

Abstract
Background
Mutations in the core promoter and precore regions of the hepatitis B virus (HBV) genome, notably the double substitution (AGG to TGA) at nt positions 1762-1764 in the core promoter, and the precore stop codon mutation G to A at nt 1896, can often explain the anti-HBe phenotype in chronic carriers. However, the A1896 mutation is restricted to HBV isolates that have T at nt 1858. The double substitution at positions 1762-1764 has been described to occur preferentially in patients infected with strains showing C instead of T at nt 1858.
Results
HBV DNAs from 29 anti-HBe Brazilian samples were characterized by nucleotide sequencing of PCR products from precore region. Among them, 18 isolates presented C at nt 1858 (mostly genotype A strains). The 11 remaining isolates (genotypes D and F) had T1858. The stop codon mutation at nt 1896 was found in seven isolates (24% of the total and 63% of the isolates that had T1858). The frequency of the double substitution at positions 1762-1764 was surprisingly low (20%) among C1858 isolates. An association between A1896 and TGA 1762-1764 mutations was observed among genotype D isolates: these showed either none of the two mutations or both. Furthermore, strains mutated at positions 1896 and/or 1762-1764 also presented an elevated number of other, less common substitutions in the core promoter and precore regions.
Conclusions
The data reported here are not in accordance with some reports from other parts of the world. In half of the isolates, none of the mutations previously described could explain the anti-HBe phenotype.
Background
Chronic hepatitis B virus (HBV) infection, defined by the persistence of the surface antigen (HBsAg) in serum for longer than six months, may lead to a wide spectrum of liver disease, including asymptomatic carrier state, chronic active hepatitis (CAH), cirrhosis, and hepatocellular carcinoma (HCC). Hepatitis B e antigen (HBeAg) is considered a marker for viral replication, whereas the presence of anti-HBe antibodies often indicates a low level of viral production. Seroconversion from HBeAg to anti-HBe normally correlates with improvement of liver disease. However, HBV variants have been described with mutations in the precore region that prevent HBeAg synthesis, despite continuing production of infectious virions. The most common of these mutations is a G to A substitution at nucleotide (nt) 1896, that prevents the production of HBeAg by introducing a premature stop codon into the open reading frame (ORF) of the precore region [1, 2]. Several studies have associated the mutation A1896 with an exacerbation of the clinical symptoms of liver disease caused by HBV [3, 4]. In other studies, however, such an association has not been observed [5, 6, 7]. The occurrence of the A1896 mutation depends upon the nucleotide (C or T) at position 1858, that forms a base pair with nt 1896 in the pregenomic RNA loop. The presence of a C at position 1858 precludes the G to A mutation at nt 1896 as this would destabilize the stem-loop structure of the RNA encapsidation signal [8]. The presence of the A1896 mutation is thus restricted to genotypes that have a T at nt 1858, as is the case for genotypes B, C, D, and E. Genotype A usually shows a C at this position [9] while genotype F may present a T or a C [10]. HBV genotypes are not uniformly distributed around the world, and the A1896 mutation has been found to be more prevalent in geographic regions where genotypes B, C and D are predominant, such as Asia and the Mediterranean area, and less prevalent in North America and Europe where genotype A is commonly found [11, 12]. In South America, where genotypes A, D, and F have been found [13, 14, 15], the frequency of the mutation is unknown.
Besides the A1896 mutation, a number of point mutations leading to initiation failure or premature termination, as well as deletions and insertions of nucleotides inducing frameshifts, have been detected in the precore region [5, 7].
Regulation of transcription and expression of the precore and core genes has been extensively studied [16, 17, 18]. Mutations in the core promoter, notably the double mutation at positions 1762 and 1764 changing AGG to TGA, have been suggested to mediate down-regulation of HBeAg production [19]. Such mutations, able to prevent or reduce the transcription of precore mRNA and HBe synthesis [20, 21, 22], have been found in anti-HBe positive chronic carriers.
In this study, we identify the mutations present in the precore and core promoter regions of HBV isolates (genotypes A, D, and F) derived from anti-HBe positive Brazilian patients.
Results
Precore stop codon mutation A1896
Nucleotide sequences of a 200-bp fragment, from the precore start codon at nt 1814, were determined for 29 HBV strains derived from anti-HBe positive chronic carriers. By phylogenetic analysis (Fig. 1), 17 isolates belonged to genotype A, nine to genotype D, and three to genotype F. These numbers were representative of the genotype distribution existing in Brazil. All isolates from genotype A showed C at position 1858 and, as expected in this case, G at nt 1896, independently of the clinical status of the patients (Table I). This C1858:G1896 pattern was also observed in one genotype F isolate derived from an asymptomatic individual. The 11 remaining isolates had T1858. Four of them, from genotype D, presented the T1858:G1896 pattern. The seven others (five from genotype D and two from genotype F) showed the 1896 G to A substitution leading to a stable T1858:A1896 base pairing and introducing a stop codon into the ORF of the precore. In the group of 11 isolates with T1858 in which the A1896 mutation could emerge (Table I), 6/7 mutated isolates were from patients with chronic liver disease (four with cirrhosis, two with CAH) whereas 3/4 isolates showing the T1858:G1896 pattern were from asymptomatic carriers (p = 0.09). This revealed a tendency towards a correlation between A1896 mutation and liver disease.
Figure 1.
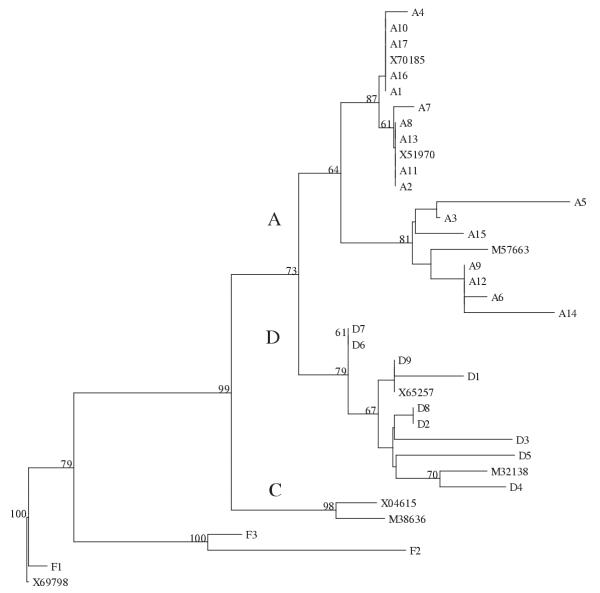
Phylogenetic tree of 37 HBV isolates, constructed with the neighbor-joining method, and based on 200 nucleotides (positions 1814-2013). Capital letters A, D, and C designate the respective genotype groupings. Bootstrap values were calculated from 100 replicates and the numbers above 60 are shown. Isolates A1 to A17, D1 to D9, and F1 to F3 are from this work. Their nucleotide sequences have been deposited in the Genbank database under accession numbers AF389988 to AF390016. The other isolates are designated by their Genbank accession numbers.
Table I.
Occurrence of the precore stop codon mutation in HBV isolates.
| Number of patients | ||||||
| Nucleotides | ||||||
| at positions | Precore stop | |||||
| Genotype | 1858:1896 | codon mutation | ||||
| With cirrhosis | With CAH | Asymptomatic | Total | |||
| A | C:G | No | 6 | 4 | 7 | 17 |
| D | T:G | No | 0 | 1 | 3 | 4 |
| T:A | Yes | 2 | 2 | 1 | 5 | |
| F | C:G | No | 0 | 0 | 1 | 1 |
| T:A | Yes | 1 | 1 | 0 | 2 | |
| Total | 9 | 8 | 12 | 29 | ||
Core promoter mutation TGA 1762-1764
In all, only seven out of 29 (24%) HBV isolates presented the precore stop codon mutation at nt 1896. Due to the low frequency of the A1896 mutation, we decided to investigate the existence of other mutations in a region of the genome covering the core promoter and precore regions, as well as the core protein initiation codon. Nucleotide sequencing was thus expanded in the 19 isolates (10 from genotype A, seven from genotype D, and two from genotype F) for which a fragment extending from nt 1720 to nt 1919 was successfully PCR amplified. Of these isolates, 14 did not show the A1896 stop mutation. No deletion or insertion was observed in any of the 19 isolates. Table II indicates that only six isolates (A5, A6, D2-D5) showed the double substitution changing AGG to TGA at positions 1762-1764 in the core promoter region, which might explain the anti-HBe seroconversion of the patients infected with isolates not showing the A1896 stop codon mutation. The triplet TGA was observed in 2/10 isolates belonging to genotype A, 4/7 D isolates, and 0/2 F strains. Remarkably, twelve isolates (A1-A4, A7-A10, D1, D6-D7, and F1) presented neither the A1896 mutation nor the 1762-1764 double substitution. Conversely, all four D isolates (D2-D5) mutated at position 1896 were also mutated at positions 1762-1764. No association was found between liver damage and mutation of the AGG 1762-1764 motif (Table II).
Table II.
Demographic and clinical data of the anti-HBe positive patients.
| Nucleotides at positions | No. of substitutionsa | |||||||
| Genotype | Patient | Age/Sex | Clinical status | 1762-1764 | 1858 | 1896 | CPMs | UPMs |
| A | A1 | 55/M | Cirrhosis | AGG | C | G | 0 | 0 |
| A2 | 41/F | Cirrhosis | AGG | C | G | 0 | 0 | |
| A3 | 45/M | Cirrhosis, HCC | AGG | C | G | 0 | 1 | |
| A4 | 40/F | Cirrhosis | AGG | C | G | 0 | 0 | |
| A5 | 10/F | Asymptomatic | TGA (mutant) | C | G | 1 | 6 | |
| A6 | 52/M | CAH | TGA (mutant) | C | G | 1 | 8 | |
| A7 | 38/M | CAH | AGG | C | G | 0 | 2 | |
| A8 | 54/M | Cirrhosis | AGG | C | G | 0 | 0 | |
| A9 | 52/M | CAH | AGG | C | G | 0 | 4 | |
| A10 | 39/M | Asymptomatic | AGG | C | G | 0 | 0 | |
| D | D1 | 36/M | CAH | AGG | T | G | 0 | 2 |
| D2 | 54/F | Asymptomatic | TGA (mutant) | T | A (stop) | 2 | 7 | |
| D3 | 56/F | CAH | TGA (mutant) | T | A (stop) | 2 | 13 | |
| D4 | 35/M | Cirrhosis | TGA (mutant) | T | A (stop) | 2 | 5 | |
| D5 | 55/M | CAH | TGA (mutant) | T | A (stop) | 2 | 8 | |
| D6 | 47/M | Asymptomatic | AGG | T | G | 0 | 6 | |
| D7 | 53/M | Asymptomatic | AGG | T | G | 0 | 6 | |
| F | F1 | 10/M | Asymptomatic | AGG | C | G | 0 | 7 |
| F2 | 35/M | CAH | AGG | T | A (stop) | 1 | 8 | |
aNumber of common (CPMs) and uncommon (UPMs) point mutations (see text) in a 200-bp genome segment, from nt 1720 to nt 1919
Other mutations
Point mutations other than those studied above were then sought in the core promoter and precore regions. For clarity, these mutations will be hereafter called uncommon point mutations in contrast to the A1896 G to A and 1762-1764 AGG to TGA substitutions referred as common point mutations . Table II (last column) shows the number of uncommon point mutations, with respect to a consensus sequence based on the genotype A isolates without common point mutations. Consequently, the number of substitutions is overestimated for isolates of genotypes D and F, since the consensus sequence for comparison is based on genotype A strains and genotype A-specific substitutions are also counted for isolates of genotypes D and F. In a general manner, it is not easy to differentiate between uncommon point mutations and the considerable natural variation in HBV sequences. Even so, it was very interesting to note that the number of uncommon point mutations was lower in the HBV isolates without common point mutations. This was evident for genotype A, since isolates A1-A4 and A7-A10 (without common point mutations) showed 0-4 uncommon point mutations (mean, 0.9), whereas isolates A5 and A6 (with one common point mutation) had 6 and 8 uncommon point mutations (mean, 7), respectively. In genotype D, the means were 4.7 for isolates (D1, D6, D7) without common point mutations, and 8.5 for isolates (D2-D5) mutated at both positions 1896 and 1762-1764.
Figure 2 shows the alignment of the nucleotide sequences from nt 1720 to nt 1919. It can be seen that several regions were conserved through all sequences, including stretches that are essential for viral replication, i.e. the AT-rich region, extending from nt 1789 to nt 1795, and the direct repeat DR1 (nt 1824 to 1834). The following substitutions appeared to be linked to the genotype (although not all the isolates of a determined genotype necessarily showed the mutation): G to A 1721 (found in genotype F), G to A 1757 (D), C to T 1802 (F), G to T 1803 (F), A to T 1850 (D and F), and C to T 1858 (D and F). Furthermore, a number of uncommon point mutations were observed that were not linked to any genotype, several of which have been previously characterized. These mutations were rarely observed in the sequences without common point mutations. Typically, substitutions in the hypervariable region from nt 1751 to nt 1755 [23] were found in 6/7 genomes with common point mutations (A5, A6, D2, D3, D5, F2) but in none of the 12 isolates without. Further, the 1727 G to A, 1740 C to T, and 1773 T to C changes were predominantly observed among the isolates otherwise mutated at crucial positions 1896 and/or 1762-1764. The isolate showing the highest number of mutations, namely D3, displayed the 1766-1768 CTT to TTA double substitution, which has been associated with increased transcription and viral encapsidation of pregenomic RNA [24, 25, 26], as well as a 1817 C to T change, introducing a stop in codon 2 of the precore (Fig. 2). The 1862 G to T point mutation, which has been proposed to contribute to the anti-HBe phenotype [27], was found in four genotype A sequences, two of them (A5, A6) with common point mutations and the two others (A3, A9) without. Additionally, the 1888 G to A mutation was present in A6 and A9 sequences. This mutation has been proposed to increase the stability of the upper stem of the encapsidation signal at the pregenomic RNA level [28]. Finally, the 1899 G to A substitution (codon 29 of the precore), previously shown to enhance the pregenomic RNA transcription [29], was found in a single isolate (D5). As a whole, 10 sequences showed neither common point mutation nor the 1862 G to T point mutation which would explain the anti-HBe phenotype.
Figure 2.

Alignment of partial nucleotide sequences of HBV isolates derived from anti-HBe carriers. The consensus sequence (based on wild-type genotype A strains) is indicated at the top. Dots represent the same nucleotides as in the consensus sequence. Letters at the left represent the genotypes of the strains. The AT-rich region (nt 1789-1795) and the direct repeat DR1 (nt 1824-1834) are underlined. The two common point mutations, i.e. the double substitution in the core promoter (nt 1762-1764) and the precore stop codon mutation (nt 1896), are indicated by arrows. The other substitutions constitute the uncommon point mutations. The initiation codons for precore and core proteins are also shown. HVR: Hypervariable region (nt 1751-1755).
Discussion
Variants of the HBV precore region have been described in anti-HBe, HBV DNA positive patients. The most common mutation was the G to A change at nt 1896 that creates a stop codon in the precore ORF. HBV strains showing this mutation cannot express the HBe antigen either on the cell membrane or in the circulation [1, 2].
In a report from Asia, 90% of the HBV isolates from anti-HBe positive, asymptomatic carriers and hepatitis patients showed the A1896 mutation [19]. Here, the mutation was observed in only 7/29 (24%) HBV isolates derived from anti-HBe positive Brazilian carriers (Table I). This low proportion was due in part to the fact that the majority of our isolates had a C at nt 1858, a feature known to preclude the stop codon mutation. In the literature, discrepancies in the frequency of the mutation can be observed, even taking in consideration only the isolates with T1858 for which the A1896 mutation may occur. Such discrepancies are observable not only between studies performed in different geographic regions, but also when carried out in a single country with isolates belonging to different genotypes. From a recent study [30], a correlation between frequency of the mutation and HBV genotype could be deduced: among isolates derived from anti-HBe positive patients and showing T at nt 1858, the mutation was present in 100%. 20%, 75%, and 100% of the isolates from genotypes B, C, D, and E, respectively. A proportion of 63% was obtained here, based on the 11 isolates showing T at nt 1858 (and belonging to genotypes D and F). In China, a country where genotypes B and C are predominant, a high prevalence (86%) of the mutation has been found [31]. Conversely, a study from Sweden revealed that none of six genotype D isolates showed the A1896 mutation during a follow-up of 17 months after anti-HBe seroconversion [32].
It has been suggested that the double substitution (AGG to TGA) at nt positions 1762-1764 in the core promoter, affecting the transcriptional regulation and protein expression of the precore gene, could explain why a precore wild-type isolate may elicit an anti-HBe phenotype [19, 23, 33]. Changes at positions 1762-1764 have been described to be preferentially selected in patients infected with genotypes showing C instead of T at nt 1858 [30, 31]. In China, indeed, these core promoter changes have been found in 91% of the isolates with C1858 compared to 27% of the isolates with T1858 [31]. However, our data demonstrated the presence of the double substitution at nt 1762-1764 in only 2/11 (18%) isolates with a C1858. Genomic characteristics other than the nucleotide present at position 1858 must therefore be involved: a notable difference between the Chinese study and ours is that Brazilian isolates with C1858 were mainly from genotype A, whereas most of the HBV strains circulating in China are from genotypes B and C. Another study, analyzing core promoter mutations in isolates with T at nt 1858 and derived from East Asian HBV carriers, showed that TGA 1762-1764 mutants were significantly more frequent in genotype C isolates than in genotype B strains [34]. Recently, Bläckberg and Kidd-Ljunggren [32] demonstrated genotypic differences in core promoter and precore sequences during seroconversion from HBeAg to anti-HBe: in samples from patients followed before and after seroconversion, it was found that 4/6 genotype A isolates showed the 1762-1764 mutation during the HBe phase. In contrast, all six genotype D isolates present in pre- and post-seroconversion samples remained wild type at positions 1896 and 1762-1764 [32].
An association between mutations A1896 and 1762-1764 (common point mutations) and severity of liver disease has been observed in several studies [3, 4, 29, 33, 35] but not in others [5, 6, 7, 36]. In our case, among seven genotype D isolates, three out of the four ones with common point mutations derived from patients with chronic liver disease, while two out of the three without common point mutations derived from asymptomatic subjects (Table II). However, no association between liver disease and mutation at positions 1762-1764 could be demonstrated.
Conclusions
The pattern of core promoter and precore mutations of HBVs derived from anti-HBe Brazilian carriers appeared to be unique among those already described. First, a low frequency (20%) of TGA 1762-1764 was noted among the genotype A isolates. Second, a strong association between A1896 and TGA 1762-1764 mutations was observed among isolates from genotype D (p < 0.05): all isolates analyzed showed either none or both mutations (Table II). Third, the common point mutations (positions 1896 and 1762-1764) were often accompanied by other mutations, notably at positions 1727, 1740, 1773, and in the hypervariable region (nt 1751-1755). Finally, only two genotype A isolates that were wild type at both positions 1896 and 1762-1764 showed other mutations which have already been associated with the anti-HBe phenotype. On the other hand, follow up of anti-HBe patients for measurement of viral load should be important to better associate the replication competence of HBV strains with anti-HBe phenotype
It has been assumed that the outcome of hepatitis B infection depends on HBV genotypes [37]. Recent studies have reported recombination events between different genotypes of HBV, which could contribute to geographical differences in the natural history of hepatitis B [38, 39]. Central and South America are the unique regions of the world where genotypes A, D and F co-circulate at a large scale. The presence of these three genotypes may account for specific variations of HBV isolates affecting the pattern of core promoter and precore mutations. To date, only a few HBV genomes from South America have been completely sequenced. Nucleotide sequencing of the whole genome of a large number of strains should help to explain the single behavior of South American HBV isolates in relation to precore-core mutations.
Materials and Methods
Patients and serological studies
Serum samples from 29 HBV chronic carriers who were referred between 1995 and 1999 to the Brazilian Reference Center for Viral Hepatitis, Rio de Janeiro, Brazil, were used in this study. All patients were tested positive for HBsAg, anti-HBe and HBV DNA, and negative for HBeAg. Twelve patients were asymptomatic HBV carriers, eight had CAH, and nine had cirrhosis.
HBsAg, HBeAg, and anti-HBe antibodies were detected in serum samples using commercially available kits in a microELISA system (Hepanostika kits, Organon Teknika, Boxtel, The Netherlands) according to the manufacturer's instructions.
DNA extraction and PCR assays
HBV DNA was extracted from serum using phenol-chloroform after treatment with proteinase K, as reported previously [15]. DNA samples were submitted to two nested PCR assays, and all PCR experiments were performed in a final volume of 50 μl under the following conditions: 94°C, 30 s; 52°C, 1 min; 72°C, 2 min; 35 cycles, followed by a final elongation of 7 min at 72°C. Sequences of the precore/ core region were amplified by nested PCR using external primers X4 (5'-AAGGT CTTACATAAGAGGAC-3', nt 1644-1663) and C2 (5'-CTAACATTGAGATTCCCG AGATTGAGA-3', nt 2458-2432), and internal primers PC1 (5'-GGCTGTAGGCA TAAATTGGTCTG-3', nt 1781-1803) and C3 (5'-TTGCCTGAGTGCAGTATGGT-3', nt 2075-2056). Core promoter region was amplified with external primers X1 (5'-ACCTCCTTTCCATGGCTGC T-3', nt 1363-1382) and C2, and internal primers X4 and C3. Amplification products (10 μl) were loaded on a 2% agarose gel, electrophoresed, stained with ethidium bromide, and visualized under UV light.
Nucleotide sequencing
PCR products were directly sequenced using the Autoload Solid Phase Sequencing system (Amersham Pharmacia Biotech, Uppsala, Sweden). This required that the second round of PCR was performed with one standard and one biotinylated primer. Dideoxy sequencing reactions (both strands) were performed using a fluorescently labeled primer whose nucleotide sequence was identical to the standard primer of the second round of PCR. The sequencing reactions were analyzed on an ALFexpress automated sequencer (Amersham Pharmacia).
Computer-Assisted Sequence analysis
Nucleotide sequences were aligned using PILEUP (Wisconsin Sequences Analysis Package GCG, Madison, WI). A phylogenetic tree was generated by neighbor-joining analysis of genetic distances, using the TREECON software package for Windows [40].
Acknowledgments
Acknowledgements
The authors are grateful to the staff of the Brazilian Reference Center for Viral Hepatitis for the gift of the serum samples. We also thank Dr. Rachel Hallett for critical reading of the manuscript and English revision.
Contributor Information
Liane De Castro, Email: decastro@gene.dbbm.fiocruz.br.
Christian Niel, Email: niel@gene.dbbm.fiocruz.br.
Selma A Gomes, Email: sagomes@gene.dbbm.fiocruz.br.
References
- Brunetto MR, Stemler M, Bonino F, Schodel F, Oliveri F, Rizzetto M, Verme G, Will H. A new hepatitis B virus strain in patients with severe anti-HBe positive chronic hepatitis B. J Hepatol. 1990;10:258–261. doi: 10.1016/0168-8278(90)90062-v. [DOI] [PubMed] [Google Scholar]
- Carman WF, Jacyna MR, Hadziyannis S, Karayiannis P, McGarvey MJ, Makris A, Thomas HC. Mutation preventing formation of hepatitis B e antigen in patients with chronic hepatitis B infection. Lancet. 1989;2(8663):588–591. doi: 10.1016/s0140-6736(89)90713-7. [DOI] [PubMed] [Google Scholar]
- Brunetto MR, Giarin MM, Oliveri F, Chiaberge E, Baldi M, Alfarano A, Serra A, Saracco G, Verme G, Will H, Bonino F. Wild-type and e antigen-minus hepatitis B viruses and course of chronic hepatitis. Proc Natl Acad Sci USA. 1991;88:4186–4190. doi: 10.1073/pnas.88.10.4186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunetto MR, Giarin M, Saracco G, Oliveri F, Calvo P, Capra G, Randone A, Abate ML, Manzini P, Capalbo M, Piantino P, Verme G, Bonino F. Hepatitis B virus unable to secrete e antigen and response to interferon in chronic hepatitis B. Gastroenterology. 1993;105:845–850. doi: 10.1016/0016-5085(93)90903-p. [DOI] [PubMed] [Google Scholar]
- Hsu H-Y, Chang M-H, Lee C-Y, Hsieh K-H, Ni Y-H, Chen P-J, Chen D-S. Precore mutant of hepatitis B virus in childhood fulminant hepatitis B: an infrequent association. J Infect Dis. 1995;171:776–781. doi: 10.1093/infdis/171.4.776. [DOI] [PubMed] [Google Scholar]
- Lindh M, Horal P, Dhillon AP, Furuta Y, Norkrans G. Hepatitis B virus carriers without precore mutations in HBeAg-negative stage show more severe liver damage. Hepatology. 1996;24:494–501. doi: 10.1002/hep.510240305. [DOI] [PubMed] [Google Scholar]
- Okamoto H, Yotsumoto S, Akahane Y, Yamanaka T, Miyazaki Y, Sugai Y, Tsuda F, Tanaka T, Miyakawa Y, Mayumi M. Hepatitis B viruses with precore region defects prevail in persistently infected hosts along with seroconversion to the antibody against e antigen. J Virol. 1990;64:1298–1303. doi: 10.1128/jvi.64.3.1298-1303.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lok AS, Akarca U, Greene S. Mutations in the pre-core region of hepatitis B virus serve to enhance the stability of the secondary structure of the pre-genome encapsidation signal. Proc Natl Acad Sci USA. 1994;91:4077–4081. doi: 10.1073/pnas.91.9.4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J-S, Tong S-P, Wen Y-M, Vitvitski L, Zhang Q, Trepo C. Hepatitis B virus genome type A rarely circulates as an HBe-minus mutant: possible contribution of a single nucleotide in the precore region. J Virol. 1993;67:5402–5410. doi: 10.1128/jvi.67.9.5402-5410.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arauz-Ruiz P, Norder H, Visoná KA, Magnius LO. Genotype F prevails in HBV infected patients of hispanic origin in Central America and may carry the precore stop mutant. J Med Virol. 1997;51:305–312. [PubMed] [Google Scholar]
- Lindh M, Andersson AS, Gusdal A. Genotypes, nt 1858 variants, and geographic origin of hepatitis B virus - large-scale analysis using a new genotyping method. J Infect Dis. 1997;175:1285–1293. doi: 10.1086/516458. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Frias F, Buti M, Jardi R, Cotrina M, Viladomiu L, Esteban R, Guardia J. Hepatitis B virus infection: precore mutants and its relation to viral genotypes and core mutations. Hepatology. 1995;22:1641–1647. doi: 10.1002/hep.1840220605. [DOI] [PubMed] [Google Scholar]
- Moraes MTB, Gomes SA, Niel C. Sequence analysis of hepatitis B virus strains of genotypes A, D, and F isolated in Brazil. Arch Virol. 1996;141:1767–1773. doi: 10.1007/BF01718299. [DOI] [PubMed] [Google Scholar]
- Moraes MTB, Niel C, Gomes SA. A polymerase chain reaction-based assay to identify genotype F of hepatitis B virus. Braz J Med Biol Res. 1999;32:45–49. doi: 10.1590/s0100-879x1999000100006. [DOI] [PubMed] [Google Scholar]
- Niel C, Moraes MTB, Gaspar AMC, Yoshida CFT, Gomes SA. Genetic diversity of hepatitis B virus strains isolated in Rio de Janeiro, Brazil . J Med Virol. 1994;44:180–186. doi: 10.1002/jmv.1890440212. [DOI] [PubMed] [Google Scholar]
- Gilbert S, Galameau L, Lamontagne A, Roy S, Belanger L. The hepatitis B virus core promoter is strongly activated by the liver nuclear receptor fetoprotein transcription factor or by ectopically expressed steroidogenic factor 1. J Virol. 2000;74:5032–5039. doi: 10.1128/jvi.74.11.5032-5039.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang WL, Su TS. The encapsidation signal of hepatitis B virus facilitates preC AUG recognition resulting in inefficient translation of the downstream genes. J Gen Virol. 1999;80:1769–1776. doi: 10.1099/0022-1317-80-7-1769. [DOI] [PubMed] [Google Scholar]
- Yuh CH, Chang YL, Ting LP. Transcriptional regulation of precore and pregenomic RNAs of hepatitis B virus. J Virol. 1992;66:4073–4084. doi: 10.1128/jvi.66.7.4073-4084.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto H, Tsuda F, Akahane Y, Sugai Y, Yoshiba M, Moriyama K, Tanaka T, Miyakawa Y, Mayumi M. Hepatitis B virus with mutations in th e core promoter for an e antigen-negative phenotype in carriers with antibody to e antigen. J Virol. 1994;68:8102–8110. doi: 10.1128/jvi.68.12.8102-8110.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckwold VE, Xu Z, Chen M, Yen TS, Ou JH. Effects of a naturally occurring mutation in the hepatitis B virus basal core promoter on precore gene expression and viral replication. J Virol. 1996;70:5845–5851. doi: 10.1128/jvi.70.9.5845-5851.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd-Ljunggren K, Oberg M, Kidd AH. The hepatitis B virus X gene: analysis of functional domain variation and gene phylogeny using multiple sequences. J Gen Virol. 1995;76:2119–2130. doi: 10.1099/0022-1317-76-9-2119. [DOI] [PubMed] [Google Scholar]
- Sato S, Suzuki K, Akahane Y, Akamatsu K, Akiyama K, Yunomura K, Tsuda F, Tanaka T, Okamoto H, Miyakawa Y, Mayumi M. Hepatitis B virus strains with mutations in the core promoter in patients with fulminant hepatitis. Ann Intern Med. 1995;122:241–248. doi: 10.7326/0003-4819-122-4-199502150-00001. [DOI] [PubMed] [Google Scholar]
- Kidd-Ljunggren K, Öberg M, Kidd AH. Hepatitis B virus X gene 1751 to 1764 mutations: implications for HBeAg status and disease. J Gen Virol. 1997;78:1469–1478. doi: 10.1099/0022-1317-78-6-1469. [DOI] [PubMed] [Google Scholar]
- Baumert TF, Rogers SA, Hasegawa K, Liang TJ. Two core promotor mutations identified in a hepatitis B virus strain associated with fulminant hepatitis result in enhanced viral replication. J Clin Invest. 1996;98:2268–2276. doi: 10.1172/JCI119037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumert TF, Marrone A, Vergalla J, Liang TJ. Naturally occurring mutations define a novel function of the hepatitis B virus core promoter in core protein expression. J Virol. 1998;72:6785–6795. doi: 10.1128/jvi.72.8.6785-6795.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Z-L, Ling R, Wang SS, Nong J, Huang CS, Harrison TJ. HBV core promoter mutations prevail in patients with hepatocellular carcinoma from Guangxi, China. J Med Virol. 1998;56:18–24. doi: 10.1002/(sici)1096-9071(199809)56:1<18::aid-jmv4>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Valliammai T, Thyagarajan SP, Zuckerman AJ, Harrison TJ. Precore and core mutations in HBV from individuals in India with chronic infection. J Med Virol. 1995;45:321–325. doi: 10.1002/jmv.1890450315. [DOI] [PubMed] [Google Scholar]
- Junker-Niepmann M, Bartenschlager R, Schaller H. A short cis-acting sequence is required for hepatitis B virus pregenome encapsidation and sufficient for packaging of foreign RNA. EMBO J. 1990;9:3389–3396. doi: 10.1002/j.1460-2075.1990.tb07540.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskus T, Radkowski M, Nowicki M, Wang L-F, Vargas H, Rakela J. Association between hepatitis B virus core promoter rearrangements and hepatocellular carcinoma. Biochem Biophys Res Commun. 1998;244:812–814. doi: 10.1006/bbrc.1998.8249. [DOI] [PubMed] [Google Scholar]
- Lindh M, Gustavson C, Mardberg K, Norkrans G, Dhillon AP, Horal P. Mutation of nucleotide 1,762 in the core promoter region during hepatitis B e seroconversion and its relation to liver damage in hepatitis B e antigen carriers. J Med Virol. 1998;55:185–190. doi: 10.1002/(sici)1096-9071(199807)55:3<185::aid-jmv1>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- Chan HLY, Hussain M, Lok AS. Different hepatitis B virus genotypes are associated with different mutations in the core promoter and precore regions during hepatitis B e antigen seroconversion. Hepatology. 1999;29:976–984. doi: 10.1002/hep.510290352. [DOI] [PubMed] [Google Scholar]
- Bläckberg J, Kidd-Ljunggren K. Genotypic differences in the hepatitis B virus core promoter and precore sequences during seroconversion from HBeAg to anti-HBe. J Med Virol. 2000;60:107–112. doi: 10.1002/(sici)1096-9071(200002)60:2<107::aid-jmv1>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- Inoue K, Yoshiba M, Sekiyama K, Okamoto H, Mayumi M. Clinical and molecular virological differences between fulminant hepatic failures following acute and chronic infection with hepatitis B virus. J Med Virol. 1998;55:35–41. doi: 10.1002/(sici)1096-9071(199805)55:1<35::aid-jmv7>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Lindh M, Hannoun C, Dhillon AP, Norkrans G, Horal P. Core promoter mutations and genotypes in relation to viral replication and liver damage in East Asian hepatitis B virus carriers. J Infect Dis. 1999;179:775–782. doi: 10.1086/314688. [DOI] [PubMed] [Google Scholar]
- Honda A, Yokosuka O, Ehata T, Tagawa M, Imazeki F, Saisho H. Detection of mutations in the enhancer 2/core promoter region of hepatitis B virus in patients with chronic hepatitis B virus infection: comparison with mutations in precore and core regions in relation to clinical status. J Med Virol. 1999;57:337–344. [PubMed] [Google Scholar]
- Pollicino T, Campo S, Raimondo G. PreS and core gene heterogeneity in hepatitis B virus (HBV) genomes isolated from patients with long-lasting HBV chronic infection. Virology. 1995;208:672–677. doi: 10.1006/viro.1995.1198. [DOI] [PubMed] [Google Scholar]
- Mayerat C, Mantegani A, Frei PC. Does hepatitis B virus (HBV) genotype influence the clinical outcome of HBV infection? J Viral Hepat. 1999;6:299–304. doi: 10.1046/j.1365-2893.1999.00174.x. [DOI] [PubMed] [Google Scholar]
- Hannoun C, Norder H, Lindh M. An aberrant genotype revealed in recombinant hepatitis B virus strains from Vietnam. J Gen Virol. 2000;81:2267–2272. doi: 10.1099/0022-1317-81-9-2267. [DOI] [PubMed] [Google Scholar]
- Morozov V, Pisareva M, Groudinin M. Homologous recombination between different genotypes of hepatitis B virus. Gene. 2000;260:55–65. doi: 10.1016/s0378-1119(00)00424-8. [DOI] [PubMed] [Google Scholar]
- Van de Peer Y, de Wachter R. TREECON for Windows: a software package for the construction and drawing of evolutionary trees for the Microsoft Windows environment. Comput Applic Biosci. 1994;10:569–570. doi: 10.1093/bioinformatics/10.5.569. [DOI] [PubMed] [Google Scholar]