

Abstract
Metastasis and chemoresistance in cancer are linked phenomena but the molecular basis for this link is unknown. We uncovered a network of paracrine signals between carcinoma, myeloid and endothelial cells that drives both processes in breast cancer. Cancer cells that overexpress CXCL1 and 2 by transcriptional hyperactivation or 4q21 amplification are primed for survival in metastatic sites. CXCL1/2 attract CD11b+Gr1+ myeloid cells into the tumor, which produce chemokines including S100A8/9 that enhance cancer cell survival. While chemotherapeutic agents kill cancer cells, these treatments trigger a parallel stromal reaction leading to TNF-α production by endothelial and other stromal cells. TNF-α heightens the expression of CXCL1/2 in cancer cells, thus amplifying the CXCL1/2-S100A8/9 loop and causing chemoresistance. CXCR2 blockers break this cycle, augmenting the efficacy of chemotherapy against breast tumors and particularly against metastasis. This network of endothelial-carcinoma-myeloid signaling interactions provides a mechanism linking chemoresistance and metastasis, with opportunities for intervention.
INTRODUCTION
Breast cancer remains the most common malignant disease in women with one million new cases diagnosed worldwide per year, causing 400,000 deaths (Gonzalez-Angulo et al., 2007). The vast majority of these deaths are due to metastatic disease. Although the five-year disease free survival rate is 89% in patients with well-treated localized breast cancer, the appearance of metastatic disease is almost always a harbinger of eventual cancer mortality. The median survival of breast cancer patients with distant metastasis is between one and two years, and only a quarter of such patients survive five or more years from diagnosis of metastases (Jones, 2008).
The two established forms of systemic therapy for metastatic disease are hormonal treatments for hormone-dependent (estrogen and/or progesterone receptor positive) cases and cytotoxic chemotherapy for cases without hormone receptors. Hormone-dependent breast cancers frequently become refractory to initially effective hormonal treatments, thus eventually requiring chemotherapy as well. Trastuzumab, an antibody to the extracellular domain of the receptor c-erbB2/HER2, often augments the chemotherapy effect in cases over-expressing this gene (Pegram et al., 2004). While tumor shrinkage is commonly accomplished on initial use of chemotherapy, the eventual emergence of tumor re-growth in the original as well as new sites is common (Jones, 2008). On developing progressive disease from initial chemotherapy, different chemotherapy drugs are usually offered to patients, but the odds of response to subsequent administrations of chemotherapy decline with each episode of response and progression. Ultimately, pan-resistance occurs, which in association with the progression of metastatic spread, an almost universally linked process, is the cause of death (Gonzalez-Angulo et al., 2007).
Drug resistance in cancer can be cell-intrinsic (Poulikakos and Rosen, 2011) or a combination of host and tumor mediated pathways (Bergers and Hanahan, 2008; Ebos et al., 2009). In the case of chemotherapeutic agents, resistance develops due to both pre-established intrinsic mechanisms as well as those acquired de-novo during the course of the treatment (Gonzalez-Angulo et al., 2007). Recent evidence points to tumor microenvironment components as potential participants in the generation of chemoresistance (Denardo et al., 2011; Gilbert and Hemann, 2010; Roodhart et al., 2011; Shaked et al., 2008; Shree et al., 2011). However, an integrated understanding of acquired drug resistance in the context of inputs from tumor and its microenvironment is lacking. Such insights could be critical for designing more effective therapies to overcome resistance and improve outcome from a palliative to curative clinical response in cancer.
Clinically as well as biologically, metastasis is intricately linked with resistance to chemotherapy (Hu et al., 2009; Morris et al., 2009). Biologically, less than 0.1% of the circulating cancer cells are estimated to withstand the harsh stresses of infiltrating and colonizing distant metastatic sites (Weiss, 2000). Similarly, a very small fraction of cells exposed to genotoxic stress can survive and outgrow under repeated cycles of chemotherapy.
This combined clinical and biological problem prompted us to ask whether both metastatic and chemotherapeutic stresses might select for cancer cells with a common set of survival advantage mechanisms. We uncovered a paracrine network, with the chemokines CXCL1 and 2 at its core that mediates lung metastasis and chemoresistance in breast cancer. We identified the signals from cancer cells that trigger this paracrine cascade, the specific stromal cell types that respond to these signals, the cancer cell survival factors delivered by the stromal cells, and the survival response of cancer cells to these stromal factors. We delineated how chemotherapy triggers a parallel reaction in the stroma and amplifies this paracrine network making therapy less effective. Blocking this axis with CXCL1/2 receptor inhibitors in combination with chemotherapy markedly reduced metastatic burden in preclinical models, addressing the key problem of why chemotherapeutic treatments fail and lead to relapse.
RESULTS
CXCL1/2 Mediate Mammary Tumor Growth and Lung Metastasis
Several observations provided a rationale to explore the potential role of CXCL1/2 in breast cancer progression and metastatic recurrence to the lungs. CXCL1 emerged among a set of genes whose expression is associated with lung relapse in breast tumors, including tumors that had not been exposed to prior chemotherapy (Minn et al., 2007; Minn et al., 2005), and as a gene that increases the aggressiveness of circulating breast tumor cells (Kim et al., 2009). CXCL1 and 2 are 90% identical by amino-acid sequence and signal through the same receptor CXCR2 (Balkwill, 2004). Indeed, CXCL1 and CXCL2 showed a similar expression pattern in a combined cohort of 615 primary breast cancers (Figure 1A). Copy number alterations at 4q21 occur in breast cancer (Beroukhim et al., 2010) and engulf fifteen genes in the amplification peak, including CXCL1-8. Fluorescence in situ hybridization (FISH) of human tissue samples showed that CXCL1 and CXCL2 were amplified in 7.5% of primary breast tumors and in 19.9% of metastases (Figures 1B and S1A). These results suggested that increased copy number contributes to the higher CXCL1/2 expression in invasive breast tumors and metastases. Additionally, high expression of CXCL1 in breast tumors without gene amplification is also possible (Bieche et al., 2007).
Figure 1. CXCL1/2 mediate mammary tumor growth and lung metastasis.
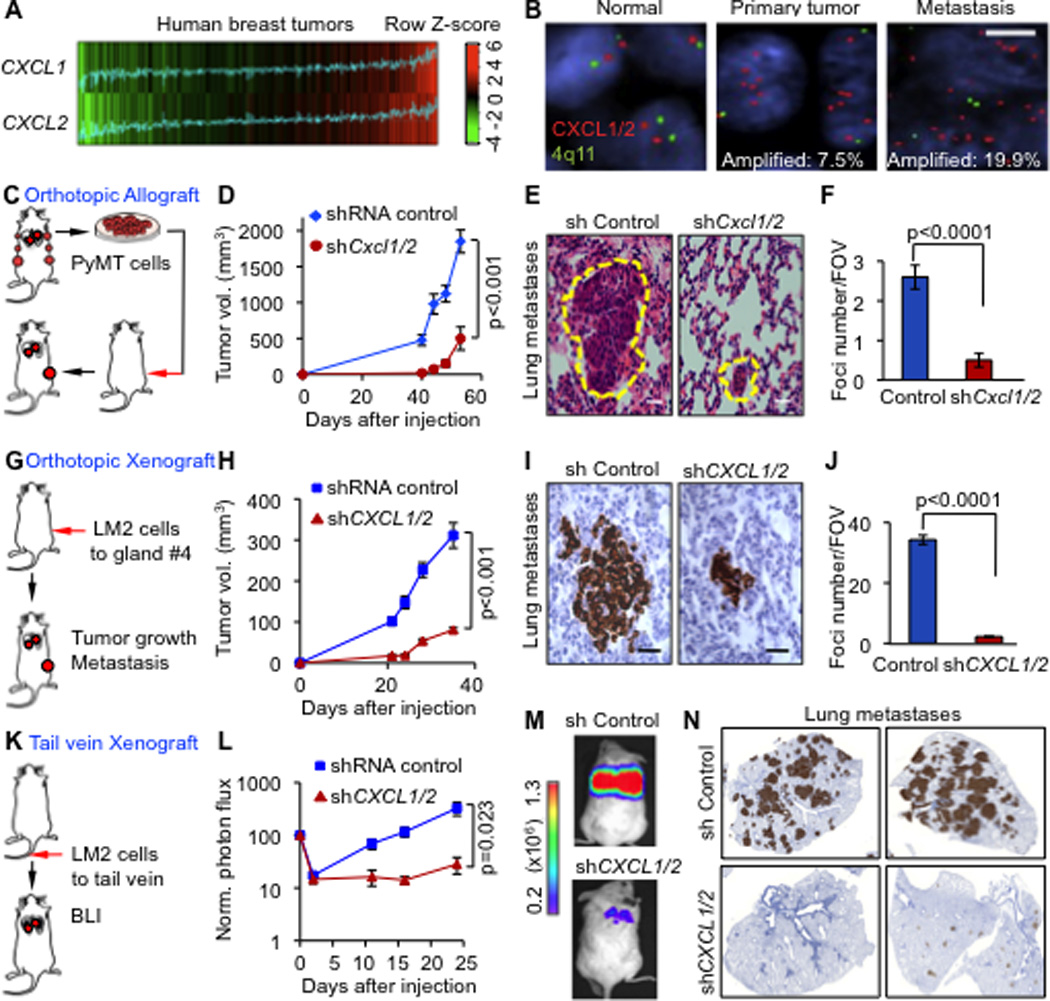
(A) CXCL1 and CXCL2 expression in 615 primary breast cancers based on microarray gene expression datasets. Correlation between the two genes was determined by Pearson’s correlation coefficient (n=615, r=0.53, p<2.2e−16). P value was determined by Student’s t-test.
(B) Breast cancer tissue microarray (TMA) samples composed of normal breast tissue, primary breast tumors and metastases (LN from lymph node and lung metastases) from patients analyzed by FISH. Signals in green correspond to 4q centromeric reference probe and signals in red correspond to CXCL1/2 probes. Scale bar, 2.5µm.
(C) Schematic representation of breast cancer progression in orthotopic allograft model. PyMT mammary cancer cells were isolated from MMTV-PyMT mammary tumors, transduced with shRNA control or shCxcl1/2 and transplanted into syngeneic mice.
(D) Growth curves of tumors from control and shCxcl1/2 PyMT-F cells. Data are averages ± SEM. n=6 mice per group.
(E) Spontaneous lung metastasis determined by H&E staining of lung sections at week 9 after tumor inoculation at endpoint. Scale bars equal 10µm.
(F) Quantitation of lung metastasis determined by automated counting of foci per field of view (FOV). Data are averages ± SEM. n=6 mice per group. P values were determined by Student’s t-test.
(G) Schematic representation of breast cancer progression in orthotopic xenograft model. LM2 metastatic breast cancer cells were implanted into immunodeficient NOD-SCID mice. Mammary tumor growth and lung metastasis were determined.
(H) Growth curves of tumors from LM2 cells transduced with control or CXCL1/2 shRNA. Data are averages ± SEM. Control n=13, shCXCL1/2 n=7.
(I) Representative histology images of spontaneous lung metastasis detected by vimentin immunostaining. Scale bars equal 60µm.
(J) Quantitation of lung metastatic burden determined by automated counting of number of foci per FOV. Shown are averages ± SEM. n=5 mice per group. P values was determined by Student’s t-test.
(K) Schematic representation of lung colonization assay in xenograft model. Luciferase labeled MDA231-LM2 cells transduced with control or shCXCL1/2 were injected intravenously and monitored over time by non-invasive bioluminescence imaging (BLI).
(L) BLI quantification of lung colonization ability of control or shCXCL1/2 LM2 cells. Data are averages ± SEM. n=7 per group. P values were determined by Student’s t-test.
(M) Representative BLI images of mice with lung metastasis
(N) Cancer cells in the lungs stained for vimentin expression.
See also Figure S1
Prompted by these findings we investigated the role of CXCL1 and 2 in breast cancer. We utilized two independent experimental systems. First, a syngeneic transplant system with cell lines derived from mammary tumors driven by a polyoma middle T transgene in two different MMTV-PyMT mouse strains, FVB/N (PyMT-F for short) and C57BL/6 (PyMT-B) (Stewart and Abrams, 2007). Second, a xenograft model to orthotopically implant LM2-4175 lung metastatic cells (LM2 for short) that were derived from the MDA-MB-231 human breast cancer cell line (Minn et al., 2005). LM2 cells showed upregulation of CXCL1/2 compared to the parental line (Figure S1B). PyMT-F and LM2 cells grew aggressively in the mammary fat pad and metastasized to the lungs. Knockdown of CXCL1 and 2 using two independent shRNA hairpins (Figure S1C–D) significantly reduced mammary tumor growth in all three models (Figures 1C–D, 1G–H and S1E–F) This decrease was associated with reduced metastasis in the lungs (Figures 1E–F, 1I–J and S1G). Similar results were obtained upon size-matching the knockdown tumors to controls (Figure S1H and I). Furthermore, lung colonization assays by tail vein injection of LM2 cells confirmed that the effect of CXCL1/2 depletion on metastasis is not solely a consequence of decreased tumor burden (Figure 1K–N). Interestingly, among other CXCR2 ligands that we tested, CXCL3 knockdown in LM2 cells contributed to tumor growth but not metastasis (Figure S1K–M). We concluded that CXCL1/2 play a prominent role in breast cancer progression and metastasis.
CXCL1/2 Recruit Myeloid Cells to Mammary Tumors
Reduction in CXCL1/2 levels in the LM2 xenograft and PyMT transplantation models was associated with a significant increase in apoptosis in the tumors (Figure 2A–B). However, CXCL1/2 knockdown was not accompanied by any visible changes in apoptosis in vitro or in angiogenesis and cell proliferation rates in tumors (Figure S2A–E). The function of CXCL1/2 is primarily mediated by binding to the G-protein coupled receptors, CXCR2 and, in some instances, CXCR1 and DARC (Balkwill, 2004). Compared to the high levels of CXCL1/2 expression in lung metastatic cell lines, the expression of CXCR1, CXCR2 and DARC was negligibly low both at the RNA and protein levels (Figure 2C and Figure S2F and (Muller et al., 2001)). Based on these results, we postulated that CXCL1/2 mediates tumor cell survival via paracrine mechanisms.
Figure 2. Carcinoma-derived CXCL1/2 supports cancer cell survival and recruit granulocytic myeloid cells to tumors.
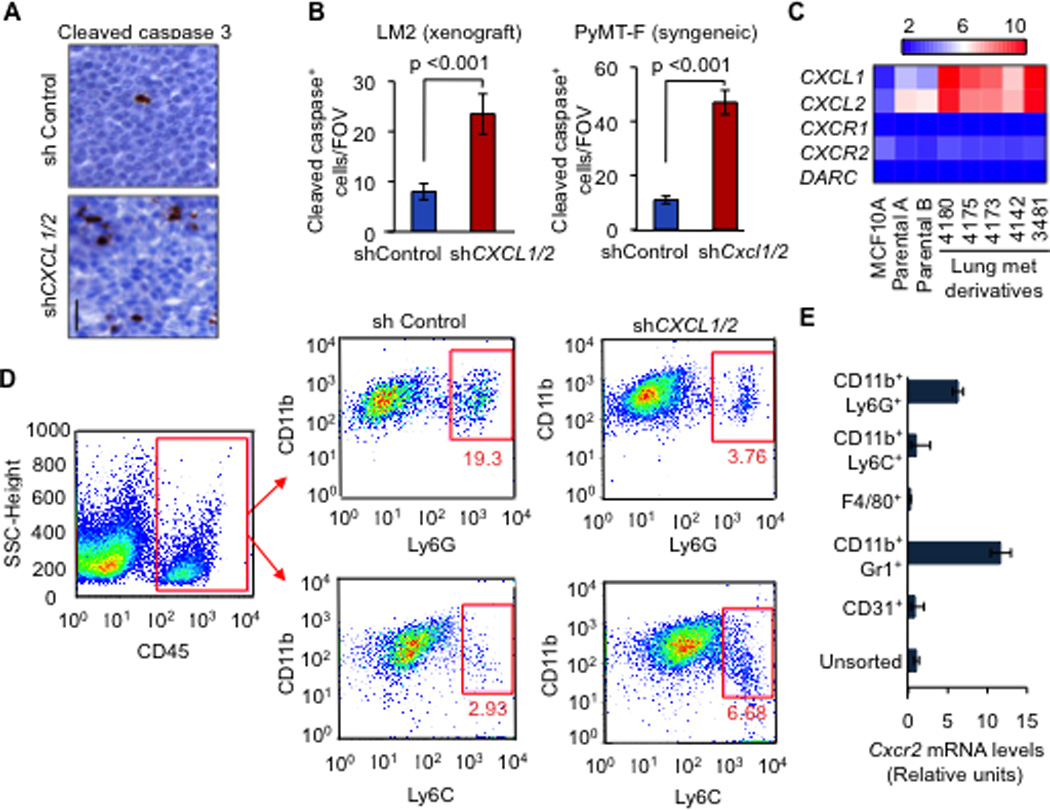
(A–B) Representative images and quantification of apoptosis in mammary tumors analyzed by Cleaved caspase-3 staining. Mouse mammary glands were injected with LM2 cells (A,B) or PyMT-F cells (B) expressing shRNA control or shCXCL1/2 and analyzed at endpoint (LM2, 6 weeks; PyMT-F, 9 weeks after tumor implantation). Scale bar equals 30µm. Data are averages ± SEM. n=4 mice per group. P values were calculated by Student’s t-test.
(C) Expression of the indicated genes from microarray gene expression analysis in non-tumor human mammary epithelial cell line MCF10A, parental MDA-MB-231 breast cancer cells and lung metastatic lines derived from MDA-MB-231 (Minn et al., 2005).
(D) Flow cytometric analysis of recruited myeloid cells in tumors formed by LM2 cells transduced with either control shRNA or shCXCL1/2 at 5 weeks after tumor inoculation. A representative gating is shown. Numbers indicate either CD11b+Ly6G+ or CD11b+Ly6C+ cells in the quadrant expressed as percentages of total CD45+ leukocytes from the same tumor. Results are representative of three independent experiments (n=3).
(E) Expression of Cxcr2 receptor in sorted subpopulations of LM2 tumors determined by qRT-PCR. Error bars represent 95% confidence interval for qRT-PCR analysis. Data is representative of two independent experiments.
See also Figure S2 and Tables S1 and S2
CXCR2 receptor is expressed by several stromal cell types such as endothelial and myeloid cells (Murdoch et al., 2008). Using a combination of immunostaining and FACS, we did a comprehensive analysis of major cell types in the tumor microenvironment whose abundance changed upon CXCL1/2 knockdown in the LM2 xenograft and PyMT-F transplant models. A significant reduction in CD11b+Gr1+ myeloid cells was observed in CXCL1/2 knockdown tumors in both models (Figure S2G–H). Myeloid-derived suppressor cells (MDSC) represent a heterogeneous group including precursors for neutrophils and monocytes that express both CD11b and Gr1 (Gabrilovich and Nagaraj, 2009). Gr1+ in mice includes cells that express the macrophage marker Ly6C and cells that express the neutrophil marker Ly6G (Ostrand-Rosenberg and Sinha, 2009). Detailed characterization of the myeloid cells showed a decrease in the CD11b+Ly6G+ granulocytic MDSC population in CXCL1/2 knockdown tumors compared to controls (Figure 2D and S2I). No appreciable decrease was observed in the CD11b+Ly6C+, F4/80+ macrophages, SMA+ myofibroblast, Ter119+ erythroid cells or endothelial cells in the CXCL1/2 depleted tumors (Figure S2J–P, Table S1 and data not shown). In line with our hypothesis, CD11b+Gr1+ cells and specifically the CD11b+Ly6G+ subpopulation expressed CXCR2 (Figure 2E, S2Q). Within the recruited CD11b+Ly6G+ population in the PyMT immunocompetent transplant tumors, 6, 12 and 25% of the cells expressed CD80, F4/80 and Sca1, respectively, and a minority expressed CD86, CD117, IL4Rα, VEGFR1 and CD34 (Table S2). Sca1, a marker of the hematopoietic stem cells, was expressed in CD11b+Ly6G+ populations, indicating potential progenitor-like phenotype of the tumor granulocytic cells recruited by CXCL1/2 (Table S2).
Interestingly, a reduction both in monocytic CD11b+Ly6C+ and granulocytic CD11b+Ly6G+ cells occurred in the lungs of mice bearing CXCL1/2 depleted tumors with minimal changes in the number of F4/80+ macrophages (Figure S2R–T). Compared to the myeloid components, lymphocytic cells represented a minority of leukocytes infiltrated in the immunocompetent PyMT transplanted tumors (Table S1). CXCL1/2 depleted tumors showed a decrease in CD4+ and CD8+ lymphocytes, no major change in γδ-TCR+ and CD25+ T-regulatory cells, and moderate increases in B220+ lymphocyte and CD49b+ NK cell numbers (Table S1). Given the low numbers and small differences within lymphocytic populations in the tumors, we chose to focus on CD11b+Gr1+ cells that are the predominant cell type recruited by CXCL1/2 in the tumor microenvironment in multiple models.
CXCL1/2 Promote Metastasis Through Myeloid Cell-derived S100A8/9
Results from our functional analysis suggested that myeloid cell types recruited by CXCL1/2 might provide paracrine factors that support the survival of cancer cells. To identify such factors, we analyzed human breast tumor gene expression datasets for genes that are expressed in association with CXCL1 (Figure 3A). Focusing on paracrine mediators, we filtered genes encoding cell surface and secreted products. Analysis of 615 breast tumors from three independent datasets yielded 43 such genes that correlated with CXCL1 with a coefficient >0.3 (Figure 3A and Table S3). These genes showed a predominance of chemokines (40%) and cytokines (21%). Similar analysis of a dataset generated from 67 metastases from breast cancer patients showed a 61% overlap in CXCL1-associated genes between primary breast tumors and metastases (Figure 3B; Tables S3, S4).
Figure 3. CXCL1/2 promote metastasis through myeloid cell-derived S100A8/9.
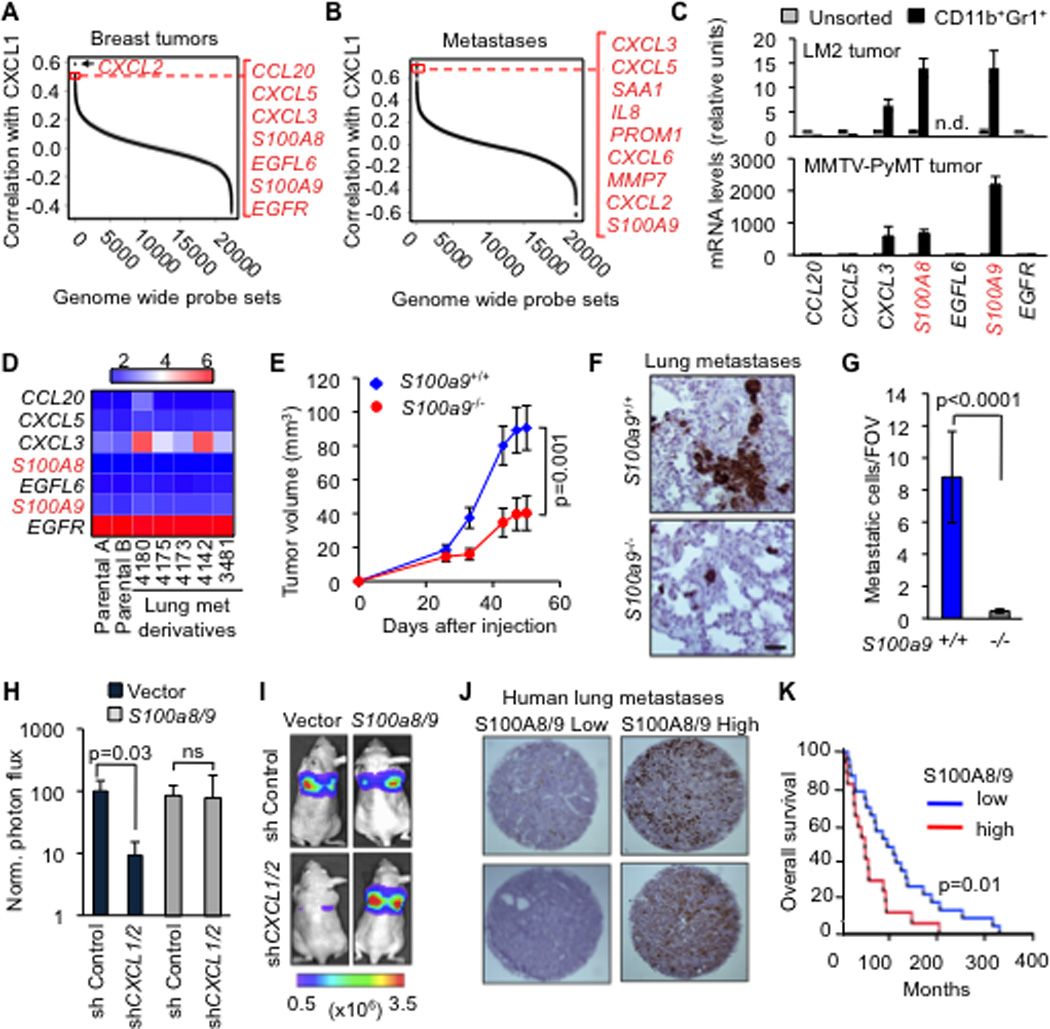
(A–B) Gene ranking according to correlation with CXCL1 expression. Expression data from breast cancer primary and metastases microarray datasets. Genes were filtered based on extracellular localization to identify paracrine mediators. The list on the right shows genes that correlate highest with CXCL1. Complete list of CXCL1 correlating genes in Tables S3 and S4.
(C) Expression of the top seven CXCL1-associated genes in (A) in the sorted CD11b+Gr1+ cells compared to unsorted tumor determined by qRT-PCR analysis from LM2 breast cancer model and MMTV-PyMT autochthonous mammary cancer model. Error bars represent 95% confidence interval. Data is representative of two independent experiments.
(D) Expression of top seven CXCL1-associated genes in breast cancer cell lines based on microarray gene expression data.
(E) LM2 tumor growth curves in mice transplanted with either S100a9+/+ or S100a9−/− bone marrow. Data points show averages ± SEM. n=19 tumors per group. P value was determined by Student’s t-test.
(F–G) Representative images and quantitation of metastatic cells in lungs detected by vimentin immunohistochemistry at 60 days after inoculation of LM2 tumors, in mice that were transplanted with S100a9+/+ or S100a9−/− bone marrow. Scale bar equals 60µm. Data points show averages ± SEM. n=4–6 mice per group. P value was determined by Student’s t-test.
(H–I) Lung colonization by LM2 cells transduced with control shRNA or shCXCL1/2, with or without ectopic expression of S100a8/9. Lung colonization was assessed by non-invasive bioluminescence imaging (BLI) at 4 weeks after tail vein injection of the cells. (H) Normalized BLI quantification (I) images represented by photon flux of lung colonization ability. Data are averages ± SEM. n=4–6 per group. ns, not significant. P value was determined by two-tailed Wilcoxon Rank Sum test.
(J) Representative TMA cores containing lung metastasis samples from breast cancer patients stained for total S100A8/9.
(K) Kaplan-Meier overall survival analysis on breast cancer patients classified by total S100A8/9 expression in lung metastasis (see panel J) n=23 for S100A8/9 low group, n=17 for S100A8/9 high group. P-values were calculated by log-rank test.
See also Figure S3 and Tables S3 and S4
To identify cancer cell survival factors derived from the recruited myeloid cells, we identified candidates that are abundantly expressed in tumor-derived CD11b+Gr1+ cells and are not of epithelial origin (Figure 3C,D and Figure S3A). S100A8 and A9 fulfilled these criteria. S100A8/9 are low molecular weight calcium-binding proteins that are associated with chronic inflammation and cancer (Gebhardt et al., 2006). S100A8/9 bind to Toll-like receptor 4 (TLR4) and RAGE (receptor for advanced glycation end products), which activate diverse signaling cascades (Gebhardt et al., 2006; Vogl et al., 2007). Both TLR4 and RAGE are expressed in breast cancer cells (Bos et al., 2009; Hsieh et al., 2003).
To determine whether myeloid S100A8/9 stimulate tumor growth and metastatic phenotype of breast cancer cells, we isolated bone marrow cells from S100A9+/+ and S100A9−/− mice (Hobbs et al., 2003) and transplanted them into irradiated immunocompromised mice. In addition to lacking S100A9, S100a9−/− bone marrow-derived cells fail to express S100A8, which is the heterodimeric partner of S100A9 (Figure S3B) (Hobbs et al., 2003). After confirming successful engraftment of S100a9+/+ or S100a9−/− bonemarrow with an efficiency of >98% (Figure S3C), we implanted LM2 cancer cells in the mammary fat pads of these mice. Mammary tumor growth and lung metastasis were significantly reduced in mice transplanted with S100a9−/− bone marrow compared to the S100a9+/+ counterpart (Figure 3E–G).
To confirm these results in a different system, we stably reduced the expression of S100a8/9 in MPRO, a murine bone marrow derived promyelocytic cell line (Figure S3D) that expresses both CD11b and Gr1 (Tsai and Collins, 1993). Coinjection of wild-type MPRO cells with LM2 cancer cells enhanced mammary tumor growth and lung metastasis, an effect that was significantly blunted by S100a8/9 knockdown in the MPRO cells (Figure S3E–F). Furthermore, the enforced expression of S100A8/9 in LM2 cells (Figure S3G–H) rescued the CXCL1/2 knockdown phenotype of reduced tumor growth and metastasis (Figures 3H,I and S3I). Together, these results suggest that S100A8/9 mediate the pro-metastatic effect of CXCL1/2 in our models.
Based on the accumulating evidence of a role of S100A8/9 in breast cancer metastasis in animal models, we sought evidence for this link in clinical samples. We immunostained tissue microarrays composed of lung metastasis samples from breast cancer patients with an antibody recognizing human S100A8/9 (Figure 3J). Kaplan Meier analysis showed that patients with high S100A8/9 in the metastatic nodules had a significantly shorter overall survival compared to low S100A8/9 (p-value=0.01) (Figure 3K). Collectively our results suggest that CXCL1/2 from breast cancer cells recruit myeloid producers of S100A8/9, which in turn promote breast cancer metastasis.
S100A8/9 Promotes Breast Cancer Cell Survival Under Chemotherapy
We investigated whether S100A8/9 in tumors are linked to enhanced cancer cell survival. Tumor-bearing mice transplanted with S100A9−/− bone marrow showed a marked increase in the number of apoptotic cells in the mammary tumors and the lung (Figure 4A). The requirement of S100A8/9 in cell survival was even more evident when tumor-bearing mice were treated with chemotherapeutic agents as an inducer of cell death (Figure 4B–C and S4A).
Figure 4. S100A8/9 promotes breast cancer cell survival under chemotherapy.
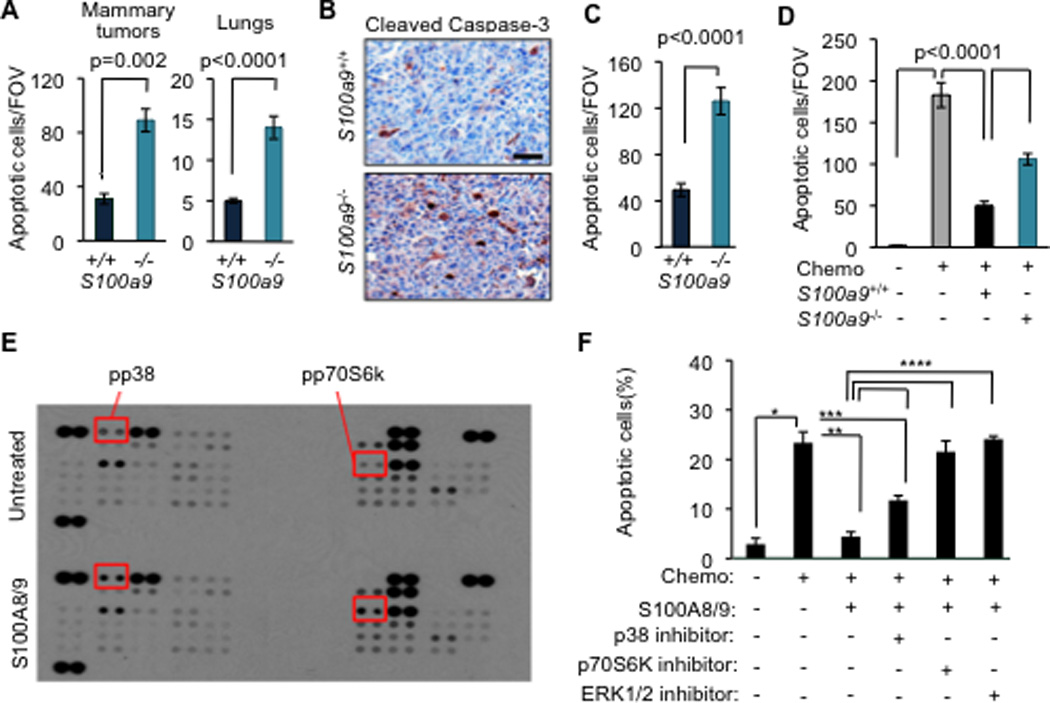
(A) Quantitation of apoptosis by cleaved caspase-3 staining in tumors and TUNEL assay in lungs in mice transplanted with S100a9+/+ or S100a9−/− bone marrow at 60 days after LM2 tumor inoculation into the mammary fat pad. Data are averages ± SEM. n=4–6 mice per group. P values were determined by Student’s t-test.
(B–C) Representative images and quantification of apoptosis by cleaved caspase 3 staining in LM2 tumors from mice transplanted with either S100a9+/+ or S100a9−/− bone marrow and subsequently treated with a combination of doxorubicin and cyclophosphamide chemotherapy (AC regimen, chemo) once weekly for 3 weeks. Scale bar represents 32µm. Data are averages ± SEM. n=5–6 tumors per group. P values were determined by Student’s t-test.
(D) TUNEL analysis detecting apoptotic cancer cells in co-culture assay. LM2 cancer cells were cultured alone or overnight in the presence of S100a9+/+ or S100a9−/− bone marrow cells and subsequently treated with chemotherapeutic drug (Chemo), doxorubicin (0.8µM). Data are average ± SEM of triplicates. P values determined by Student’s t-test.
(E) Screening of signaling pathways activated by S100A8/9 in LM2 metastatic cancer cells by probing human phospho-kinase array with lysates from cells treated with either PBS or 10µg/ml recombinant S100A8/9. Shorter exposure of the same blot shown in S4. Proteins showing increased phosphorylation upon S100A8/9 treatment are highlighted.
(F) TUNEL analysis detecting apoptotic cancer cells in co-culture assay upon pharmacological inhibition of p38, p70S6K and ERK pathways in the presence of recombinant S100A8/9. LM2 cancer cells were pretreated with 10µg/ml of S100A8/9 for 1 h and subsequently treated with doxorubicin (0.8µM, chemo) either alone or in the presence of 5µM of p38 inhibitor SB203580 or 10µM of p70S6K inhibitor PF4708671 or 10µM of ERK inhibitor FR180204 for 16h. LM2 cells treated with saline were used as controls. Quantification of apoptosis was done by calculating the percentage of TUNEL+/DAPI+ cells per FOV. Data are shown as average ± SEM from triplicates. *, **, ***, **** denote p=0.004, 0.02, 0.04, 0.01, respectively. P values were determined by Student’s t-test.
See also Figure S4
Similar numbers of CD11b+ and CD68+ myeloid cells were present in tumors and lung nodules from mice transplanted with either S100A9−/− or S100A9+/+ bone marrow cells (Figure S4B and data not shown). This result ruled out the possibility that higher apoptotic rates in the S100A9−/− group were due to a defective accumulation of myeloid cells (Hiratsuka et al., 2008). Consistent with the survival advantage provided by S100A8/9 in vivo, co-culture of cancer cells with bone marrow derived CD11b+Gr1+ cells protected cancer cells from doxorubicin-induced apoptosis (Figure 4D). However, this protection was more limited in co-culture with CD11b+Gr1+ cells derived from S100A9−/− marrow (Figure 4D), confirming that the pro-survival properties of myeloid factors under stress chemotherapy conditions are partly due to S100A8/9.
Binding of S100A8/9 to receptors RAGE and TLR4 can activate diverse signaling cascades (Gebhardt et al., 2006; Ichikawa et al., 2011; Vogl et al., 2007). S100A8/9 addition only weakly activated NFκB in LM2 breast cancer cells and we detected no activation of the Akt or STAT3 pathways (Figure S4C and data not shown). Using blot arrays that detect phosphorylation of 46 different protein kinases, we found that S100A8/9 cause activation of ERK1/2, p38 MAPK, and p70S6K in LM2 cells (Figures 4E and S4D), as confirmed by immunoblotting (Figures S4E–F). Addition of an ERK1/2 inhibitor (FR180204) or a p70S6K inhibitor (PF4708671) abolished the chemo-protective effect of S100A8/9 whereas addition of a p38 inhibitor (SB203580) showed partial effect (Figure 4F), suggesting that the ERK1/2 and p70S6K signaling mediate the pro-survival effect of S100A8/9 in metastatic cells.
The CXCL1/2–S100A8/9 Survival Axis Is Hyperactivated by Chemotherapy
Most patients who develop metastatic disease receive chemotherapy at some point in the management of their illness. Tumor shrinkage—partial or less commonly, complete remissions—is usually accomplished, but these benefits are transient and most patients eventually develop chemotherapy-resistant, widely disseminated cancer (Gonzalez-Angulo et al., 2007; Jones, 2008). We hypothesized that the CXCL1/2-S100A8/9 survival axis could nurture tumor cells under chemotherapeutic stress thereby selecting for aggressive metastatic progeny. To address this question, we treated mice bearing LM2 tumors with doxorubicin and cyclophosphamide (AC regimen), a commonly used chemotherapy combination in the clinic. MDA-MB-231, the parental breast cancer cell line from which LM2 was derived, was originally isolated from pleural effusion of a patient who was resistant to AC and 5-fluorouracil chemotherapy and had relapsed (Cailleau et al., 1974). Chemotherapy treatment of mice bearing LM2 tumors initially resulted in significant apoptosis and a concomitant delay in tumor growth (Figure 5A–B). However, after subsequent rounds of chemotherapy, tumors became refractory as evidenced by reduction in apoptosis and resumed tumor growth (Figure 5A–B).
Figure 5. CXCL1/2 paracrine axis is hyperactivated upon chemotherapy treatment.
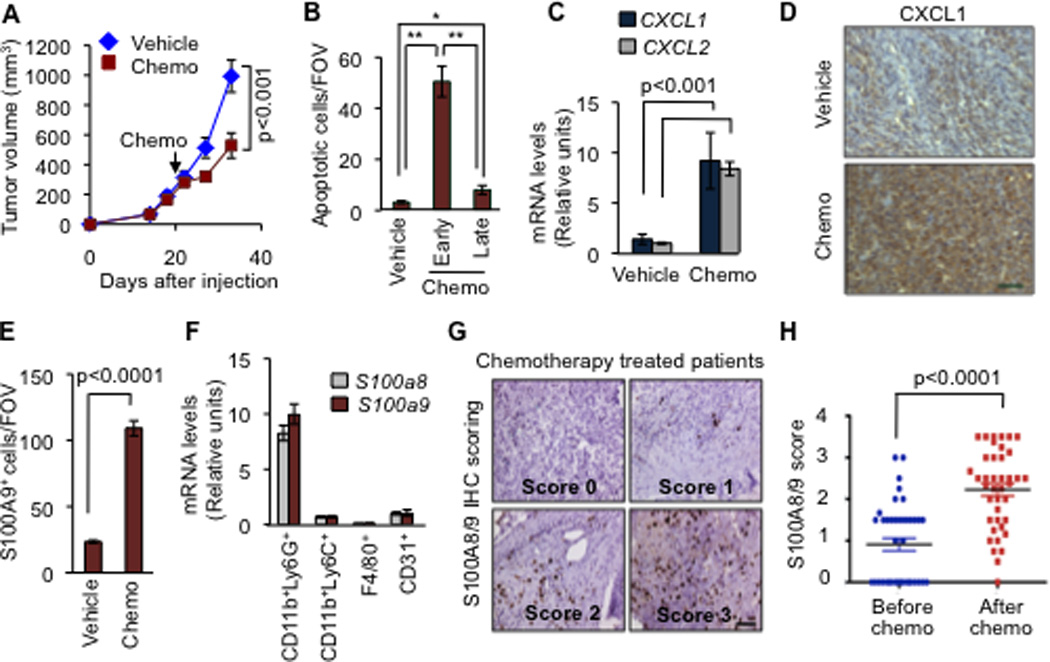
(A) Tumor growth in mice treated with saline vehicle or a combination of doxorubicin and cyclophosphamide chemotherapy (AC chemo). The treatment was initiated once LM2 tumors reached 300 mm3 and was repeated once weekly. Data represent averages ± SEM. n=6–8 mice per group. P values were determined by Student’s t-test.
(B) Apoptosis determined by TUNEL staining in tumors treated with vehicle or AC chemotherapy for 3 days (early) or 8 days (late) both using the same treatment regimen. Data represent averages ± SEM. n=3–5 mice per group. P values were calculated by Student’s t-test. *p=0.02, **p<0.0001.
(C) CXCL1/2 expression in whole tumors harvested from mice treated with saline vehicle or AC chemotherapy for 8 days. Data represent averages±SEM. n=6–8 mice per group. P values were determined by Student’s t-test.
(D) Representative CXCL1 expression in whole tumors analyzed by immunohistochemistry harvested from mice treated with saline vehicle or AC chemotherapy (prolonged treatment) from two independent cohorts of three mice each. Scale bar represents 70µm.
(E) Quantitation of S100A9 positive cells in tumors from control and AC chemotherapy treated mice (prolonged treatment). Data presented are average numbers of S100A9 positive cells per FOV ± SEM. n=4–5 mice/group. Data representative of three independent experiments.
(F) Expression of S100a8 and S100a9 in sorted subpopulations of LM2 tumors after chemotherapy treatment determined by qRT-PCR. Error bars represent 95% confidence interval. Data representative of two independent experiments.
(G) Immunohistochemical analysis of S100A8/9 in tumors from breast cancer patients before and after AC chemotherapy treatment. Representative images of scored S100A8/9 sections are shown. Scale bar equals 60µm.
(H) S100A8/9 expression score in paired patient tumor samples, before and after chemotherapy. Data represent expression score. n=40. P values determined by Wilcoxon’s paired test, comparing pre and post-treatment levels from each patient.
To investigate the involvement of the CXCL1/2-S100A8/9 in cancer cell survival during chemotherapy challenge, we analyzed the expression of CXCL1 and CXCL2 in AC chemotherapy treated tumors. AC treated tumors significantly upregulated CXCL1/2 expression (Figure 5C–D) and this effect was associated with increased recruitment of S100A9-expressing cells (Figure 5E) and CD11b+Ly6G+ granulocytic cells (Figure 5F). CXCL1/2 upregulation in the LM2 tumors was but was also observed with another commonly used chemotherapeutic agent, paclitaxel (Figure S5A). In addition to CXCL1/2 and S100A8/9, the CXCL1-associated chemokine genes CCL20 and CXCL3 were also highly expressed in response to chemotherapy (Figure S5B). Although induction of CXCL chemokines occurs during chemotherapy-induced senescence (Coppe et al., 2008), there was no discernible increase in senescence in LM2 tumors upon AC chemotherapy treatment (Figure S5C–D). Together, these results suggest that chemotherapy activates a burst of paracrine factors including the cancer cell survival axis CXCL1/2-S100A8/9 that selects for cancer cells that can resist chemotherapy.
S100A8/9 association with resistance to perioperative chemotherapy
Neoadjuvant chemotherapy—the use of cytotoxic drugs prior to surgery for primary breast cancer—is an option for patients with operable disease. This has long been the standard approach for patients with locally advanced primary disease in an effort to shrink the tumor and thereby make complete tumor removal possible. While these treatments usually cause tumor volume regression, some cases are chemotherapy-resistant de novo (Gonzalez-Angulo et al., 2007). To address whether the CXCL1/2-S100A8/9 survival loop is activated in cancer patients with primary disease, we stained matched breast tumor sections from a cohort of patients before and after chemotherapy treatment. Consistent with our experimental models, a significant increase in S100A9 expressing cells was observed in breast cancers after chemotherapy treatment (Figure 5G–H and Table S5). In contrast, Fascin that is part of a lung metastasis gene signature (Minn et al., 2005) did not show the same trend upon chemotherapy treatment as S100A8/9 (Figure S5E), further confirming the specificity of the association of S100A8/9 with chemotherapy resistance.
Chemotherapy Induces TNF-α to Boost the CXCL1/2–S100A8/9 Axis
Hyperactivation of the CXCL-S100A8/A9 loop upon chemotherapy treatment prompted us to explore the mechanism behind the therapy-induced CXCL1/2 upregulation. In our experimental models, enhanced expression of CXCL1/2 in response to chemotherapy was not due to additional amplification of the locus as determined by FISH analysis (Figure S6A). Direct treatment of LM2 cells with the chemotherapeutic agents did not induce CXCL1/2 expression (Figure S6B and data not shown). However, LM2 tumor cells incubated with conditioned media from chemotherapy-treated primary endothelial cells or primary bone marrow-derived cells showed a significant increase in CXCL1/2 expression (Figures 6A and S6B).
Figure 6. TNF-α from chemotherapy-activated stroma boosts the CXCL1/2 survival axis.
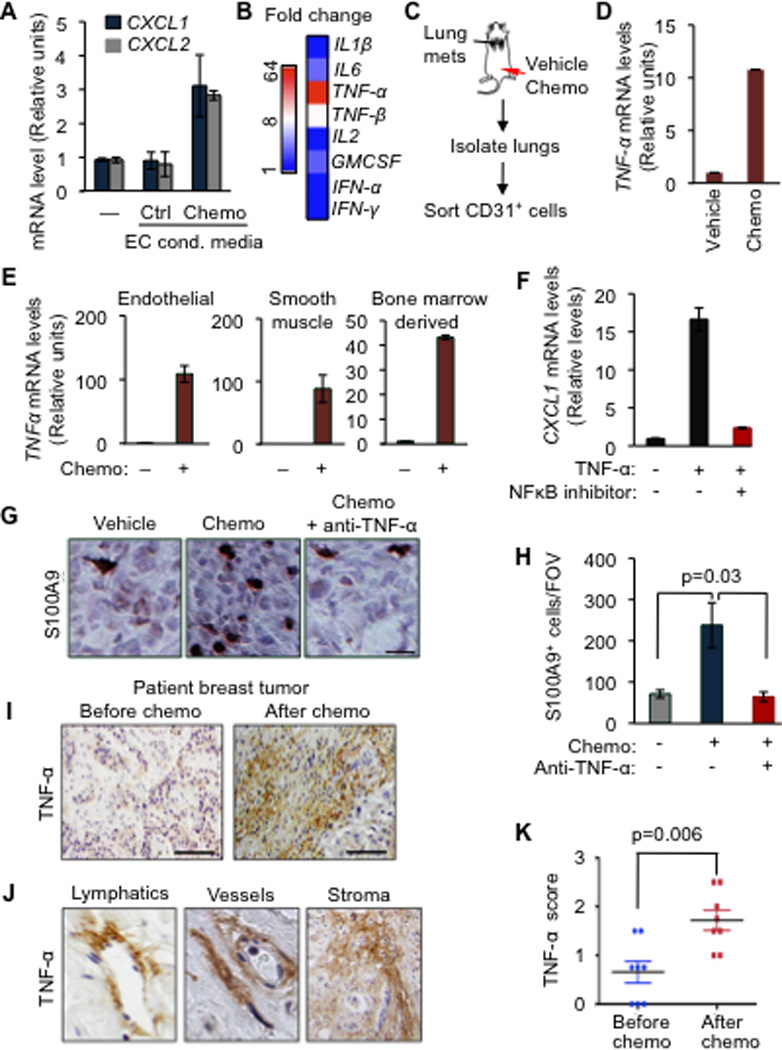
(A) CXCL1/2 expression in MDA231-LM2 cancer cells either alone (–) or in the presence of conditioned media from primary human umbilical vein endothelial cells (HUVEC) that were either untreated (control) or treated with 0.8µM doxorubicin (chemo), as determined by qRT-PCR. Data represent average expression ± SEM.
(B) Heatmap representing expression changes in inflammatory cytokines in HUVEC treated with doxorubicin (0.8µM). Heatmap generated by conversion of qRT-PCR data that was normalized to β2M as housekeeping gene control.
(C) Schematic representation of experimental procedures. Lung endothelial cells were purified from LM2 tumor-bearing mice that were treated with chemotherapy. Mice showing established lung metastasis 7 weeks after tail-vein injection of LM2 cells were either treated with vehicle (saline) or AC chemotherapy. CD31+ endothelial cells were purified from dissociated lung tissue by flow cytometry.
(D) TNF-α expression in isolated CD31+ lung endothelial cells from doxorubicin treated tumor-bearing mice. n=2–4 mice per group. Data represent averages ± SEM.
(E) TNF-α expression in the indicated primary cells upon doxorubicin chemotherapy treatment for 16h as determined by qRT-PCR analysis. Error bars represent 95% confidence interval for qRT-PCR analysis. Data is representative of three independent experiments.
(F) CXCL1 expression in LM2 cancer cells treated with vehicle or TNF-α for 2h in the presence of a 100µM NBD (NEMO-binding domain) inhibitory peptide of the NF-κB pathway. Data represent averages ± SEM.
(G–H) Representative images of S100A9 expression by immunohistochemistry and quantitation of S100A9 positive cells in tumors from control or AC chemotherapy treated mice, with or without anti-TNF-α blocking antibody (infliximab). Mice bearing CN34LM1 tumors were treated once weekly for five weeks starting at 10 weeks after tumor inoculation, with PBS vehicle, AC chemotherapy (chemo), or chemotherapy plus anti-TNF-α antibody. Scale bar, 32µm. Data are averages ± SEM. n=3–5 mice/group. P values were calculated by Student’s t-test.
(I) Immunohistochemical analysis of TNF-α expression in human primary breast tumor before and after chemotherapy treatment. Scale bar equals 120µm.
(J) Stromal rich areas containing lymphatic vessels, blood vessels and fibroblasts with high TNF-α staining from primary breast tumors after AC chemotherapy treatment. Magnified fields from images taken at 40× magnification.
(K) Comparison of stromal TNF-α expression score in paired breast tumors before and after chemotherapy. n=8 patients. P value was determined by Wilcoxon’s paired test, comparing pre and post-treatment levels from each patient.
See also Figure S6
CXCL1/2 are target genes of the NFκB/STAT1 pathway (Amiri and Richmond, 2003). Among a panel of prototypical activators of the NFκB/STAT1 pathway that we assayed by qRT-PCR, TNF-α was strikingly induced in endothelial cells upon doxorubicin chemotherapy treatment (Figure 6B). Moreover, we observed a ten-fold increase in TNF-α expression in purified lung endothelial cells from LM2 tumor bearing mice systemically treated with AC chemotherapy (Figure 6C,D). TNF-α induction in response to chemotherapy also occurred in smooth muscle and bone marrow derived cells (Figure 6E) suggesting that TNF-α release is a general response to chemotherapy in different stromal cell types. NFκB activation by TNF-α stimulated the expression of CXCL1/2 in LM2 tumor cells, as determined with the use of NFκB pathway inhibitor (Nemo binding domain peptide or NBD) (Figure 6F). Thus, TNF-α from chemotherapy-activated stroma can boost the CXCL1/2-S100A8/9 loop. Treatment with anti-TNF-α antibody infliximab reduced recruitment of S100A9-expressing cells in chemotherapy-treated mammary tumors (Figure 6G–H). These tumors were formed with cell line CN34-LM1 derived from the pleural fluid of a stage IV breast cancer patient whose disease had progressed on therapy (Tavazoie et al., 2008). CN34-LM1 is more indolent than LM2 and allows for longer-term tumor growth experiments.
Consistent with our hypothesis, a significant increase in TNF-α immunostaining was observed in patient samples after neoadjuvant AC chemotherapy treatment (Figure 6I–K) and in LM2 mammary tumors in mice treated with AC chemotherapy (Figure S6C). Importantly, histopathological analysis revealed that cells from the tumor microenvironment specifically lymphatic and blood vessels and fibroblast-rich stroma showed strong TNF-α staining after chemotherapy (Figure 6J and S6D).
Blocking CXCL1 Signaling Increases the Effectiveness of Chemotherapy
Our findings suggest that a self-defeating consequence of at least some chemotherapy drugs is the release of potent pro-inflammatory cytokines such as TNF-α from stromal sources. Such pro-inflammatory bursts can boost the CXCL1/2-S100A8/9 survival axis and facilitates the expansion of chemoresistant breast cancer cells. These results presented us with an option of targeting the tumor microenvironment in order to sensitize breast cancer cells to chemotherapy and to the stress of invading and colonizing distant tissues. Therefore, we utilized antagonists of CXCR2, the primary receptor for CXCL1/2, since derivatives of these pharmacological inhibitors are in clinical trials for chronic inflammatory diseases and show no major toxicity with long-term usage (Chapman et al., 2009). Furthermore, targeting the immune microenvironment would be an attractive option because of the potentially low selective pressure for mutations and epigenetic changes on the stroma compared to the cancer cell genomes.
Based on this rationale, we designed preclinical trials in xenograft mice implanted with either of the two independent metastatic breast cancer cell lines, MDA231-LM2 and CN34-LM1. The mice were treated with a combination of AC chemotherapy and CXCR2 antagonist starting after lung metastasis was detectable by BLI (data not shown). Tumor-bearing mice treated with AC chemotherapy alone showed a reduction in tumor growth (Figures 7A,B and S7A,B). However metastatic cells were not completely eliminated and micrometastases were detected throughout the lungs (Figure 7C,D). As a single agent CXCR2 inhibitor had partial to no effect depending on the model system. However, when AC was combined with the CXCR2 inhibitor, lung metastatic burden was markedly reduced and the interaction between the drugs exhibited synergism (Figure 7C,D). Together, these results highlight the potential of targeting the CXCL1/2-S100A8/9 axis thereby sensitizing distant metastases to standard of care chemotherapy.
Figure 7. Pharmacological inhibition of CXCL1 signaling sensitizes cancer cells to chemotherapy in metastatic breast cancer.
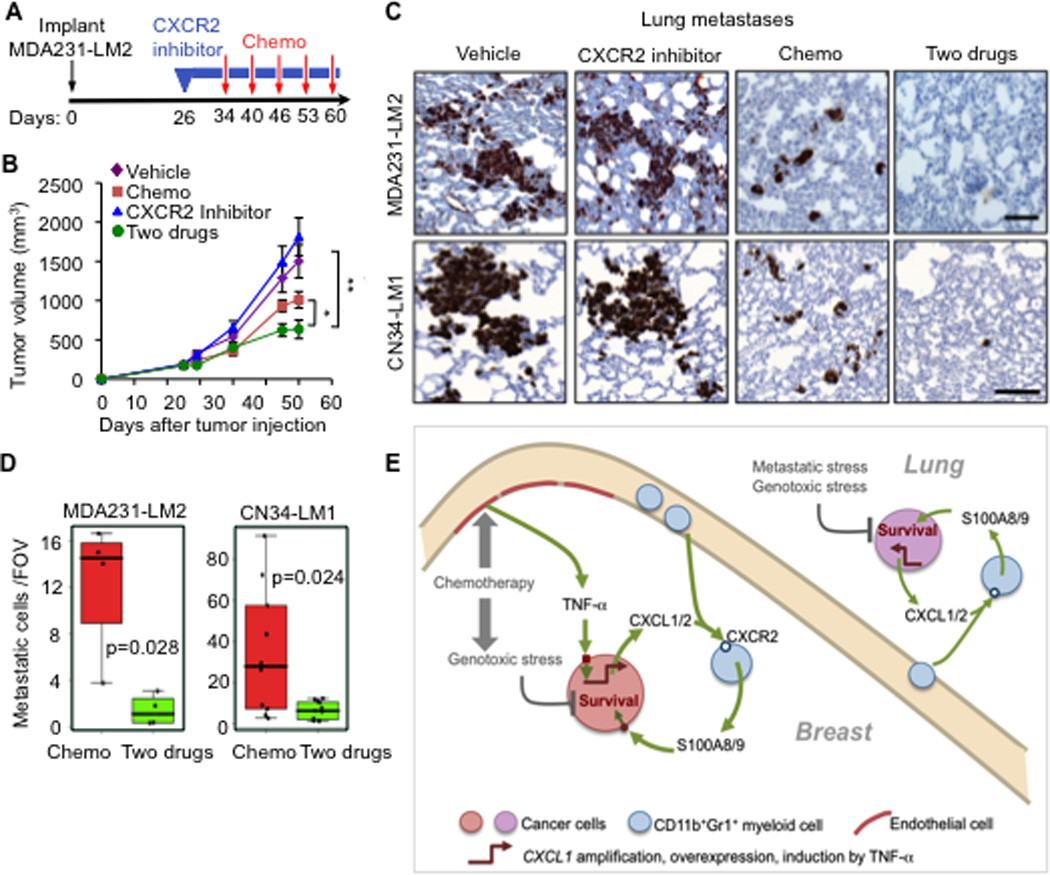
(A–B) Schematic treatment flow (A), and tumor growth (B) of LM2 tumors in mice treated with PEG vehicle or CXCR2 inhibitor for the indicated periods (blue boxes) and treatment with saline vehicle or AC chemotherapy at the indicated days (red arrows). Data represent average expression ± SEM. n=10–13 mice per group. P values were determined by Student’s t-test. *p=0.02, **p=0.007.
(C–D) Lung metastasis in MDA231-LM2 and CN34-LM1 orthotopic xenograft models undergoing treatment (C) Representative images of lung sections stained for vimentin expression marking metastatic cancer cells. Scale bars equal 100µm. (D) Quantitation of metastasis based on number of cancer cells in lung sections. Data are average foci per FOV ± SEM. n=5–10 mice per group. Whiskers represent minimum and maximum values. P values were determined by two-tailed Wilcoxon rank-sum test.
(E) Model showing how CXCL1 paracrine interactions promote resistance to chemotherapy and metastasis in breast tumors and lung microenvironment. Genotoxic agents such as doxorubicin, cyclophosphamide and paclitaxel limit the survival of cancer cells but also increase TNF-α production from endothelial cells. TNF-α enhances CXCL1/2 expression in cancer cells. Other modes of CXCL1/2 upregulation in cancer cells include 4q21 amplification and overexpression. CXCL1/2 from cancer cells recruit CD11b+Gr1+ myeloid cells that express CXCR2 (receptors for CXCL1/2). Myeloid cells recruited by CXCL1/2 thereby enhance viability of cancer cells through S100A8/9 factors.
See also Figure S7
DISCUSSION
A Three-Way Paracrine Axis Underlying Chemoresistance and Metastasis
The major impediments to cure advanced breast cancer are the emergence of pan-resistance to all known chemotherapy drugs and the development of widely metastatic disease, two phenomena that are closely linked clinically (Gonzalez-Angulo et al., 2007). In addressing this challenge, our work links CXCL1/2 and S100A8/9 as functional partners of a paracrine loop between breast cancer cells and CD11b+Gr1+ myeloid cells that supports the survival of cancer cells facing the rigors of invading new microenvironments or the impact of chemotherapy (Figure 7E). Therapeutically targeting such common mediators of chemoresistance and distant relapse would be of interest because these are the two main clinical challenges after primary tumor resection.
The critical role of the microenvironment in tumor progression and response to therapy is being increasingly recognized (Condeelis and Pollard, 2006; Denardo et al., 2011; Gilbert and Hemann, 2010; Shree et al., 2011; Tan et al., 2011). The present work sheds light on how the tumor microenvironment responds to chemotherapy with hyperactivation of a TNF-α-CXCL1/2-S100A8/9 paracrine axis for cancer cell survival under stress. Recent reports (Denardo et al., 2011; Gilbert and Hemann, 2010) and the present work show that chemotherapy induces a storm of cytokines and chemokines in the tumor microenvironment many of which might be linked to chemoresistance. Our current findings suggest that this cytokine burst includes TNF-α induced in several components of the tumor microenvironment after chemotherapy treatment. An undesirable consequence of the stromal TNF-α is to further boost CXCL1/2 expression in breast cancer cells. A higher level of CXCL1/2 then drives the paracrine loop involving myeloid cell-derived S100A8/9 to enhance cancer cell survival (Figure 7E). An adverse cycle involving TNF-α-CXCL1/2-S100A8/9 could thus be expanded in response to chemotherapy. Such a paracrine survival network could be beneficial to cancer cells under stress both in the primary tumor and at distant metastatic sites. Once initiated, this chemo-protective program could become self-sustaining, leading to the enrichment of residual aggressive clones able to resist chemotherapy and thrive in the lung parenchyma and elsewhere.
Biological and Clinical Implications
Several additional insights emerge from this work. CD11b+Gr1+ myeloid cells are a heterogeneous group with previously identified roles in tumor angiogenesis and T cell immunosuppression (Gabrilovich and Nagaraj, 2009; Ostrand-Rosenberg and Sinha, 2009; Shojaei et al., 2007). Our study delineates a new role for the CD11b+Gr1+ cells in mediating metastatic breast cancer cell survival through the production of S100A8/9. In addition to activating MAPK pathways (Gebhardt et al., 2006; Ichikawa et al., 2011), we find that S100A8/9 activate p70S6K as contributors to the pro-survival effect of S100A8/9 in these cells. In line with our findings, recent Phase 2 study in breast cancer patients showed that non-responders of neoadjuvant chemotherapy and patients with residual disease had significantly higher circulating MDSC levels than did responders (Montero et al., 2011). These findings accentuate the clinical relevance of CD11b+Gr1+ in rendering chemotherapy ineffective and promoting metastasis.
Our findings argue that although therapy induced inflammation is a predominant feature of the use of chemotherapy, disrupting the CXCL1 driven paracrine axis could improve therapeutic response in existing lesions and also suppress metastasis, even at an advanced stage of tumor progression. CXCR2 receptor antagonists are in clinical trials for chronic inflammatory diseases (Chapman et al., 2009), and we show that these agents are a promising pharmacological approach in metastatic breast cancer when combined with standard chemotherapeutic regimen. The effective combination of chemotherapy with CXCR2 inhibitors at the metastatic site in our preclinical models underscores the potential application of this therapy to limit disseminated tumor burden. Moreover, the important role of CXCR2 in pancreatic adenocarcinoma models (Ijichi et al., 2011) and of S100A8/9 in colorectal cancer (Ichikawa et al., 2011) suggest that the relevance of targeting the CXCL1/2–S100A8/9 axis may extend beyond breast cancer.
In conclusion, our results provide mechanistic insights into the link between two major hurdles in treating breast cancer: chemoresistance and metastasis. Our findings functionally unify three important inflammatory modulators, TNF-α, CXCL1/2 and S100A8/9, in a tumor-stroma paracrine axis that provides a survival advantage to metastatic cells in stressed primary and metastatic microenvironments. This raises the possibility of clinically targeting this axis both to limit the dissemination of cancer cells and to diminish drug resistance.
EXPERIMENTAL PROCEDURES
Flow cytometry analysis
Whole tumors or lung tissues were dissected, cut into small pieces and dissociated using 0.5% collagenase Type III (Worthington Biochemical) and 1% Dispase II (Roche) in PBS for 1–2 h. Resulting single cell suspensions were washed in PBS with 2% heat-inactivated fetal calf-serum and filtered through 70 µm nylon mesh. Cell fractions were incubated for 10 mins at 4°C with anti-mouse Fc block CD16/32 antibody (2.4G2 BD) in PBS containing 1% BSA to avoid non-specific antibody binding. Cells were subsequently washed in PBS/BSA and stained with either Ig controls or fluorophore conjugated antibodies in MACS buffer (0.5%BSA, 2 mM EDTA in PBS). Data acquisition was performed on a FACS Calibur (BD Biosciences) or Cytomation CyAn (Beckman Coulter) and analysis was done using Flowjo version 9 (Tree star, Inc.).
Animal studies
All experiments using animals were done in accordance to a protocol approved by MSKCC Institutional Animal Care and Use Committee (IACUC). S100a9+/+ and S100a9−/− mice (Hobbs et al., 2003), NOD-SCID NCR (NCI), athymic NCR nu/nu (Harlan), NIHIII homozygous nu/nu (Charles River), C57BL/6 (Jackson Labs), FVB/N (Charles River) female mice aged between 5–7 weeks were used. Autochthonous MMTV driven-polyoma virus middle T transgenic mice (Kim et al., 2009) were utilized for isolation of primary PyMT and bone marrow derived cells. Refer to Extended Experimental Procedures for detailed assays, drug treatment and bone marrow transplantation.
Patient samples
Paraffin embedded tissue microarrays containing primary breast cancer samples (IMH-364) and lymph node metastases (BRM481) were purchased from Imgenex and Pantomics, respectively. Paraffin embedded tissue microarrays and sections from lung metastases and primary breast tumor cores before and after chemotherapy treatment were acquired from the MSKCC Department of Pathology in compliance with protocols approved by the MSKCC Institutional Review Board (IRB). Staining details provided in Extended Experimental Procedures.
Bioinformatic and statistical analysis
All bioinformatic analyses were conducted in R. Microarray data from human tumor data sets were processed as described (Zhang et al., 2009). Survival curves for patients were calculated using Kaplan-Meier method and differences between the curves were determined by log rank test. All other experiments were analyzed using two-sided Wilcoxon rank-sum test or unpaired two-sided t-test without unequal variance assumption unless specified. P values ≤ 0.05 were considered significant. See Extended Experimental Procedures for details.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank J. Joyce, J. Bromberg, E. Pamer, HG Wendel, I. Ferrero, Z. Granot, R. Downey and members of Massague laboratory for insightful discussions, J. Howard for her help with clinical cases, D. Macalinao, W. Shu, M. Akram, K. Chadalavada, T. Tong, M. Turkekul, A. Barlas, E. de Stanchina for technical advice and support. This work was funded by NIH grant CA94060 and the Alan and Sandra Gerry Metastasis Research Initiative. S.A. is supported by a Department of Defense Era of Hope postdoctoral fellowship. J.M. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Amiri KI, Richmond A. Fine tuning the transcriptional regulation of the CXCL1 chemokine. Prog Nucleic Acid Res Mol Biol. 2003;74:1–36. doi: 10.1016/s0079-6603(03)01009-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004;4:540–550. doi: 10.1038/nrc1388. [DOI] [PubMed] [Google Scholar]
- Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8:592–603. doi: 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieche I, Chavey C, Andrieu C, Busson M, Vacher S, Le Corre L, Guinebretiere JM, Burlinchon S, Lidereau R, Lazennec G. CXC chemokines located in the 4q21 region are up-regulated in breast cancer. Endocr Relat Cancer. 2007;14:1039–1052. doi: 10.1677/erc.1.01301. [DOI] [PubMed] [Google Scholar]
- Bos PD, Zhang XH, Nadal C, Shu W, Gomis RR, Nguyen DX, Minn AJ, van de Vijver MJ, Gerald WL, Foekens JA, et al. Genes that mediate breast cancer metastasis to the brain. Nature. 2009;459:1005–1009. doi: 10.1038/nature08021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cailleau R, Young R, Olive M, Reeves WJ., Jr Breast tumor cell lines from pleural effusions. J Natl Cancer Inst. 1974;53:661–674. doi: 10.1093/jnci/53.3.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman RW, Phillips JE, Hipkin RW, Curran AK, Lundell D, Fine JS. CXCR2 antagonists for the treatment of pulmonary disease. Pharmacol Ther. 2009;121:55–68. doi: 10.1016/j.pharmthera.2008.10.005. [DOI] [PubMed] [Google Scholar]
- Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124:263–266. doi: 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, Gallagher WM, Wadhwani N, Keil SD, Junaid SA, et al. Leukocyte Complexity Predicts Breast Cancer Survival and Functionally Regulates Response to Chemotherapy. Cancer Discov. 2011;1:54–67. doi: 10.1158/2159-8274.CD-10-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebos JM, Lee CR, Kerbel RS. Tumor and host-mediated pathways of resistance and disease progression in response to antiangiogenic therapy. Clin Cancer Res. 2009;15:5020–5025. doi: 10.1158/1078-0432.CCR-09-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebhardt C, Nemeth J, Angel P, Hess J. S100A8 and S100A9 in inflammation and cancer. Biochem Pharmacol. 2006;72:1622–1631. doi: 10.1016/j.bcp.2006.05.017. [DOI] [PubMed] [Google Scholar]
- Gilbert LA, Hemann MT. DNA damage-mediated induction of a chemoresistant niche. Cell. 2010;143:355–366. doi: 10.1016/j.cell.2010.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Angulo AM, Morales-Vasquez F, Hortobagyi GN. Overview of resistance to systemic therapy in patients with breast cancer. Adv Exp Med Biol. 2007;608:1–22. doi: 10.1007/978-0-387-74039-3_1. [DOI] [PubMed] [Google Scholar]
- Hiratsuka S, Watanabe A, Sakurai Y, Akashi-Takamura S, Ishibashi S, Miyake K, Shibuya M, Akira S, Aburatani H, Maru Y. The S100A8-serum amyloid A3-TLR4 paracrine cascade establishes a pre-metastatic phase. Nat Cell Biol. 2008;10:1349–1355. doi: 10.1038/ncb1794. [DOI] [PubMed] [Google Scholar]
- Hobbs JA, May R, Tanousis K, McNeill E, Mathies M, Gebhardt C, Henderson R, Robinson MJ, Hogg N. Myeloid cell function in MRP-14 (S100A9) null mice. Mol Cell Biol. 2003;23:2564–2576. doi: 10.1128/MCB.23.7.2564-2576.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh HL, Schafer BW, Sasaki N, Heizmann CW. Expression analysis of S100 proteins and RAGE in human tumors using tissue microarrays. Biochem Biophys Res Commun. 2003;307:375–381. doi: 10.1016/s0006-291x(03)01190-2. [DOI] [PubMed] [Google Scholar]
- Hu G, Chong RA, Yang Q, Wei Y, Blanco MA, Li F, Reiss M, Au JL, Haffty BG, Kang Y. MTDH activation by 8q22 genomic gain promotes chemoresistance and metastasis of poor-prognosis breast cancer. Cancer Cell. 2009;15:9–20. doi: 10.1016/j.ccr.2008.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa M, Williams R, Wang L, Vogl T, Srikrishna G. S100A8/A9 activate key genes and pathways in colon tumor progression. Mol Cancer Res. 2011;9:133–148. doi: 10.1158/1541-7786.MCR-10-0394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ijichi H, Chytil A, Gorska AE, Aakre ME, Bierie B, Tada M, Mohri D, Miyabayashi K, Asaoka Y, Maeda S, et al. Inhibiting Cxcr2 disrupts tumor-stromal interactions and improves survival in a mouse model of pancreatic ductal adenocarcinoma. J Clin Invest. 2011;121:4106–4117. doi: 10.1172/JCI42754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SE. Metastatic breast cancer: the treatment challenge. Clin Breast Cancer. 2008;8:224–233. doi: 10.3816/CBC.2008.n.025. [DOI] [PubMed] [Google Scholar]
- Kim MY, Oskarsson T, Acharyya S, Nguyen DX, Zhang XH, Norton L, Massague J. Tumor self-seeding by circulating cancer cells. Cell. 2009;139:1315–1326. doi: 10.1016/j.cell.2009.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minn AJ, Gupta GP, Padua D, Bos P, Nguyen DX, Nuyten D, Kreike B, Zhang Y, Wang Y, Ishwaran H, et al. Lung metastasis genes couple breast tumor size and metastatic spread. Proc Natl Acad Sci U S A. 2007;104:6740–6745. doi: 10.1073/pnas.0701138104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, Viale A, Olshen AB, Gerald WL, Massague J. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436:518–524. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montero AJ, Diaz-Montero CM, Deutsch YE, Hurley J, Koniaris LG, Rumboldt T, Yasir S, Jorda M, Garret-Mayer E, Avisar E, et al. Phase 2 study of neoadjuvant treatment with NOV-002 in combination with doxorubicin and cyclophosphamide followed by docetaxel in patients with HER-2 negative clinical stage II-IIIc breast cancer. Breast Cancer Res Treat. 2011 doi: 10.1007/s10549-011-1889-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris PG, McArthur HL, Hudis CA. Therapeutic options for metastatic breast cancer. Expert Opin Pharmacother. 2009;10:967–981. doi: 10.1517/14656560902834961. [DOI] [PubMed] [Google Scholar]
- Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–56. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer. 2008;8:618–631. doi: 10.1038/nrc2444. [DOI] [PubMed] [Google Scholar]
- Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol. 2009;182:4499–4506. doi: 10.4049/jimmunol.0802740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegram MD, Konecny GE, O'Callaghan C, Beryt M, Pietras R, Slamon DJ. Rational combinations of trastuzumab with chemotherapeutic drugs used in the treatment of breast cancer. J Natl Cancer Inst. 2004;96:739–749. doi: 10.1093/jnci/djh131. [DOI] [PubMed] [Google Scholar]
- Poulikakos PI, Rosen N. Mutant BRAF melanomas--dependence and resistance. Cancer Cell. 2011;19:11–15. doi: 10.1016/j.ccr.2011.01.008. [DOI] [PubMed] [Google Scholar]
- Roodhart JM, Daenen LG, Stigter EC, Prins HJ, Gerrits J, Houthuijzen JM, Gerritsen MG, Schipper HS, Backer MJ, van Amersfoort M, et al. Mesenchymal stem cells induce resistance to chemotherapy through the release of platinum-induced fatty acids. Cancer Cell. 2011;20:370–383. doi: 10.1016/j.ccr.2011.08.010. [DOI] [PubMed] [Google Scholar]
- Shaked Y, Henke E, Roodhart JM, Mancuso P, Langenberg MH, Colleoni M, Daenen LG, Man S, Xu P, Emmenegger U, et al. Rapid chemotherapy-induced acute endothelial progenitor cell mobilization: implications for antiangiogenic drugs as chemosensitizing agents. Cancer Cell. 2008;14:263–273. doi: 10.1016/j.ccr.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shojaei F, Wu X, Malik AK, Zhong C, Baldwin ME, Schanz S, Fuh G, Gerber HP, Ferrara N. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat Biotechnol. 2007;25:911–920. doi: 10.1038/nbt1323. [DOI] [PubMed] [Google Scholar]
- Shree T, Olson OC, Elie BT, Kester JC, Garfall AL, Simpson K, Bell-McGuinn KM, Zabor EC, Brogi E, Joyce JA. Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Genes Dev. 2011;25:2465–2479. doi: 10.1101/gad.180331.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart TJ, Abrams SI. Altered immune function during long-term host-tumor interactions can be modulated to retard autochthonous neoplastic growth. J Immunol. 2007;179:2851–2859. doi: 10.4049/jimmunol.179.5.2851. [DOI] [PubMed] [Google Scholar]
- Tan W, Zhang W, Strasner A, Grivennikov S, Cheng JQ, Hoffman RM, Karin M. Tumour-infiltrating regulatory T cells stimulate mammary cancer metastasis through RANKL-RANK signalling. Nature. 2011;470:548–553. doi: 10.1038/nature09707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavazoie SF, Alarcon C, Oskarsson T, Padua D, Wang Q, Bos PD, Gerald WL, Massague J. Endogenous human microRNAs that suppress breast cancer metastasis. Nature. 2008;451:147–152. doi: 10.1038/nature06487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai S, Collins SJ. A dominant negative retinoic acid receptor blocks neutrophil differentiation at the promyelocyte stage. Proc Natl Acad Sci U S A. 1993;90:7153–7157. doi: 10.1073/pnas.90.15.7153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogl T, Tenbrock K, Ludwig S, Leukert N, Ehrhardt C, van Zoelen MA, Nacken W, Foell D, van der Poll T, Sorg C, et al. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med. 2007;13:1042–1049. doi: 10.1038/nm1638. [DOI] [PubMed] [Google Scholar]
- Weiss L. Metastasis of cancer: a conceptual history from antiquity to the 1990s. Cancer Metastasis Rev. 2000;19:I–XI. 193–383. [PubMed] [Google Scholar]
- Zhang XH, Wang Q, Gerald W, Hudis CA, Norton L, Smid M, Foekens JA, Massague J. Latent bone metastasis in breast cancer tied to Src-dependent survival signals. Cancer Cell. 2009;16:67–78. doi: 10.1016/j.ccr.2009.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.