

Uracil is an undesired component of DNA, as it arises from spontaneous deamination of cytosine.[1] This hydrolysis reaction promotes mutations, since the resulting U-G pair can be misread during DNA replication. As a result, multiple cellular enzymes have evolved to detect uracil in DNA and remove it prior to replication.[2] In E. coli uracil DNA glycosylase (UDG) enzyme functions to guard the bacterial genome. In humans, similar enzyme activities exist, including the proteins UNG1/2, SMUG, and TDG.[3] These enzymes flip uracil out of the DNA helix and cleave it from its deoxyribose sugar, leaving an abasic site in its place.[4]
Chemical sensors of UDG enzymes could be useful in multiple respects. Mechanistic studies of the enzymes can benefit from such probes.[5] In addition, if such probes could function in cells, they could be used to screen activity of enzyme mutants, and to probe biological regulation of DNA repair pathways. Finally, inhibitors of DNA repair enzymes are currently receiving intense interest in anticancer therapeutic approaches;[6] thus simple light-up reporters of such enzymes could be also useful in screening drug candidates.
Previous efforts to develop fluorescent sensors for uracil-glycosylase enzymes have been limited by large size, by their indirect readout and by poor performance. Designs have generally relied on large duplex DNA constructs of 28-39 nucleotides.[7] Signals were generated by changes in conformation (loss of duplex structure) which results after uracil is removed and the DNA strand is ultimately cleaved. Signal enhancements of ca. 4- to 8-fold were reported; these signals report only indirectly on the uracil deglycosylation because they require subsequent steps of DNA degradation and/or unwinding before the signal is seen. Although such DNA constructs were able to function in vitro, function of such structures in biological media such as cell extracts can be further complicated by false signals from DNA-unwinding activities in the cell, from nuclease degradation, and from other DNA-binding proteins (such as single-strand-binding proteins) that can all lead to loss of duplex integrity. Although one previous report describes signals generated in mammalian cells with a 39-base DNA construct,[7a] control experiments to rule out these likely sources of background signal were not performed. Finally, the large size of such constructs adds cost, complexity and lowers the likelihood of intracellular uptake.
It has been known for some time that UDG enzymes also show activity on single-stranded DNAs.[8] One earlier report described an in vitro kinetics assay for E. coli UDG making use of double- and single-stranded DNAs containing the fluorescent dye 2-aminopurine (2AP), which is quenched in DNA; increases in fluorescence of 3- to 8-fold were reported with UDG.[9] 2AP deoxynucleoside is a convenient probe in DNA as it stacks like a DNA base; however, it has low fluorescence efficiency and emits in the UV region.[10]
Our aim was to construct sensors for UDG activity that are simpler, smaller and more effective than previous examples. As part of a program to build enzyme sensors from small synthetic DNA oligomers containing fluorescent DNA base replacements,[11] we observed that the fluorophore pyrene is especially well quenched by the DNA base thymine, a phenomenon that occurs by the photoinduced charge transfer (PICT) mechanism.[12] Pyrene is especially useful as a fluorophore because it has a high quantum yield, robust brightness, and is shifted to the red by ca. 40 nm relative to 2AP, allowing for detection in the visible region. Pyrene deoxyriboside (Y), an unnatural DNA nucleoside, has been used broadly as a reporter of DNA structure and interactions.[13,14] Both beta and alpha anomers of Y are known to stack strongly with neighboring DNA bases and stabilize DNA helices in which they are substituted.[15] Other pyrene-substituted nucleosides and nucleobase analogues are known in the literature as well.[16]
In preliminary experiments we found that, like thymine, uracil also effectively quenches pyrene deoxyriboside when adjacent to it. This led us to conceive of the possibility of a direct enzyme sensing strategy: if UDG activity were able to remove uracil next to pyrene, the fluorescence of this reporter would be strongly enhanced in real time as the reaction proceeds. However, it was not clear whether this large, hydrophobic unnatural nucleobase would unfavorably interact near the enzyme active site.
To test this possibility we prepared a set of short single-stranded DNA oligomers containing pyrene α-deoxyriboside (see probes 1-9 in Fig. 1). They were designed to contain the minimum DNA-like structure that might retain enzymatic activity, and to place one or two uracils directly adjacent to the chromophore. Probe 2 was identical to 1 but contained thymine residues instead of uracil as a control for UDG activity, which is specific to uracil. The candidate probes were characterized by MALDI-mass spectrometry (see SI) and by their absorption and fluorescence spectra. We found that all were quenched by the incorporated DNA bases by at least 90% (and some by 98.7%; Table S2). Although all contain the same pyrene fluorophore, quantum yields vary by a factor of 9, ranging from probes 4 and 5 (Φfl = 0.032), which contained one uracil, to probes 1, 6, and 7, (0.004) which contained two. Thus it appeared that uracil is highly effective at quenching pyrene, yielding an intense quenching effect of 98.7% in probes 1, 6 and 7 relative to reference compound 10. Comparison of 1 and 2 shows that uracil is at least as effective at quenching pyrene as thymine, which quenches via PICT from pyrene to the nucleobase.[12] Given the similar electronic properties of uracil, we presume the same mechanism is active in our probes. Adenine is known not to quench pyrene significantly.[17]
Figure 1.
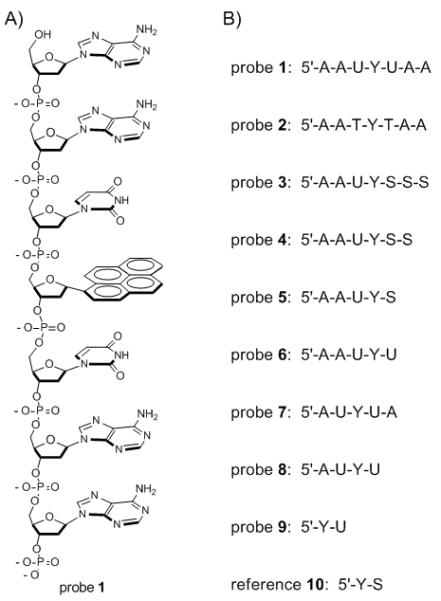
A) Structure of probe 1; B) Sequences of probes in this study. Y = α-pyrene deoxyriboside; S = tetrahydrofuran spacer
Next we tested the ability of the probes to act as substrates of Escherichia coli UDG (Ec UDG) in buffer at 37 °C. Probes were tested at 400 nM concentration, and enzymatic activity was evaluated by fluorescence increases over time (see Fig. 2). Apparent initial rates of the resulting reactions were evaluated by the slopes taken from early timepoints; these are given in the SI. Note that apparent rates for 1, 6, 7, 8 are likely for the second uracil removal and are thus somewhat slowed by this fact. We found that 1 and 3-8 were in fact substrates for the enzyme, but with apparent rates varying by a factor of ca. 500. Compounds 6-8 (4mer and 5mer length) proved suboptimal in size and were relatively slow substrates, requiring several hours to reach maximum signal (SI). Not surprisingly, control thymine-containing probe 2 showed no measurable activity.[8] Probes 1, 3, 4 and 5 were all quite good substrates. Comparison of probes 3-5 with 1 shows that only four bases (including pyrene) appear to be necessary for strong activity. Interestingly, it also appears that additional phosphodiester backbone interactions contribute favorably to the activity, as added abasic monomers (S, Fig. 1B) maintain a high level of substrate activity relative to shorter compounds.
Figure 2.
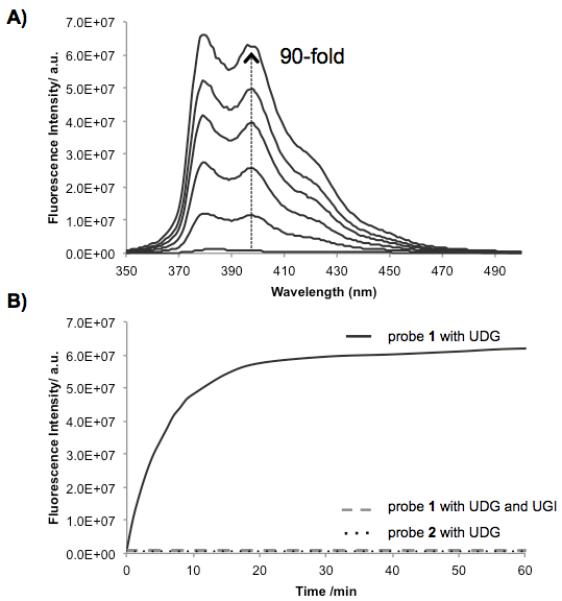
In vitro enzymatic assay of probes incubated with E. coli UDG. (A) Fluorescence spectra over a 60-min timecourse (excitation 340 nm). (B) Increase in emission over time at 395 nm, along with data for thymine-containing control (2) and with UDG inhibitor (UGI). [probe] = 400 nM; [UDG] = 1 unit/mL; [UGI] = 1 unit/mL at pH 8.0, 37 °C.
Of the probes that displayed high enzyme substrate activity, probe 1 also showed the strongest internal quenching. As a result, the probe shows a robust 90-fold light-up signal after one hour with the enzyme (Fig. 2). Probe 5 was the simplest compound with strong enzymatic activity; it also proved to be a useful sensor, although its light-up response (due to the presence of only one quenching uracil) is smaller, at 9-fold (see SI). To confirm that probes are responding specifically to uracil deglycosylase signal rather than nonspecific nuclease activity or some other fluorescence-producing mechanism, we measured timecourses with 1, control 2, and 1 with a known UDG inhibitor (UGI) added (Fig. 2B). Results showed the rapid and strong light-up behavior of 1, but no detectable signal from 2 (which differs by only two methyl groups). No signal was observed when the specific inhibitor was present. To gain further evidence as to the mechanism of fluorogenic behavior in the probes, we incubated probes 3 and 1 (which contain one or two uracil residues, respectively) with UDG and analyzed them by MALDI-mass spectrometry. The results confirmed the loss of these uracils (mass 95) with replacement by water, yielding abasic sites in both cases (Fig. 3), as expected for the UDG enzyme mechanism.[4]
Figure 3.
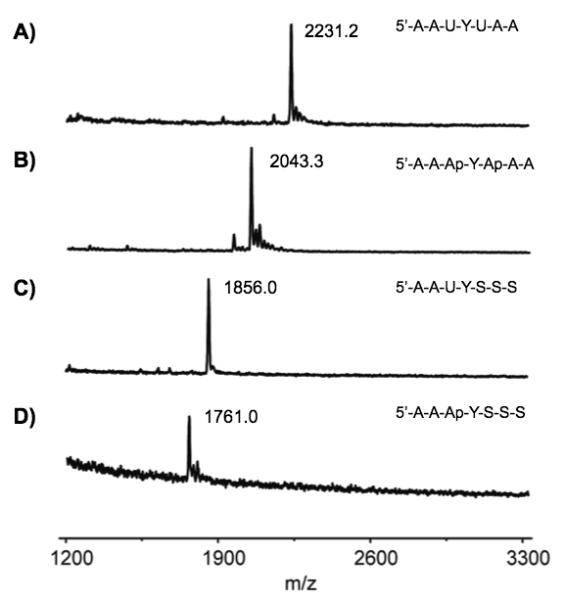
MALDI-TOF mass spectra of (A) probe 1, (B) probe 1 after treatment with UDG, (C) probe 3 and (D) probe 3 after treatment with UDG, confirming loss of uracil in the enzymatic reaction.
To obtain a measure of the efficiency of probe 1 as a UDG substrate, we performed steady-state kinetics measurements with varied probe concentrations. We found that the compound exhibited Michaelis-Menten saturation behavior, and was readily fit to a Lineweaver-Burk plot. This revealed a Km(app) of 210 nM, which is in the same range as native DNA substrates,[9] establishing that the unnatural pyrene residue does not adversely affect enzyme substrate activity for E. coli UDG. We also showed that a thermostable UDG from A. fulgidus[18] (Afu UDG) which is closely homologous to E. coli UDG exhibits similar activity with probe 1 as the E. coli enzyme (see SI).
Given the observed success of compound 1 in reporting with bacterial UDG enzymes in vitro, we wondered whether it might be possible to observe such activity directly associated with cells. To carry out preliminary experiments in this direction, we used an E. coli strain in which the Ec UDG had been deleted and transformed with a plasmid encoding a thermostable Afu UDG[18] under the control of an IPTG inducible promoter. We grew the bacteria and then treated them with probe (5 μM) in buffer at 20 °C or at 65 °C (where Afu UDG enzyme activity is optimal) for 1-4 h. Observations by fluorescence microscopy clearly revealed blue signals for compound 1 for the bacteria expressing Afu UDG (Fig. 4). Importantly, little or no signal was seen when control probe 2 was instead used, establishing that the fluorescence arises not from random nuclease activity, but instead from uracil-specific activity. Similarly, no signal was observed for the bacteria that have Ec UDG deleted (see SI). We also observed that probe 1 could signal apparent changes in the relative amounts of activity: signals were high after addition of IPTG, an inducer of protein expression, whereas without IPTG, signals were observable but considerably lower.
Figure 4.

Images and spectra of sensors with bacterial cells. (A,B) Bright field (left) and fluorescence microscope images (right) of Escherichia coli (BW(310)DE3udg(-)) cells transformed with the pET28a plasmid containing the Afu UDG gene. Cells were incubated in a solution containing (A) probe 1 or (B) thymine control probe 2 at 5 μM for 4 h (65 °C). Excitation 330-380 nm; emission >420 nm. (C,D) Spectra of bacterial solutions diluted tenfold (excitation 340 nm). (C) Probe 1 with bacteria expressing Afu UDG, incubated with probe for 1 or 4 h; (D) Control probe 2 with bacteria under the same conditions.
We confirmed that these signals were indeed originating from pyrene by measuring fluorescence spectra of the bacterial solutions (Fig. 4C), which revealed the characteristic bands of pyrene emission. Intensity of the emission measured in this way corroborated our visual observations: higher signals were seen with bacteria treated with IPTG, and higher signals were seen at 65 °C relative to 20 °C, consistent with the Afu enzyme’s optimal activity at the higher temperature. Only very small levels of emission were seen with control probe 2 or with bacteria lacking UDG activity.
Taken together, the data show that this small modified DNA probe design yields highly efficient chemosensors for probing an enzymatic glycosylase activity in real time. Our design yields a very strongly quenched starting probe, yielding low background. The use of pyrene, as a robust fluorophore, yields a bright positive signal after reaction, and its longer wavelength provides signals in the visible range which can be imaged by microscopy directly in cells. The direct mechanism yielding signal allows for real-time fluorogenic reporting on UDG activity, which is not the case with previous sensor constructs built from a considerably larger duplex DNA architecture.[7]
A further advantage of probes such as 1 or 5 is their small size and simplicity, which allows them to be synthesized easily and may aid in cellular uptake. It is worth noting that some of the known uracil glycosylases (such as TDG) have been shown to prefer double-stranded DNA as a substrate, rather than single-stranded DNA.[2,3] For such enzymes, single-stranded oligomers such as 1 should be considered useful reporters of the presence of enzymatic activity and of relative levels of this activity, rather than as native-like substrates. However, some UDG enzymes (e.g. UNG2 and SMUG in humans) are known to readily accept single-stranded DNAs as substrates as well; it is not yet clear whether such enzymes in the cellular setting act primarily on double-stranded DNA or on single-stranded replication or transcription intermediates. To the extent that such single-stranded intermediates are biological targets of the enzyme, the current chemosensors could be considered native-like substrates and might be directly useful for mechanistic studies. Previous studies have, in this light, made use of short ssDNA substrates containing 2-AP;[9] the stronger light-up response, higher quantum yield and longer wavelength of pyrene may aid in such studies in the future. It will also be of interest in the future to build the pyrene fluorophore Y into native duplex DNA reporters for mechanistic studies of enzymes that act primarily on double-stranded substrates.[19]
In summary, our data shows that very small modified DNA oligomers containing the unnatural fluorescent base pyrene can act as highly efficient reporters of UDG activity both in vitro with purified enzymes and in the context of bacterial cells as well. The favorable fluorescence properties of pyrene make it possible for the small chemosensors to be imaged by microscopy, thus allowing for direct observation of a cellular repair activity. Future work will be directed to use of compounds such as 1 in screening enzyme mutants as well as in studying multiple classes of uracil glycosylase enzymes, including those from eukaryotic cells.
Supplementary Material
Footnotes
This work was supported by the U.S. National Institutes of Health (GM067201 to E.T.K. and CA67985 to S.S.D.). T.O. acknowledges a postdoctoral fellowship from JSPS.
Supporting Information Available: Characterization data of modified DNAs; details of optical measurements; additional data from enzymatic studies and bacterial experiments. Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
Contributor Information
Dr. Toshikazu Ono, Department of Chemistry Stanford University Stanford, CA 94305, USA
Dr. Shenliang Wang, Department of Chemistry Stanford University Stanford, CA 94305, USA
Dr. Chi-Kin Koo, Department of Chemistry Stanford University Stanford, CA 94305, USA
Lisa Engstrom, Department of Chemistry University of California Davis, CA 95616, USA.
Prof. Dr. Sheila S. David, Department of Chemistry University of California Davis, CA 95616, USA
Dr. Eric T. Kool, Department of Chemistry Stanford University Stanford, CA 94305, USA.
References
- [1] a).Hayakawa H, Sekiguchi M. Biochem. Biophys. Res. Commun. 1978;83:1312. doi: 10.1016/0006-291x(78)91364-5. [DOI] [PubMed] [Google Scholar]; b) Duncan BK, Miller JH. Nature. 1980;287:560. doi: 10.1038/287560a0. [DOI] [PubMed] [Google Scholar]
- [2].Pearl LH. Mutat. Res. 2000;460:165. doi: 10.1016/s0921-8777(00)00025-2. [DOI] [PubMed] [Google Scholar]
- [3].Sousa MM, Krokan HE, Slupphaug G. Mol. Aspects Med. 2007;28:276. doi: 10.1016/j.mam.2007.04.006. [DOI] [PubMed] [Google Scholar]
- [4] a).Parikh SS, Walcher G, Jones GD, Slupphaug G, Krokan HE, Blackburn GM, Tainer JA. Proc. Natl. Acad. Sci. USA. 2000;97:5083. doi: 10.1073/pnas.97.10.5083. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Stivers JT, Jiang YL. Chem. Rev. 2003;103:2729. doi: 10.1021/cr010219b. [DOI] [PubMed] [Google Scholar]
- [5].Stivers JT. Nucleic Acids Res. 1998;26:3837. doi: 10.1093/nar/26.16.3837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6] a).Anders CK, Winer EP, Ford JM, Dent R, Silver DP, Sledge GW, Carey LA. Clin. Cancer Res. 2010;16:4702. doi: 10.1158/1078-0432.CCR-10-0939. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Moeller BJ, Arap W, Pasqualini R. Curr. Drug Targets. 2008;11:1336. doi: 10.2174/1389450111007011336. [DOI] [PubMed] [Google Scholar]
- [7] a).Maksimenko A, Ishchenko AA, Sanz G, Laval J, Elder RH, Saparbaev MK. Biochem. Biophys. Res. Commun. 2004;19:240. doi: 10.1016/j.bbrc.2004.04.179. [DOI] [PubMed] [Google Scholar]; b) Liu B, Yang X, Wang K, Tan W, Li H, Tang H. Anal. Biochem. 2007;366:237. doi: 10.1016/j.ab.2007.04.049. [DOI] [PubMed] [Google Scholar]; c) Kim Y, Hong IS. Bull. Korean Chem. Soc. 2009;30:2149. [Google Scholar]
- [8].Lindahl T. Proc. Natl. Acad. Sci. USA. 1974;71:3649–3653. doi: 10.1073/pnas.71.9.3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Drohat AC, Jagadeesh J, Ferguson E, Stivers JT. Biochemistry. 1999;38:11866. doi: 10.1021/bi9910878. [DOI] [PubMed] [Google Scholar]
- [10].Ward DC, Reich E, Stryer L. J. Biol. Chem. 1969;244:1228. [PubMed] [Google Scholar]
- [11] a).Dai N, Teo YN, Kool ET. Chem. Commun. 2010;46:1221–1223. doi: 10.1039/b926338a. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Dai N, Guo J, Teo YN, Kool ET. Angew. Chem. Int. Ed. 2011;50:5105–5109. doi: 10.1002/anie.201007805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Telser J, Cruickshank KA, Morrison LE, Netzel TL, Chan C-K. J. Am. Chem. Soc. 1989;111:7226. [Google Scholar]
- [13].Ren RX-F, Chaudhuri NC, Paris PL, Rumney S, Kool ET. J. Am. Chem. Soc. 1996;118:7671–7678. doi: 10.1021/ja9612763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14] a).Paris PL, Langenhan J, Kool ET. Nucleic Acids Res. 1998;26:3789. doi: 10.1093/nar/26.16.3789. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Matray TJ, Kool ET. J. Am. Chem. Soc. 1998;120:6191. doi: 10.1021/ja9803310. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Matray TJ, Kool ET. Nature. 1999;399:704. doi: 10.1038/21453. [DOI] [PubMed] [Google Scholar]; d) Sun L, Wang M, Kool ET, Taylor J-S. Biochemistry. 2000;39:14603. doi: 10.1021/bi001446v. [DOI] [PubMed] [Google Scholar]; e) Jiang YL, Stivers JT, Song F. Biochemistry. 2002;41:11248. doi: 10.1021/bi026227j. [DOI] [PubMed] [Google Scholar]; f) Guo J, Wang S, Dai N, Teo YN, Kool ET. Proc. Natl. Acad. Sci. USA. 2011;108:3493. doi: 10.1073/pnas.1017349108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15] a).Guckian KM, Schweitzer BA, Ren RX-F, Sheils CJ, Tahmassebi DC, Kool ET. J. Am. Chem. Soc. 2000;122:2213. doi: 10.1021/ja9934854. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cuppoletti A, Cho Y, Park JS, Strässler C, Kool ET. Bioconj. Chem. 2005;16:528. doi: 10.1021/bc0497766. [DOI] [PubMed] [Google Scholar]
- [16] a).Yamana K, Iwase R, Furutani S, Tsuchida H, Zako H, Yamaoka T, Murakami A. Nucleic Acids Res. 1999;27:2387. doi: 10.1093/nar/27.11.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Amann N, Pandurski E, Fiebig T, Wagenknecht HA. Chemistry. 2002;8:4877. doi: 10.1002/1521-3765(20021104)8:21<4877::AID-CHEM4877>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]; c) Langenegger SM, Häner R. Chem. Commun. 2004:2792. doi: 10.1039/b412831a. [DOI] [PubMed] [Google Scholar]; d) Babu BR, Wengel J. Chem. Commun. 2001:2114. doi: 10.1039/b106065c. [DOI] [PubMed] [Google Scholar]; e) Seo YJ, Kim BH. Chem. Commun. 2006:150. doi: 10.1039/b514079j. [DOI] [PubMed] [Google Scholar]; f) Nakamura M, Ohtoshi Y, Yamana K. Chem. Commun. 2005:5163. doi: 10.1039/b507808c. [DOI] [PubMed] [Google Scholar]; g) Ostergaard ME, Hrdlicka PJ. Chem. Soc. Rev. 2011 Apr 13; [Epub ahead of print] [Google Scholar]
- [17].Wilson JN, Cho Y, Tan S, Cuppoletti A, Kool ET. ChemBioChem. 2008;9:279. doi: 10.1002/cbic.200700381. [DOI] [PubMed] [Google Scholar]
- [18].Sandigursky M, Franklin WA. J. Biol. Chem. 2000;275:19146. doi: 10.1074/jbc.M001995200. [DOI] [PubMed] [Google Scholar]
- [19].Jiang YL, Kwon K, Stivers JT. J. Biol. Chem. 2001;276:42347. doi: 10.1074/jbc.M106594200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.