

Abstract
Historically, mice and guinea pigs have been the rodent models of choice for therapeutic and prophylactic countermeasure testing against Ebola virus disease (EVD). Recently, hamsters have emerged as a novel animal model for the in vivo study of EVD. In this review, we discuss the history of the hamster as a research laboratory animal, as well as current benefits and challenges of this model. Availability of immunological reagents is addressed. Salient features of EVD in hamsters, including relevant pathology and coagulation parameters, are compared directly with the mouse, guinea pig and nonhuman primate models.
Keywords: Ebola, filovirus, hamster model, rodent model, pathogenesis
1. Introduction
Ebola virus (EBOV), a member of the family Filoviridae, is the etiologic agent of Ebola virus disease (EVD), a severe hemorrhagic fever syndrome with unusually high case-fatality rates, ranging between 65–90%. Filoviruses are emerging/reemerging zoonotic agents that are highly virulent in primates, and the frequency of outbreaks in Africa and Asia and impact on ape populations have been increasing in recent years. Introduction of filoviruses into human populations leads to serious, albeit limited, epidemics. Interhuman transmission occurs by direct person-to-person contact and possibly by fomites and droplets. Filoviruses infect, among others, monocytes, macrophages, dendritic cells, hepatocytes, and endothelial cells. In the infected primate, these viral infections lead to severe cytokine imbalances that impair the innate and adaptive immune responses, disseminated intravascular coagulation (e.g., hemorrhages, thrombi), and organ necroses that result in multi-organ failure and shock. No approved vaccines or effective therapeutics are currently available. Because of the high case-fatality rates of EVD and the lack of an approved vaccine or therapy, EBOV is classified as a category A pathogen requiring biosafety level-4 (BSL-4) biocontainment.
2. Ebola Virus Disease (Humans): Clinical Presentation and Pathogenesis
Ebolaviruses likely enter the body via direct contact (skin abrasions, mucous membranes) or contact with bodily fluids to directly access the vascular system or indirectly access the lymphatic system [1]. Limited human data indicate that monocytes/macrophages and dendritic cells are primary sites of virus replication [2]. Ebolaviruses spread from initial infection sites via macrophages and dendritic cells trafficking to regional lymph nodes, liver, and spleen [1]. After an incubation period of 4–16 days, patients initially present with influenza-like symptoms such as abdominal pain, anorexia, arthralgia, asthenia, back pain, diarrhea, fever, headaches, enlarged lymph nodes, myalgia, nausea, or vomiting [3,4,5]. After approximately 5–7 days, a maculopapular rash usually develops on the face, buttocks, trunk, or arms and later generalizes over almost the entire body. As EVD progresses, more severe and multisystem symptoms include respiratory (e.g., chest pain, terminal tachypnea, cough), vascular (e.g., conjunctival injection, postural hypotension, disseminated intravascular coagulation), urinary (e.g., anuria), and neurologic (e.g., headache, confusion, coma) manifestations [5,6]. In fatal EVD cases, hemorrhagic manifestations are usually striking with hematemesis, hemoptysis, melena, hematuria, epistaxis, and/or widespread petechiae and ecchymoses involving skin, mucous membranes, and internal organs [7,8]. Hemorrhagic manifestations occur approximately 3–4 days postonset of influenza-like symptoms and progress, in fatal cases, to disseminated intravascular coagulation (decrease of clotting factors, thrombocytopenia, increased fibrin degradation products, prolonged thrombin and activated partial thromboplastin times) [5,9,10].
Death occurs 3–21 days after disease onset from shock after multi-organ failure (liver, spleen, kidney). Liver damage is characterized by hepatocellular necrosis, sinusoidal dilation and congestion (hepatomegaly), and elevated concentrations of aspartate transaminase (AST), alanine aminotransferase (ALT), alkaline phosphatase (ALP), and γ-glutamyl transferase (GGT) [2,3,4,10]. Within the spleen, marked hyperemia and splenomegaly, cellular depletion of the red pulp, and/or marked atrophy of the lymphoid follicles are observed [8,11]. Lymphoid hypoplasia or depletion has been noted in patients with EVD [2,8,11]. Leukopenia (lymphopenia) and granulocytosis are present initially in patients with EVD, and as the disease progresses, leukocytosis results from an increase in immature granulocytes and atypical lymphocytes [4,12,13]. Acute renal tubular necrosis and calcification of renal tubules and glomerular tufts are noted [2,3,14]. Creatinine and urea concentrations increase prior to renal failure, and hypokalemia is typical due to diarrhea and/or vomiting. Lung hemorrhage (hemoptysis) progresses to focal atelectasis and is accompanied by interstitial pneumonitis [2,11].
Infected monocytes/macrophages release soluble mediators including proinflammatory cytokines and vasoactive substances [15]. These cytokines recruit additional macrophages to infected areas and could increase the number of target cells available for virus infection, further amplifying an already dysregulated host response [16]. EBOV-infected patients with a fatal outcome exhibited increased concentrations of interferon (IFN)-γ, IFN-α, interleukin (IL)-2, IL-6, IL-8, IL-10, IL-1 receptor antagonist, macrophage inflammatory protein-1β, neopterin, or tumor necrosis factor (TNF)-α, although differences in cytokine release were apparent between variants of EBOV [15,17,18].
3. Existing Animal Models of EVD
The development of animal models that accurately reflect human disease is critical to our understanding of the pathogenesis of EVD and evaluation of countermeasures against filoviruses. Because of the sporadic and geographically isolated nature of EVD outbreaks, clinical efficacy studies may not be feasible. Clinical data and access to human tissues from fatal cases are limited.
Another option for licensing new drugs and vaccines for EVD is extrapolation of data derived from accurate, validated animal models supported by human safety evaluation data and pharmacokinetic information. The “Animal Rule” from the U.S. Food and Drug Administration [19] requires that a countermeasure be evaluated in animal models in which the route and dose of virus administration, time to onset of disease, and time course/progression of disease optimally mimic the pathophysiology of human disease. One of the challenges with this regulatory pathway is the development of animal models that recapitulate human disease, as data on the clinical presentation of EVD disease in humans are quite limited.
The nonhuman primate (NHP) model of EVD is the gold-standard for the study of EVD pathogenesis that most closely resembles what we currently know regarding human disease. Guinea pigs and mice are regarded as models for preliminary evaluation of therapeutic interventions against EVD. As wild-type EBOV does not produce appreciable disease in these rodent models, EBOV was adapted by serial passage to produce fatal infection following intraperitoneal (IP) inoculation [20,21]. The pathogenesis of EVD from adapted rodent viruses differs in a number of aspects from EVD in humans and NHPs. Important clinical signs of EVD in humans and NHPs such as fever and maculopapular rash are not present in mice infected with mouse-adapted Ebola virus (MA-EBOV) [20,22]. Fever is present in guinea pigs infected with guinea pig-adapted Ebola virus (GPA-EBOV), but maculopapular rash does not develop in these animals [21]. Mice infected with MA-EBOV do not consistently display coagulation abnormalities (Table 1) [20,23,24]. Compared to mice, guinea pigs infected with GPA-EBOV develop coagulation defects, including a drop in platelet counts and an increase in coagulation time, but fibrin deposition and coagulopathy (i.e., disseminated intravascular coagulation) are not as marked as that observed in NHPs [21,25].
Table 1.
Coagulation parameters in animal models of Ebola virus disease a.
| Coagulation Parameter | Rhesus Macaque b | Syrian Hamster c | Guinea Pig d | Mouse c |
|---|---|---|---|---|
| Increased prothrombin time (PT) | (++) [26,27] | (+++) | (+++) | (-) [20,27] |
| Increased activated partial thromboplastin time (aPTT) | (++) [26,27] | (+++) | (++) | (-)[20,27] |
| Increased thrombin time (TT) | (++) [26,27] | (++) | ND | ND |
| Late hypofibrinogenemia | (+++) [26,27] | (++) | (-) (increased fibrinogen) | (-/+) [20,27] |
| Decreased protein C activity % | (+++) [26,27] | (+++) | ND | ND |
| Thrombocytopenia | (++) [26,27] | (++) | (++) e [20] / (+++) f [21] | (++) [20,28] |
a: From Ebihara [27], unless otherwise noted
b: Infected with wild-type EBOV
c: Infected with MA-EBOV
d: Hartley guinea pigs infected with GPA-EBOV, unless otherwise noted
e: Inbred strain 2 United States Army Medical Research Institute for Infectious Diseases (USAMRIID) guinea pig
colony infected with MA-EBOV
f: Inbred strain 13 USAMRIID guinea pig colony infected with GPA-EBOV
ND: no data
Further, bystander lymphocyte apoptosis, an important feature in primates and mice, has not been determined in guinea pigs infected with GPA-EBOV. Mice infected with MA-EBOV differ from guinea pigs infected with GPA-EBOV and monkeys infected with wild-type EBOV in that they display a decrease in blood urea nitrogen (BUN), rather than an increase [20]. Because of these differences in EVD in rodent models, a number of therapeutic interventions that are effective in rodents challenged with adapted EBOV fail to protect NHPs challenged with wild-type EBOV from EVD (Table 2 and Table 3, see Supplemental Tables 1–2 for unabridged versions). Of the five equivalent vaccines tested in rodents and NHPs with a comparable degree of immunocompetence, two vaccines had equivalent protection in all animal models tested, and three vaccines that provided some protection in rodents were not protective in NHPs. In evaluation of peri-exposure treatment of EVD, vesicular stomatitis virus (VSV) viral vectors provided roughly the same percentage of protection in guinea pigs and NHPs. Transfer of immune serum or equivalent polyclonal or monoclonal antibodies to naïve infected animals provided no protection to NHPs and some protection to rodents. Administration of equivalent antisense phosphorodiamidate morpholino oligomers to NHPs provided less protection against EVD than rodents.
Table 2.
Efficacy of vaccines in animal models of Ebola virus disease.
| Vaccines | Immunization Schedule | Mouse Model | Guinea Pig Model | NHP Model |
|---|---|---|---|---|
| Virus Vectors | ||||
| HPIV3 Immunogens | Guinea Pigs: |
|
||
| HPIV3- NHPs: | ||||
| ||||
| VSV ∆GP Immunogens | Immunocompetent Mice: |
|
||
| EBOV GP attenuated [32,33,34,35,36,37,38] | ||||
| NOD-SCID Mice: IP 2 × 105 PFU of VSV∆GP/EBOV GP | ||||
| Guinea Pigs: IP 2 × 105 PFU-1–2 doses of VSV∆GP/EBOV GP [32] | ||||
| HIV + NHPs: IM 1 × 107 PFU [34] | ||||
| Immunocompetent NHPs: | ||||
| VV Immunogens | Guinea Pigs: SC 107 of VV/EBOV GP–3 doses [39] |
|
|
|
| NHPs: SC of VV/EBOV GP–3 doses [24] | ||||
| Virus-like Particles (VLPs) | ||||
| VEEV RNA (VRP) encoding: | Mice: |
|
|
|
| Guinea Pigs: SC 107 IU of VRP EBOV GP, NP, or GP+NP–2 or 3 doses [41,44] | ||||
| NHPs: SC 2 x 106 FFU of VRP EBOV GP, NP or GP+NP–3 doses [24] | ||||
| Ebola Virus Vaccines | ||||
| EBOV | Mice |
|
||
| NHPs: | ||||
Abbreviations: EBOV: Zaire ebolavirus species; FFU: focus-forming units; GP: glycoprotein; HIV: human immunodeficiency virus; GPA: guinea pig adapted; HPIV3: human parainfluenza virus type 3; IM: intramuscular; IN: intranasal; INA: 1,5-iodonaphthylazide; IP: intraperitoneal; IT: intratracheal; IV: intravenous; MA: mouse adapted; NHP: nonhuman primate; NOD: nonobese diabetic; NP: nucleoprotein; PFU: plaque-forming units; PO: oral; RNA: ribonucleic acid; SC: subcutaneous; SCID: severe combined immunodeficiency; TCID: tissue culture infective dose; VEEV: Venezuelan equine encephalitis virus; VLP: virus-like particles; VP: viral protein; VRPs: VEEV RNA replicon particles; VSV: vesicular stomatitis virus: VV: vaccinia virus
Table 3.
Efficacy of peri-exposure treatment in animal models of Ebola virus disease.
| Peri- exposure Treatment | Dose and Route of Administration | Mouse Model | Guinea Pig Model | NHP Model |
|---|---|---|---|---|
| Virus Vectors | ||||
| VSV ∆GP Immunogens | Mice: IP 2 x 105 VSV ∆GP/EBOV GP PFU -1 day before or +30 minutes or +1 day after challenge [51] |
|
|
|
| ||||
| Guinea pigs: IP 2 x 105 VSV ∆GP/EBOV GP PFU -24 hours or +1 or 24 hours [51] | ||||
| NHPs: IM 2 x 107 PFU of VSV ∆GP/EBOV GP [51] +20–30 minutes | ||||
| Passive Immunity | ||||
| Pooled immune serum to live EBOV [46,52] | Mice: IP 1 mL of antisera (anti-EBOV IgG titers of ≥6,400) -1 day or + 1 day [46] |
|
|
|
| NHPs: IV 6 mL/kg whole blood immediately after challenge and +3 or 4 days (anti-EBOV IgG ELISA titers of 100,000) [52] | ||||
| Passive Immunity | ||||
| Purified polyclonal IgG antibody against: | Mice: |
|
|
|
| ||||
| Guinea Pigs: IM 1 mL/kg of horse IgG + several minutes and +3 days, or +4 days only [54] | ||||
| NHPs: | ||||
| mAb EBOV GP-specific | Guinea Pigs: |
|
|
|
|
||||
| NHPs: IV 50 mg/kg -1 day and +4 days [56] | ||||
| Antiviral Agents | ||||
| Antisense Phosphorodiamidate morpholino oligomers (PMO) [57,58,59,60] | Mice: |
|
|
|
| Guinea Pigs: IP 10 mg of each PMO targeting VP24, VP35 or L -1 day or +1 or 6 days after challenge [58] | Reduced viremia and release of IL-6 and MCP-1 with PMOs targeting VP24 and VP35 | |||
| Antiviral Agents | ||||
| Antisense PMOs (continued) | s: |
|
||
| ||||
AAbbreviations: EBOV: Zaire ebolavirus species; GP: glycoprotein; Ig: immunoglobulin; IL-6: interleukin-6; IM: intramuscular; IP: intraperitoneal; IV: intravenous; L: L polymerase; mAb: monoclonal antibody; MCP-1: monocyte chemotactic protein-1; NHP: nonhuman primate; PFU: plaque-forming units; PMO: antisense phosphorodiamidate morpholino oligomers; RNA: ribonucleic acid; SC: subcutaneous; VP: viral protein; VSV: vesicular stomatitis virus
4. History of Outbred Strains of Syrian Hamster in U.S.
Syrian hamsters (Mesocricetus auratus) are used in research studies of infectious diseases and cancer. In particular, Syrian hamsters are recognized as valuable animal models for studying emerging, acute human viral diseases caused by highly pathogenic RNA viruses. Outbred strains of Syrian hamsters in the U.S. are currently available from 3 sources: Simonsen Laboratories, Charles River Laboratories, and Harlan Laboratories. Most or all of these sources obtained Syrian hamster stock from Jerusalem. In 1930, lengthy experiments on leishmaniasis at the Hebrew University of Jerusalem were hampered by limitations of the only animal model known for the disease, the Chinese hamster (Cricetus griseus) [61]. Continuous replenishment of Chinese hamster stocks from China was needed as conditions for successful breeding of captive Chinese hamsters were not known, and such hamsters succumbed to a Pasturella epidemic in the region. Instead, an endemic species of hamster, the Syrian or golden hamster was substituted for the Chinese hamster. Syrian hamsters are easily bred in captivity, relatively free from natural hamster diseases, but are susceptible to experimental pathogens and have a short life cycle [61,62]. With the success of the domesticated Syrian hamster as a model for leishmaniasis, tuberculosis, and brucellosis, Jerusalem scientists sent shipments of Syrian hamsters to U.S. research institutions (Western Reserve University, Rockefeller Foundation, U.S. Public Health Service) in 1938 and 1942. Leishmaniasis-infected hamsters were sent to the military during World War II (probably not bred) for evaluation of therapeutic interventions [61]. U.S. research studies utilizing the Syrian stocks originally from Jerusalem were first published the 1940s [63]. In the mid 1940’s, Albert F. Marsh obtained stock probably from U.S. Public Health Service to found Gulf Hamstery [63,64]. Simonsen Laboratories acquired stock originating with Gulf Hamstery in 1962 and interbred their hamsters with stock from ARS/Sprague Dawley in 1978 [65]. In 1983, Simonsen Laboratories derived the Sim: BR golden strain that is still commercially available. Similarly, Engle Laboratory Animals and Lakeview Hamstery purchased stock from Gulf Hamstery in 1949 [63,64]. Lakeview Hamstery became a subsidiary of Charles River in 1969, and stock (Crl: LVG) is still available today [66,67]. Engle Laboratory Animals was acquired by Harlan Laboratories in 1984, and the hamster stock originally from Engle Laboratory Animals is no longer commercially available [64]. Syrian hamster stock from ARS/Sprague Dawley was sent to Central Institute for Laboratory Breeding in Hanover, Germany (HAN: AURA) in 1973 [68,69]. Harlan Laboratories purchased the Central Institute for Laboratory Breeding in 1994, and stock (HsdHan: AURA) is currently available.
Currently available inbred strains of Syrian hamsters include Bio 1.5, Bio 14.6, Bio 15.16, Bio F1B, Bio HT, and Bio To-2 [70]. These strains are used as disease models for carcinogenicity, dental caries, cardiomyopathy, muscular dystrophy, diabetes, atherosclerosis, and hypertension.
5. Syrian Hamsters as a Model of Viral Hemorrhagic Fever Diseases
Approximately 145,895 hamsters were used in research in 2010 in the U.S. comprising 13% of total animal usage [71]. Although hamsters are still widely used, the number of hamsters currently used in research is well below peak usage of over 500,000 in the 1980s [72]. However, the total number of papers published from 1971 to 2011 has steadily increased (Figure 1) with the greatest increase in studies of viral infections. Availability, size, ease of care and breeding in laboratory conditions, and cost contribute to the popularity of Syrian hamster as an alternative to nonhuman primates for viral research.
Figure 1.

Number of publications utilizing hamsters between 1971 through 2011. An increase in use of hamsters as an animal model for parasitic, bacterial, and viral diseases is noted in the literature. During the 10-year period of 2001-2011, the largest increase in publications was observed, the majority of which were in the virology sector.
Hamsters are valuable animal models for studying viral hemorrhagic fevers, including EVD [27], Marburg virus disease [73], hantavirus cardio-pulmonary syndrome [74,75,76,77,78,79], arenavirus hemorrhagic fevers [80], yellow fever hemorrhagic fever [80,81,82], and phlebovirus models of Rift Valley fever (Table 4) [83]. All of these viruses cause hemorrhagic fevers in hamsters, but infections caused by some of these viruses (e.g., Pirital, Maporal) that serve as disease models (e.g., hantavirus cardiopulmonary syndrome, Lassa fever) are not pathogenic in humans [78,84]. A number of these viruses were hamster adapted (Marburg, yellow fever, Pichinde) [73,80,81,85] or mouse adapted (Ebola) [27].
Table 4.
Hamsters as a model for viral hemorrhagic fevers.
| Pathology and laboratory abnormalities | Virus |
|---|---|
| Spleen | |
|
Punta Toro [83] |
| Pirital [84,86,87] | |
| Marburg [73] | |
|
Ebola [27] |
|
Gabek Forest [83] |
| Yellow fever [81,82,85] | |
|
Ebola [27] |
|
Andes [75] |
|
Marburg [73] |
|
Ebola [27] |
| Liver | |
|
Punta Toro [83] |
|
Ebola [27] |
|
Gabek Forest [83] |
|
Yellow fever [81,82,85] |
|
Pirital [84,86,87] |
| Andes [75,76,79] | |
|
Maporal [78] |
|
Yellow Fever [81,85] |
| Pirital [84,86] | |
|
Yellow Fever [81] |
|
Punta Toro [83] |
| Gabek Forest [83] | |
| Yellow Fever [81,85] | |
| Pichinde [80] | |
| Pirital [84,86] | |
| Lymph nodes | |
|
Punta Toro [83] |
|
Ebola [27] |
|
Ebola [27] |
|
Andes [76] |
|
Marburg [73] |
|
Ebola [27] |
|
Ebola [27] |
| Lung | |
|
Yellow fever[81] |
|
Andes[75,79] |
| Maporal [78] | |
|
Punta Toro [83] |
| Pirital [87] | |
|
Gabek Fores [83] |
| Pirital [84,86] | |
| Andes [76] | |
|
Marburg [73] |
| Kidney | |
|
Yellow Fever [82,85] |
|
Marburg [73] |
|
Gabek Forest [83] |
|
Maporal [78] |
|
Pirital [84,86] |
| Vascular dysregulation | |
|
Andes [75,79] |
| Pichinde [80] | |
| Yellow fever [80] | |
| Maporal [78] | |
|
Pichinde [80] |
| Yellow fever [81] | |
| Pirital [86] | |
| Coagulopathy | |
|
Yellow Fever [81] |
| Pirital [84,86] | |
| Andes [76] | |
| Ebola [27] | |
|
Pirital [86] |
| Ebola [27] | |
|
Yellow fever [81] |
| Ebola [27] | |
| Andes [76] | |
| Pirital [84] | |
|
Pirital [84] |
|
Ebola [27] |
|
Andes [76] |
|
Pirital [84,86] |
|
Andes [75] |
| Yellow fever [81] | |
| Hematological abnormalities | |
|
Yellow fever [81] |
|
Pichinde [80] |
| Pirital [86] | |
| Andes [75] | |
|
Andes [75] |
| Cytokines | |
|
Pichinde [80] |
| Andes [76] | |
|
Andes [76] |
| Yellow fever [85] | |
| Ebola [27] | |
Viral hemorrhagic fever is a syndrome characterized by fever, malaise, increased vascular permeability, and coagulation abnormalities that may lead to hemorrhage [80,88]. A number of factors contribute to alterations in vascular function such as direct cytolytic infection of the endothelium, changes in tight junctions between endothelial cells, alterations in coagulation pathways, disruption of hematopoiesis, and/or the release of cytokines and other permeability factors (e.g., tissue factor, TNF-α, nitric oxide) from endothelial cells, neutrophils, macrophages, and/or monocytes [1,80,89,90,91,92]. Although EBOV does infect endothelial cells of NHPs, infection occurs late in the disease course after the development of disseminated intravascular coagulation [89]. Rather, researchers consider the release of cytokines and other vasoactive mediators to disrupt the endothelial barrier, causing plasma volume loss, hypovolemic shock, multi-organ failure, and death [93]. Impairment of liver function may alter the production of vitamin K-associated coagulation factors (e.g., factor VII) and coagulation inhibitors (e.g., protein C) that could contribute to coagulopathy [26,27,92]. Thrombocytopenia occurs in part due to consumptive coagulopathy, but evidence from bone marrow aspirates in EBOV-infected nonhuman primates also reveals damaged megakaryocytes and atypical platelets [91].
Among the Syrian hamster models of viral hemorrhagic fevers, liver and lungs are the commonly affected organs. Signs of tachycardia and tachypnea and results of hematological, blood chemistry, and coagulation tests indicative of vascular leakage or shock are noted in Syrian hamsters infected with Andes virus, [77] Pichindé or Pirital viruses, [80,84,86] or yellow fever virus [80]. Upregulation of cytokines in one study of hamsters challenged with Pichindé virus preceded vascular leakage [80]. The search for an animal model that more closely resembles human EVD than other rodent models to date led to the development of the Syrian hamster model.
6. Syrian Hamsters as an Ebola Virus Disease Model
Data from a study of Syrian hamsters challenged IP or SC with MA-EBOV or wild-type EBOV indicate that only MA-EBOV given IP causes EVD reminiscent of human disease including, severe coagulopathy, lymphocyte apoptosis, cytokine dysregulation (e.g., suppression of early type I IFN responses), target organ necrosis and/or apoptosis (i.e., lymph nodes, spleen, liver), and lethal outcome (Table 5, Table 6) (Figure 2) [27]. Such suppression/non-induction of type-I IFN response and aberrant pro-inflammatory responses are suggested as some of the key pathogenic processes that lead to lethal outcome [20,27]. In contrast to MA-EBOV challenge, wild-type EBOV given IP in hamsters causes activation of early type-I IFN responses, mild inflammatory responses, induction of less-prominent apoptosis, and minimal pathological changes [27].
Table 5.
Comparison of current animal models of Ebola virus disease.
| Macaque | Guinea pig | Hamster | Mouse | |
|---|---|---|---|---|
| Hallmarks of Human Disease | ||||
| Target cells/organs | + | + | + | + |
| Cytokine dysregulation | + | ND a | + | +/- |
| Lymphocyte apoptosis | + | ND a | + | + |
| Coagulation dysfunction | ++ | + | ++ | +/- |
| Advantages/Disadvantages | ||||
| Availability | - | +/- | + | + |
| Ease of handling | - | +/- | + | + |
| Research reagents | ++ | - | +/++ | +++ |
| Pathogenicity of MA-EBOV | ++ [20] b | + | +++ | +++ |
a: ND = no data
b: MA-EBOV = mouse-adapted EBOV
Table 6.
Comparison of pathological changes in different animal models of Ebola virus disease.
| Liver | Spleen | |||||
|---|---|---|---|---|---|---|
| Hepatocellular degeneration and necrosis | Inflammation | Fibrin | Lymphoid necrosis and loss | Inflammation | Fibrin | |
| Mouse | Diffuse, random | Neutrophilic | Little | Multifocal, mild | Neutrophilic | Little |
| Guinea pig | Diffuse, random | Neutrophilic and histiocytic | Little to moderate | Diffuse, severe | Neutrophilic | Little to moderate |
| Hamster | Diffuse, midzonal | Neutrophilic | Little | Diffuse, moderate to severe | Neutrophilic | Little |
| NHP | Diffuse, random | Neutrophilic | Abundant | Diffuse, severe | Neutrophilic | Abundant |
Figure 2.

Temporal progression of disease in the Syrian hamster model of Ebola virus disease. Following exposure to 1000 focus-forming units of MA-EBOV IP, hamsters begin showing signs of illness around day 3. Changes in the innate immune response, coagulation parameters, and pathology are observed as early as days 1 and 2.
The severity of coagulopathy observed in Syrian hamsters infected with MA-EBOV is similar to that observed in rhesus macaques following challenge with wild-type EBOV (Table 1). Hepatic changes in Syrian hamsters closely resemble those of rhesus macaques, including disseminated hepatocellular degeneration and necrosis with infiltration of moderate numbers of neutrophils and fewer macrophages than neutrophils (Table 6). In contrast to macaques, little fibrin deposition occurs within hepatic sinusoids of hamsters (Figure 3). Likewise, splenic lesions in hamsters are also similar to those observed in macaques and are characterized by necrosis of lymphocytes and marked loss of white pulp. Additionally, multifocal acute splenitis is characterized by moderate numbers of viable and degenerate neutrophils and fewer macrophages than neutrophils mixed with necrotic debris within the red pulp. Lymph nodes also display diffuse lymphoid necrosis and loss along with acute lymphadenitis and draining hemorrhage (Figure 3). In terminal Syrian hamsters, all cytokines tested (IL-1β, IL-2, IL-4, IL-6, and IL-12p35; tumor growth factor [TGF]-β; IFN-γ induced protein [IP]-10 and IFN-γ; TNF-α) are upregulated in the spleen, liver, and blood, indicating potentially uncontrolled immune responses.
Figure 3.
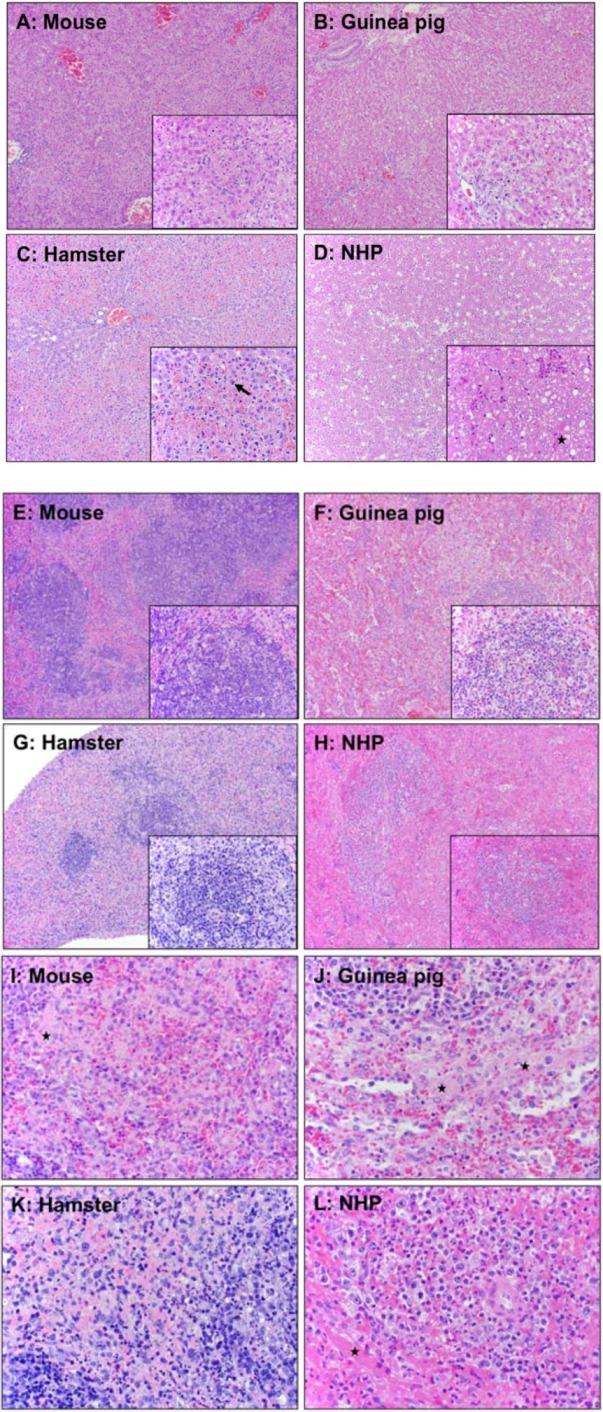
Comparison of pathology in mouse, guinea pig, hamster, and nonhuman primate. A Balb/c mouse and a Syrian hamster were infected IP with MA-EBOV; a Hartley strain of guinea pig was infected with GPA-EBOV; and a macaque was infected with wild-type EBOV. (A-D) Pathological changes in liver of different animal models. (A) Mouse: Multifocal, random hepatocellular degeneration and necrosis (10x and 40x inset). (B) Guinea pig: Diffuse, random hepatocellular degeneration and necrosis. Inflammatory cells are nearly absent (10x and 40x inset). (C–D) Hamsters: Liver. (C) Diffuse, midzonal hepatocellular degeneration, necrosis, and congestion. Inflammatory cells are nearly absent (10x). Solid arrow: prominent intracytoplasmic filovirus inclusion bodies in hepatocytes (40x). (D) Diffuse, random hepatocellular degeneration and necrosis (10x). Solid star: fibrin deposition (40x inset). (E-L) Pathological changes in spleen of different animal models. (E and I) Mouse: White and red pulp. White pulp (E); diffuse lymphoid necrosis and loss (10x and 40x inset). Red pulp (I); mild to moderate acute splenitis and small amounts of fibrin (solid star) (40x). (F and J) Guinea pig: White and red pulp. White pulp (F); multifocal lymphoid necrosis (10x and 40x inset). Red pulp (J); multifocal, mild to moderate acute splenitis with necrosis. Solid star: small amounts of fibrin at marginal zone (40x). (G and K) Hamster: White and red pulp. White pulp (G); diffuse lymphoid necrosis (10x and 40x inset). Red pulp (K); mild to moderate acute splenitis with monocytic degeneration and necrosis (40x). (H and L) NHP: White and red pulp. White pulp (H); diffuse lymphoid necrosis (10x and 20x inset). Red pulp (L); diffuse, moderate acute splenitis (40x). Solid star: fibrin.
7. Reagent and Assay Development
Until recently, lack of available reagents and specific assays to monitor host responses in hamsters (including early innate immune responses) limited investigators to studies on disease progression, humoral immune responses, and pathology. The lack of a complete genome sequence of the Syrian hamster has retarded the development of molecular, genetic, and antibody-based assays. In lieu of a complete genome, a number of studies evaluated the cross reactivity of antibody-based (ELISA, Luminex®) assays developed for other rodents against Syrian hamster cytokines, chemokines, adherins, growth factors, and other serum factors (Table 7) [76,80,94]. Data from most of these studies indicate limited cross reactivity of Syrian hamster proteins to other rodent antibodies. However, monoclonal antibodies from rats, mice, goats, and rabbits specific for hamsters successfully identified hamster surface markers of immune cells (T cells, B cells, dendritic cells, macrophages) via flow cytometry [74,95,96,97]. Microarray proteome expression studies have quantified hamster responses to disease through cross species hybridization of Syrian hamster RNA to cDNA from other species (e.g., rat, mouse, human) [98,99,100,101,102]. The complete transcriptome of the Syrian hamster has been determined but is not yet publicly available, and a microarray chip is currently under development. As hamster-specific antibodies have not been made against cytokines/chemokines, gene expression of these factors during infection is tracked through quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) [103,104,105,106,107,108,109]. Recently, use of qRT-PCR has been extended to include 51 registered hamster gene sequences targeting apoptosis, cell junction integrity, cell proliferation, and coagulation in addition to immunological responses [94]. qRT-PCR assays were utilized to profile host responses in hamsters infected with yellow fever virus, Andes virus, and EBOV [27,85,94,109]. Use of qRT-PCR assays will also contribute to identification of host response factors needed for survival in animals treated with antiviral drugs and of protective immune response in vaccinated animals. Such assays will be used until the full genome sequence is available for the development of large scale microarrays.
Table 7.
Cross-reactive or hamster-specific reagents.
| Cross-reactive antibodies | |
| Mouse anti-mouse/rat MHC II [74] | Rat anti-mouse CD4 [74,96,97] |
| Mouse anti-mouse Thy1.2 [74] | |
| Mouse anti-rat CD8, CD8β [74,97] | |
| Mouse, rabbit, and goat anti-hamster antibodies | |
| Mouse dendritic cell marker [95] | Mouse CD18 [95] |
| Mouse pan lymphocyte [95] | Mouse MHC II [95] |
| Mouse T cell [95] | Rabbit IgG [95] |
| Mouse B cell [95] | Rabbit IgM [74] |
| Mouse CD45 [95] | Goat IgG [74,78,96,97] |
| Cross-reactive cytokine, chemokine, and serum factor assays | |
| Rat GM-CSF [94] | Mouse / Rat VCAM-1 [76,80] |
| Rat Leptin [94] | Mouse / Rat vWF [80] |
| Rat GRO/KC [94] | Mouse / Rat VEGF [80] |
| Rat / Mouse IL-1α [80,94] | Mouse / Rat MDC [80] |
| Mouse MIG [94] | Mouse / Rat SCF [80] |
| Mouse IL-13 [94] | Mouse GCP-2 [80] |
| Mouse / Rat IP-10 [76,80] | Mouse MCP-3 [80] |
| Mouse /Rat M-CSF [76,80] | Mouse MIP-2 [80] |
| Mouse /Rat MCP-1 [76,80] | Mouse MIP-3β [80] |
| Mouse Fibrinogen [80] | Mouse AST [80] |
| Cross-reactive microarray hybridization | |
| DNA Microarrays | MicroRNA Microarrays |
|
|
| Mouse genes [100,101] | |
8. Future Perspectives
Although the predictive value of the hamster model for efficacy testing of vaccines and therapeutics remains to be shown, numerous research tools are now available that will facilitate the use of this animal model in future research on Ebola virus pathogenesis. The newly developed hamster EVD model will certainly augment and perhaps may one day replace mice and guinea models as an alternative model for pathogenesis studies and efficacy testing. Hamsters infected with MA-EBOV currently exhibit EVD manifestations most similar to primates, particularly with respect to coagulation abnormalities. Of particular interest is employment of this hamster model to confirm the efficacy of drugs used in NHPs to control coagulopathy (e.g., recombinant activated protein C, recombinant nematode anticoagulant protein c2).
Acknowledgments
The authors are grateful to Deborah A. Baer for assistance with formatting and Donna Perry for helpful discussions. VWJ performed this work as an employee of Tunnell Consulting, Inc., a subcontractor to Battelle Memorial Institute under its prime contract with NIAID, under Contract No. HHSN272200200016I. FdKM and LB are contractors to Battelle Memorial Institute under Contract No. HHSN272200200016I.
Supplementary Files
Conflict of Interest
The authors declare no conflict of interest.
References
- 1.Schnittler H.J., Feldmann H. Viral hemorrhagic fever--a vascular disease? Thromb. Haemostasis. 2003;89:967–972. [PubMed] [Google Scholar]
- 2.Zaki S.R., Goldsmith C.S. Pathologic features of filovirus infections in humans. Curr. Top. Microbiol. Immunol. 1999;235:97–116. doi: 10.1007/978-3-642-59949-1_7. [DOI] [PubMed] [Google Scholar]
- 3.Bwaka M.A., Bonnet M.J., Calain P., Colebunders R., De Roo A., Guimard Y., Katwiki K.R., Kibadi K., Kipasa M.A., Kuvula K.J., et al. Ebola hemorrhagic fever in Kikwit, Democratic Republic of the Congo: clinical observations in 103 patients. J. Infect. Dis. 1999;179:S1–S7. doi: 10.1086/314568. [DOI] [PubMed] [Google Scholar]
- 4.Formenty P., Hatz C., Le Guenno B., Stoll A., Rogenmoser P., Widmer A. Human infection due to Ebola virus, subtype Cote d'Ivoire: Clinical and biologic presentation. J. Infect. Dis. 1999;179:S48–S53. doi: 10.1086/514285. [DOI] [PubMed] [Google Scholar]
- 5.Kuhn J.H. Filoviruses. A compendium of 40 years of epidemiological, clinical, and laboratory studies. Arch. Virol. Suppl. 2008;20:13–360. [PubMed] [Google Scholar]
- 6.Smith D.H., Francis D.P., Simpson D.I. African Haemorrhagic Fever in the Southern Sudan, 1976: The Clinical Manifestations. In: Pattyn S.R., editor. Ebola Virus Haemorrhagic Fever—Proceedings of an International Colloquium on Ebola Virus Infection and Other Haemorrhagic Fevers; Antwerp, Belgium. December 6–8, 1977; Amsterdam, Netherlands: Elsevier North Holland Biomedical Press; 1978. [(accessed on 14 June 2012)]. pp. 28–31. Available online: http://www.itg.be/internet/ebola/default.htm. [Google Scholar]
- 7.Sureau P.H. Firsthand clinical observations of hemorrhagic manifestations in Ebola hemorrhagic fever in Zaire. Rev. Infect. Dis. 1989;11:S790–S793. doi: 10.1093/clinids/11.Supplement_4.S790. [DOI] [PubMed] [Google Scholar]
- 8.World Health Organization. Ebola haemorrhagic fever in Sudan, 1976. [(accessed on 19 June 2012)];Bull. World Health Organ. 1978 52:247–250. Available online: http://whqlibdoc.who.int/bulletin/1978/Vol56-No2/bulletin_1978_56%282%29_247–270.pdf. [Google Scholar]
- 9.Rollin P.E., Bausch D.G., Sanchez A. Blood chemistry measurements and D-Dimer levels associated with fatal and nonfatal outcomes in humans infected with Sudan Ebola virus. J. Infect. Dis. 2007;196:S364–S371. doi: 10.1086/520613. [DOI] [PubMed] [Google Scholar]
- 10.Richards G.A., Murphy S., Jobson R., Mer M., Zinman C., Taylor R., Swanepoel R., Duse A., Sharp G., De La Rey I.C., et al. Unexpected Ebola virus in a tertiary setting: clinical and epidemiologic aspects. Crit. Care Med. 2000;28:240–244. doi: 10.1097/00003246-200001000-00041. [DOI] [PubMed] [Google Scholar]
- 11.Dietrich M., Schumacher H.H., Peters D., Knobloch J. Human pathology of Ebola (MARIDI) virus Infection in the Sudan. In: Pattyn S.R., editor. Ebola Virus Haemorrhagic Fever—Proceedings of an International Colloquium on Ebola Virus Infection and Other Haemorrhagic Fevers; Antwerp, Belgium. December 6–8, 1977; Amsterdam, Netherlands: Elsevier North Holland Biomedical Press; 1978. [(accessed on 14 June 2012)]. pp. 37–41. Available online: http://www.itg.be/internet/ebola/default.htm. [Google Scholar]
- 12.Sanchez A., Lukwiya M., Bausch D., Mahanty S., Sanchez A.J., Wagoner K.D., Rollin P.E. Analysis of human peripheral blood samples from fatal and nonfatal cases of Ebola (Sudan) hemorrhagic fever: Cellular responses, virus load, and nitric oxide levels. J. Virol. 2004;78:10370–10377. doi: 10.1128/JVI.78.19.10370-10377.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kortepeter M.G., Bausch D.G., Bray M. Basic clinical and laboratory features of filoviral hemorrhagic fever. J. Infect. Dis. 2011;204:S810–S816. doi: 10.1093/infdis/jir299. [DOI] [PubMed] [Google Scholar]
- 14.Murphy F.A. Pathology of Ebola virus infection. In: Pattyn S.R., editor. Ebola Virus Haemorrhagic Fever—Proceedings of an International Colloquium on Ebola Virus Infection and Other Haemorrhagic Fevers; Antwerp, Belgium. December 6–8, 1977; Amsterdam, Netherlands: Elsevier North Holland Biomedical Press; 1978. [(accessed on 14 June 2012)]. pp. 41–53. Available online: http://www.itg.be/internet/ebola/default.htm. [Google Scholar]
- 15.Baize S., Leroy E.M., Georges A.J., Georges-Courbot M.C., Capron M., Bedjabaga I., Lansoud-Soukate J., Mavoungou E. Inflammatory responses in Ebola virus-infected patients. Clin. Exp. Immunol. 2002;128:163–168. doi: 10.1046/j.1365-2249.2002.01800.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wauquier N., Becquart P., Padilla C., Baize S., Leroy E.M. Human fatal zaire ebola virus infection is associated with an aberrant innate immunity and with massive lymphocyte apoptosis. PLoS Neglected Tropical Diseases. 2010;4:e837. doi: 10.1371/journal.pntd.0000837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Villinger F., Rollin P.E., Brar S.S., Chikkala N.F., Winter J., Sundstrom J.B., Zaki S.R., Swanepoel R., Ansari A.A., Peters C.J. Markedly elevated levels of interferon (IFN)-gamma, IFN-alpha, interleukin (IL)-2, IL-10, and tumor necrosis factor-alpha associated with fatal Ebola virus infection. J. Infect. Dis. 1999;179:S188–S191. doi: 10.1086/514283. [DOI] [PubMed] [Google Scholar]
- 18.Hutchinson K.L., Rollin P.E. Cytokine and chemokine expression in humans infected with Sudan Ebola virus. J. Infect. Dis. 2007;196:S357–S363. doi: 10.1086/520611. [DOI] [PubMed] [Google Scholar]
- 19.Food and Drug Administration. New Drug and Biological Drug Products; Evidence Needed to Demonstrate Effectiveness of New Drugs when Human Efficacy Studies are not Ethical or Feasible. Federal Register. 2002;67:37988–37998. [PubMed] [Google Scholar]
- 20.Bray M. The role of the Type I interferon response in the resistance of mice to filovirus infection. J. Gen. Virol. 2001;82:1365–1373. doi: 10.1099/0022-1317-82-6-1365. [DOI] [PubMed] [Google Scholar]
- 21.Connolly B.M., Steele K.E., Davis K.J., Geisbert T.W., Kell W.M., Jaax N.K., Jahrling P.B. Pathogenesis of experimental Ebola virus infection in guinea pigs. J. Infect. Dis. 1999;179:S203–S217. doi: 10.1086/514305. [DOI] [PubMed] [Google Scholar]
- 22.Bray M., Davis K., Geisbert T., Schmaljohn C., Huggins J. A mouse model for evaluation of prophylaxis and therapy of Ebola hemorrhagic fever. J. Infect. Dis. 1998;178:651–661. doi: 10.1086/515386. [DOI] [PubMed] [Google Scholar]
- 23.Cilloniz C., Ebihara H., Ni C., Neumann G., Korth M.J., Kelly S.M., Kawaoka Y., Feldmann H., Katze M.G. Functional genomics reveals the induction of inflammatory response and metalloproteinase gene expression during lethal Ebola virus infection. J. Virol. 2011;85:9060–9068. doi: 10.1128/JVI.00659-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Geisbert T.W., Pushko P., Anderson K., Smith J., Davis K.J., Jahrling P.B. Evaluation in nonhuman primates of vaccines against Ebola virus. Emerg. Infect. Dis. 2002;8:503–507. doi: 10.3201/eid0805.010284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reed D.S., Mohamadzadeh M. Status and challenges of filovirus vaccines. Vaccine. 2007;25:1923–1934. doi: 10.1016/j.vaccine.2006.11.037. [DOI] [PubMed] [Google Scholar]
- 26.Ebihara H., Rockx B., Marzi A., Feldmann F., Haddock E., Brining D., LaCasse R.A., Gardner D., Feldmann H. Host response dynamics following lethal infection of rhesus macaques with Zaire ebolavirus. J. Infect. Dis. 2011;204:S991–S999. doi: 10.1093/infdis/jir336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ebihara H., Zivcec M., Gardner D., Falzarano D., Lacasse R., Rosenke R., Long D., Haddock E., Fischer E., Kawaoka Y., et al. A Syrian golden hamster model recapitulating Ebola hemorrhagic fever. J. Infect. Dis. 2012 doi: 10.1093/infdis/jis626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Michelow I.C., Lear C., Scully C., Prugar L.I., Longley C.B., Yantosca L.M., Ji X., Karpel M., Brudner M., Takahashi K., et al. High-dose mannose-binding lectin therapy for Ebola virus infection. J. Infect. Dis. 2011;203:175–179. doi: 10.1093/infdis/jiq025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bukreyev A., Marzi A., Feldmann F., Zhang L., Yang L., Ward J.M., Dorward D.W., Pickles R.J., Murphy B.R., Feldmann H., et al. Chimeric human parainfluenza virus bearing the Ebola virus glycoprotein as the sole surface protein is immunogenic and highly protective against Ebola virus challenge. Virology. 2009;383:348–361. doi: 10.1016/j.virol.2008.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bukreyev A., Rollin P.E., Tate M.K., Yang L., Zaki S.R., Shieh W.J., Murphy B.R., Collins P.L., Sanchez A. Successful topical respiratory tract immunization of primates against Ebola virus. J. Virol. 2007;81:6379–6388. doi: 10.1128/JVI.00105-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bukreyev A., Yang L., Zaki S.R., Shieh W.J., Rollin P.E., Murphy B.R., Collins P.L., Sanchez A. A single intranasal inoculation with a paramyxovirus-vectored vaccine protects guinea pigs against a lethal-dose Ebola virus challenge. J. Virol. 2006;80:2267–2279. doi: 10.1128/JVI.80.5.2267-2279.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marzi A., Ebihara H., Callison J., Groseth A., Williams K.J., Geisbert T.W., Feldmann H. Vesicular stomatitis virus-based Ebola vaccines with improved cross-protective efficacy. J. Infect. Dis. 2011;204:S1066–S10674. doi: 10.1093/infdis/jir348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garbutt M., Liebscher R., Wahl-Jensen V., Jones S., Moller P., Wagner R., Volchkov V., Klenk H.D., Feldmann H., Stroher U. Properties of replication-competent vesicular stomatitis virus vectors expressing glycoproteins of filoviruses and arenaviruses. J. Virol. 2004;78:5458–5465. doi: 10.1128/JVI.78.10.5458-5465.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Geisbert T.W., Daddario-Dicaprio K.M., Lewis M.G., Geisbert J.B., Grolla A., Leung A., Paragas J., Matthias L., Smith M.A., Jones S.M., et al. Vesicular stomatitis virus-based ebola vaccine is well-tolerated and protects immunocompromised nonhuman primates. PLoS Pathogens. 2008;4:e1000225. doi: 10.1371/journal.ppat.1000225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jones S.M., Stroher U., Fernando L., Qiu X., Alimonti J., Melito P., Bray M., Klenk H.D., Feldmann H. Assessment of a vesicular stomatitis virus-based vaccine by use of the mouse model of Ebola virus hemorrhagic fever. J. Infect. Dis. 2007;196:S404–S412. doi: 10.1086/520591. [DOI] [PubMed] [Google Scholar]
- 36.Qiu X., Fernando L., Alimonti J.B., Melito P.L., Feldmann F., Dick D., Stroher U., Feldmann H., Jones S.M. Mucosal immunization of cynomolgus macaques with the VSVDeltaG/ZEBOVGP vaccine stimulates strong ebola GP-specific immune responses. PloS One. 2009;4:e5547. doi: 10.1371/journal.pone.0005547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Geisbert T.W., Daddario-Dicaprio K.M., Geisbert J.B., Reed D.S., Feldmann F., Grolla A., Stroher U., Fritz E.A., Hensley L.E., Jones S.M., et al. Vesicular stomatitis virus-based vaccines protect nonhuman primates against aerosol challenge with Ebola and Marburg viruses. Vaccine. 2008;26:6894–6900. doi: 10.1016/j.vaccine.2008.09.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jones S.M., Feldmann H., Stroher U., Geisbert J.B., Fernando L., Grolla A., Klenk H.D., Sullivan N.J., Volchkov V.E., Fritz E.A., et al. Live attenuated recombinant vaccine protects nonhuman primates against Ebola and Marburg viruses. Nat. Med. 2005;11:786–790. doi: 10.1038/nm1258. [DOI] [PubMed] [Google Scholar]
- 39.Gilligan K., Geisbert J., Jahrling P., Anderson K. Assessment of Protective Immunity Conferred by Recombinant Vaccinia Viruses to Guinea Pigs Challenged with Ebola Virus. In: Brown F., Burton D., Doherty P., Mekalanos J., Norrby E., editors. Vaccines 97: Molecular Approaches to the Control of Infectious Diseases. Cold Spring Harbor Laboratory Press; Plainview, NY, USA: 1997. pp. 87–92. [Google Scholar]
- 40.Wilson J.A., Hart M.K. Protection from Ebola virus mediated by cytotoxic T lymphocytes specific for the viral nucleoprotein. J. Virol. 2001;75:2660–2664. doi: 10.1128/JVI.75.6.2660-2664.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pushko P., Bray M., Ludwig G.V., Parker M., Schmaljohn A., Sanchez A., Jahrling P.B., Smith J.F. Recombinant RNA replicons derived from attenuated Venezuelan equine encephalitis virus protect guinea pigs and mice from Ebola hemorrhagic fever virus. Vaccine. 2000;19:142–153. doi: 10.1016/S0264-410X(00)00113-4. [DOI] [PubMed] [Google Scholar]
- 42.Olinger G.G., Bailey M.A., Dye J.M., Bakken R., Kuehne A., Kondig J., Wilson J., Hogan R.J., Hart M.K. Protective cytotoxic T-cell responses induced by venezuelan equine encephalitis virus replicons expressing Ebola virus proteins. J. Virol. 2005;79:14189–14196. doi: 10.1128/JVI.79.22.14189-14196.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Phoolcharoen W., Dye J.M., Kilbourne J., Piensook K., Pratt W.D., Arntzen C.J., Chen Q., Mason H.S., Herbst-Kralovetz M.M. A Nonreplicating Subunit Vaccine Protects Mice Against Lethal Ebola Virus Challenge; Proceedings of the National Academy of Sciences of the United States of America; 2011. pp. 20695–20700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pushko P., Geisbert J., Parker M., Jahrling P., Smith J. Individual and bivalent vaccines based on alphavirus replicons protect guinea pigs against infection with Lassa and Ebola viruses. J. Virol. 2001;75:11677–11685. doi: 10.1128/JVI.75.23.11677-11685.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wilson J.A., Bray M., Bakken R., Hart M.K. Vaccine potential of Ebola virus VP24, VP30, VP35, and VP40 proteins. Virology. 2001;286:384–390. doi: 10.1006/viro.2001.1012. [DOI] [PubMed] [Google Scholar]
- 46.Gupta M., Mahanty S., Bray M., Ahmed R., Rollin P.E. Passive transfer of antibodies protects immunocompetent and imunodeficient mice against lethal Ebola virus infection without complete inhibition of viral replication. J. Virol. 2001;75:4649–4654. doi: 10.1128/JVI.75.10.4649-4654.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gupta M., Mahanty S., Greer P., Towner J.S., Shieh W.J., Zaki S.R., Ahmed R., Rollin P.E. Persistent infection with ebola virus under conditions of partial immunity. J. Virol. 2004;78:958–967. doi: 10.1128/JVI.78.2.958-967.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Warfield K.L., Bosio C.M., Welcher B.C., Deal E.M., Mohamadzadeh M., Schmaljohn A., Aman M.J., Bavari S. Ebola virus-like particles protect from lethal Ebola virus infection; Proceedings of the National Academy of Sciences of the United States of America; 2003. pp. 15889–15894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rao M., Bray M., Alving C.R., Jahrling P., Matyas G.R. Induction of immune responses in mice and monkeys to Ebola virus after immunization with liposome-encapsulated irradiated Ebola virus: protection in mice requires CD4(+) T cells. J. Virol. 2002;76:9176–9185. doi: 10.1128/JVI.76.18.9176-9185.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Warfield K.L., Swenson D.L., Olinger G.G., Kalina W.V., Viard M., Aitichou M., Chi X., Ibrahim S., Blumenthal R., Raviv Y., et al. Ebola virus inactivation with preservation of antigenic and structural integrity by a photoinducible alkylating agent. J. Infect. Dis. 2007;196:S276–S283. doi: 10.1086/520605. [DOI] [PubMed] [Google Scholar]
- 51.Feldmann H., Jones S.M., Daddario-DiCaprio K.M., Geisbert J.B., Stroher U., Grolla A., Bray M., Fritz E.A., Fernando L., Feldmann F., et al. Effective post-exposure treatment of Ebola infection. PLoS Pathogens. 2007;3:e2. doi: 10.1371/journal.ppat.0030002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jahrling P.B., Geisbert J.B., Swearengen J.R., Larsen T., Geisbert T.W. Ebola hemorrhagic fever: Evaluation of passive immunotherapy in nonhuman primates. J. Infect. Dis. 2007;196:S400–S403. doi: 10.1086/520587. [DOI] [PubMed] [Google Scholar]
- 53.Jahrling P.B., Geisbert J., Swearengen J.R., Jaax G.P., Lewis T., Huggins J.W., Schmidt J.J., LeDuc J.W., Peters C.J. Passive immunization of Ebola virus-infected cynomolgus monkeys with immunoglobulin from hyperimmune horses. Arch. Virol. Suppl. 1996;11:135–140. doi: 10.1007/978-3-7091-7482-1_12. [DOI] [PubMed] [Google Scholar]
- 54.Jahrling P.B., Geisbert T.W., Geisbert J.B., Swearengen J.R., Bray M., Jaax N.K., Huggins J.W., LeDuc J.W., Peters C.J. Evaluation of immune globulin and recombinant interferon-alpha2b for treatment of experimental Ebola virus infections. J. Infect. Dis. 1999;179:S224–S234. doi: 10.1086/514310. [DOI] [PubMed] [Google Scholar]
- 55.Parren P.W., Geisbert T.W., Maruyama T., Jahrling P.B., Burton D.R. Pre- and postexposure prophylaxis of Ebola virus infection in an animal model by passive transfer of a neutralizing human antibody. J. Virol. 2002;76:6408–6412. doi: 10.1128/JVI.76.12.6408-6412.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oswald W.B., Geisbert T.W., Davis K.J., Geisbert J.B., Sullivan N.J., Jahrling P.B., Parren P.W., Burton D.R. Neutralizing antibody fails to impact the course of Ebola virus infection in monkeys. PLoS Pathogens. 2007;3:e9. doi: 10.1371/journal.ppat.0030009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Enterlein S., Warfield K.L., Swenson D.L., Stein D.A., Smith J.L., Gamble C.S., Kroeker A.D., Iversen P.L., Bavari S., Muhlberger E. VP35 knockdown inhibits Ebola virus amplification and protects against lethal infection in mice. Antimicrob. Agents Chemother. 2006;50:984–993. doi: 10.1128/AAC.50.3.984-993.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Warfield K.L., Swenson D.L., Olinger G.G., Nichols D.K., Pratt W.D., Blouch R., Stein D.A., Aman M.J., Iversen P.L., Bavari S. Gene-specific countermeasures against Ebola virus based on antisense phosphorodiamidate morpholino oligomers. PLoS Pathogens. 2006;2:e1. doi: 10.1371/journal.ppat.0020001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Warren T.K., Warfield K.L., Wells J., Swenson D.L., Donner K.S., Van Tongeren S.A., Garza N.L., Dong L., Mourich D.V., Crumley S., et al. Advanced antisense therapies for postexposure protection against lethal filovirus infections. Nat. Med. 2010;16:991–994. doi: 10.1038/nm.2202. [DOI] [PubMed] [Google Scholar]
- 60.Swenson D.L., Warfield K.L., Warren T.K., Lovejoy C., Hassinger J.N., Ruthel G., Blouch R.E., Moulton H.M., Weller D.D., Iversen P.L., Bavari S. Chemical modifications of antisense morpholino oligomers enhance their efficacy against Ebola virus infection. Antimicrob. Agents Chemothe.r. 2009;53:2089–2099. doi: 10.1128/AAC.00936-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Adler S. Origin of the golden hamster Cricetus auratus as a laboratory animal. Nature. 1948;162:256. doi: 10.1038/162256b0. [DOI] [PubMed] [Google Scholar]
- 62.Boschert K. Hamsters: Biology, care, diseases & models. [(accessed on 26 June 2012)]. Available online: http://netvet.wustl.edu/species/hamsters/hamstbio.txt.
- 63.Murphy M.R. History of the Capture and Domestication of the Syrian Golden Hamster (Mesocricetus auratus Waterhouse) In: Siegel H.I., editor. The Hamster: Reproduction and Behavior. Plenum Press; New York, NY, USA: 1985. pp. 3–20. [Google Scholar]
- 64.Smith G.D. Taxonomy and History. In: Suckow M.A.; Stevens, K.A.; Wilson R.P., editors. The Laboratory Rabbit, guinea pig, hamster, and other rodents. Acedemic Press; Waltham MA: 2012. [Google Scholar]
- 65.Simonsen Laboratories. Origin and Strain Designation: Syrian Hamsters. [(accessed on 20 March 2012)]. Available online: http://www.simlab.com/products.html.
- 66.Charles River Laboratories International. Charles River History. [(accessed on 20 March 2012)]. Available online: http://www.criver.com/en-us/aboutus/history/Pages/home.aspx.
- 67.Charles River Laboratories International. LVG golden syrian hamsters 2012 US pricing. [(accessed on 20 March 2012)]. Available online: http://www.criver.com/sitecollectiondocuments/rm_rm_c_lvg_golden_syrian_hamsters.pdf.
- 68.Harlan Laboratories. Syrian Hamster. 3248 Harlan Research Models and Services (RMS) Datasheets v4. [(accessed on 20 March 2012)]. Available online: http://www.harlan.com/products_and_services/research_models_and_services/research_models.
- 69.Kunstyr I., Schoeneberg U., Friedhoff K.T. Host specificity of Giardia muris isolates from mouse and golden hamster. Parasitol. Res. 1992;78:621–622. doi: 10.1007/BF00936463. [DOI] [PubMed] [Google Scholar]
- 70.Bio Breeders. Hamster models and applications for human diseases. [(accessed on 26 June 2012)]. Available online: http://www.biobreeders.com/hamstermodels.html.
- 71.U.S. Department of Agriculture. Animals Used in Research: Animal Care Annual Report of Activities, Fiscal Year 2010. [(accessed 30 June 2012)]. Available online: http://www.aphis.usda.gov/animal_welfare/efoia/downloads/2010_Animals_Used_In_Research.pdf.
- 72.Hankenson F.C., Van Hoosier G.L. Biology and Diseases of Hamsters. In: Fox J.G., Anderson L.C., Loew F.L., Quimby F.W., editors. Laboratory Animal Medicine. Academic Press; San Diego, CA, USA: 2002. pp. 167–202. [Google Scholar]
- 73.Zlotnik I. "Marburg disease". The Pathology of Experimentally Infected Hamsters. In: Martini G.A., Siegert R., editors. Marburg Virus Disease. Springer-Verlag; Berlin, Germany: 1971. pp. 129–135. [Google Scholar]
- 74.Hammerbeck C.D., Hooper J.W. T cells are not required for pathogenesis in the Syrian hamster model of hantavirus pulmonary syndrome. J. Virol. 2011;85:9929–9944. doi: 10.1128/JVI.05356-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wahl-Jensen V., Chapman J., Asher L., Fisher R., Zimmerman M., Larsen T., Hooper J.W. Temporal analysis of Andes virus and Sin Nombre virus infections of Syrian hamsters. J. Virol. 2007;81:7449–7462. doi: 10.1128/JVI.00238-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Safronetz D., Zivcec M., Lacasse R., Feldmann F., Rosenke R., Long D., Haddock E., Brining D., Gardner D., Feldmann H., et al. Pathogenesis and host response in Syrian hamsters following intranasal infection with Andes virus. PLoS Pathogens. 2011;7:e1002426. doi: 10.1371/journal.ppat.1002426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Campen M.J., Milazzo M.L., Fulhorst C.F., Obot Akata C.J., Koster F. Characterization of shock in a hamster model of hantavirus infection. Virology. 2006;356:45–49. doi: 10.1016/j.virol.2006.07.044. [DOI] [PubMed] [Google Scholar]
- 78.Milazzo M.L., Eyzaguirre E.J., Molina C.P., Fulhorst C.F. Maporal viral infection in the Syrian golden hamster: A model of hantavirus pulmonary syndrome. J. Infect. Dis. 2002;186:1390–1395. doi: 10.1086/344735. [DOI] [PubMed] [Google Scholar]
- 79.Hooper J.W., Larsen T., Custer D.M., Schmaljohn C.S. A lethal disease model for hantavirus pulmonary syndrome. Virology. 2001;289:6–14. doi: 10.1006/viro.2001.1133. [DOI] [PubMed] [Google Scholar]
- 80.Gowen B.B., Julander J.G., London N.R., Wong M.H., Larson D., Morrey J.D., Li D.Y., Bray M. Assessing changes in vascular permeability in a hamster model of viral hemorrhagic fever. Virol. J. 2010;7:240. doi: 10.1186/1743-422X-7-240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sbrana E., Xiao S.Y., Popov V.L., Newman P.C., Tesh R.B. Experimental yellow fever virus infection in the golden hamster (Mesocricetus auratus) III. Clinical laboratory values. Am. J. Trop. Med. Hyg. 2006;74:1084–1089. [PubMed] [Google Scholar]
- 82.Xiao S.Y., Zhang H., Guzman H., Tesh R.B. Experimental yellow fever virus infection in the Golden hamster (Mesocricetus auratus). II. Pathology. J. Infect. Dis. 2001;183:1437–1444. doi: 10.1086/320200. [DOI] [PubMed] [Google Scholar]
- 83.Fisher A.F., Tesh R.B., Tonry J., Guzman H., Liu D., Xiao S.Y. Induction of severe disease in hamsters by two sandfly fever group viruses, Punta toro and Gabek Forest (Phlebovirus, Bunyaviridae), similar to that caused by Rift Valley fever virus. Am. J. Trop. Med. Hyg. 2003;69:269–276. [PubMed] [Google Scholar]
- 84.Vela E.M., Knostman K.A., Mott J.M., Warren R.L., Garver J.N., Vela L.J., Stammen R.L. Genistein, a general kinase inhibitor, as a potential antiviral for arenaviral hemorrhagic fever as described in the Pirital virus-Syrian golden hamster model. Antivir. Res. 2010;87:318–328. doi: 10.1016/j.antiviral.2010.06.007. [DOI] [PubMed] [Google Scholar]
- 85.Li G., Duan T., Wu X., Tesh R.B., Soong L., Xiao S.Y. Yellow fever virus infection in Syrian golden hamsters: Relationship between cytokine expression and pathologic changes. Int. J. Clin. Exp. Pathol. 2008;1:169–179. [PMC free article] [PubMed] [Google Scholar]
- 86.Sbrana E., Mateo R.I., Xiao S.Y., Popov V.L., Newman P.C., Tesh R.B. Clinical laboratory, virologic, and pathologic changes in hamsters experimentally infected with Pirital virus (Arenaviridae): A rodent model of Lassa fever. Am. J. Trop. Med. Hyg. 2006;74:1096–1102. [PubMed] [Google Scholar]
- 87.Xiao S.Y., Zhang H., Yang Y., Tesh R.B. Pirital virus (Arenaviridae) infection in the syrian golden hamster, Mesocricetus auratus: a new animal model for arenaviral hemorrhagic fever. Am. J. Trop. Med. Hyg. 2001;64:111–118. doi: 10.4269/ajtmh.2001.64.111. [DOI] [PubMed] [Google Scholar]
- 88.Lima E.Q., Nogueira M.L. Viral hemorrhagic fever-induced acute kidney injury. Semin. Nephrol. 2008;28:409–415. doi: 10.1016/j.semnephrol.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 89.Geisbert T.W., Young H.A., Jahrling P.B., Davis K.J., Larsen T., Kagan E., Hensley L.E. Pathogenesis of Ebola hemorrhagic fever in primate models: Evidence that hemorrhage is not a direct effect of virus-induced cytolysis of endothelial cells. Am. J. Pathol. 2003;163:2371–2382. doi: 10.1016/S0002-9440(10)63592-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Geisbert T.W., Young H.A., Jahrling P.B., Davis K.J., Kagan E., Hensley L.E. Mechanisms underlying coagulation abnormalities in ebola hemorrhagic fever: Overexpression of tissue factor in primate monocytes/macrophages is a key event. J. Infect. Dis. 2003;188:1618–1629. doi: 10.1086/379724. [DOI] [PubMed] [Google Scholar]
- 91.Ryabchikova E.I., Kolesnikova L.V., Netesov S.V. Animal pathology of filoviral infections. Curr. Top Microbiol. Immunol. 1999;235:145–173. doi: 10.1007/978-3-642-59949-1_9. [DOI] [PubMed] [Google Scholar]
- 92.Feldmann H., Geisbert T.W. Ebola haemorrhagic fever. Lancet. 2011;377:849–862. doi: 10.1016/S0140-6736(10)60667-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Aleksandrowicz P., Wolf K., Falzarano D., Feldmann H., Seebach J., Schnittler H. Viral haemorrhagic fever and vascular alterations. Hamostaseologie. 2008;28:77–84. [PubMed] [Google Scholar]
- 94.Zivcec M., Safronetz D., Haddock E., Feldmann H., Ebihara H. Validation of assays to monitor immune responses in the Syrian golden hamster (Mesocricetus auratus) J. Immunol. Meth. 2011;368:24–35. doi: 10.1016/j.jim.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.McNees A.L., Vilchez R.A., Heard T.C., Sroller V., Wong C., Herron A.J., Hamilton M.J., Davis W.C., Butel J.S. SV40 lymphomagenesis in Syrian golden hamsters. Virology. 2009;384:114–124. doi: 10.1016/j.virol.2008.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dondji B., Sun T., Bungiro R.D., Vermeire J.J., Harrison L.M., Bifulco C., Cappello M. CD4 T cells mediate mucosal and systemic immune responses to experimental hookworm infection. Parasite Immunol. 2010;32:406–413. doi: 10.1111/j.1365-3024.2010.01204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dondji B., Bungiro R.D., Harrison L.M., Vermeire J.J., Bifulco C., McMahon-Pratt D., Cappello M. Role for nitric oxide in hookworm-associated immune suppression. Infect. Immun. 2008;76:2560–2567. doi: 10.1128/IAI.00094-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yang K., Zhang G., Mei J., Chen D., Wu M. Screening and analysis of pathogenic genes during DMBA-induced buccal mucosa carcinogenesis in golden hamsters. Oncol. Rep. 2010;23:1619–1624. doi: 10.3892/or_00000803. [DOI] [PubMed] [Google Scholar]
- 99.Yu T., Wang X.Y., Gong R.G., Li A., Yang S., Cao Y.T., Wen Y.M., Wang C.M., Yi X.Z. The expression profile of microRNAs in a model of 7,12-dimethyl-benz[a]anthrance-induced oral carcinogenesis in Syrian hamster. J. Exp. Clin. Canc. Res. 2009;28:64. doi: 10.1186/1756-9966-28-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hohlweg U., Hosel M., Dorn A., Webb D., Hilger-Eversheim K., Remus R., Schmitz B., Buettner R., Schramme A., Corzilius L., et al. Intraperitoneal dissemination of Ad12-induced undifferentiated neuroectodermal hamster tumors: De novo methylation and transcription patterns of integrated viral and of cellular genes. Virus Res. 2003;98:45–56. doi: 10.1016/j.virusres.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 101.Maywood E.S., Chahad-Ehlers S., Garabette M.L., Pritchard C., Underhill P., Greenfield A., Ebling F.J., Akhtar R.A., Kyriacou C.P., Hastings M.H., et al. Differential testicular gene expression in seasonal fertility. J. Biol. Rhythm. 2009;24:114–125. doi: 10.1177/0748730409332029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bradley K.C., Boulware M.B., Jiang H., Doerge R.W., Meisel R.L., Mermelstein P.G. Changes in gene expression within the nucleus accumbens and striatum following sexual experience. Gene Brain Behav. 2005;4:31–44. doi: 10.1111/j.1601-183X.2004.00093.x. [DOI] [PubMed] [Google Scholar]
- 103.Matsui M., Rouleau V., Bruyere-Ostells L., Goarant C. Gene expression profiles of immune mediators and histopathological findings in animal models of leptospirosis: comparison between susceptible hamsters and resistant mice. Infect Immun. 2011;79:4480–4492. doi: 10.1128/IAI.05727-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gowen B.B., Fairman J., Dow S., Troyer R., Wong M.H., Jung K.H., Melby P.C., Morrey J.D. Prophylaxis with cationic liposome-DNA complexes protects hamsters from phleboviral disease: importance of liposomal delivery and CpG motifs. Antivir. Res. 2009;81:37–46. doi: 10.1016/j.antiviral.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gowen B.B., Judge J.W., Wong M.H., Jung K.H., Aylsworth C.F., Melby P.C., Rosenberg B., Morrey J.D. Immunoprophylaxis of Punta Toro virus (Phlebovirus, Bunyaviridae) infection in hamsters with recombinant Eimeria profilin-like antigen. Int. Immunopharm. 2008;8:1089–1094. doi: 10.1016/j.intimp.2008.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mendez S., Valenzuela J.G., Wu W., Hotez P.J. Host cytokine production, lymphoproliferation, and antibody responses during the course of Ancylostoma ceylanicum infection in the Golden Syrian hamster. Infect. Immun. 2005;73:3402–3407. doi: 10.1128/IAI.73.6.3402-3407.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rockx B., Brining D., Kramer J., Callison J., Ebihara H., Mansfield K., Feldmann H. Clinical outcome of henipavirus infection in hamsters is determined by the route and dose of infection. J. Virol. 2011;85:7658–7671. doi: 10.1128/JVI.00473-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rama Iniguez S., Dea-Ayuela M.A., Sanchez-Brunete J.A., Torrado J.J., Alunda J.M., Bolas-Fernandez F. Real-time reverse transcription-PCR quantification of cytokine mRNA expression in golden Syrian hamster infected with Leishmania infantum and treated with a new amphotericin B formulation. Antimicrob. Agents Chemother. 2006;50:1195–1201. doi: 10.1128/AAC.50.4.1195-1201.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Brown K.S., Safronetz D., Marzi A., Ebihara H., Feldmann H. Vesicular stomatitis virus-based vaccine protects hamsters against lethal challenge with Andes virus. J. Virol. 2011;85:12781–12791. doi: 10.1128/JVI.00794-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.