

Abstract
Protein sulfenylation is a post-translational modification of emerging importance in higher eukaryotes. However, investigation of its diverse roles remains challenging, particularly within a native cellular environment. Herein we report the development and application of DYn-2, a new chemoselective probe for detecting sulfenylated proteins in human cells. These studies show that epidermal growth factor receptor–mediated signaling results in H2O2 production and oxidation of downstream proteins. In addition, we demonstrate that DYn-2 has the ability to detect differences in sulfenylation rates within the cell, which are associated with differences in target protein localization. We also show that the direct modification of epidermal growth factor receptor by H2O2 at a critical active site cysteine (Cys797) enhances its tyrosine kinase activity. Collectively, our findings reveal sulfenylation as a global signaling mechanism that is akin to phosphorylation and has regulatory implications for other receptor tyrosine kinases and irreversible inhibitors that target oxidant-sensitive cysteines in proteins.
H2O2 not only is a source of oxidative stress but also acts as an essential second messenger in signal transduction networks of normal, healthy cells, wherein growth factors, cytokines and a variety of other ligands trigger its production through the activation of their corresponding receptors1,2. Indeed, H2O2 has been demonstrated to regulate many basic cellular processes including proliferation, differentiation, growth, migration and survival. For example, binding of epidermal growth factor (EGF) to the extracellular domain of the EGF receptor (EGFR) results in the assembly and activation of NADPH oxidase (Nox) complexes, which generate H2O2 (refs. 3,4) (Fig. 1a). Once formed, H2O2 modulates signaling cascades by reaction with specific biomolecular targets.
Figure 1. Cellular redox status affects EGF-mediated signaling.

(a) EGF binding to EGFR and subsequent dimerization induce receptor autophosphorylation on specific tyrosine residues within the cytoplasmic domain. The newly phosphorylated (P) sites serve as interaction platforms for proteins involved in key prosurvival pathways, such as the PI 3K-AKT and Ras-ERK pathways. Receptor-ligand interaction also stimulates the production of ROS and oxidation of intracellular biomolecules, leading to modulation of the signaling cascade. (b) Confocal fluorescence images of EGFR localization in A431 cells before (T0) and after stimulation with 100 ng ml−1 EGF for 2 min, 15 min, 30 min and 60 min (T1, T2, T3 and T4, respectively). White arrows highlight changes in receptor localization (T0, plasma membrane; T1, membrane ruffles; T2, cell stretching and migration; T3, perinuclear and endosomal membranes; T4, cell surface and ruffles). Nuclei were counterstained with DAPI (blue). Scale bars, 10 μm. (c) EGF-induced ROS generation in A431 cells as revealed by DC F fluorescence. Where specified, cells were treated with gefitinib (Gefit), afatinib (Afat), apocynin (Apo), wortmannin (Wort), NAC or L-NAME before EGF stimulation. Data are representative of three independent readings and were normalized to the vehicle control. Error bars, ± s.e.m. *P < 0.05, **P < 0.001 when compared against cells treated with EGF only. (d–f) Western blots showing phosphorylated (p) and total EGFR, AKT and/or ERK. A431 cells were stimulated with the indicated concentrations of EGF, H2O2 or vehicle for 5 min (d) or with 100 ng ml−1 EGF or vehicle for 5 min (e,f). Where specified, cells were treated with the indicated concentrations of PEG-catalase (e), apocynin (f) or gefitinib (e,f) before EGF stimulation. Full western blots for all experiments are shown in Supplementary Figure 2.
There is now a wealth of evidence indicating that protein cysteine residues are sensitive targets of H2O2, both by direct oxidation and through the action of thiol peroxidases5,6. The product of the reaction between H2O2 and a thiolate is sulfenic acid (–SOH). Known as sulfenylation, this modification is reversible (either directly or indirectly by disulfide formation) and provides a mechanism by which changes in cellular redox state can be exploited to regulate protein function, as in phosphorylation7,8. Recent studies shed new light on the role of sulfenic acid and expand the repertoire of proteins that can undergo sulfenylation9–13, hinting at the regulatory potential and importance of these modifications. Nonetheless, the scope of sulfenylation in biological processes, particularly in eukaryotic signal transduction, remains virtually unknown.
Investigating the role of sulfenylation remains challenging, particularly in the context of the native cellular environment14. We now present the development and application of DYn-2, a chemoselective probe for detecting sulfenylated proteins directly in cells with improved sensitivity. These studies show that DYn-2 is capable of monitoring global changes in protein sulfenylation generated by Nox-mediated growth factor signaling. In addition, we demonstrate that DYn-2 has the ability to detect differences in sulfenylation rates within the cell due to differences in target protein localization. Finally, we show that modification of EGFR by H2O2 at a critical cysteine (Cys797) in its catalytic site stimulates its kinase activity, thereby demonstrating that sulfenylation, as well as phosphorylation, can regulate receptor tyrosine kinase (RTK) function.
RESULTS
EGF modulates cell morphology and EGFR trafficking
To investigate events after the interaction of EGF with its receptor, we used the human epidermoid carcinoma A431 cell line, which naturally expresses high concentrations of EGFR. As shown by phase-contrast microscopy, EGF stimulation induces rapid changes in cell shape (Supplementary Results, Supplementary Fig. 1). Additionally, we used immunofluorescence to determine whether EGF-dependent changes in morphology coincide with receptor mobilization (Fig. 1b). EGFR localized to the plasma membrane without EGF stimulation and concentrated at sites of membrane ruffling within 2 min of mitogen treatment. By 30 min, the majority of EGFR had accumulated in punctate foci throughout the peripheral cytoplasm, and after 1 h, internalized receptors had recycled back to the cell surface. These data show that EGF stimulation markedly changes cell morphology and receptor localization, setting the stage to probe oxidant-mediated signal transduction.
Cellular redox balance affects EGF-mediated signaling
Next, we examined the relationship between EGFR signaling and reactive oxygen species (ROS) in A431 cells. Intracellular generation of ROS was measured via the conversion of 2′,7′-dihydrodichlorofluorescein diacetate (H2DCF-DA) to the fluorescent product dichlorofluorescein (DCF). Coincident with membrane ruffling, EGF-stimulated cells showed an increase in DCF fluorescence intensity (Fig. 1c). Moreover, reversible and irreversible inhibitors of EGFR (gefitinib and afatinib, respectively), Nox (apocynin), phosphatidylinositol-3-OH kinase (PI3K, wortmannin) and the antioxidant N-acetylcysteine (NAC) attenuated EGF-dependent ROS generation. Control experiments with an NO synthase inhibitor (L-NAME) had no significant (P > 0.05) impact on ROS concentrations, as expected (Fig. 1c). These experiments support and extend previous observations3,15 that EGF-mediated ROS production requires activation of both EGFR and Nox.
We then investigated the effect of exogenous H2O2 on the phosphorylation of EGFR and the downstream kinases AKT and ERK. In the absence of EGF, treatment with H2O2 was sufficient to trigger a dose-dependent increase in phosphorylation of each kinase (Fig. 1d, and see Supplementary Fig. 2). Control experiments showed that each protein became phosphorylated in response to EGF and that EGFR or PI3K inhibitors attenuated this effect, as expected (Fig. 1d and Supplementary Fig. 3a). Subsequently, we examined the role of H2O2 produced by EGF stimulation (that is, endogenous H2O2) on pathway activation. Scavenging of growth factor–induced H2O2 with polyethylene glycol–catalase (PEG-catalase, Sigma) or NAC suppressed EGFR phosphorylation (Fig. 1e), global tyrosine phosphorylation and AKT and ERK activation (Supplementary Fig. 3b–e). Additionally, the Nox inhibitors apocynin and diphenyleneiodium (DPI) blunted protein phosphorylation, whereas L-NAME had no apparent effect (Fig. 1f and Supplementary Fig. 3f,g). Collectively, these data underscore the importance of endogenous H2O2 for EGFR signaling as a result of Nox activation.
The requirement for protein sulfenylation in yeast H2O2 sensing16 and T-cell activation17 has been shown through inhibition with 5,5-dimethyl-1,3-cyclohexadione (dimedone), a small molecule that reacts selectively with sulfenic acid under aqueous conditions18–20 (Fig. 2a). Along these lines, treatment of cells with dimedone before EGF stimulation inhibited phosphorylation of EGFR, AKT and ERK (Supplementary Fig. 3h), consistent with an essential role for protein sulfenylation in EGFR signaling.
Figure 2. Development and validation of probes for detecting sulfenic acid.

(a) Selective reaction between sulfenic acid and dimedone. (b) Chemical structures of chemical reporters for sulfenic acid. (c) Design and synthesis of DYn-2 (2). LD A, lithium diisopropylamide; HMPA, hexamethyl-phosphoramide. (d) Comparison of DAz-2 and DYn-2 detection of sulfenic acid in recombinant Gpx3. 50 μM protein was untreated or exposed to 100 μM H2O2 and incubated in the presence or absence of 1 mM probe for 15 min at 37 °C. Labeled proteins were detected by streptavidin–horseradish peroxidase (Strep-HRP) western blot. Comparable protein loading was confirmed by reprobing the blot with His tag–specific antibody. (e) Western blots showing DAz-2 and DYn-2 detection of protein sulfenic acids and total GAPDH in A431 cells. Cells were stimulated with 100 ng ml−1 EGF or vehicle for 5 min and then incubated with 5 mM probe or vehicle for 1 h at 37 °C.
Synthesis and evaluation of DYn-1 and DYn-2
Chemical probes directly conjugated to biotin or a fluorophore often have limited cell permeability owing to their bulky detection tags. Accordingly, protocols involving such reagents typically involve the homogenization of cells before labeling, which disrupts the native environment, including the redox balance. To address this issue, we have developed a strategy for detecting protein sulfenic acids directly in cells11,20,21, wherein the dimedone warhead is functionalized with a small azide chemical handle that does not impede membrane permeability (DAz-1 and DAz-2; Fig. 2b). An alkyne-functionalized detection tag is then appended after homogenization using the Staudinger ligation or click chemistry.
Recent studies demonstrate that alkynyl-chemical reporters, in combination with azide-bearing detection tags, offer superior sensitivity relative to the reverse combination of azide reporter and alkynyl detection tags22. In light of this observation, we designed and synthesized the alkyne-modified analogs DYn-1 (1) and DYn-2 (2) (Fig. 2b,c and Supplementary Methods). The synthesis began with ethyl protection of the reactive diketone. Alkylation of 3-ethoxycyclohex-2-enone with 3-bromopropyne to afford DYn-1 proceeded smoothly; however, low yields were obtained in analogous reactions for 5-iodopent-1-yne. As a result, we examined monoalkylation of the dianion of 1,3-cyclohexadione. Using this strategy, DYn-2 was prepared without protecting groups in a single step from commercially available materials in 96% yield (Fig. 2c).
With DYn-1 and DYn-2 in hand, we performed comparative studies to determine their utility for detecting protein sulfenic acid modifications alongside DAz-2. To this end, we used a recombinant thiol peroxidase from budding yeast, known as Gpx3, that has an active site cysteine (Cys36) that is readily oxidized to sulfenic acid16. Analysis of these reactions by western blotting revealed robust, H2O2-dependent labeling of Gpx3 by DYn-2, with increased intensity relative to labeling by DAz-2 (Fig. 2d). Control reactions, performed in the absence of probe, showed no detectable signal by western blotting (Fig. 2d). Conversely, DYn-1 showed a marked reduction in sulfenic acid labeling compared to DYn-2 (Supplementary Fig. 4a). Therefore, DYn-1 was not pursued further.
Next, we verified the nature of the covalent adduct formed between oxidized Gpx3 and DYn-2 by ESI-MS (Supplementary Fig. 4b,c). Analysis of the intact protein afforded a single major species with a molecular weight of 22,916.39 Da, consistent with a single DYn-2 adduct. Detailed examination of trypsin cleavage products confirmed Cys36 as the site of modification as indicated by the doubly charged peptide ion at m/z 551.52, which corresponds to H2N-C-(2)GFTPQYK-OH and the series of b- and y-type ions observed in the MS/MS spectrum. Overall, western blot and MS analyses establish that DYn-2 selectively targets protein sulfenic acid modifications.
We then evaluated DYn-2 for its ability to detect sulfenic acids in cells using the strategy outlined in Supplementary Figure 5a. Analysis of probe labeling by western blotting revealed sulfenylated proteins in both A431 and HeLa cells (Fig. 2e and Supplementary Fig. 5b,c). The qualitative profile of DYn-2 labeling was similar to that of DAz-2 labeling, suggesting that the probes reacted with the same protein targets. Notably, the total signal from DYn-2 labeling was greater than that from DAz-2 labeling under identical conditions, and the signal ratio of EGF-stimulated and unstimulated A431 cells was almost 40% greater than that for DAz-2. Detection of sulfenylated proteins by DYn-2 was also dependent on probe dose and incubation time (Supplementary Fig. 5d,e). Control reactions performed with or without catalase in lysis buffer further confirmed that DYn-2 labeling did not occur after cell homogenization (Supplementary Fig. 5f). Addition of DYn-2, before or after EGF treatment, did not affect phosphorylation of EGFR or downstream targets (Supplementary Figs. 3h and 5g), most likely because of the decrease in reactivity inherent in many dimedone analogs. Probe-treated cells showed no loss of viability and maintained redox balance (Supplementary Fig. 6). Collectively, these results validate DYn-2 as a robust chemical reporter for protein sulfenylation in cells and for our general approach of tagging oxidized proteins in situ.
Dynamic, global protein sulfenylation in response to EGF
Our preceding studies reveal EGF-dependent changes in protein sulfenylation. To our knowledge, this observation is the first of its kind, and thus we investigated this discovery in greater detail. Addition of EGF to A431 cells increased intracellular ROS (Fig. 3a) and protein sulfenylation (Fig. 3b,c) in a dose-dependent manner. The maximal increase in sulfenic acid modification was apparent at 100 ng ml−1 EGF, and it fell to the basal level at 500 ng ml−1 EGF. ROS generation (Fig. 3d) and protein sulfenylation (Fig. 3e,f) were also dynamic temporal events that peaked 5 min after EGF stimulation (100 ng ml−1) and declined thereafter. Furthermore, pharmacological studies indicated that EGF-dependent changes in protein sulfenylation required EGFR, PI3K and Nox activation, as well as intracellular H2O2 (Supplementary Fig. 7).
Figure 3. EGF-mediated ROS production and protein sulfenylation.

(a–f) ROS production, as indicated by DCF fluorescence (a,d) or western blotting for protein sulfenylation with Strep-HRP (b,e), in A431 cells incubated with EGF at the indicated concentrations (a,b) or for the indicated times (d,e). In d and e, all cells were stimulated with 500 (d) or 100 ng ml−1 EGF (e) or vehicle. Panels c and f show densitometric quantification of b and e, respectively. Throughout, GAPDH was used as a loading control. Data are representative of three independent readings for ROS measurements and four independent experiments for western blots. Error bars, mean ± s.e.m. *P < 0.05, **P < 0.01 and ***P < 0.001 compared to vehicle control. (g) Fluorescence images of sulfenylation (red) in A431 cells before (T0) and after stimulation with 100 ng ml−1 EGF for 0.5 min, 1 min, 1.5 min or 2 min, followed by treatment with 5 mM dimedone for 5 min at 37 °C in EGF-containing medium; total EGF exposure was 5.5 min, 6 min, 6.5 min and 7 min (T1–4, respectively). Nuclei were counterstained with DAPI (blue). Scale bars, 10 μm. (h) A431 cells were stimulated with 100 ng ml−1 EGF or vehicle for 0.5 min and treated with dimedone as in g. Cells were stained for the dimedone-protein adduct (green) and Nox2 (red). Scale bars, 10 μm.
Fluorescence microscopy with antibodies specific for the protein-dimedone adduct21 further highlighted the dynamic nature of EGF-mediated protein sulfenylation (Fig. 3g). Relative to unstimulated cells, cells treated with EGF had markedly increased signal intensity and peaked at 6 min, whereas control samples without primary antibody showed no signal (Supplementary Fig. 8). The slight difference in sulfenylation peak times observed by western blot (and ROS concentrations by DCF) and immunofluorescence analyses is most likely due to variations in sample handling inherent to each procedure.
There are seven isoforms of nonphagocytic NADPH oxidase (Nox1, Nox2, Nox3, Nox4, Nox5, Duox1 and Duox2) that show unique activation mechanisms and tissue-specific expression23. Western blot and immunofluorescence analyses revealed that Nox2 is a major isoform in A431 cells (Supplementary Fig. 9a,b). Because proteins in the vicinity of Nox are prime targets for oxidation, we wondered whether Nox2 might colocalize with sites of protein sulfenylation. Immunofluorescence analysis indicated the distribution of Nox2 at the plasma membrane and perinuclear area (Fig. 3h and Supplementary Fig. 9c). Remarkably, the merged image of Nox2 and protein sulfenylation revealed a high degree of colocalization (Fig. 3h). Together, these data show that EGF stimulation results in dynamic changes in protein sulfenylation, which occurs in cells at sites overlapping with Nox2 localization.
Differential oxidation of tyrosine phosphatases
We next sought to identify targets of H2O2 within the EGFR pathway. Growth factor–induced ROS generation is commonly attributed to oxidation and inactivation of an essential active site cysteine residue in protein tyrosine phosphatases (PTPs). Though the analysis of cysteine oxidation in cell extracts indicates that PTP inhibition promotes kinase signaling17,24,25, the rates of these reactions are orders of magnitude slower than those of H2O2-metabolizing enzymes, making their physiological relevance uncertain26. Because direct evidence of PTP oxidation in cells has not yet been reported, we used DYn-2 to investigate sulfenylation in three signaling phosphatases, PTEN, PTP1B and SHP2. PTEN is predominantly cytoplasmic and functions reciprocally to PI3K, and PTP1B downregulates endocytosed receptors within the endoplasmic reticulum, whereas SHP2 interacts directly with autophosphorylated EGFR at the plasma membrane through its SH2 domains.
Western blot analysis of immunoprecipitated PTPs showed that each protein underwent EGF-dependent sulfenylation in A431 cells (Fig. 4a–c). Moreover, each PTP showed a distinct oxidation profile with respect to growth factor concentration (Fig. 4a–c): sulfenylation of SHP2 peaked at a relatively low concentration of EGF (20 ng ml−1), followed by PTEN (500 ng ml−1) and PTP1B (750 ng ml−1). Subsequently, we investigated PTP localization in cells before and after EGF treatment. Immunofluorescence staining shows that SHP2 underwent a dramatic change in localization, concentrating at sites of plasma membrane ruffling, whereas EGF had no apparent effect on PTEN or PTP1B (Fig. 4d–f). Overall, these data demonstrate that PTPs undergo EGF-dependent oxidation and suggest that the extent of sulfenylation in the cell may be related to differences in target protein localization.
Figure 4. Differential sulfenylation of PTPs in EGF-treated cells.

(a–c) Western blots (WB) showing sulfenylated and total immunoprecipitated PTEN, PTP1B and SHP2. A431 cells were stimulated with EGF or vehicle for 2 min at the indicated concentrations and then incubated with 5 mM DYn-2 or vehicle for 1 h at 37 °C. Lysates were immunoprecipitated with mouse PTEN- (a), mouse PTP1B- (b) or rabbit SHP2-specific antibody (c). Sulfenylation of PTPs was detected by Strep-HRP western blot. Western blots were reprobed for total PTP as indicated to verify equivalent recovery. (d–f) Confocal fluorescence images of A431 cells stimulated with vehicle or 100 ng ml−1 EGF for 5 min. Cells were stained with PTEN- (d), PTP1B- (e) or SHP2-specific antibody (f). Nuclei were counterstained with DAPI (blue). Scale bars, 10 μm. The white arrows in f highlight the changes in subcellular localization of SHP2 after stimulation with EGF.
Identification of EGFR as a sensitive target of H2O2
The overall amount of EGFR autophosphorylation reflects the balance between kinase and phosphatase activities. H2O2-induced PTP inhibition would shift the balance toward phosphorylation; however, the increase in EGFR phosphorylation could similarly be accounted for by H2O2-mediated enhancement of intrinsic kinase activity. To examine this possibility, we first tested whether EGFR is a target of H2O2 in cells. Remarkably, these studies revealed that EGF stimulation leads to robust sulfenic acid modification of EGFR (Fig. 5a), a result that was recapitulated with exogenous H2O2 (Fig. 5b). EGFR sulfenylation peaked at the lowest concentration of EGF used in this study (4 ng ml−1) and at ~10 μM exogenous H2O2 (Supplementary Fig. 9d). Given the marked increase in EGFR oxidation at low EGF concentrations, we wondered whether the receptor might form a complex with Nox2. This proposal was confirmed by coimmunoprecipitation, which demonstrated EGF- and time-dependent association of Nox2 with EGFR and vice versa (Fig. 5c,d). In addition, we found that the EGFR–Nox complex immunoprecipitated together with SHP2, consistent with its propensity for oxidation in cells (Supplementary Fig. 9e).
Figure 5. EGF-mediated sulfenylation of EGFR Cys797 in cells.
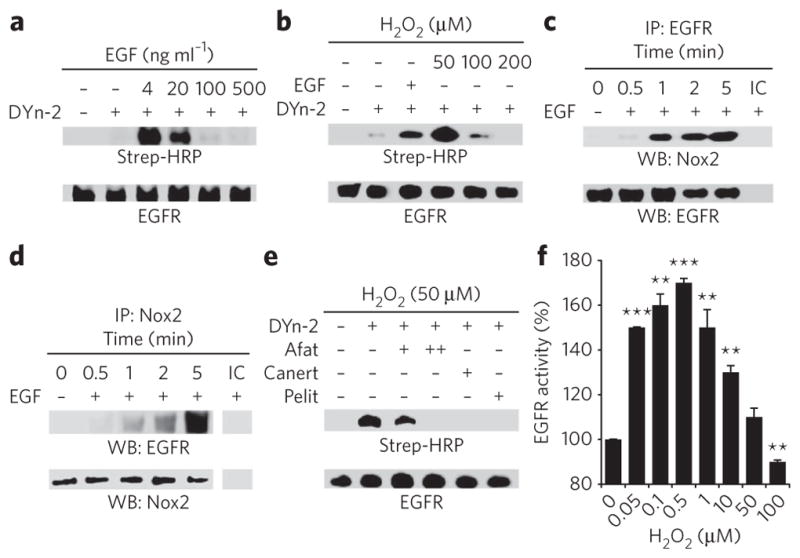
(a,b) Western blots showing sulfenylated and total EGFR. A431 cells were stimulated with EGF at the indicated concentrations or vehicle for 2 min (a) or H2O2 for 10 min (b), and sulfenic acids were detected by Strep-HRP western blot. (c,d) Western blots showing coimmunprecipitation of Nox2 and EGFR. A431 cells were stimulated with 100 ng ml−1 EGF or vehicle for the indicated times and immunoprecipitated using EGFR-specific (c) or Nox2-specific antibodies (d). Comparable recovery of immunoprecipitated (IP ) EGFR was confirmed by probing the blot with EGFR-specific antibody in c and with Nox2-specific antibody in d. IC, isotype control. (e) Western blot showing sulfenylated and total EGFR. A431 cells were incubated with 1 μM or 5 μM afatinib (+ and ++, respectively), 10 μM canertinib (Canert), 1 μM pelitinib (Pelit) or vehicle before treatment with H2O2, and sulfenylation was detected as in b. (f) Measurement of EGFR tyrosine kinase activity in vitro. Data are representative of three independent readings and represent the mean ± s.e.m. **P < 0.01 and ***P < 0.001 compared to vehicle control.
Oxidation of the EGFR active site modulates kinase activity
The kinase domain of EGFR contains six cysteine residues. Of these, a conserved cysteine within the ATP binding site (Cys797; Supplementary Fig. 9f) is a major target for irreversible inhibitors in cancer clinical trials27,28. On this basis, we hypothesized that Cys797 is the site of oxidation. This proposal was supported by studies with irreversible inhibitors, which blocked exogenous H2O2-mediated EGFR sulfenylation (Fig. 5e). Next, we mapped the site of EGF-induced oxidation in cells using dimedone. ESI-LC/MS/MS analysis of pepsin-digested EGFR confirmed Cys797 as the site of covalent modification on the basis of the doubly charged peptide ion at m/z 402.80, which corresponds to H2N-MPFGCL-OH, with dimedone attached to the cysteine residue, and the series of b- and y-type ions observed in the MS/MS spectrum (Supplementary Fig. 9g). The reduced EGFR peptide was also detected, and the ratio of peak areas of the dimedone-modified peptide ion relative to those in the unmodified version was approximately 6:1 (Supplementary Fig. 9h).
Given the proximity of Cys797 to the ATP binding site (Supplementary Fig. 9f), it is plausible that its oxidation modulates enzymatic activity. To test this hypothesis, we performed activity assays using the recombinant EGFR kinase domain. First, we verified that enzyme activity increased as a function of EGFR concentration and decreased with inhibitor treatment (Supplementary Fig. 9i–k). Subsequent studies revealed that tyrosine kinase activity was enhanced, relative to the untreated control, by moderate H2O2 concentrations (0.05–10 μM) and was decreased at concentrations greater than 50 μM (Fig. 5f). Incubation with the reducing agent dithiothreitol mitigated inhibition by H2O2 (Supplementary Fig. 9l), indicating that the decline in EGFR activity at high oxidant concentrations involves reversible thiol oxidation. Control experiments also showed that H2O2 had no significant (P > 0.05) effect on other components of the assay system (Supplementary Fig. 9m). Collectively, these data demonstrate that EGFR Cys797 is a direct target of endogenous H2O2, apparently through its association with Nox2, and that the amount of H2O2 signaling enhances EGFR kinase activity.
DISCUSSION
Historically, protein cysteine oxidation has been investigated using indirect methods of detection14 (Supplementary Fig. 10a,b). As these approaches require comprehensive blocking of free thiols at the outset of the procedure, their application is restricted to the analysis of oxidation within purified proteins or cell lysates. Alternatively, oxidative cysteine modifications can be detected on the basis of their distinct chemical attributes using selective probes (Supplementary Fig. 10c), which enable the modifications to be detected directly in cells. This is not a trivial consideration as redox potentials differ markedly among subcellular compartments29, and when the redox balance of the cell is disrupted during lysis, proteins undergo a massive amount of artifactual oxidation. This fundamental but often ignored issue increases the challenge involved in detecting modifications in low-abundance proteins and in interpreting biological significance.
With the development of DYn-2, we expand the chemical toolbox used to probe protein sulfenic acid formation in cells. The discovery that protein sulfenylation is a dynamic process during EGFR-mediated signaling most likely has broader implications for other receptor-mediated processes. Consistent with this proposal, alterations in protein sulfenic acid modifications have been observed in lysates generated from HEK293 cells treated with the cytokine TNFα (ref. 30) and from CD8+ T cells stimulated with CD3- and CD28- specific antibodies17. Though the changes in global protein sulfenylation observed in our study generally showed a strong positive correlation with the amount of ROS, cells treated with 500 ng ml−1 EGF were an exception to this rule. Interestingly, the apparent lack of sulfenylation is consistent with the absence of global disulfide bond formation in A431 cells under these conditions31 and may reflect oxidation of sulfenic to sulfinic acid, upregulation of efflux transporters, dissociation of EGFR clusters from lipid rafts, activation of alternate pathways that function independently of cysteine oxidation, or both.
Each protein analyzed in this study had a unique sulfenylation profile in cells (Fig. 6a,b). For PTPs, differential susceptibility to oxidation is particularly notable because their active site cysteines are deprotonated at physiological pH and their rates of reaction with H2O2 are almost identical in biochemical studies32,33. One possible explanation for this apparent paradox is that the proximity of target proteins to the source of ROS (such as Nox) has a considerable influence on rates of oxidation within the cell. Consistent with this model, we observed that EGFR and SHP2 form a complex with Nox2. On the other hand, oxidation of PTP1B, a phosphatase in the endoplasmic reticulum, was not observed until much higher concentrations of EGF were applied. Along these lines, a recent study in aortic endothelial cells showed that PTP1B oxidation by Nox4 requires the localization of both proteins to the endoplasmic reticulum34. Alternatively, the absence of PTP1B oxidation at lower EGF concentrations might result from sulfenyl amide formation outcompeting the DYn-2 trap (Supplementary Fig. 10d). However, this scenario seems unlikely as sulfenyl amide condensation in PTP1B is expected to be slower than intramolecular disulfide formation in PTEN and SHP2 by a factor of at least two orders of magnitude35,36.
Figure 6. Model for redox regulation of EGFR signaling.

(a) Densitometric quantification of EGFR and PTP sulfenylation from blots in Figures 4a–c and 5a. Data are representative of four independent experiments and represent the mean ± s.e.m. for each protein. *P < 0.05, **P < 0.01 and ***P < 0.001 compared to vehicle control. (b) The mitogen EGF binds to EGFR and induces the production of ROS in A431 cells via Nox2. The proximity of target proteins to Nox2 has an impact on the rate of cysteine oxidation within the cell. Dashed lines are relevant to EGFR internalization. TK, tyrosine kinase. (c) Model for H2O2-mediated increase in EGFR kinase activity. Nox-generated H2O2 directly modifies EGFR at a critical cysteine (Cys797) in the active site, which enhances its tyrosine kinase activity. Endogenous H2O2 also oxidizes and deactivates PTPs, which serves to maintain EGFR phosphorylation. Collectively, these events lead to an increase in receptor autophosphorylation, which promotes signaling through downstream pathways.
Another central finding of this study is that EGFR becomes sulfenylated at Cys797 in EGF-stimulated cells, and this modification enhances its intrinsic tyrosine kinase activity (Fig. 6c). Interestingly, the decline in EGFR activity at higher H2O2 concentrations may reflect disulfide bond formation of Cys797 with another cysteine in the kinase domain; however, future studies will be required to fully address this possibility. The biphasic response of recombinant EGFR kinase activity to H2O2 paralleled that of receptor sulfenic acid modification in cells; however, the H2O2 concentration required for maximal rate enhancement was lower by a factor of approximately 20 relative that needed for sulfenylation in cells. One likely explanation for this difference is that antioxidant enzymes and other biomolecular targets consume the H2O2 applied to cells. Another noteworthy aspect of this biphasic behavior is that cellular sulfenylation and kinase activity decreased at concentrations of H2O2 above 50 μM, whereas EGFR phosphorylation continued to increase at peroxide concentrations above 500 μM. These findings suggest a complex interplay between EGFR kinase activity and PTP inhibition at different concentrations of H2O2, wherein low concentrations stimulate catalysis, an effect that at higher doses may be lost but is compensated for by PTP inactivation. Additionally, oxidation of Cys797 could positively regulate other aspects of EGFR function, including protein-protein interactions.
It is intriguing to consider the possibility that cysteine oxidation may serve as a general mechanism to regulate RTK activity. Of the ~95 receptor and non-receptor protein tyrosine kinases (PTKs) in the human genome, nine additional members, including two additional EGFR family members, Her2 and Her4 (ref. 28), have a cysteine residue at the structural position that corresponds to Cys797. Another subfamily of PTKs, which includes cytoplasmic Src as well as FGFR1, share a cysteine residue within a conserved glycine-rich loop that interacts with the γ-phosphate of ATP. Interestingly, cellular studies implicate cysteine oxidation in Src regulation37–39, albeit with apparently contradictory results. Furthermore, biochemical analysis of Src shows that the glycine-loop cysteine is reactive and that addition of dithiothreitol to recombinant FGFR1 stimulates kinase activity40. To date, however, it has not been ascertained whether Src is a direct target of signaling-mediated H2O2 in cells, nor has the reaction of peroxide with FGFR1 been reported.
The mutation or amplification of EGFR in a number of human carcinomas, including breast and lung cancers, has motivated the development of inhibitors such as analogs that covalently modify Cys797, which are currently under evaluation in clinical trials27,28. Recently, we have reported that overexpression of EGFR and Her2 in breast cancer cell lines correlates with elevated H2O2 and global protein sulfenylation21. Coupled with the discovery that EGFR Cys797 undergoes sulfenic acid modification, these findings raise several fundamental questions vis-à-vis cysteine oxidation and thiol-targeted irreversible inhibitors. For example, how does sulfenylation affect the potency of irreversible inhibitors designed to target the thiol form of EGFR Cys797? Conversely, could the propensity for EGFR Cys797 to undergo sulfenylation be exploited to develop a new class of irreversible inhibitors that incorporate a nucleophilic warhead? Taking this one step further, whether this strategy could be exploited to selectively target EGFR in cancer cells with abnormally high concentrations of reactive oxygen species remains to be investigated. This approach need not be restricted to kinases, however, and could be applied to other enzymes or noncatalytic proteins with redox-sensitive cysteine residues, as we have demonstrated recently for PTPs41. These topics represent new and exciting avenues for future research.
In summary, we have developed the new chemoselective probe DYn-2 for detecting sulfenylated proteins in cells. Using this reagent, we have shown that growth factor–mediated signal transduction leads to oxidation of key signaling proteins, including EGFR. From a broader perspective, our findings highlight sulfenylation as a signaling mechanism analogous to phosphorylation and allude to new redox-based strategies for therapy development. In conjunction with new tools for ROS detection2,42,43 and quantitative proteomic analysis41,44–46, these results presage a bright future for the dissection of the regulatory mechanisms that underlie redox regulation of cell signaling.
METHODS
DYn-2 (2)
Lithium diisopropylamide (LDA) was prepared by the dropwise addition of 2.5 M solution of n-butyllithium (15.7 ml, 39.2 mmol) to a solution of diisopropylamine (3.97 g, 39.2 mmol) in tetrahydrofuran (THF; 40 ml), and the resulting pale-yellow mixture was stirred at −78 °C for 30 min in a 250-ml flask equipped with a magnetic stir bar under N2 pressure. A solution of 1,3-cyclohexadione (2.0 g, 17.8 mmol) in THF (20 ml) and hexamethylphosphoramide (HMPA; 10 ml) was added dropwise to the LDA solution at −78 °C. The resulting mixture was stirred at −78 °C for 1.5 h. The temperature was increased to 0 °C briefly to facilitate the stirring and then cooled again to −78 °C. To this dianion slurry, a solution of 5-iodopent-1-yne (3.81 g, 19.6 mmol) in THF (20 ml) was added dropwise at −78 °C. The reaction was stirred and allowed to warm to 25 °C over 2 h. The mixture was then neutralized with 1.0 M HCl (22 ml) and concentrated under reduced pressure. The residue was diluted with H2O and extracted with ethyl acetate (3 × 50 ml). The organic phase was then washed with brine, dried over anhydrous MgSO4 and concentrated. Purification by column chromatography (gradient: dichloromethane/methanol from 100:0 to 98:2) afforded compound 2 as a mixture of the keto and enol forms (3.0 g, 96% yield). The product was further purified by reversed-phase preparative HPLC (Varian Polaris 5 C18-A 150 × 21.2 mm column) using a gradient of water/acetonitrile from 95:5 to 5:95 over 30 min. 1H-NMR (400 MHz, CDCl3): δ 5.42 (s, 1H), 3.41 (d, J = 4.0 Hz, 2H), 2.75–1.72 (m, 16H), 1.68–1.45 (m, 6H). 13C-NMR (100 MHz, CDCl3): δ 204.9, 204.4, 197.0, 189.2, 104.2, 84.4, 84.1, 69.1, 68.9, 58.5, 49.1, 41.8, 39.9, 30.1, 29.6, 28.5, 26.4, 26.2, 26.1, 24.7, 18.8, 18.7. ESI-MS, m/z for C11H14O2: calculated, 178.23; observed, 179.1 [M + H]+.
Cell culture
HeLa cells were cultured as previously described11. A431 cells (American Type Culture Collection) were maintained at 37 °C in a 5% CO2, humidified atmosphere. Unless indicated otherwise, cells were cultured in high-glucose DMEM medium (Invitrogen) containing 10% FBS (Invitrogen), 1% GlutaMax (Invitrogen), 1% MEM nonessential amino acids (Invitrogen), and 1% penicillin-streptomycin (Invitrogen). For EGF treatment, cells were cultured until 80–90% confluent, rinsed with PBS and placed in high-glucose DMEM medium without serum for 16–18 h. After serum deprivation, cells were treated with the indicated concentration of EGF for the indicated time period. EGF treatment was stopped by removing the medium and washing with PBS.
Sulfenic acid labeling in cells
HeLa cells were labeled as previously described11. A431 cells were lifted with 0.25% trypsin-EDTA, harvested by centrifugation at 1,500g for 2 min, washed and resuspended in serum-free DMEM at a density of 3–4 × 106 cells ml−1. Intact cells in suspension were incubated with DMSO vehicle (2% v/v) or the indicated concentration of sulfenic acid probe (DYn-2, DAz-2 or dimedone) at 37 °C in a 5% CO2, humidified atmosphere with periodic gentle agitation. Following treatment for the indicated time, cells were collected and washed with PBS. The resulting cells were routinely counted using a hemocytometer and uniformly showed greater than 90% viability by trypan blue exclusion.
Click chemistry
Cell lysate (200 μg, 1 mg ml−1) was pretreated with 75 μl NeutrAvidin-agarose (Pierce) to remove endogenously biotinylated proteins. The precleared lysate was incubated with 100 μM azide- or alkyne-biotin, 1 mM tris(2-carboxyethyl)phosphine hydrochloride, 100 μM tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine ligand and 1 mM CuSO4 for 1 h at 25 °C with gentle rocking (final reaction volume of 200 μl). The reaction was quenched by 40 mM EDTA, and the proteins were subjected to methanol precipitation. The resulting protein precipitate was then resolubilized in Laemmli sample buffer containing 5% (w/v) SDS in PBS. To analyze immunoprecipitated proteins, the resin was treated with 20 μl click chemistry mix (100 μM azide-biotin, 1 mM TCEP, 100 μM TBTA, 1 mM CuSO4 in PBS) as above; reactions were quenched by boiling with 20 μl Laemmli sample buffer for 10 min.
Immunostaining and fluorescence imaging
A431 cells were seeded on collagencoated coverslips (BD Biosciences) and cultured as described above. The cells were then fixed with 4% paraformaldehyde in PBS for 15 min, and washed three times with PBS and then blocked in 5% horse serum, 0.1% Triton X-100 (Sigma) in PBS (blocking solution) for 30 min at 25 °C. The cells were then treated with rabbit EGFR-specific antibody (1005, Santa Cruz Biotechnology), mouse PTEN-specific antibody (A2B1, Santa Cruz Biotechnology), mouse PTP1B-specific antibody (FG6, Calbiochem) or rabbit SHP2-specific antibody (Santa Cruz Biotechnology) at 2 μg ml−1 in blocking solution for 1 h at 25 °C. Control cells were treated with PBS only. The cells were washed three times in PBS and incubated with Alexa 594–conjugated goat rabbit-specific (Invitrogen), Alexa488-conjugated goat rabbit-specific (Invitrogen) or Alexa 488–conjugated goat mouse-specific (Invitrogen) secondary antibodies diluted to 1:1,000 in blocking solution for 1 h at 25 °C in the dark. For experiments involving dimedone, cells were fixed in cold methanol:acetone (1:1), blocked and treated with rabbit 2-thiodimedone–specific antibody (1:3000) as previously described21. The cells were then washed three times with PBS and stained by Alexa 594–conjugated goat rabbit-specific secondary antibody (1:1,000) for 1 h at 25 °C in the dark. To visualize Nox2, cells were double stained with rabbit 2-thiodimedone–specific antibody (1:3,000) and phycoerythrin (PE)-conjugated mouse Nox2-specific antibody (7D5, MBL International, 1:1,000) followed by Alexa 488–conjugated goat rabbit-specific secondary antibody (1:1,000). Cells were then washed three times with blocking solution, counter-stained with 0.1 mg ml−1 DAPI, washed with PBS and mounted with Fluoromount G (Southern Biotech). Confocal fluorescence imaging studies on A431 cells were performed with an Olympus FV1000 microscope and a 100× oil-immersion objective lens. Excitation of Alexa 488 conjugates was carried out with an argon laser and emission was collected using a 488-nm to 515-nm filter set. Excitation of Alexa 594– or PE-conjugate was carried out with a helium-neon laser, and emission was collected using a 548-nm to 644-nm filter set.
Supplementary Material
Acknowledgments
The authors acknowledge funding from the Camille Henry Dreyfus Teacher Scholar Award (to K.S.C.) and the American Heart Association Scientist Development Award (0835419N to K.S.C.). The authors also wish to thank R. Petter for helpful discussions.
Footnotes
Author contributions
C.E.P., T.H.T. and A.H. performed cell culture and immunostaining experiments. F.J.G. performed mass spectrometry experiments. V.G. and S.E.L. performed synthetic experiments. K.S.C. designed experimental strategies with help from C.E.P. and V.G. K.S.C. and C.E.P. wrote the paper with input from all coauthors.
Competing financial interests
The authors declare no competing financial interests.
Additional information
Supplementary information and chemical compound information are available online at http://www.nature.com/naturechemicalbiology/. Reprints and permissions information is available online at http://www.nature.com/reprints/index.html. Correspondence and requests for materials should be addressed to C.E.P. or K.S.C.
References
- 1.Finkel T. Signal transduction by reactive oxygen species. J Cell Biol. 2011;194:7–15. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dickinson BC, Chang CJ. Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat Chem Biol. 2011;7:504–511. doi: 10.1038/nchembio.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miller EW, Tulyathan O, Isacoff EY, Chang CJ. Molecular imaging of hydrogen peroxide produced for cell signaling. Nat Chem Biol. 2007;3:263–267. doi: 10.1038/nchembio871. erratum 3 349 2007. [DOI] [PubMed] [Google Scholar]
- 4.Woo HA, et al. Inactivation of peroxiredoxin I by phosphorylation allows localized H2O2 accumulation for cell signaling. Cell. 2010;140:517–528. doi: 10.1016/j.cell.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 5.Paulsen CE, Carroll KS. Orchestrating redox signaling networks through regulatory cysteine switches. ACS Chem Biol. 2010;5:47–62. doi: 10.1021/cb900258z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Winterbourn CC, Hampton MB. Thiol chemistry and specificity in redox signaling. Free Radic Biol Med. 2008;45:549–561. doi: 10.1016/j.freeradbiomed.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 7.Reddie KG, Carroll KS. Expanding the functional diversity of proteins through cysteine oxidation. Curr Opin Chem Biol. 2008;12:746–754. doi: 10.1016/j.cbpa.2008.07.028. [DOI] [PubMed] [Google Scholar]
- 8.Roos G, Messens J. Protein sulfenic acid formation: from cellular damage to redox regulation. Free Radic Biol Med. 2011;51:314–326. doi: 10.1016/j.freeradbiomed.2011.04.031. [DOI] [PubMed] [Google Scholar]
- 9.Charles RL, et al. Protein sulfenation as a redox sensor: proteomics studies using a novel biotinylated dimedone analogue. Mol Cell Proteomics. 2007;6:1473–1484. doi: 10.1074/mcp.M700065-MCP200. [DOI] [PubMed] [Google Scholar]
- 10.Depuydt M, et al. A periplasmic reducing system protects single cysteine residues from oxidation. Science. 2009;326:1109–1111. doi: 10.1126/science.1179557. [DOI] [PubMed] [Google Scholar]
- 11.Leonard SE, Reddie KG, Carroll KS. Mining the thiol proteome for sulfenic acid modifications reveals new targets for oxidation in cells. ACS Chem Biol. 2009;4:783–799. doi: 10.1021/cb900105q. [DOI] [PubMed] [Google Scholar]
- 12.Takanishi CL, Wood MJ. A genetically encoded probe for the identification of proteins that form sulfenic acid in response to H2O2 in Saccharomyces cerevisiae. J Proteome Res. 2011;10:2715–2724. doi: 10.1021/pr1009542. [DOI] [PubMed] [Google Scholar]
- 13.Wani R, et al. Isoform-specific regulation of Akt by PDGF-induced reactive oxygen species. Proc Natl Acad Sci USA. 2011;108:10550–10555. doi: 10.1073/pnas.1011665108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leonard SE, Carroll KS. Chemical ‘omics’ approaches for understanding protein cysteine oxidation in biology. Curr Opin Chem Biol. 2011;15:88–102. doi: 10.1016/j.cbpa.2010.11.012. [DOI] [PubMed] [Google Scholar]
- 15.Bae YS, et al. Epidermal growth factor (EGF)-induced generation of hydrogen peroxide. Role in EGF receptor-mediated tyrosine phosphorylation. J Biol Chem. 1997;272:217–221. [PubMed] [Google Scholar]
- 16.Paulsen CE, Carroll KS. Chemical dissection of an essential redox switch in yeast. Chem Biol. 2009;16:217–225. doi: 10.1016/j.chembiol.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 17.Michalek RD, et al. The requirement of reversible cysteine sulfenic acid formation for T cell activation and function. J Immunol. 2007;179:6456–6467. doi: 10.4049/jimmunol.179.10.6456. [DOI] [PubMed] [Google Scholar]
- 18.Benitez LV, Allison WS. The inactivation of the acyl phosphatase activity catalyzed by the sulfenic acid form of glyceraldehyde 3-phosphate dehydrogenase by dimedone and olefins. J Biol Chem. 1974;249:6234–6243. [PubMed] [Google Scholar]
- 19.Poole LB, et al. Fluorescent and affinity-based tools to detect cysteine sulfenic acid formation in proteins. Bioconjug Chem. 2007;18:2004–2017. doi: 10.1021/bc700257a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reddie KG, Seo YH, Muse WB, III, Leonard SE, Carroll KS. A chemical approach for detecting sulfenic acid-modified proteins in living cells. Mol Biosyst. 2008;4:521–531. doi: 10.1039/b719986d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seo YH, Carroll KS. Profiling protein thiol oxidation in tumor cells using sulfenic acid-specific antibodies. Proc Natl Acad Sci USA. 2009;106:16163–16168. doi: 10.1073/pnas.0903015106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Charron G, et al. Robust fluorescent detection of protein fatty-acylation with chemical reporters. J Am Chem Soc. 2009;131:4967–4975. doi: 10.1021/ja810122f. [DOI] [PubMed] [Google Scholar]
- 23.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 24.Lee SR, Kwon KS, Kim SR, Rhee SG. Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J Biol Chem. 1998;273:15366–15372. doi: 10.1074/jbc.273.25.15366. [DOI] [PubMed] [Google Scholar]
- 25.Meng TC, Fukada T, Tonks NK. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol Cell. 2002;9:387–399. doi: 10.1016/s1097-2765(02)00445-8. [DOI] [PubMed] [Google Scholar]
- 26.Tanner JJ, Parsons ZD, Cummings AH, Zhou H, Gates KS. Redox regulation of protein tyrosine phosphatases: structural and chemical aspects. Antioxid Redox Signal. 2011;15:77–97. doi: 10.1089/ars.2010.3611. [DOI] [PubMed] [Google Scholar]
- 27.Singh J, Petter RC, Baillie TA, Whitty A. The resurgence of covalent drugs. Nat Rev Drug Discov. 2011;10:307–317. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- 28.Singh J, Petter RC, Kluge AF. Targeted covalent drugs of the kinase family. Curr Opin Chem Biol. 2010;14:475–480. doi: 10.1016/j.cbpa.2010.06.168. [DOI] [PubMed] [Google Scholar]
- 29.Go YM, Jones DP. Redox compartmentalization in eukaryotic cells. Biochim Biophys Acta. 2008;1780:1273–1290. doi: 10.1016/j.bbagen.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nelson KJ, et al. Use of dimedone-based chemical probes for sulfenic acid detection methods to visualize and identify labeled proteins. Methods Enzymol. 2010;473:95–115. doi: 10.1016/S0076-6879(10)73004-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cuddihy SL, Winterbourn CC, Hampton MB. Assessment of redox changes to hydrogen peroxide-sensitive proteins during EGF signaling. Antioxid Redox Signal. 2011;15:167–174. doi: 10.1089/ars.2010.3843. [DOI] [PubMed] [Google Scholar]
- 32.Chen CY, Willard D, Rudolph J. Redox regulation of SH2-domain-containing protein tyrosine phosphatases by two backdoor cysteines. Biochemistry. 2009;48:1399–1409. doi: 10.1021/bi801973z. [DOI] [PubMed] [Google Scholar]
- 33.Denu JM, Tanner KG. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry. 1998;37:5633–5642. doi: 10.1021/bi973035t. [DOI] [PubMed] [Google Scholar]
- 34.Chen K, Kirber MT, Xiao H, Yang Y, Keaney JF., Jr Regulation of ROS signal transduction by NADPH oxidase 4 localization. J Cell Biol. 2008;181:1129–1139. doi: 10.1083/jcb.200709049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee C, et al. Redox regulation of OxyR requires specific disulfide bond formation involving a rapid kinetic reaction path. Nat Struct Mol Biol. 2004;11:1179–1185. doi: 10.1038/nsmb856. [DOI] [PubMed] [Google Scholar]
- 36.Lee JW, Soonsanga S, Helmann JD. A complex thiolate switch regulates the Bacillus subtilis organic peroxide sensor OhrR. Proc Natl Acad Sci USA. 2007;104:8743–8748. doi: 10.1073/pnas.0702081104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Giannoni E, Buricchi F, Raugei G, Ramponi G, Chiarugi P. Intracellular reactive oxygen species activate Src tyrosine kinase during cell adhesion and anchorage-dependent cell growth. Mol Cell Biol. 2005;25:6391–6403. doi: 10.1128/MCB.25.15.6391-6403.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tang H, Hao Q, Rutherford SA, Low B, Zhao ZJ. Inactivation of SRC family tyrosine kinases by reactive oxygen species in vivo. J Biol Chem. 2005;280:23918–23925. doi: 10.1074/jbc.M503498200. [DOI] [PubMed] [Google Scholar]
- 39.Cunnick JM, et al. Role of tyrosine kinase activity of epidermal growth factor receptor in the lysophosphatidic acid-stimulated mitogen-activated protein kinase pathway. J Biol Chem. 1998;273:14468–14475. doi: 10.1074/jbc.273.23.14468. [DOI] [PubMed] [Google Scholar]
- 40.Kemble DJ, Sun G. Direct and specific inactivation of protein tyrosine kinases in the Src and FGFR families by reversible cysteine oxidation. Proc Natl Acad Sci USA. 2009;106:5070–5075. doi: 10.1073/pnas.0806117106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leonard SE, Garcia FJ, Goodsell DS, Carroll KS. Redox-based probes for protein tyrosine phosphatases. Angew Chem Int Edn Engl. 2011;50:4423–4427. doi: 10.1002/anie.201007871. [DOI] [PubMed] [Google Scholar]
- 42.Dickinson BC, Huynh C, Chang CJ. A palette of fluorescent probes with varying emission colors for imaging hydrogen peroxide signaling in living cells. J Am Chem Soc. 2010;132:5906–5915. doi: 10.1021/ja1014103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dickinson BC, Peltier J, Stone D, Schaffer DV, Chang CJ. Nox2 redox signaling maintains essential cell populations in the brain. Nat Chem Biol. 2011;7:106–112. doi: 10.1038/nchembio.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Truong TH, Garcia FJ, Seo YH, Carroll KS. Isotope-coded chemical reporter and acid-cleavable affinity reagents for monitoring protein sulfenic acids. Bioorg Med Chem Lett. 2011;21:5015–5020. doi: 10.1016/j.bmcl.2011.04.115. [DOI] [PubMed] [Google Scholar]
- 45.Weerapana E, et al. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature. 2010;468:790–795. doi: 10.1038/nature09472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Seo YH, Carroll KS. Quantification of protein sulfenic acid modifications using isotope-coded dimedone and iododimedone. Angew Chem Int Edn Engl. 2011;50:1342–1345. doi: 10.1002/anie.201007175. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.