

Abstract
Telomerase is present in most human cancers, and proliferative stem cells including germline cells. Telomerase plays an essential role in tumorigenesis by maintaining/elongating telomeric DNA, and thus preventing the telomere shortening that results in replicative senescence. Understanding telomerase action in vivo has important implication for both cancer and aging, but there are not robust methods for monitoring telomerase action. By combining a series of cell biological and biochemical approaches, and taking advantage of the enzyme DSN that specifically cuts double-stranded DNA and releases the telomeric overhangs, we have developed a method to monitor telomerase action during one cell cycle. Here, we describe this method using Hela carcinoma cells as an example.
Keywords: Telomere, telomerase, overhang, C-rich Fill-in, cell synchronization, BrdU, CsCl gradients, Duplex Specific Nuclease (DSN)
1. Introduction
Human telomeres are comprised of several kilobases of double-stranded TTAGGG/AATCCC DNA terminating in a 12 to 300nt single-stranded G-rich sequence (the G-rich overhang) (1–2). In normal somatic cells, telomeres shorten at the rate of 50–200bp/PD (population doubling) due to the end replication problem of linear chromosome, processing and stochastic events. Eventually, critically short telomeres trigger replicative senescence or apoptosis(3). To counteract this continuous telomere loss, most cancer and proliferative stem cells express telomerase, a ribonucleoprotein complex that exhibits reverse transcriptase activity that can extend telomeres by adding hexameric repeats of GGTTAG to telomeric overhangs (4–5). This extension is finished by a C-rich strand fill-in that makes some of the extended G-overhang double-stranded.
Most cancer cells maintain a constant telomere length, where telomerase is only adding enough to compensate for the rate of shortening. Techniques to measure telomere length, such as Telomere Restriction Fragment assay (TRF)(6), Telomere FISH(7), and Single Telomere Length Analysis (STELA)(8), can not measure telomerase action if the length is not changing. Telomerase action will extend the G-rich overhangs until C-rich fill-in makes the extended overhangs shorter. By monitoring the change of G-overhang length during the cell cycle, one should be able to tell when and how telomerase extension and C-rich fill-in occur. Using Duplex Specific Nuclease (DSN), that specifically digests double stranded DNA to <10bp fragments while keeping single stranded G-overhang intact, we have developed a new method for directly studying the dynamics of the G-overhang during cell cycle.
We have found that telomerase extension is coupled to telomere replication, which occurs throughout S phase in human cells, whereas C-rich fill-in is delayed until S/G2 (9–10). The time-lag between these two events allowed us to investigate telomerase action during S phase and C-rich fill-in at S/G2. Cells synchronized at G1/S are released into S phase in the presence of BrdU. Telomere replication incorporates BrdU into both leading and lagging daughters. The leading daughter’s G-strand is newly synthesized, and its overhangs will be fully BrdU substituted regardless of whether or not telomerase has acted (Fig 1). However, the parental G-strand provides the template for lagging strand synthesis, and lagging daughters initially have overhangs that contain only thymidine. If telomerase acts it will incorporate BrdU into the newly synthesized GGTTAG repeats, thus the overhangs of lagging daughters will contain a mixture of thymidine and BrdU containing repeats and should have an intermediate density on CsCl gradients (Fig 1). At S/G2, C-rich fill-in makes the thymidine-containing portion of the overhang double-stranded, leaving a fully BrdU substituted overhang that has the highest density. During S phase, by monitoring what fraction of lagging overhangs are of intermediate density versus low density, one can determine the percentage of telomeres extended by telomerase. In addition, the intermediate density per se indicates the ratio of thymidine to BrdU in an overhang that can be used to calculate the average size of telomerase extension (the BrdU part of the overhang) once the total length of the overhang has been determined.
Figure 1.

Strategy for detecting telomerase extension and C-rich Fill-in on lagging daughter telomere.
Figure 2 shows an outline of the different steps needed for this analysis. A brief explanation is provided for each step prior to a detailed description of the protocols. Hela cells are used as an example.
Figure 2.
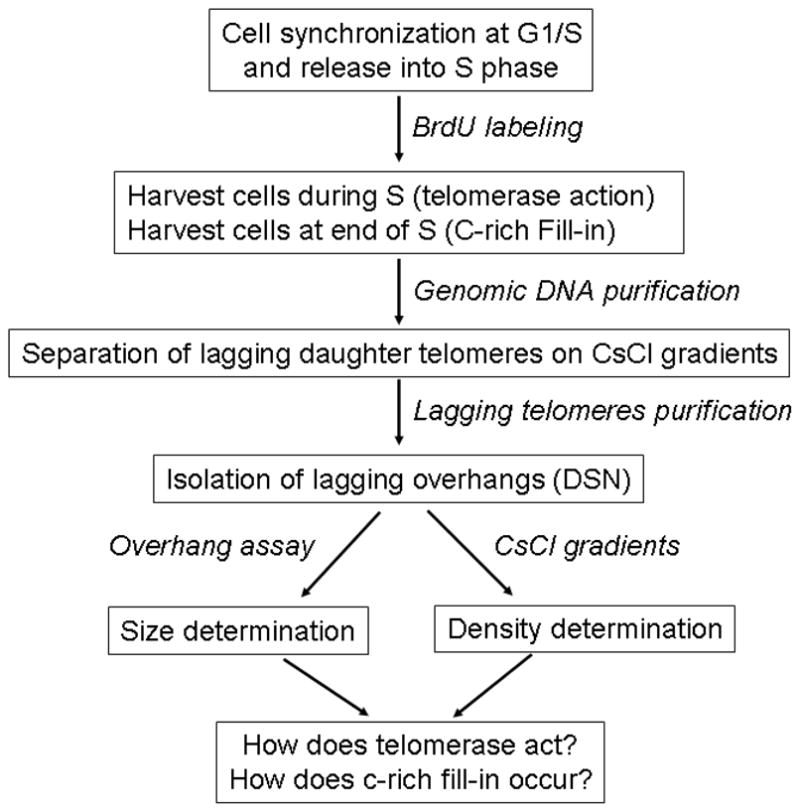
Outline of steps for study of telomerase action and C-rich fill-in.
2. Materials
2.1 Cell culture and synchronization
2.2 Genomic DNA purification
Blood & cell culture DNA midi kit (Qiagen, Inc) (see Note 3)
Ethyl Alcohol (70%)
Millipore water with 5mM Tris-HCl, pH 8.0,
Water bath, 37°C
2.3 Restriction enzyme digestion and CsCl gradients
10×NEB buffer 2 (NEB Inc)
Restriction enzymes: Hinf I, Rsa I, Msp I, Hae III, Alu I, Hha I (NEB)
CsCl (USB Inc)
1M Tris-HCl (pH 8.0)
0.5M EDTA (pH 8.0)
Ultracentrifuge L8-M with VTi80 vertical rotor (Beckman Inc) (see Note 4)
Quick-seal centrifuge tubes (13×51mm) (Beckman)
Light mineral oil (Sigma-Aldrich)
27guage 1/2″ and 21guage 1″ needles, 5ml syringe and 0.5ml tube
Refractometer (Milto Roy)
2.4 Slot blot and hybridization with C-rich telomeric probe
20×SSC: 3M NaCl, 0.3M sodium citrate, pH 7.0
1M NaOH
Protran BA 85 Nitrocellulose Transfer Membrane (Watman)
Minifold® II Slot blot (Schleicher & Schuell, Inc) (see Note 5)
80°C oven
Hybridization buffer: 6×SSC, 5×Denhardt’s solution, 0.5%(w/v) SDS
C-rich telomeric probe made as described in 3.4 of chapter “Telomere G-overhang Length Measurement: DSN method”
Washing buffer 1: 2×SSC and 0.1% SDS
Washing buffer 2: 0.5×SSC and 0.1% SDS
Hybridization oven (Hybridiser HB-1D, Techne, Inc)
Phosphor screen (Molecular Dynamics)
Typhoon™ (GE Healthcare)
2.5 Purification of lagging daughter telomeres from CsCl gradients
Ultrapure™ agarose (Invitrogen Inc)
Ethanol (100%, 70%)
NaCl (5M)
Centrifuge with temperature control
DSN 1×buffer: 50mM Tris-HCl, pH 8.0, 5mM MgCl2, 1mM DTT
2.6 Second CsCl gradients, slot blot and hybridization
Materials required are the same as listed in 2.3, 2.4.
2.7 Data analysis
Software requirement: ImageQuant 5.2 and IQTools 3.0 for quantification of slot blot and software that can perform Gaussian distribution analysis, e.g Graphpad prism 5.
3. Methods
(all experiments need to be performed in the absence of fluorescent light, which damages BrdU)
3.1 Cell synchronization at G1/S and BrdU labeling (see Note 6)
Telomeres replicate and telomerase acts throughout S phase whereas C-rich fill-in only happens at the end of S and G2. Cells are synchronized at G1/S and released into S phase in the presence of BrdU to label DNA. In Hela cells, S phase lasts 6–8hours, so cells are harvested4h after release (during S phase) to study telomerase action or 8h after release(S/G2) to study C-rich fill-in.
Grow cells to 30–40% confluence in DMEM medium with 10% calf serum.
Add thymidine to 2mM final and incubate for 19h.
Wash cells three times with pre-warmed solution A and re-feed with fresh serum-rich DMEM medium.
Incubate for 9h before adding 2mM thymidine.
Incubate for 16h and wash three times with pre-warmed solution A, re-feed with fresh medium, add BrdU to a final concentration of 100μM.
Harvest cells after 4 or 8 hours.
3.2 Genomic DNA purification
Purify genomic DNA following the manufacture’s instruction.
Wash the DNA pellets twice with 70% ethanol.
Let DNA dry in air.
Dissolve genomic DNA in Millipore water with 5mM Tris-HCl (pH 8.0), overnight incubation at 37°C ensure that all DNA is completely solubilized and any 3′ end structures are resolved.
3.3 Separation of lagging daughter telomeres on CsCl gradients
Due to the absence of restriction enzyme recognition sites within the TTAGGG tandem repeat sequence, telomeric DNA can be released by restriction enzyme digestion which cleaves genomic DNA into <300bp fragments while leaving telomere DNA uncut. In the presence of the thylmidine analogue BrdU, DNA replication incorporates BrdU into the daughter strands. Lagging synthesis of CCCTAA only incorporate one BrdU per six nucleotides, wheres leading synthesis of TTAGGG incorporates two BrdU per six nucleotides. Leading telomeres thus incorporate more of the heavier BrdU nucleotide and thus have a greater density compared to lagging telomeres, permitting separation on CsCl density gradients(11).
Digest genomic DNA with 6 restriction enzymes (materials 2.3). Start with at least 400μg DNA. DNA is incubated with 6 enzymes in 1×NEB buffer 2 at 37°C overnight in order to get complete digestion (or at least 8hours). (see Note 7)
Make CsCl solution (~8M, density ~1.788) containing 5mM Tris-HCl (pH 8.0) and 1mM EDTA.
Mix digested genomic DNA with CsCl solution and adjust the final density to be ~1.766 using CsCl powder or water. Inject mixed solution into centrifugation tube and seal the tube.
Place tube into rotor and spin at 55K RPM (RCF=120443g) for 20h at 25°C, let rotor slow down without braking (usually it takes 45min to completely stop).
Make a small hole on the bottom of tube using 27G 1/2″ needle, push the sample out by injecting mineral oil into tube from top of tube (see Fig 3).
Collect the fractions at 100μl per fraction. (approximately 50 fractions total)
-
Measure the density of each fraction. Read the refractive index from refractometer following the instruction provided by the manufacture, convert the refractive index of each fraction to density using the following formula:
where RI is the refrective index.
Figure 3.

Illustration of how to collect the fractions from CsCl gradients.
3.4 Slot blot to determine fractions containing lagging strand telomeres
After spinning, DNA with different densities will shift to the corresponding densities of CsCl. To determine which fractions contain lagging telomeres, the aliquots of DNA are denatured by NaOH, neutralized with SSC and loaded on a slot blot. After hybridizing to a telomeric specific C-rich probe, the fractions that have telomere DNA will be located.
Dilute aliquots of DNA (5μl) from each fraction with water to 50μl, add the same volume (50 μl) of 0.2M NaOH and incubate the mixture at 37 °C for 10min.
Neutralize the sample by adding 100μl 12×SSC.
Assemble slot blot apparatus according to manufacture’s instructions. The membrane needs to be prewetted in 20×SSC.
Apply vacuum, wash the wells with 400μl 20×SSC, and load treated samples to wells in the sequence with which they were collected.
Wash each well with 400μl 20×SSC twice, let vacuum run for additional 10min.
Disassemble the slot blot apparatus and carefully remove the membrane.
Bake the membrane at 80°C for 2h to immobilize DNA on membrane. Do not use UV to crosslink the DNA to the membrane since BrdU is much more reactive to UV than thymidine.
Prehybridize membranes at 42°C for at least 30min using hybridization buffer.
Add 5–10μl high specific activity telomere C-rich probe to hybridization buffer to a concentration of 1μl/ml.
Hybridize at 42°C overnight.
After thybridization, wash the blots at 42°C for 15min in washing buffer 1, then 15min in washing buffer 2.
Remove the blot from the last wash, drain and wrap in plastic wrap such as SaranWrap®, expose to PhosphoImager screens for appropriate time (30min to 2h)
Scan screens on the Typhoon PhosphoImager®.
Quantify the signal intensity, draw graph by using the density of each fraction (3.3.7) as the X-axis and the intensity as the Y-axis.
3.5 Purification of lagging strand telomere DNA from CsCl fraction
Leading daughter telomeres are located at a density of ~1.788, lagging daughter DNA is at a density of 1.766 while unreplicated telomeres are at a density of ~1.744 (see Note 8). To purify lagging telomere DNA, the fractions containing lagging DNA are pooled together, surface dialyzed to reduce salt concentration and then ethanol precipitated. (see Note 9)
Pool 4–5 fractions of lagging peak together. (see Note 10)
Melt 25ml 2% agarose in water containing 5mM Tris-HCl (pH 8.0) and pour gel into 50ml tube.
Load pooled DNA fractions on the top of gel, rock the tube for 1h at room temperature on a rocking platform as used for western blots. The tube is maintained in a “vertical” position so the DNA solution is maximally exposed to the agarose surface.
Remove dialyzed DNA to a 2ml tube, mix with 1/25 volume of 5M NaCl and 2 volumes of ice-cold ethanol, store at −20 °C for at least 1h.
Centrifuge at 13000RPM (18000g) for 30min at 4°C.
Discard the supernatant, wash the DNA pellet with 70% ice-cold ethanol twice.
Dry pellet in air, usually it takes 10–15min at room temperature.
Dissolve DNA in 50μl 1×DSN buffer. Incubation at 37 °C for 10minhelps DNA dissolve.
Measure DNA concentration, usually 10–20μg DNA containing the lagging telomere daughters can be recovered from 400μg genomic starting DNA.
3.6 Determine the length of lagging daughter overhang
The overhang length of the lagging daughter can be measured by following the method described in the chapter “Telomere G-overhang Length Measurement: The DSN method”, starting with step 2.
3.7 Determine density of lagging overhangs extended by telomerase
Lagging overhangs containing only thymidine have a density of ~1.744, whereas the density of overhangs with thymidine fully substituted by BrdU is ~1.831. For lagging daughters, the initial overhang contains only thymidine, but telomerase incorporates BrdU into each new synthesized repeat, making the overhang heavier. Therefore, depending on the initial overhang length and the number of repeats telomerase added, the overhang’s density should be between ~1.744 and ~1.830. Assuming that the density shift is proportional to the percentage of incorporated BrdU in the overhang, one can calculate the ratio of thymidine to BrdU by measuring its density.
Add 1–2U DSN (0.2U/5μg) to purified lagging DNA, and incubate at 37 °C for 2h.
Make CsCl solution (density ~1.783) containing 1mM EDTA (pH 8.0). (see Note 11)
Mix DSN digested DNA with CsCl solution, inject them into tube and seal tube.
Spin at 60K RPM (RCF=143337g) for 20hours at 25°C, let rotor slow down without braking.
Make a small hole on the bottom of tube using 27G 1/2″ needle, push the sample out by injecting mineral oil into tube from top of tube.
Collect the fractions at 100μl per fraction. (approximately 50 fraction in total)
Measure density of each fraction as described above.
Assemble slot blot, prewash with 20×SSC, load each fraction into slots, and wash with 20×SSC twice. It is not necessary to denature the samples since only single-stranded DNA survives DSN digestion.
Let membrane dry in air for 2–3 h.
UV-crosslink DNA on membrane by setting energy to be 70mj/cm2 in a commercial UV crosslinker (The crossliking efficiency following baking is not as good as with UV, possibly because the DNA is single-stranded).
Prehybridize membrane at 42°C for at least 30min using hybridization buffer.
Add 5–10μl high specific activity telomere C-rich probe to hybridization buffer to a concentration of 1μl/ml. (see Note 12)
Hybridize at 42°C overnight.
After the hybridization, wash the blots with washing buffer 1 twice, 15min each, followed by washing buffer 2 for 15min at 42°C.
Remove the blot from the last wash, drain and wrap in SaranWrapR, expose to PhosphoImager screens for several hours to overnight.
Scan screen on Typhoon PhosphoImager®.
3.8 Data analysis
The central questions regarding telomerase action are the following: first, how many telomeres in cells have been extended by telomerase during one cell cycle; second, on average how many repeats of GGTTAG did telomerase add to each telomere? To answer these questions, three parameters need to be extracted from the experimental data: the relative amount of unextended and extended overhangs, expressed in these experiments as the relative signal intensity of the thymidine overhang peak versus the intermediate peak; the density of intermediate peak— this is extremely useful for determining the percentage of BrdU in lagging overhangs; and the lagging overhangs length as measured in 3.6. In this section, methods to extract these parameters from the raw data are described.
Quantify the signal intensity for each fraction on slot blot.
Using density as the X-axis and intensity as the Y-axis, draw the graph.
-
Fit the data to the function of “sum of two Gaussian distributions” using the built-in equation in software GraphPad Prism:
where, AREA1 and AREA2 are peak areas, Mean1 and 2 represent the densities at peaks, SD1 and 2 are standard deviations, pi is a constant.
From fitting, the density of the intermediate peak will be given (Mean2), as well as the relative amounts of un-extended and extended overhangs that were expressed as AREA1 and AREA2.
-
Draw a linear regression line between the highest density (1.831, 100% BrdU) and the lowest density (1.744, 0% BrdU) using density and percentage of BrdU as X-and Y-axis respectively, then the following linear regression equation can be obtained:
where, BrdU% is the percentage of BrdU in the overhang and D is density of intermediate peak.
Input intermediate density (Mean2) to linear regression equation and export the percentage of BrdU (BrdU%).
-
Calculate the average length of extension (ALE) using the following expressions:
where, OL is overhang length determined in 3.6.
- Calculate the percentage of extended telomeres (PEE) using the following expressions:
Footnotes
DMEM medium with 10% calf serum works well for Hela cell synchronization and BrdU incorporation. It is also suitable for other cancer cell lines such as H1299 and A549. However, different medium may be used if DMEM does not work well on your cells.
1×PBS as wash buffer can also be used.
Any genomic DNA purification kit should be good for this purpose.
The vertical rotor (VTi80, 90) significantly shortens spinning time. One can also use a swinging bucket rotor, but it will take several days to form gradients.
Other slot blot equipment may work but the one described in this method gives a low background and a high reproducibility.
The method described here are optimized for synchronization of normal HeLa cells, which produced synchronization of >90% cells at G1/S. If double thymidine blocking strategy does not work well for your cells, different approaches such as aphidicolin should be tried. If a high percentage of synchronization cannot be obtained, a minimum of 50% cells synchronized should be good enough as long as there are no cells leaking from S phase to G2 before the end of S.
The concentration of each enzyme used is 0.5unit/μg. Use of just two enzymes (HinfI and RsaI), works almost as well as 6 enzyme digestion.
The density of each peak may change from time to time, but the variation shouldn’t be beyond ±0.0055. It is also worth noticing that the distance (in density) between leading and lagging peaks should be equal to the distance between lagging and unreplicated peaks.
This method will give a recovery of ~ 40% in purifying DNA from CsCl.
With more fractions pooled, there is an increased probabilityof contamination by leading telomeres and unreplicated DNA.
Overhangs may locate on any density ranging from ~1.744 (pure thymidine overhang) to 1.831 (fully BrdU substituted overhang) depending on how many repeats were added by telomerase. The CsCl gradients generated for overhang analysis should thus cover all possible densities. To this end, increase the initial density of CsCl (~1.783) and the spinning speed (60K).
Using fresh isotope to make C-rich telomeric probe is recommended. Isotope exceeding one half-life should not be used.
References
- 1.Zhao Y, Hoshiyama H, Shay JW, Wright WE. Quantitative telomeric overhang determination using a double-strand specific nuclease. Nucleic Acids Res. 2008;36:e14. doi: 10.1093/nar/gkm1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wright WE, Tesmer VM, Huffman KE, Levene SD, Shay JW. Normal human chromosomes have long G-rich telomeric overhangs at one end. Genes Dev. 1997;11:2801–2809. doi: 10.1101/gad.11.21.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smogorzewska A, de Lange T. Regulation of telomerase by telomeric proteins. Annu Rev Biochem. 2004;73:177–208. doi: 10.1146/annurev.biochem.73.071403.160049. [DOI] [PubMed] [Google Scholar]
- 4.Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL, Shay JW. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266:2011–2015. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- 5.Greider CW, Blackburn EH. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell. 1985;43:405–413. doi: 10.1016/0092-8674(85)90170-9. [DOI] [PubMed] [Google Scholar]
- 6.Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature. 1990;345:458–460. doi: 10.1038/345458a0. [DOI] [PubMed] [Google Scholar]
- 7.Meyne J, Moyzis RK. In situ hybridization using synthetic oligomers as probes for centromere and telomere repeats. Methods Mol Biol. 1994;33:63–74. doi: 10.1385/0-89603-280-9:63. [DOI] [PubMed] [Google Scholar]
- 8.Baird DM, Rowson J, Wynford-Thomas D, Kipling D. Extensive allelic variation and ultrashort telomeres in senescent human cells. Nat Genet. 2003;33:203–207. doi: 10.1038/ng1084. [DOI] [PubMed] [Google Scholar]
- 9.Wright WE, Tesmer VM, Liao ML, Shay JW. Normal human telomeres are not late replicating. Exp Cell Res. 1999;251:492–499. doi: 10.1006/excr.1999.4602. [DOI] [PubMed] [Google Scholar]
- 10.Zhao Y, Sfeir AJ, Zou Y, Buseman CM, Chow TT, Shay JW, Wright WE. Telomere extension occurs at most chromosome ends and is uncoupled from fill-in in human cancer cells. Cell. 2009;138:463–475. doi: 10.1016/j.cell.2009.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chai W, Du Q, Shay JW, Wright WE. Human telomeres have different overhang sizes at leading versus lagging strands. Mol Cell. 2006;21:427–435. doi: 10.1016/j.molcel.2005.12.004. [DOI] [PubMed] [Google Scholar]