic signaling cascade, which can mediate “oncogenic dependence,” which promotes oncogene-addicted tumor cell proliferation and survival, and “oncogenic expedience,” which promotes tumor cell invasion and metastasis. Furthermore, many of the MET targeting agents under clinical investigations are multitargeted agents, for example, cabozantinib (XL184 targeting MET, VEGFR2, RET, and AXL), with perhaps the exception of tivantinib and onartuzumab.
LKB1 (STK11)
LKB1 gene (also known as STK11) is a tumor suppressor gene located on chromosome 19p13.3 (Fig. 6). The LKB1 gene was initially identified as the causative agent behind Peutz-Jeghers syndrome through a germline inactivating mutation.
Figure 6.

Spectrum of LKB1 oncogenic mutations among different racial groups with NSCLC.100-102,104 The different color shades represent LKB1 mutational rates reported by different studies.
LKB1 mutation is typically rare in most types of cancer, with the exception of pancreatic cancer, where it is present in 4% of cell lines and primary tumors and also NSCLC. It was discovered that LKB1 possesses inactivating mutations in NSCLC tumors and was found to be a fairly common event.98 Interestingly, mutations in the LKB1 tumor suppressor gene were found to widely vary in frequency across different racial groups. LKB1 mutational frequency as identified in multiple studies was reported in approximately 17% to 35% of NSCLC in the white population compared to only 3% to 7% in the Asian NSCLC population.99-101
Early studies on NSCLC primary tumors and cell lines of undetermined racial background reported an average LKB1 mutation rate of 33% to 39%.98,99 A study by Koivunen et al.100 observed the LKB1 mutation to be higher in NSCLC tumors in the US population (17%) compared with 5% of NSCLC in the Korean population. In another study conducted on a sample of Chinese lung adenocarcinomas, LKB1 mutational frequency was found to be similar at 6.9%, while another study conducted in Japan observed a rate of 3%.101,102 Another interesting finding reported in the literature was the F354L mutation that occurred in 6.1% of Korean lung cancer patients.103 Similarly, a study conducted by Suzuki et al.104 in Chiba, Japan, observed 14.4% LKB1 mutations in lung adenocarcinoma samples, all of which expressed F354L substitutions. In another recent study, Gill et al.105 reported that the occurrence of a homozygous deletion of LKB1 was significantly more frequent in whites (35%) than in African American patients (6%).
Unlike EGFR, co-existing KRAS activating mutations were found with LKB1 inactivating mutations in lung cancer samples.98 Mutations associated with smoking history and KRAS mutations were found to be almost mutually exclusive with EGFR mutations.100 The LKB1 mutations also tended to occur more commonly in adenocarcinomas than in squamous cell carcinomas, where the inactivation of LKB1 was observed at a rate of 34% and 19%, respectively, but remains a common event in both histological subtypes.106
BRAF
BRAF kinase belongs to a family of serine-threonine protein kinases that includes ARAF, BRAF, and CRAF (RAF1). Mutant BRAF has been implicated in the pathogenesis of several cancers, including melanoma, NSCLC, ovarian cancer, papillary thyroid cancer, and colorectal cancer. The most commonly identified BRAF mutation is V600E, which accounts for 90% of BRAF mutations in melanoma. In NSCLC, BRAF gene mutations were identified in 1% to 3% of all samples.12,107-109 In a recent study that sampled 697 patients with lung adenocarcinoma, 18 patients tested positive for BRAF mutations, all of whom were white.110 The identified BRAF mutations were V600E (50%), G469A (39%), and D594G (11%). It is also noteworthy that no patient with a BRAF mutation had a concomitant mutation in EGFR or KRAS or a translocation in ALK. Most recently, a mutated BRAF-specific inhibitor, vemurafenib, has been approved for clinical use in V600E BRAF-mediated cutaneous melanoma. This raises the possibility of matching the BRAF inhibitor to mutated BRAF expressing NSCLC in the future and would be worth investigating.
PIK3CA
The PIK3CA gene encodes p110α, one of the catalytic subunits of phosphatidylinositol 3-kinases (PI3Ks), which belongs to a family of lipid kinases involved in many cellular processes, including cell growth, proliferation, differentiation, motility, and survival. PI3K is a heterodimer composed of 2 subunits: an 85-kDa regulatory subunit (p85) and a 110-kDa catalytic subunit. PI3K converts PI(4,5)P2 to PI(3,4,5)P3 on the inner leaflet of the cell membrane. PI(3,4,5)P3 recruits important downstream signaling proteins, such as AKT, to the cell membrane, resulting in increased activity of these proteins. PIK3CA was found to be mutated in over 30% of colorectal cancers.111 Somatic mutations in PIK3CA have been also found in 1% to 3% of all NSCLCs.111,112 Most of the mutations tended to cluster within 2 mutational hot spots. They also tended to occur more commonly in squamous cell carcinoma.112 PIK3CA shows significant potential as a candidate in cancer-targeted drug therapy. Currently, there are several ongoing clinical trials using PI3K inhibitors. Of interest, the PI3K inhibitor may also have a role to overcome acquired EGFR TKI–resistant disease since PIK3CA mutations have been identified in these tumor tissues in a rebiopsy study.52
Oncogenic Chromosomal Gene Rearrangements
ALK: Oncogenic Chromosomal Translocations
ALK is another tyrosine kinase receptor that is abnormal in various types of malignancies (Fig. 7). While the role of ALK in human cancer has long been recognized in NPM-ALK fusion in non-Hodgkin lymphoma,113 EML4-ALK fusion was documented in the literature for the first time in NSCLC only recently by Soda et al.114 in 2007 as a novel potential oncogenic driver mutant kinase.115 Approximately 3% to 7% of lung tumors harbor ALK fusions.114-116 Multiple different EML4-ALK fusion variants have been described in NSCLC, typically with varying fusion sites at EML4 but with a constant fusion site within ALK.78,114,115,117-119 EML4-ALK fusions are usually found in light (<10 pack years) or never smokers who tend to be of a younger age.116,119-121 The EML4-ALK oncogenic rearrangement was also found to be different across different racial groups. In the Asian cohort, several studies determined the incidence of the oncogenic translocation to be in the range of 2.3% to 6.7% with no significant difference compared to Asian never smokers.31-33 On the other hand, the rate of the EML4-ALK rearrangement was found to be much lower in whites, with most studies supporting a range between 1% to 3%.114,119,120,122,123 Interestingly, according to one study conducted on a cohort of NSCLC specimens collected from Italy and Spain, the incidence rate was found to be more similar to the Asian cohort at 7.5%.124
Figure 7.

Spectrum of EML4-ALK oncogenic driver fusions among different racial groups with NSCLC.114,116,119,120,122,124,146 The different color shades represent EML4-ALK rates reported by different studies. Data among human populations other than white and Asian are lacking thus far.
In most cases, EML4-ALK fusions were nonoverlapping with other oncogenic mutations of EGFR or KRAS.119-122 The presence of EML4-ALK fusions is also associated with EGFR TKI resistance.119,121,122 Although the relationship between EML4-ALK and MET is not well established yet, crizotinib, a drug initially developed as a MET inhibitor, has recently been approved by the US Food and Drug Administration (FDA) for the treatment of EML4-ALK–positive NSCLC and can now be prescribed as a first-line treatment.125 In a recent study by Shaw et al.,126 ALK-positive NSCLC patients on crizotinib therapy were associated with improved survival compared to crizotinib-naive controls. Unfortunately, as with other targeted cancer drugs, patients eventually develop a resistance against crizotinib. Novel resistance mutations are continually identified. Choi et al.127 reported 4374G→A and 4493C→A in the ALK gene, followed by Sasaki et al.,128 who reported the F1174L mutation in the ALK kinase domain. With NGS platform analysis, more novel forms of ALK fusion have recently been uncovered, such as C2orf44-ALK in colorectal cancer.129 Further fusion variants of human oncogenic translocations in lung cancer would be expected to be generated with more efficient and affordable NGS efforts as applied in different human populations in the near future.
RET
The RET proto-oncogene encodes a RTK for the glial cell line–derived neurotropic factor family of ligands; RET signaling is essential for neural development and maintenance.130 RET mutations are known to be present in thyroid cancers. In fact, 10% to 20% of all sporadic papillary thyroid carcinomas contain RET fusions in which 60% to 70% of them are attributed to RET-PTC1 fusion.131,132 In lung cancer, KIF5B-RET fusion was recently discovered in a small cohort of NSCLC patients, with 0.8% (1/121) of the patients who tested positive being of European ancestry and 2% (9/405) of Asian descent. Interestingly, there were no known oncogenic mutations detected in RET-positive patients, suggesting that RET fusion might be the driving force behind the oncogenic process. In another study by Kohno et al.,133 they reported the presence of KIF5B-RET fusion in 1% to 2% of lung adenocarcinomas in both Asians and non-Asians. The relation between smoking and KIF5B-RET fusion remains unclear. Worthy of note, PTC-RET–positive thyroid cancers are sensitive to sorafenib, a RET inhibitor; this raises the notion to test RET kinase inhibitors in patients with KIF5B-RET–positive NSCLC to determine potential clinical benefits.129 Moreover, RET can also be targeted by a novel kinase inhibitor cabozantinib (XL184), which also has inhibitory activities against MET, VEGFR2, and AXL besides RET and will be formally tested in an upcoming phase 2 NSCLC study with RET fusion patients.
ROS1
ROS1 is a RTK of the insulin receptor family. The gene was originally found to be fused to the adjacent FIG gene in glioblastomas. Later on, ROS1 fusions were identified in approximately 2% of NSCLCs and postulated as a potential driver mutation.134,135 These fusions resulted in RTK activation, although the details of the downstream signaling transduced by ROS1 fusion are not fully understood yet. In a recent study by Bergethon et al.,136 they reported that patients with ROS1 rearrangements were significantly younger, more likely to be never smokers, and were overrepresented in the Asian race. Interestingly, they were also able to show that ROS-positive status was associated with sensitivity towards TKIs, specifically crizotinib, with a patient demonstrating prompt and durable complete response. Overall, ROS1 as a potential therapeutic target in lung cancer, and its molecular alterations and racial differences and determinants of inhibitor sensitivity and resistance, would warrant further definition and investigation.
Genetic Polymorphisms in Lung Cancer: Impact on Targeted Therapy
Studies show that genetic polymorphic variations of EGFR have an association with EGFR mRNA expression.137 Interestingly, the –216G/T polymorphic variant either alone or with –191C/A located in the EGFR gene promoter transcriptional start site was associated with either better clinical outcome or increased gefitinib toxicity or both.137,138 These genetic polymorphic variations were both found in white patients and were relatively rare in Asians.139 Other studies investigating the intron 1 enhancer element among different racial groups tried to correlate the CA repeat length to outcome, EGFR expression, and gene copy number along with EGFR TKI therapeutic efficacy and toxicity.140 Unfortunately, these gene polymorphism studies were found to be difficult to interpret and not very reliable because they were underpowered with multiple confounding factors.
Lung Cancer Genome Analysis: NGS and Future Directions
In this review, we highlighted several recent examples of novel lung cancer genome alterations such as KIF5B-RET fusions129,133 that were uncovered using high-throughput NGS of tumor samples. In a study by Chmielecki et al.,141 they were able to map out tyrosine kinase fusions in the conserved GXGXXG kinase motif in a NGS study. In another recent study by Pleasance et al.,142 investigators identified 22,910 somatic substitutions in a single SCLC cell line, “NCI-H209,” in whole genome sequencing analysis. These mutations were found to represent the effects of carcinogens associated with tobacco smoking. NGS and high-throughput cancer genome decoding in recent years have undeniably brought forth a genomic revolution in cancer research and clinical personalized cancer therapeutics. The new generation of non-Sanger–based sequencing technologies has delivered on its promise of sequencing DNA at unprecedented speeds, thereby enabling impressive scientific achievements and novel biological application. As the cost of NGS declines at a rapid pace, and more novel and faster NGS platforms continue to emerge in the market, the process of cancer genome sequencing is already undergoing a rapid “democratization process.” NGS of cancer genomes is becoming more readily available beyond just a handful of large academic or industrial genome centers. Global genome analysis is also underway and would ultimately facilitate a deeper understanding of human cancer genome variations, including lung cancer, among different human racial populations.
Although all these advancements are very exciting, many challenges still lie ahead. First, the enormous bioinformatics output will require an extensive amount of talent and expertise that is likely the “bottleneck” for further progress in genome science. Second, the digital and global sharing of patient genomic information poses many ethical, legal, and socioeconomic issues and concerns that transcend territories of patient privacy and health care insurability. Nonetheless, there are accelerated efforts from the government (National Institutes of Health/National Human Genome Research Institute [NIH/NHGRI]) to summon resources to research the legal and social impact of clinical cancer genome sequencing to keep pace with the technological advances but also to facilitate their translational leap into clinical patient care benefits. On the other hand, as the technological advances bring forth more sophisticated than ever sequencing platforms and capacity, it cannot be overemphasized that the source and quality of the tumors to be sequenced still hold the key to the validity of the resultant data and integrity of the published literature ultimately. Detailed and accurate annotation of the source of tumor DNA, as well as processing methodology, should be presented in such studies. For instance, the use of laser microdissection or whole genome amplification and the specific sequencing method should be reported. Moreover, it would be crucially important to specify in studies whether the sequencing was performed from primary versus metastatic tumor sites59-62 and, if the latter, which specific organ site of metastasis.
Finally, despite all the challenges, it is perhaps now not unrealistic to anticipate that implementing tumor genomic profiling using NGS across different human populations in lung cancer would enormously expand our knowledge base in lung cancer biology and racial disparities in the disease outcome. It is also expected that a more wide adoption of clinical cancer genome sequencing would open new fronts into the development of molecularly personalized cancer therapy.
Acknowledgments
Patrick C. Ma is supported by the Department of Defense Congressionally Directed Medical Research Program: Lung Cancer Research Program, a Lung Cancer Promising Clinician Research Award (W81XWH-10-1-0690), and the Taussig Cancer Institute of the Cleveland Clinic. The authors apologize that they are not able to cite all of the original references of all the relevant studies under discussion in this review, owing to the scope of the review topic and the space limitation.
Footnotes
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received the following financial support for the research, authorship, and/or publication of this article: Patrick C. Ma received research support from Daiichi Sankyo Inc. and ArQule Inc. in sponsored clinical trial funding and has received consultant honoraria as an advisory board member.
References
- 1. Siegel R, Ward E, Brawley O, Jemal A. Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin. 2011;61:212-36 [DOI] [PubMed] [Google Scholar]
- 2. Choong NW, Ma PC, Salgia R. Therapeutic targeting of receptor tyrosine kinases in lung cancer. Expert Opin Ther Targets. 2005;9:533-59 [DOI] [PubMed] [Google Scholar]
- 3. Pao W, Kris MG, Iafrate AJ, et al. Integration of molecular profiling into the lung cancer clinic. Clin Cancer Res. 2009;15:5317-22 [DOI] [PubMed] [Google Scholar]
- 4. Hardy D, Liu CC, Xia R, et al. Racial disparities and treatment trends in a large cohort of elderly black and white patients with nonsmall cell lung cancer. Cancer. 2009;115:2199-211 [DOI] [PubMed] [Google Scholar]
- 5. Gadgeel SM, Severson RK, Kau Y, Graff J, Weiss LK, Kalemkerian GP. Impact of race in lung cancer: analysis of temporal trends from a surveillance, epidemiology, and end results database. Chest. 2001;120:55-63 [DOI] [PubMed] [Google Scholar]
- 6. Schwartz AG, Swanson GM. Lung carcinoma in African Americans and whites: a population-based study in metropolitan Detroit, Michigan. Cancer. 1997;79:45-52 [DOI] [PubMed] [Google Scholar]
- 7. Weir BA, Woo MS, Getz G, et al. Characterizing the cancer genome in lung adenocarcinoma. Nature. 2007;450:893-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tang X, Kadara H, Behrens C, et al. Abnormalities of the TITF-1 lineage-specific oncogene in NSCLC: implications in lung cancer pathogenesis and prognosis. Clin Cancer Res. 2011;17:2434-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Broet P, Dalmasso C, Tan EH, et al. Genomic profiles specific to patient ethnicity in lung adenocarcinoma. Clin Cancer Res. 2011;17:3542-50 [DOI] [PubMed] [Google Scholar]
- 10. Job B, Bernheim A, Beau-Faller M, et al. Genomic aberrations in lung adenocarcinoma in never smokers. PLoS One. 2010;5:e15145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Modrek B, Ge L, Pandita A, et al. Oncogenic activating mutations are associated with local copy gain. Mol Cancer Res. 2009;7:1244-52 [DOI] [PubMed] [Google Scholar]
- 12. Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949-54 [DOI] [PubMed] [Google Scholar]
- 13. Ding L, Getz G, Wheeler DA, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129-39 [DOI] [PubMed] [Google Scholar]
- 15. Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497-500 [DOI] [PubMed] [Google Scholar]
- 16. Pao W, Miller V, Zakowski M, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101:13306-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Choong NW, Dietrich S, Seiwert TY, et al. Gefitinib response of erlotinib-refractory lung cancer involving meninges: role of EGFR mutation. Nat Clin Pract Oncol. 2006;3:50-7, quiz 1 p following 57. [DOI] [PubMed] [Google Scholar]
- 18. Tang Z, Jiang S, Du R, et al. Disruption of the EGFR E884-R958 ion pair conserved in the human kinome differentially alters signaling and inhibitor sensitivity. Oncogene. 2009;28:518-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gazdar AF, Shigematsu H, Herz J, Minna JD. Mutations and addiction to EGFR: the Achilles ‘heal’ of lung cancers? Trends Mol Med. 2004;10:481-6 [DOI] [PubMed] [Google Scholar]
- 20. Shigematsu H, Gazdar AF. Somatic mutations of epidermal growth factor receptor signaling pathway in lung cancers. Int J Cancer. 2006;118:257-62 [DOI] [PubMed] [Google Scholar]
- 21. Eberhard DA, Johnson BE, Amler LC, et al. Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J Clin Oncol. 2005;23:5900-9 [DOI] [PubMed] [Google Scholar]
- 22. Bell DW, Lynch TJ, Haserlat SM, et al. Epidermal growth factor receptor mutations and gene amplification in non-small-cell lung cancer: molecular analysis of the IDEAL/INTACT gefitinib trials. J Clin Oncol. 2005;23:8081-92 [DOI] [PubMed] [Google Scholar]
- 23. Zhou W, Christiani DC. East meets West: ethnic differences in epidemiology and clinical behaviors of lung cancer between East Asians and Caucasians. Chin J Cancer. 2011;30:287-92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kosaka T, Yatabe Y, Endoh H, Kuwano H, Takahashi T, Mitsudomi T. Mutations of the epidermal growth factor receptor gene in lung cancer: biological and clinical implications. Cancer Res. 2004;64:8919-23 [DOI] [PubMed] [Google Scholar]
- 25. Tokumo M, Toyooka S, Kiura K, et al. The relationship between epidermal growth factor receptor mutations and clinicopathologic features in non-small cell lung cancers. Clin Cancer Res. 2005;11:1167-73 [PubMed] [Google Scholar]
- 26. Sonobe M, Manabe T, Wada H, Tanaka F. Mutations in the epidermal growth factor receptor gene are linked to smoking-independent, lung adenocarcinoma. Br J Cancer. 2005;93:355-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huang SF, Liu HP, Li LH, et al. High frequency of epidermal growth factor receptor mutations with complex patterns in non-small cell lung cancers related to gefitinib responsiveness in Taiwan. Clin Cancer Res. 2004;10:8195-203 [DOI] [PubMed] [Google Scholar]
- 28. Soung YH, Lee JW, Kim SY, et al. Mutational analysis of EGFR and K-RAS genes in lung adenocarcinomas. Virchows Arch. 2005;446:483-8 [DOI] [PubMed] [Google Scholar]
- 29. Shigematsu H, Lin L, Takahashi T, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst. 2005;97:339-46 [DOI] [PubMed] [Google Scholar]
- 30. Sugio K, Uramoto H, Ono K, et al. Mutations within the tyrosine kinase domain of EGFR gene specifically occur in lung adenocarcinoma patients with a low exposure of tobacco smoking. Br J Cancer. 2006;94:896-903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li C, Fang R, Sun Y, et al. Spectrum of oncogenic driver mutations in lung adenocarcinomas from East Asian never smokers. PLoS One. 2011;6:e28204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kim HR, Shim HS, Chung JH, et al. Distinct clinical features and outcomes in never-smokers with nonsmall cell lung cancer who harbor EGFR or KRAS mutations or ALK rearrangement. Cancer. 2012;118:729-39 [DOI] [PubMed] [Google Scholar]
- 33. Ren S, Kuang P, Zheng L, et al. Analysis of driver mutations in female non-smoker Asian patients with pulmonary adenocarcinoma. Cell Biochem Biophys. Epub 2012 Jun 17. [DOI] [PubMed] [Google Scholar]
- 34. Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947-57 [DOI] [PubMed] [Google Scholar]
- 35. Keedy VL, Temin S, Somerfield MR, et al. American Society of Clinical Oncology provisional clinical opinion: epidermal growth factor receptor (EGFR) mutation testing for patients with advanced non-small-cell lung cancer considering first-line EGFR tyrosine kinase inhibitor therapy. J Clin Oncol. 2011;29:2121-7 [DOI] [PubMed] [Google Scholar]
- 36. Yatabe Y, Takahashi T, Mitsudomi T. Epidermal growth factor receptor gene amplification is acquired in association with tumor progression of EGFR-mutated lung cancer. Cancer Res. 2008;68:2106-11 [DOI] [PubMed] [Google Scholar]
- 37. Varella-Garcia M. Stratification of non-small cell lung cancer patients for therapy with epidermal growth factor receptor inhibitors: the EGFR fluorescence in situ hybridization assay. Diagn Pathol. 2006;1:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cappuzzo F, Hirsch FR, Rossi E, et al. Epidermal growth factor receptor gene and protein and gefitinib sensitivity in non-small-cell lung cancer. J Natl Cancer Inst. 2005;97:643-55 [DOI] [PubMed] [Google Scholar]
- 39. Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786-92 [DOI] [PubMed] [Google Scholar]
- 40. Pao W, Miller VA, Politi KA, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mulloy R, Ferrand A, Kim Y, et al. Epidermal growth factor receptor mutants from human lung cancers exhibit enhanced catalytic activity and increased sensitivity to gefitinib. Cancer Res. 2007;67:2325-30 [DOI] [PubMed] [Google Scholar]
- 42. Kosaka T, Yatabe Y, Endoh H, et al. Analysis of epidermal growth factor receptor gene mutation in patients with non-small cell lung cancer and acquired resistance to gefitinib. Clin Cancer Res. 2006;12:5764-9 [DOI] [PubMed] [Google Scholar]
- 43. Greulich H, Chen TH, Feng W, et al. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med. 2005;2:e313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tokumo M, Toyooka S, Ichihara S, et al. Double mutation and gene copy number of EGFR in gefitinib refractory non-small-cell lung cancer. Lung Cancer. 2006;53:117-21 [DOI] [PubMed] [Google Scholar]
- 45. Balak MN, Gong Y, Riely GJ, et al. Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin Cancer Res. 2006;12:6494-501 [DOI] [PubMed] [Google Scholar]
- 46. Leidner RS, Fu P, Clifford B, et al. Genetic abnormalities of the EGFR pathway in African American patients with non-small-cell lung cancer. J Clin Oncol. 2009;27:5620-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Harada T, Lopez-Chavez A, Xi L, Raffeld M, Wang Y, Giaccone G. Characterization of epidermal growth factor receptor mutations in non-small-cell lung cancer patients of African-American ancestry. Oncogene. 2011;30:1744-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Asahina H, Yamazaki K, Kinoshita I, Yokouchi H, Dosaka-Akita H, Nishimura M. Non-responsiveness to gefitinib in a patient with lung adenocarcinoma having rare EGFR mutations S768I and V769L. Lung Cancer. 2006;54:419-22 [DOI] [PubMed] [Google Scholar]
- 49. Chen YR, Fu YN, Lin CH, et al. Distinctive activation patterns in constitutively active and gefitinib-sensitive EGFR mutants. Oncogene. 2006;25:1205-15 [DOI] [PubMed] [Google Scholar]
- 50. Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039-43 [DOI] [PubMed] [Google Scholar]
- 51. Bean J, Brennan C, Shih JY, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A. 2007;104:20932-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3:75ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Abidoye O, Ferguson MK, Salgia R. Lung carcinoma in African Americans. Nat Clin Pract Oncol. 2007;4:118-29 [DOI] [PubMed] [Google Scholar]
- 54. Yang SH, Mechanic LE, Yang P, et al. Mutations in the tyrosine kinase domain of the epidermal growth factor receptor in non-small cell lung cancer. Clin Cancer Res. 2005;11:2106-10 [DOI] [PubMed] [Google Scholar]
- 55. Cote ML, Haddad R, Edwards DJ, et al. Frequency and type of epidermal growth factor receptor mutations in African Americans with non-small cell lung cancer. J Thorac Oncol. 2011;6:627-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Reinersman JM, Johnson ML, Riely GJ, et al. Frequency of EGFR and KRAS mutations in lung adenocarcinomas in African Americans. J Thorac Oncol. 2011;6:28-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Arrieta O, Cardona AF, Federico Bramuglia G, et al. Genotyping non-small cell lung cancer (NSCLC) in Latin America. J Thorac Oncol. 2011;6:1955-9 [DOI] [PubMed] [Google Scholar]
- 58. Bacchi CE, Ciol H, Queiroga EM, Benine LC, Silva LH, Ojopi EB. Epidermal growth factor receptor and KRAS mutations in Brazilian lung cancer patients. Clinics (Sao Paulo). 2012;67:419-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Han HS, Eom DW, Kim JH, et al. EGFR mutation status in primary lung adenocarcinomas and corresponding metastatic lesions: discordance in pleural metastases. Clin Lung Cancer. 2011;12:380-6 [DOI] [PubMed] [Google Scholar]
- 60. Han C, Zou H, Ma J, Zhou Y, Zhao J. [Comparison of EGFR and KRAS status between primary non-small cell lung cancer and corresponding metastases: a systematic review and meta-analysis]. Zhongguo Fei Ai Za Zhi. 2010;13:882-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kalikaki A, Koutsopoulos A, Trypaki M, et al. Comparison of EGFR and K-RAS gene status between primary tumours and corresponding metastases in NSCLC. Br J Cancer. 2008;99:923-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Monaco SE, Nikiforova MN, Cieply K, Teot LA, Khalbuss WE, Dacic S. A comparison of EGFR and KRAS status in primary lung carcinoma and matched metastases. Hum Pathol. 2010;41:94-102 [DOI] [PubMed] [Google Scholar]
- 63. Shepherd FA. Molecular selection trumps clinical selection. J Clin Oncol. 2011;29:2843-4 [DOI] [PubMed] [Google Scholar]
- 64. Aviel-Ronen S, Blackhall FH, Shepherd FA, Tsao MS. K-ras mutations in non-small-cell lung carcinoma: a review. Clin Lung Cancer. 2006;8:30-8 [DOI] [PubMed] [Google Scholar]
- 65. Marchetti A, Martella C, Felicioni L, et al. EGFR mutations in non-small-cell lung cancer: analysis of a large series of cases and development of a rapid and sensitive method for diagnostic screening with potential implications on pharmacologic treatment. J Clin Oncol. 2005;23:857-65 [DOI] [PubMed] [Google Scholar]
- 66. Toyooka S, Tokumo M, Shigematsu H, et al. Mutational and epigenetic evidence for independent pathways for lung adenocarcinomas arising in smokers and never smokers. Cancer Res. 2006;66:1371-5 [DOI] [PubMed] [Google Scholar]
- 67. Toyooka S, Yatabe Y, Tokumo M, et al. Mutations of epidermal growth factor receptor and K-ras genes in adenosquamous carcinoma of the lung. Int J Cancer. 2006;118:1588-90 [DOI] [PubMed] [Google Scholar]
- 68. Pao W, Wang TY, Riely GJ, et al. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005;2:e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Riely GJ, Kris MG, Rosenbaum D, et al. Frequency and distinctive spectrum of KRAS mutations in never smokers with lung adenocarcinoma. Clin Cancer Res. 2008;14:5731-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sun Y, Ren Y, Fang Z, et al. Lung adenocarcinoma from East Asian never-smokers is a disease largely defined by targetable oncogenic mutant kinases. J Clin Oncol. 2010;28:4616-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Linardou H, Dahabreh IJ, Kanaloupiti D, et al. Assessment of somatic k-RAS mutations as a mechanism associated with resistance to EGFR-targeted agents: a systematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. Lancet Oncol. 2008;9:962-72 [DOI] [PubMed] [Google Scholar]
- 72. Mao C, Qiu LX, Liao RY, et al. KRAS mutations and resistance to EGFR-TKIs treatment in patients with non-small cell lung cancer: a meta-analysis of 22 studies. Lung Cancer. 2010;69:272-8 [DOI] [PubMed] [Google Scholar]
- 73. Hunt JD, Strimas A, Martin JE, et al. Differences in KRAS mutation spectrum in lung cancer cases between African Americans and Caucasians after occupational or environmental exposure to known carcinogens. Cancer Epidemiol Biomarkers Prev. 2002;11:1405-12 [PubMed] [Google Scholar]
- 74. Ma PC, Maulik G, Christensen J, Salgia R. c-Met: structure, functions and potential for therapeutic inhibition. Cancer Metastasis Rev. 2003;22:309-25 [DOI] [PubMed] [Google Scholar]
- 75. Wojta J, Nakamura T, Fabry A, et al. Hepatocyte growth factor stimulates expression of plasminogen activator inhibitor type 1 and tissue factor in HepG2 cells. Blood. 1994;84:151-7 [PubMed] [Google Scholar]
- 76. Dunsmore SE, Rubin JS, Kovacs SO, Chedid M, Parks WC, Welgus HG. Mechanisms of hepatocyte growth factor stimulation of keratinocyte metalloproteinase production. J Biol Chem. 1996;271:24576-82 [DOI] [PubMed] [Google Scholar]
- 77. Besser D, Bardelli A, Didichenko S, et al. Regulation of the urokinase-type plasminogen activator gene by the oncogene Tpr-Met involves GRB2. Oncogene. 1997;14:705-11 [DOI] [PubMed] [Google Scholar]
- 78. Rikova K, Guo A, Zeng Q, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131:1190-203 [DOI] [PubMed] [Google Scholar]
- 79. Guo A, Villen J, Kornhauser J, et al. Signaling networks assembled by oncogenic EGFR and c-Met. Proc Natl Acad Sci U S A. 2008;105:692-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Tang Z, Du R, Jiang S, et al. Dual MET-EGFR combinatorial inhibition against T790M-EGFR-mediated erlotinib-resistant lung cancer. Br J Cancer. 2008;99:911-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Schmidt L, Duh FM, Chen F, et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat Genet. 1997;16:68-73 [DOI] [PubMed] [Google Scholar]
- 82. Schmidt L, Junker K, Nakaigawa N, et al. Novel mutations of the MET proto-oncogene in papillary renal carcinomas. Oncogene. 1999;18:2343-50 [DOI] [PubMed] [Google Scholar]
- 83. Schmidt L, Junker K, Weirich G, et al. Two North American families with hereditary papillary renal carcinoma and identical novel mutations in the MET proto-oncogene. Cancer Res. 1998;58:1719-22 [PubMed] [Google Scholar]
- 84. Ma PC, Kijima T, Maulik G, et al. c-MET mutational analysis in small cell lung cancer: novel juxtamembrane domain mutations regulating cytoskeletal functions. Cancer Res. 2003;63:6272-81 [PubMed] [Google Scholar]
- 85. Ma PC, Jagadeeswaran R, Jagadeesh S, et al. Functional expression and mutations of c-Met and its therapeutic inhibition with SU11274 and small interfering RNA in non-small cell lung cancer. Cancer Res. 2005;65:1479-88 [DOI] [PubMed] [Google Scholar]
- 86. Zaffaroni D, Spinola M, Galvan A, et al. Met proto-oncogene juxtamembrane rare variations in mouse and humans: differential effects of Arg and Cys alleles on mouse lung tumorigenesis. Oncogene. 2005;24:1084-90 [DOI] [PubMed] [Google Scholar]
- 87. Kong-Beltran M, Seshagiri S, Zha J, et al. Somatic mutations lead to an oncogenic deletion of met in lung cancer. Cancer Res. 2006;66:283-9 [DOI] [PubMed] [Google Scholar]
- 88. Lee JH, Gao CF, Lee CC, Kim MD, Vande Woude GF. An alternatively spliced form of Met receptor is tumorigenic. Exp Mol Med. 2006;38:565-73 [DOI] [PubMed] [Google Scholar]
- 89. Krishnaswamy S, Kanteti R, Duke-Cohan JS, et al. Ethnic differences and functional analysis of MET mutations in lung cancer. Clin Cancer Res. 2009;15:5714-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Graveel CR, DeGroot JD, Sigler RE, Vande Woude GF. Germline met mutations in mice reveal mutation- and background-associated differences in tumor profiles. PLoS One. 2010;5:e13586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Okuda K, Sasaki H, Yukiue H, Yano M, Fujii Y. Met gene copy number predicts the prognosis for completely resected non-small cell lung cancer. Cancer Sci. 2008;99:2280-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Knudsen BS, Vande Woude G. Showering c-MET-dependent cancers with drugs. Curr Opin Genet Dev. 2008;18:87-96 [DOI] [PubMed] [Google Scholar]
- 93. Feng Y, Thiagarajan PS, Ma PC. MET signaling: novel targeted inhibition and its clinical development in lung cancer. J Thorac Oncol. 2012;7:459-67 [DOI] [PubMed] [Google Scholar]
- 94. Smolen GA, Sordella R, Muir B, et al. Amplification of MET may identify a subset of cancers with extreme sensitivity to the selective tyrosine kinase inhibitor PHA-665752. Proc Natl Acad Sci U S A. 2006;103:2316-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Xie Q, Bradley R, Kang L, et al. Hepatocyte growth factor (HGF) autocrine activation predicts sensitivity to MET inhibition in glioblastoma. Proc Natl Acad Sci U S A. 2012;109:570-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Berthou S, Aebersold DM, Schmidt LS, et al. The Met kinase inhibitor SU11274 exhibits a selective inhibition pattern toward different receptor mutated variants. Oncogene. 2004;23:5387-93 [DOI] [PubMed] [Google Scholar]
- 97. Gherardi E, Birchmeier W, Birchmeier C, Vande Woude G. Targeting MET in cancer: rationale and progress. Nat Rev Cancer. 2012;12:89-103 [DOI] [PubMed] [Google Scholar]
- 98. Sanchez-Cespedes M, Parrella P, Esteller M, et al. Inactivation of LKB1/STK11 is a common event in adenocarcinomas of the lung. Cancer Res. 2002;62:3659-62 [PubMed] [Google Scholar]
- 99. Matsumoto S, Iwakawa R, Takahashi K, et al. Prevalence and specificity of LKB1 genetic alterations in lung cancers. Oncogene. 2007;26:5911-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Koivunen JP, Kim J, Lee J, et al. Mutations in the LKB1 tumour suppressor are frequently detected in tumours from Caucasian but not Asian lung cancer patients. Br J Cancer. 2008;99:245-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Gao B, Sun Y, Zhang J, et al. Spectrum of LKB1, EGFR, and KRAS mutations in Chinese lung adenocarcinomas. J Thorac Oncol. 2010;5:1130-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Onozato R, Kosaka T, Achiwa H, et al. LKB1 gene mutations in Japanese lung cancer patients. Cancer Sci. 2007;98:1747-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Launonen V, Avizienyte E, Loukola A, et al. No evidence of Peutz-Jeghers syndrome gene LKB1 involvement in left-sided colorectal carcinomas. Cancer Res. 2000;60:546-8 [PubMed] [Google Scholar]
- 104. Suzuki Y, Oonishi T, Kudo T, Doi H. LKB1, TP16, EGFR, and KRAS somatic mutations in lung adenocarcinomas from a Chiba Prefecture, Japan cohort. Drug Discov Ther. 2012;6:24-30 [PubMed] [Google Scholar]
- 105. Gill RK, Yang SH, Meerzaman D, et al. Frequent homozygous deletion of the LKB1/STK11 gene in non-small cell lung cancer. Oncogene. 2011;30:3784-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Ji H, Ramsey MR, Hayes DN, et al. LKB1 modulates lung cancer differentiation and metastasis. Nature. 2007;448:807-10 [DOI] [PubMed] [Google Scholar]
- 107. Naoki K, Chen TH, Richards WG, Sugarbaker DJ, Meyerson M. Missense mutations of the BRAF gene in human lung adenocarcinoma. Cancer Res. 2002;62:7001-3 [PubMed] [Google Scholar]
- 108. Brose MS, Volpe P, Feldman M, et al. BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res. 2002;62:6997-7000 [PubMed] [Google Scholar]
- 109. Pratilas CA, Hanrahan AJ, Halilovic E, et al. Genetic predictors of MEK dependence in non-small cell lung cancer. Cancer Res. 2008;68:9375-83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Paik PK, Arcila ME, Fara M, et al. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J Clin Oncol. 2011;29:2046-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Samuels Y, Wang Z, Bardelli A, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. [DOI] [PubMed] [Google Scholar]
- 112. Kawano O, Sasaki H, Endo K, et al. PIK3CA mutation status in Japanese lung cancer patients. Lung Cancer. 2006;54:209-15 [DOI] [PubMed] [Google Scholar]
- 113. Morris SW, Kirstein MN, Valentine MB, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science. 1994;263:1281-4 [DOI] [PubMed] [Google Scholar]
- 114. Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561-6 [DOI] [PubMed] [Google Scholar]
- 115. Takeuchi K, Choi YL, Soda M, et al. Multiplex reverse transcription-PCR screening for EML4-ALK fusion transcripts. Clin Cancer Res. 2008;14:6618-24 [DOI] [PubMed] [Google Scholar]
- 116. Koivunen JP, Mermel C, Zejnullahu K, et al. EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer. Clin Cancer Res. 2008;14:4275-83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Choi YL, Takeuchi K, Soda M, et al. Identification of novel isoforms of the EML4-ALK transforming gene in non-small cell lung cancer. Cancer Res. 2008;68:4971-6 [DOI] [PubMed] [Google Scholar]
- 118. Takeuchi K, Choi YL, Togashi Y, et al. KIF5B-ALK, a novel fusion oncokinase identified by an immunohistochemistry-based diagnostic system for ALK-positive lung cancer. Clin Cancer Res. 2009;15:3143-9 [DOI] [PubMed] [Google Scholar]
- 119. Wong DW, Leung EL, So KK, et al. The EML4-ALK fusion gene is involved in various histologic types of lung cancers from nonsmokers with wild-type EGFR and KRAS. Cancer. 2009;115:1723-33 [DOI] [PubMed] [Google Scholar]
- 120. Inamura K, Takeuchi K, Togashi Y, et al. EML4-ALK fusion is linked to histological characteristics in a subset of lung cancers. J Thorac Oncol. 2008;3:13-7 [DOI] [PubMed] [Google Scholar]
- 121. Inamura K, Takeuchi K, Togashi Y, et al. EML4-ALK lung cancers are characterized by rare other mutations, a TTF-1 cell lineage, an acinar histology, and young onset. Mod Pathol. 2009;22:508-15 [DOI] [PubMed] [Google Scholar]
- 122. Shinmura K, Kageyama S, Tao H, et al. EML4-ALK fusion transcripts, but no NPM-, TPM3-, CLTC-, ATIC-, or TFG-ALK fusion transcripts, in non-small cell lung carcinomas. Lung Cancer. 2008;61:163-9 [DOI] [PubMed] [Google Scholar]
- 123. Jin G, Jeon HS, Lee EB, et al. EML4-ALK fusion gene in Korean non-small cell lung cancer. J Korean Med Sci. 2012;27:228-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Martelli MP, Sozzi G, Hernandez L, et al. EML4-ALK rearrangement in non-small cell lung cancer and non-tumor lung tissues. Am J Pathol. 2009;174:661-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Shaw AT, Yasothan U, Kirkpatrick P. Crizotinib. Nat Rev Drug Discov. 2011;10:897-8 [DOI] [PubMed] [Google Scholar]
- 126. Shaw AT, Yeap BY, Solomon BJ, et al. Effect of crizotinib on overall survival in patients with advanced non-small-cell lung cancer harbouring ALK gene rearrangement: a retrospective analysis. Lancet Oncol. 2011;12:1004-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Choi YL, Soda M, Yamashita Y, et al. , et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med. 2010;363:1734-9 [DOI] [PubMed] [Google Scholar]
- 128. Sasaki T, Okuda K, Zheng W, et al. The neuroblastoma-associated F1174L ALK mutation causes resistance to an ALK kinase inhibitor in ALK-translocated cancers. Cancer Res. 2010;70:10038-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Lipson D, Capelletti M, Yelensky R, et al. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat Med. 2012;18:382-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Knowles PP, Murray-Rust J, Kjaer S, et al. Structure and chemical inhibition of the RET tyrosine kinase domain. J Biol Chem. 2006;281:33577-87 [DOI] [PubMed] [Google Scholar]
- 131. Ciampi R, Nikiforov YE. RET/PTC rearrangements and BRAF mutations in thyroid tumorigenesis. Endocrinology. 2007;148:936-41 [DOI] [PubMed] [Google Scholar]
- 132. Nikiforov YE. Thyroid carcinoma: molecular pathways and therapeutic targets. Mod Pathol. 2008;21 Suppl 2:S37-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Kohno T, Ichikawa H, Totoki Y, et al. KIF5B-RET fusions in lung adenocarcinoma. Nat Med. 2012;18:375-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Birchmeier C, Sharma S, Wigler M. Expression and rearrangement of the ROS1 gene in human glioblastoma cells. Proc Natl Acad Sci U S A. 1987;84:9270-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Birchmeier C, O’Neill K, Riggs M, Wigler M. Characterization of ROS1 cDNA from a human glioblastoma cell line. Proc Natl Acad Sci U S A. 1990;87:4799-803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Bergethon K, Shaw AT, Ignatius Ou SH, et al. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol. 2012;30:863-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Liu G, Gurubhagavatula S, Zhou W, et al. Epidermal growth factor receptor polymorphisms and clinical outcomes in non-small-cell lung cancer patients treated with gefitinib. Pharmacogenomics J. 2008;8:129-38 [DOI] [PubMed] [Google Scholar]
- 138. Cusatis G, Gregorc V, Li J, et al. Pharmacogenetics of ABCG2 and adverse reactions to gefitinib. J Natl Cancer Inst. 2006;98:1739-42 [DOI] [PubMed] [Google Scholar]
- 139. Ichihara S, Toyooka S, Fujiwara Y, et al. The impact of epidermal growth factor receptor gene status on gefitinib-treated Japanese patients with non-small-cell lung cancer. Int J Cancer. 2007;120:1239-47 [DOI] [PubMed] [Google Scholar]
- 140. John T, Liu G, Tsao MS. Overview of molecular testing in non-small-cell lung cancer: mutational analysis, gene copy number, protein expression and other biomarkers of EGFR for the prediction of response to tyrosine kinase inhibitors. Oncogene. 2009;28 Suppl 1:S14-23 [DOI] [PubMed] [Google Scholar]
- 141. Chmielecki J, Peifer M, Jia P, et al. Targeted next-generation sequencing of DNA regions proximal to a conserved GXGXXG signaling motif enables systematic discovery of tyrosine kinase fusions in cancer. Nucleic Acids Res. 2010;38:6985-96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Pleasance ED, Stephens PJ, O’Meara S, et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature. 2010;463:184-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Bae NC, et al. EGFR, ERBB2, and KRAS mutations in Korean non-small cell lung cancer patients. Cancer Genet Cytogenet. 2007;173(2):107-113 [DOI] [PubMed] [Google Scholar]
- 144. Xu J, et al. Somatic mutation analysis ofEGFR, KRAS, BRAF and PIK3CA in 861 patients with non-small cell lung cancer. Cancer Biomark. 2011;10(2)639. [DOI] [PubMed] [Google Scholar]
- 145. Wu CC, et al. Reversed mutation rates of KRAS and EGFR genes in adenocarcinoma of the lung in Taiwan and their implications. Cancer. 2008;113(11):3199-208 [DOI] [PubMed] [Google Scholar]
- 146. Pemer S, et al. EML4-ALK fusion lung cancer: a rare acquired event. Neoplasia. 2008;10(3):298-302 [DOI] [PMC free article] [PubMed] [Google Scholar]