

Abstract
The emerging demand for programmable functionalization of existing base nanocarriers necessitates development of an efficient approach for cargo loading that avoids nanoparticle redesign for each individual application. Herein, we demonstrate in vivo a postformulation strategy for lipidic nanocarrier functionalization with the use of a linker peptide, which rapidly and stably integrates cargos into lipidic membranes of nanocarriers after simple mixing through a self-assembling process. We exemplified this strategy by generating a VCAM-1-targeted perfluorocarbon nanoparticle for in vivo targeting in atherosclerosis (ApoE-deficient) and breast cancer (STAT-1-deficient) models. In the atherosclerotic model, a 4.1-fold augmentation in binding to affected aortas was observed for targeted vs. nontargeted nanoparticles (P<0.0298). Likewise, in the breast cancer model, a 4.9-fold increase in the nanoparticle signal from tumor vasculature was observed for targeted vs. nontargeted nanoparticles (P<0.0216). In each case, the nanoparticle was registered with fluorine (19F) magnetic resonance spectroscopy of the nanoparticle perfluorocarbon core, yielding a quantitative estimate of the number of tissue-bound nanoparticles. Because other common nanocarriers with lipid coatings (e.g., liposomes, micelles, etc.) can employ this strategy, this peptide linker postformulation approach is applicable to more than half of the available nanosystems currently in clinical trials or clinical uses.—Pan, H., Myerson, J. W., Hu, L., Marsh, J. N., Hou K., Scott, M. J., Allen, J. S., Hu, G., San Roman, S., Lanza, G. M., Schreiber, R. D., Schlesinger, P. H., Wickline, S. A. Programmable nanoparticle functionalization for in vivo targeting.
Keywords: postformulation, molecular targeting, inflammation, atherosclerosis, cancer
Targeted nanoscale delivery systems represent a promising approach for selective molecular imaging and site-specific therapy. In current practice, cargo moieties are incorporated into the nanocarriers during the initial formulation process, which necessitates a new design and formulation process for each individual application (1–3). Hypothetically, a more economical and efficient method would use a generic base carrier, to which selected cargos could be added as needed in a postformulation mixing step, which might facilitate both the design and preclinical testing of such combination agents. Moreover, recent programmatic interest has emerged for the development of adaptable nanotechnologies that enable programmable cargo loading for diagnosis and therapy (4). Attention already has turned toward postformulation functionalization approaches that operate either through physical association or chemical conjugation (5). Prior physical association methods have been described for liposomes, but they are temperature dependent, require incubation of liposomes with micelles at 60°C for 2 h, and necessitate incorporation of DSPE-PEG to ensure liposome transfer (6, 7). Alternatively, chemical conjugation processes require the introduction of active conjugation sites to the base carriers, the use of catalytic species, and a purification step to remove reactants (5, 8).
Our goal is to develop a generalizable postformulation strategy without aforementioned prerequirements for functionalization of lipidic nanocarriers, which could find a broad clinical utilities in view of the fact that lipidic delivery systems account for more than half of such systems now in preclinical investigation or in clinic application (9). In recent work, we noted that the cytolytic peptide melittin (10), which comprises 50% of the honeybee Apis mellifera venom, spontaneously inserts and binds tightly into lipid monolayers surrounding perfluorocarbon (PFC) nanoparticles without affecting their integrity, whereas the same peptide results in rapid destruction of liposomal or cellular membranes (11, 12). We have also reported previously that melittin can be engineered to extensively attenuate its lytic activity, while still retaining its ability to stably integrate into a lipid monolayered or bilayered membrane in an α-helical secondary structure that preserves hydrophobic interactions with membrane phospholipid tails (13). Separation of melittin's lytic activity from its membrane integration property yields a “linker peptide”, VLTTGLPALISWIKRKRQQ, which is a truncated version of melittin, capable of loading cargos onto the lipid nanocarriers, with conjugation options at either the carboxyl or amino terminus of the linker peptide (13, 14). Such a linker peptide postformulation strategy takes advantages of the biophysical properties of these biomaterials to permit rapid swapping of functional moieties in lipidic vehicles through a self-assembling process without requiring reformulation of the base carrier itself (13–15). However, the stability and performance of such a system for targeting applications have not been assessed in vivo in a complex biological environment that would offer proof of concept for potential clinical applications.
Accordingly, we sought to exemplify in vivo applications of this postformulation strategy by generating vascular cell adhesion molecule 1 (VCAM-1)-targeted perfluorocarbon (PFC) nanoparticles for molecular biomarker detection in cardiovascular disease and cancer, since VCAM-1 plays a pivotal role in the progression of numerous inflammatory diseases (16–22). For in vivo VCAM-1 detection, we first confirmed the VCAM-1 targeting function of the nanoparticles with the use of the ApoE-deficient mouse model for atherosclerosis, in which VCAM-1 expression is well established (23–27). Subsequently, we applied VCAM-1-targeted PFC nanoparticles to a breast cancer mouse model featuring STAT-1-deficient tumor cells implanted into mammary fat pads, which we show herein express VCAM-1 biomarkers in the tumor vascular. Our results demonstrated this postformulation strategy could enable functionalization of preformed lipidic nanostructures for in vivo application by mimicking the lipid membrane inserting potential of native membrane active peptides.
MATERIALS AND METHODS
PFC nanoparticle formulation
PFC nanoparticles were formulated by utilizing previously described methods (15). Briefly, a lipid/surfactant comixture of 99 mol% egg lecithin and 1 mol% dipalmitoyl-phosphatidylethanolamine (DPPE; Avanti Polar Lipids, Piscataway, NJ, USA) was dissolved in methanol:chloroform (1:3, v/v). Solvent was evaporated under reduced pressure to produce a lipid film, which was dried in a 50°C vacuum oven overnight to obtain the surfactant. Then the surfactant (2.0%, w/v), perfluorocarbon: either perfluorooctyl bromide (PFOB) or perfluoro-15-crown-5-ether (CE; 20%, w/v; Gateway Specialty Chemicals, St. Peters, MO, USA), and distilled, deionized water were blended and emulsified at 20,000 psi for 4 min in an ice bath (S110 Microfluidics emulsifier; Microfluidics, Newton, MA, USA).
Peptide synthesis
The targeting combination peptide (TCP), linker peptide VCAM-1-targeting ligand, was produced by solid-phase synthesis utilizing a peptide synthesizer (model no. CS136; CS Bio Co., Menlo Park, CA, USA). A cocktail containing 88% trifluoroacetic acid (TFA), 5% phenol, 5% water, and 2% triisopropylsilane (TIPS) (v/v) was used for peptide-resin cleavage. Crude peptides were then dissolved in 0.1% acetic acid for purification by reversed phase HPLC (C18 column, flow rate 1 ml/min) in 30 to 50% gradient of acetonitrile (containing 10% water and 0.2% acetic acid)/water (containing 0.1% acetic acid) over 80 min. The identity of the peptide was validated by mass spectrometry (Washington University Proteomics Center, St. Louis, MO, USA) and the purity (>95%) was confirmed by analytical HPLC. All chemicals were obtained from Sigma (St. Louis, MO, USA). Amino acids and resins were obtained from EMD Biosciences (Philadelphia, PA, USA).
Incorporation of VCAM-1 targeting ligand onto PFC nanoparticles
VCAM-1-targeted PFC nanoparticles were generated by mixing PFC nanoparticles with a known amount of TCP, which was dissolved in MilliQ H2O (Millipore, Billerica, MA, USA) at 10 mM. Every 15 μl 10 mM TCP was mixed with 30 μl of PFC nanoparticles. After incubation at 4°C overnight, the mixture was centrifuged at 970 rpm (100 g equivalent) for 10 min to remove unincorporated peptides. The peptide in the supernatant was quantified by measuring intrinsic tryptophan fluorescence (described below) compared with a standard curve.
Size distribution and ζ potential of nanoparticles
The size distributions of the nanoparticles with or without targeting ligand incorporation were analyzed by dynamic light scattering (Brookhaven Instruments, Holtsville, NY, USA) and plotted according to intensity. The ζ-potential values were determined with a phase-analysis light scattering (PALS) ζ-potential analyzer (Brookhaven Instruments). Data were acquired in the PALS mode following solution equilibration at 25°C. All samples were diluted in Milli-Q water.
Far-UV circular dichroism (CD) spectroscopy
CD spectra measurements were acquired with a Jasco J-810 spectropolarimeter (Jasco, Eastern, MD). Spectra were scanned in a 1-mm path length quartz cuvette in the far-UV range from 190 to 260 nm at a scan rate of 100 nm/min. An average of 10 scans was collected for all spectra. Measurement buffer was 10 mM potassium phosphate (pH 7.0).
Fluorescence spectroscopy for peptide quantification
Tryptophan fluorescence emission spectra (300–500 nm) were measured after excitation at 280 nm in a fluorescent spectrofluorometer (Varian Inc, Palo Alto, CA, USA) and used for peptide loading quantification against a standard curve.
In vivo stability of VCAM-1-targeted nanoparticles
Peptides were chemically conjugated with 99mTc. To produce 99mTc-labeled peptides, 99mTc tricarbonyl complexes were prepared by adding 0.02 ml of 8.19 mCi 99mTcO4− (Cardinal Health Nuclear Pharmacy, St. Louis, MO, USA) into 0.6 ml saline, and then the mixture was transferred into an isolink kit (Mallinckrodt, St. Louis, MO, USA) and heated at 100°C for 20 min to form a Tc-99m tricarbonyl precursor. After reequilibration to atmospheric pressure, the pH of the reaction mixture was adjusted to around 10 with 120 μl of 1 M HCl, after which the mixture (0.78 ml) rested for 5 min. Then 69 μl of a 3.5-mM peptide solution was added to the 0.78-ml mixture followed by incubation in a water bath for 30 min at 50–55°C to allow transchelation. After 99mTc-labeled peptide formation, the radiolabeling efficiency was determined by thin-layer paper chromatography performed on Whatman paper and developed in 0.2 M ammonium acetate:methanol:water (20:100:200). The paper was cut into 10 pieces, each 1 cm in length, and each was counted individually with a γ counter to measure radioactivity (counts per minute), which was used to determine the radiolabel yield and which was shown to exceed 95%. For pharmacokinetic analysis, 1 ml/kg radioactive VCAM-1-targeted PFC nanoparticles generated by using 99mTc-labeled peptide or free 99mTc-labeled peptide were injected by tail vein into C57BL/6 mice, and blood samples were collected at various time points up to 2 h after injection for radioactivity analysis. To allow for radioactive decay, at 7 d after blood collection, the same samples were assessed by 19F magnetic resonance spectroscopy (MRS) at 11.7 T to quantify particle numbers based on the known linear relationship between the 19F signal and particle concentration (28, 29).
19F MRS at 11.7 T
19F MRS of tissues was performed on a Varian 11.7 T scanner by utilizing a custom-designed 0.5-cm 4-turn solenoid radio-frequency coil and standard procedures published previously (29). Tissues were placed in Eppendorf centrifuge tubes and scanned together with an internal standard comprising PFOB nanoparticles. 19F MRS [spin echo sequence; number of averages 1024 (tumors) or 2048 (aortas); flip angle 90°] was performed for quantitative evaluation of site-specific delivery of PFC nanoparticles.
Cell culture and preparation for tumor implantation
All cells were maintained in a humidified atmosphere of 95% air and 5% CO2. The murine tumor cell line was generated from STAT-1−/− female mice that spontaneously develop mammary adenocarcinomas (SSM 2 cell line; Dr. Robert D. Schreiber's laboratory, Washington University School of Medicine). SSM 2 cells were maintained in SSM 2 cell culture medium containing 10% (v/v) heart-inactivated FBS, 50/50 (v/v) Ham's F12/DMEM, 2% (v/v) l-glutamine, 1% (v/v) sodium pyruvate, 1% (v/v) Pen/Strep, and 0.1% (v/v) B-mercaptoethanol (Washington University Tissue Culture Support Center, St. Louis, MO, USA). Stock SSM 2 cells were thawed, split twice after reaching ∼80% confluency, and then harvested for implantation at 106 cells in 10 μl of phenol red-free F12/DMEM (Washington University Tissue Culture Support Center).
Animal experimental protocol
The experimental animal protocols were approved by the Animal Care Committee of the Washington University School of Medicine. For the atherosclerosis animal model, male ApoE−/− mice and age-controlled male C57BL/6 mice at age of 6 wk were obtained from Jackson Laboratory (Bar Harbor, ME, USA). ApoE−/− mice were fed with an adjusted-calories diet (42% from fat; Harlan Laboratories, Madison, WI, USA) for 8 wk before receiving an intravenous injection of 1 ml/kg VCAM-1-targeted nanoparticles (n=7) or nontargeted nanoparticles (n=4). Age-matched C57BL6 mice receiving either VCAM-1-targeted nanoparticles (n=3) or nontargeted nanoparticles (n=5) served as controls. The body weights of ApoE−/− (34±3 g) and control (38±3 g) mice were comparable. Nanoparticles were allowed to circulate for 2 h. After systemic saline perfusion, aortas were excised from the base of heart to the diaphragm in exactly the same manner for each animal and saved in saline for MRS analysis at 11.7 T. For the breast cancer model, each 129SVE-F (129S6/SvEvTac, Taconic, Hudson, NY, USA) mouse was implanted in the mammary fat pad with 106 SSM2 cells. At 30 d after the implantation, mice were randomly divided into 2 groups (n=5). One group of mice received VCAM-1-targeted CE nanoparticles, and the other received nontargeted CE nanoparticles. After 2 h of circulation, tumors were excised following systemic saline perfusion for ex vivo MRS analysis at 11.7 T.
Histological assessment
Frozen tissues were sectioned and acetone fixed before staining. Expression of VCAM-1 in mouse aortas was delineated with purified rat anti-mouse CD106 and FITC goat anti-rat Ig (cat. no. 553330 and 554016, respectively; BD Pharmingen, San Diego, CA, USA). For detecting VCAM-1 expression in SSM tumor sections, purified rat anti-mouse CD106 antibody for immunohistochemistry (550547; BD Pharmingen) was used; and ABC kit and VIP substrate Kit (Vector Laboratories, Burlingame, CA, USA) were used for developing along with methyl green nuclear counterstaining.
Statistics
Nonparametric statistical analyses as noted in the text were performed with SAS (SAS Institute, Cary, NC, USA) to evaluate differences among test groups. The nonparametric 1-way Wilcoxon rank sum test with continuity correction (Z=0.5) was performed to evaluate 2-sided significance between groups of mice, as noted in Results. Significance was assigned at P < 0.05. Data are presented as means ± se.
RESULTS
Generation and physical characterization of VCAM-1-targeted PFC nanoparticles
Figure 1 diagrams the production of VCAM-1-targeted PFC nanoparticles. We synthesized two components separately: base PFC nanoparticles as carrier vehicles, and the TCPs of anti-VCAM-1 targeting peptide on the C terminus of the peptide linker as the functional targeting entity (13). Base carrier PFC nanoparticles were produced by standard formulation methods, as described previously (15). The PFC nanoparticles were composed of a hydrophobic PFC core and a surrounding lipid monolayer, which provided the incorporation site for the targeting ligands. To incorporate the VCAM-1-targeting ligand (VHPKQHR; ref. 26) onto the PFC nanoparticles, we first fused the VCAM-1-targeting ligand onto the C terminus of the linker peptide (VLTTGLPALISWIKRKRQQ; ref. 13) with the use of 2 glycines as a spacer to generate TCPs with solid-phase peptide synthesis (SPPS; Fig. 1A). Controlled incorporation of anti-VCAM-1-targeting peptide into PFC nanoparticles is then achieved by adjusting the amount of TCP mixed with PFC nanoparticles (13). After mixing and incubating TCP and PFC nanoparticles, the VCAM-1-targeted nanoparticles self-assembled (Fig. 1B). After overnight incubation at 4°C to maximize loading, unloaded TCPs were removed through gentle centrifugation at 970 rpm (100 g) for 10 min. The supernatant was measured by tryptophan fluorescence to determine the amount of peptide that was retained on the nanoparticle. In this study, 41,096 copies of TCP were incorporated onto each nanoparticle.
Figure 1.

Schematic of postformulation functionalization of PFC nanoparticles with a linker peptide. A) By using solid-phase peptide synthesis, TCP was produced with a VCAM-1-targeting ligand on the C terminus of linker peptide, with 2 glycines between them as a spacer. B) Base PFC nanoparticles were mixed with TCPs and incubated at 4°C overnight to generate VCAM-1-targeted nanoparticles through a self-assembling process.
The hydrodynamic diameter of the PFC nanoparticles was 227.0 ± 1.4 nm, without TCP loading (Fig. 2A). After TCP loading, the hydrodynamic diameter of targeted PFC nanoparticles remained essentially unchanged at 231.2 ± 1.5 nm (Fig. 2A). The ζ potentials were measured to evaluate the nanoparticle surface charge. The base PFC nanoparticles exhibited negative ζ potentials of −15.4 ± 0.9 mV (Fig. 2A). The VCAM-1-targeted PFC nanoparticles exhibited positive ζ potential of 22.8 ± 0.6 mV (Fig. 2A). Because of the net positive charge of the TCP, the observed positive ζ potential of the nanoparticle-peptide complex confirmed successful integration of the TCP into the PFC nanoparticles. The morphology of the nanoparticles also was illustrated in transmission electron microscopy images (Fig. 2B).
Figure 2.
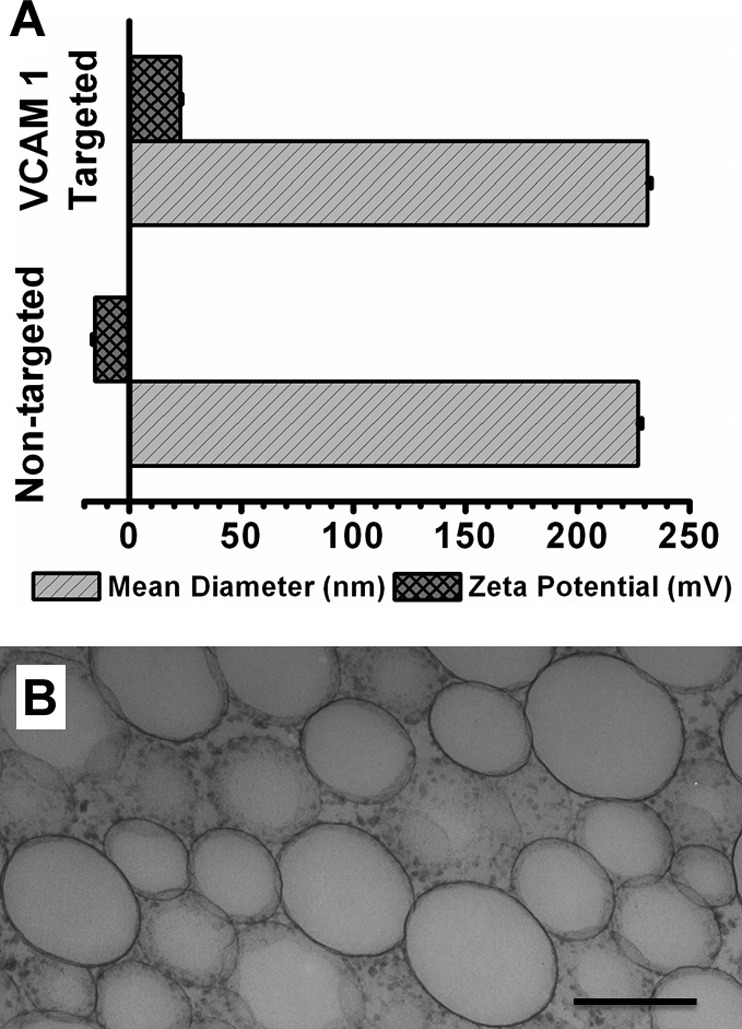
Physical characterization of VCAM-1-targeted nanoparticles. A) Mean hydrodynamic diameter and ζ potential of PFC nanoparticles with or without insertion of TCP, respectively. Addition of VCAM-1-targeting ligands does not affect the mean hydrodynamic diameter of base PFC nanoparticles, but shifts the ζ potential to a more positive voltage range. B) Transmission electron micrograph illustrating the morphology of the nanoparticles.
Biophysical characterization of TCP self-integration into PFC nanoparticle lipid monolayer
The far-UV CD spectra can provide secondary structural information for peptides. The CD spectra of TCP either free in solution or incorporated into the PFC nanoparticles were acquired. As shown in Fig. 3A, free TCP in solution exhibited a strong negative peak near 200 nm in the CD spectrum, demonstrating the unordered random coil structure. The CD spectrum of TCP incorporated into the PFC nanoparticles presented two minima at 222 and 208 nm, suggesting that TCP formed α-helical secondary structures after incorporating into the lipid membrane of the PFC nanoparticles (Fig. 3B). The α-helical structure formed by the hydrophobic section of the TCP thus enables strong hydrophobic interactions with surrounding membrane lipids.
Figure 3.

Biophysical characterization of targeting ligand incorporation into the lipid membrane of PFC nanoparticles. A, B) Secondary structure of TCP is altered after lipid insertion. A) Free TCP in solution exhibits a random coil. B) After integration into the lipid membrane of PFC nanoparticles, TCP adopts an α-helical secondary structure. C) Representative tryptophan emission spectra from TCP before and after addition of CE nanoparticles. Blue shift of 8 nm (triangle vs. square) demonstrates that the tryptophan residue moves from a hydrophilic environment into a more hydrophobic environment. D) Representative tryptophan emission spectra from TCP before and after addition of PFOB nanoparticles. Fluorescence quenching (circle vs. square) demonstrates that the tryptophan residue is positioned within the Förster distance of the PFOB core of the nanoparticles.
Furthermore, fluorescence spectra readouts were used to evaluate peptide-PFC nanoparticle complex formation and illustrate the relative proximity of the TCP to the hydrophobic perfluorocarbon core of the PFC nanoparticles. For the fluorescence spectra study, we formulated two types of PFC nanoparticles with the same lipid monolayer shell but different hydrophobic PFC core. One formulation contained a PFOB core, the other CE core. The TCP possesses one tryptophan residue, whose emission spectrum should undergo a blue shift when situated in a hydrophobic environment as compared to the hydrophilic environment (30). Following the addition of TCP conjugates to CE nanoparticles, the tryptophan fluorescence emission spectra of TCP exhibited a clear blue shift of 8 nm (Fig. 3C), suggesting that TCP had integrated into the lipid membrane of the nanoparticles. Moreover, the tryptophan fluorescence emission spectra of TCP also exhibit an expected quenching effect, which occurred after mixing TCP with PFOB nanoparticles (Fig. 3D, circles). The bromine of PFOB should quench intrinsic tryptophan fluorescence by Förster resonance energy transfer (FRET), when they are close enough for intermolecular interactions (31, 32), because the Förster distance of 8 Å yields 50% quenching efficiency (32). These results confirm that the hydrophobic segment of TCP formed an α-helical structure and inserted into the lipid membrane of PFC nanoparticles, with its tryptophan region located within 8 Å of the core of the PFC nanoparticles.
In vivo stability of the targeted nanoparticles
In a pilot evaluation of the in vivo stability of the targeted nanoparticles generated by the linker peptide postformulation strategy, we labeled the TCP with 99mTc, and loaded 99mTc-labeled TCP onto PFC nanoparticles to generate VCAM-1-targeted PFC nanoparticles before intravenous injection. The time-activity profiles of TCP and the 19F core of the PFC nanoparticles were assessed in sequential blood samples in 2 mice up to 2 h postinjection to observe any early release of the peptide cargo from the nanoparticle. The radioactivity measurements represent the blood profile of the labeled peptide that have integrated into the lipid membrane of PFC nanoparticles, whereas the MRS measurements represent the blood profile of the base nanoparticles by indicating the fluorine core components of the nanoparticles. Assuming first-order decay kinetics, a conventional noncompartmental monoexponential fit of the form y = y0 +Ae−xτ was obtained (33), where τ is a time constant and y0 is an asymptote indicating terminal differences in clearance for the two moieties. The paired data points and fits (R2>0.988 for all regressions) are comparable, suggesting that the peptides in the lipid membrane and nanoparticles track together. At all time points, the 19F signal decay remained slightly higher than 99mTc signal, and the difference between these two signals was <10% at the end of the experiments, and <20% at any given time. These results suggest that >80% of loaded peptides remained in the particle but that a small amount of peptide could have leached from the peptide-nanoparticle complex in vivo (Fig. 4), which would then have dissipated more quickly than does the nanoparticle itself. These pilot results indicated that the targeted nanoparticles could be stable enough to remain functional for biomarker targeting.
Figure 4.

Representative blood 99mTc and 19F profiles from two different mice. Data are normalized to the first time point measurement to show that 99mTc and 19F profiles tracked with each other. 99mTc signal decay was only slightly faster than 19F signal decay, suggesting minimal potential peptide leaching from the nanoparticle complex.
VCAM-1 detection and quantification in atherosclerosis
For in vivo VCAM-1 detection, VCAM-1-targeted CE nanoparticles were used for quantitative measurements of activated aortic endothelium in cholesterol-fed ApoE−/− mice with 19F MRS. After feeding with a high cholesterol diet for 8 wk, ApoE−/− and age-matched C57BL/6 mice were injected intravenously with VCAM-1-targeted or nontargeted CE nanoparticles. Following 2 h of circulation, aortas were excised after systemic saline perfusion for ex vivo MRS analysis at 11.7 T, and all signals were compensated for body weight in grams. The MRS signal from the aortas of ApoE−/− mice receiving VCAM-1-targeted CE nanoparticles indicated binding of 12.3 ± 3.1 × 105 nanoparticles (NP)/g, while that from the aorta of ApoE−/− mice receiving nontargeted CE nanoparticles indicated the presence of only 3.0 ± 2.0 × 105 NP/g. For the age-controlled C57-BL6 mice, MRS signals from the aorta of the mice receiving either targeted or nontargeted CE nanoparticles demonstrated 2.5 ± 0.6 × 105 or 2.8 ± 0.9 × 105 NP/g, respectively. The results revealed a 4.1-fold increase in the amount of CE nanoparticles detected in aortas from the ApoE−/− mice receiving VCAM-1-targeted nanoparticles as compared with other groups (Fig. 5). The 19F signal detected from the ApoE−/− mice receiving VCAM-1-targeted nanoparticles was significantly higher than that from the ApoE−/− mice receiving nontargeted nanoparticles (P=0.0298), or C57BL/6 mice receiving VCAM-1-targeted nanoparticles (P=0.0402) or C57BL/6 mice receiving nontargeted nanoparticles (P=0.0230). Also, VCAM-1 staining of the aortic sections of ApoE−/− mice or age-matched C57BL/6 control mice demonstrated substantially more VCAM-1 expression on the endothelium of the ApoE−/− aortas than that of the controls. Representative images are shown in Fig. 6.
Figure 5.

Validation of in vivo targeting in atherosclerotic mouse. Aortas from ApoE−/− mice receiving VCAM-1-targeted nanoparticles manifested significantly higher 19F signals than did all other groups: ApoE−/− mice receiving nontargeted nanoparticles or control mice receiving either VCAM-1-targeted or nontargeted nanoparticles. *P < 0.05.
Figure 6.

Histological depiction of in vivo targeting in atherosclerosis. A) Representative VCAM-1 staining on the aorta of control mice, where only autofluorescence signals were detected. B) Higher-magnification view of the boxed area in panel A. C) Representative VCAM-1 staining (green) in the aorta of ApoE−/− mice (FITC-labeled secondary antibody). Green signal in the lumen of the aorta demonstrates strong VCAM-1 expression. D) Higher-magnification view of the boxed area in panel C. Blue represents DAPI nuclear staining.
VCAM-1 detection and quantification in breast cancer
At 30 d after implantation, SSM 2 mice were randomly divided into 2 groups. One group of mice received VCAM-1-targeted CE nanoparticles, and the other group received nontargeted CE nanoparticles. After 2 h in circulation, tumors were excised following systemic saline perfusion for ex vivo MRS analysis at 11.7 T. The results indicate significantly more 19F signal detectable in the tumors from the mice injected with VCAM-1 targeted CE nanoparticles (17.2±3.1×108 NP/g tumor) than that from the mice injected with nontargeted CE nanoparticles (3.5±1.9×108 NP/g tumor) and the differences are statistically significant (P=0.0216; Fig. 7A). The results revealed a 4.9-fold increase in the amount of CE nanoparticles detected in tumors from SSM 2 mice receiving VCAM-1-targeted nanoparticles as compared with mice receiving nontargeted nanoparticles. To validate the MRS results, the tumor tissue was sectioned and stained with VCAM-1 antibody. Positive staining was observed, which delineated VCAM-1 expression primarily on the tumor vasculature (Fig. 7B, C). These results also suggested localized VCAM-1 expression as a potential biomarker in this breast tumor mouse model.
Figure 7.

Validation of in vivo targeting in breast cancer. A) Tumors from SSM mice receiving VCAM-1-targeted nanoparticles exhibited significantly higher 19F signals than did those from mice receiving nontargeted nanoparticles. *P < 0.05. B, C) Representative VCAM-1 staining for SSM tumor sections. C) Higher-magnification view of the boxed area in panel B. Purple staining illustrates VCAM-1 expression in the vasculature of SSM tumor. Green represents nuclear counterstaining.
DISCUSSION
In this study, for the first time, we have validated in vivo targeting applications of targeted PFC nanoparticles that were generated through a postformulation self-assembling process by using a linker peptide construct capable of programmable cargo incorporation. PFC nanoparticles were chosen for this study because of their multifunctional diagnostic and therapeutic utility. These nanoparticles exhibit unique 19F magnetic resonance (MR) contrast and ultrasound contrast potential for imaging (34–37). Moreover, preformulated versions of the PFC nanoparticles targeted to αvβ3-integrins are in FDA-approved clinical trials (IND 108320), which is indicative of acceptable safety, efficacy, and scale-up potential. However, other lipidic substrates including liposomes could be utilized as well (13). Indeed, the potential clinical relevance and the high priority for developing a facile postformulation strategy for targeting and multiplexing nanosystems are evident from recent programmatic opportunities to develop flexible individualized nanodelivery systems (4).
Consistent with previous studies (13, 14), incorporation of functional entities onto the PFC nanoparticles through linker peptide strategy did not affect the size of the PFC nanoparticles, while positively shifting the ζ potential of the peptide-PFC nanoparticle complex, which confirmed the integration of the targeting entities incorporated into the lipid membrane of PFC nanoparticles, because the peptide itself exhibits a net positive charge. Prior surface plasmon resonance studies in vitro (13) have demonstrated that targeting ligands tightly bind to lipid membrane of PFC nanoparticles through the linker peptide. Here, we have evaluated the mechanisms responsible for the tight binding by biophysical characterization with far-UV CD and tryptophan fluorescence emission spectrum measurements. CD spectra were acquired to define the secondary structure of TCP either free in solution (i.e., in an aqueous environment), or after incorporation into the PFC nanoparticles (in a hydrophobic environment). The CD measurements demonstrated conclusively that the peptide forms an α-helical secondary structure when incorporated into the lipid membrane of PFC nanoparticles. The hydrophobic tail of the peptide assumes an α-helical structure, which then undergoes hydrophobic interactions with surrounding lipids. These hydrophobic interactions are known to exert a negative free energy change (38), which contributes to the maintenance of a stable peptide-PFC nanoparticle complex by hydrophobic interactions. Also, the tryptophan fluorescence emission spectra indicate that the tryptophan region of the peptide is situated deeply in the lipid membrane, where it is <8 Å from the hydrophobic PFOB core. These observations are consistent with prior results for melittin-PFC nanoparticle complexes (12), which illustrate the biomimicking behavior of this linker peptide strategy for postformulation functionalization of lipidic nanocarriers.
To exemplify the potential utility of the system, we investigated applications of the targeted nanoparticles in vivo in two animal models. In the well-established ApoE-deficient mouse model for atherosclerosis study (39–41), VCAM-1 promotes firm adhesion of monocytes to activated endothelium at atherosclerotic sites, (24, 42–45) and triggers a shape change of activated endothelial cells to facilitate leukocyte transendothelial migration (46). Although prior results have shown that targeting and in vivo imaging of VCAM-1 expression in ApoE-deficient mice can be performed with standard preformulated nanoparticle preparations (23–27), we sought principally to demonstrate stability, specificity, and active targeting for the postformulation preparations rather than to focus on imaging per se. Here, we show that ApoE−/− mice fed a high-cholesterol diet for 8 wk already manifest up-regulated VCAM-1 in the endothelium of the aorta, as compared to age-matched control mice. The results indicate that a significantly higher 19F signal is detectable from aortas in the high-cholesterol-fed ApoE−/− mice as compared with the control groups, which is consistent with previously reported findings on VCAM-1 expression in ApoE−/− mice (24). We also illustrated that through linker peptide postformulation functionalization, targeting functionality can be added to PFC nanoparticles, while retaining the special advantage of this PFC nanocarrier in its ability to enable quantification of the MR signal in terms of absolute amount of bound particles.
In the breast cancer model of STAT-1 deficiency (47), the tumor vascular VCAM-1 expression was targeted effectively with our targeted nanoparticle. STAT-1 suppresses c-erbB2 (i.e., HER-2/neu)-controlled cellular transformation (48), and STAT-1 deficiency enhances c-erbB2-mediated breast tumor growth (49). Also, c-erbB2-positive cells exhibit elevated VCAM-1 expression in cocultured endothelial cells (50). These reported results suggest that VCAM-1 may be expressed in this breast tumor model, which is consistent with our in vivo VCAM-1-targeting results. Moreover, we confirmed vasculature VCAM-1 expression in this tumor model by immunohistochemistry staining. Because STAT-1-deficient mice up-regulate estrogen receptor-α (ERα) and progesterone receptors (PRs) and closely resemble the phenotype of human luminal breast cancers (51), we propose that these targeted PFC nanoparticles could facilitate evaluation of VCAM-1-related molecular drivers in this particular tumor.
Regarding targeting ligand retention on the base particles, targeting peptides may leach from the particles to a minor extent in the circulation, but this will not affect the specific targeting functionality of the nanoparticles. Whether the peptide will transfer to serum proteins remains to be illustrated, but if it did occur, there would be no contribution to the specifically bound 19F signal, because any serum protein binding would not be measureable, as these elements contain no fluorine for signaling. The upshot would be only a slight diminution in 19F signal due to competition for binding sites. Although it is possible that nanoparticles could be ingested by neutrophils or monocytes after intravenous administration that then home to a site, it seems less likely that sufficient cellular uptake and localization would occur within 2 h to produce the signals we are seeing. Even if this is possible, monocyte-ingested particles will contribute only to the nonspecific signal. Therefore, our results clearly demonstrated the specific in vivo targeting of the targeted nanoparticles generated through postformulation functionalization by utilizing linker peptide.
For proof of concept, specific targeting of the targeted PFC nanoparticles generated by using the linker peptide was validated in the tissues after systemic saline perfusion to remove all nonspecific blood pool signal, which was done to derive a clear quantitative estimate of specific binding. We note that for in vivo applications where blood pool particles may be present at the time of imaging, any signal originating from unbound circulating nanoparticles can be suppressed by using 19F diffusion-sensitive MRI/MRS techniques to selectively eliminate the mobile 19F signal in the bloodstream (29).
It has been reported that melittin induces IgE responses in about one-third of patients with bee sting allergies (53). Also, the immunogenicity of melittin may be mouse strain specific: in some strains of mice, melittin residue 20–26 positive-charged hydrophilic segment is responsible for antibody responses (54), while melittin residue 11–19 α-helical amphiphilic segment induces T-cell responses (55). The linker peptide is an N-terminal truncation of melittin and includes the aforementioned melittin segments. However, melittin's immunogenicity depends on its oligomerization in the cell membrane (56), which is intentionally more limited for the truncated linker peptide. Although the potential for immunogenicity remains a concern most notably to patients with bee sting allergies, one solution would be to synthesize the linker peptide with d-amino acids, since d-melittin is not immunogenic in mice (56). It has been also reported that melittin could inhibit NF-κB- and Akt-signaling pathways (57–59), although the molecular mechanism by which melittin modulates cellular signaling pathways remains to be elucidated. However, in our previous report (14), the results suggested that linker peptide does not affect these two cellular signaling pathways in the same manner or extent as melittin itself. Because nanoparticles might be taken up by circulating immune cells, understanding the potential effects on signaling in immune cells would be pertinent for viable product candidates, especially when carrying cargos that could modulate certain cellular signaling pathways.
In summary, we have validated in vivo targeting applications of PFC nanoparticles that were generated through a postformulation self-assembling process by using a linker peptide construct capable of programmable cargo incorporation. Unlike other postformulation functionalization methods, this biomimicking strategy requires no modification of the base lipidic nanoparticle formulation, avoids the use of catalytic species, and requires no specialized lipidic species. Because the peptide linker is generic and can be modified with numerous cargos through standard chemistries, the insertion of a variety of different cargos should be reasonably straightforward, because the biophysical interactions between the base carrier and the linker peptide are equivalent in each case (13–15). In this study, PFC nanoparticles were chosen because of their unique 19F MR contrast and ultrasound contrast potential for imaging (34–37) and their therapeutic utility (11, 12). Moreover, preformulated versions of the PFC nanoparticles targeted to αvβ3-integrins are in FDA-approved clinical trials (IND 108320), which is indicative of acceptable safety, efficacy, and scale-up potential. However, other lipidic substrates, including liposomes, micelles, living cells, lipoproteins, or coated viruses could be utilized as carrier candidates for functionalization (13).
Acknowledgments
The authors thank Ralph Fuhrhop for PFC nanoparticle formulation, and Suqin Yao for the statistical analysis.
This work was supported by U.S. National Institutes of Health grants (HL-073646 and CA-119342) to S.A.W.
Footnotes
- CD
- circular dichroism
- CE
- perfluoro-15-crown-5-ether
- MR
- magnetic resonance
- MRS
- magnetic resonance spectroscopy
- PFC
- perfluorocarbon
- PFOB
- perfluorooctyl bromide
- SSM
- STAT-1−/− female mice that spontaneously develop mammary adenocarcinomas
- TCP
- targeting combination peptide
- VCAM-1
- vascular cell adhesion molecule 1
REFERENCES
- 1. Masotti A. (2010) Multifuntional nanoparticles: preparation and applications in biomedicine and in non-invasive bioimaging. Recent Patents Nanotechnol. 4, 53–62 [DOI] [PubMed] [Google Scholar]
- 2. Petros R. A., DeSimone J. M. (2010) Strategies in the design of nanoparticles for therapeutic applications. Nat. Rev. 9, 615–627 [DOI] [PubMed] [Google Scholar]
- 3. Cavalieri F., Zhou M., Ashokkumar M. (2010) The design of multifunctional microbubbles for ultrasound image-guided cancer therapy. Curr. Topics Med. Chem. 10, 1198–1210 [DOI] [PubMed] [Google Scholar]
- 4. Defense Advanced Projects Research Agency (2012) DARPA-BAA-12-34: in vivo nanosensors for therapeutics (IVN:Tx). Solicitation notice, Contracts Management Office, DARPA, Arlington, VA, USA [Google Scholar]
- 5. Perrier T., Saulnier P., Benoit J. P. (2010) Methods for the functionalisation of nanoparticles: new insights and perspectives. Chemistry 16, 11516–11529 [DOI] [PubMed] [Google Scholar]
- 6. Perrier T., Saulnier P., Fouchet F., Lautram N., Benoit J. P. (2010) Post-insertion into lipid NanoCapsules (LNCs): from experimental aspects to mechanisms. Int. J. Pharmaceut. 396, 204–209 [DOI] [PubMed] [Google Scholar]
- 7. Moreira J. N., Ishida T., Gaspar R., Allen T. M. (2002) Use of the post-insertion technique to insert peptide ligands into pre-formed stealth liposomes with retention of binding activity and cytotoxicity. Pharmaceut. Res. 19, 265–269 [DOI] [PubMed] [Google Scholar]
- 8. Von Maltzahn G., Ren Y., Park J. H., Min D. H., Kotamraju V. R., Jayakumar J., Fogal V., Sailor M. J., Ruoslahti E., Bhatia S. N. (2008) In vivo tumor cell targeting with “click” nanoparticles. Bioconj. Chem. 19, 1570–1578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Etheridge M., Campbell S., Erdman A., Haynes C., Wolf S., McCullough J. (2012) The big picture on nanomedicine: the state of investigational and approved nanomedicine products. [E-pub ahead of print] Nanomed. Nanotechnol. Biol. Med. doi: 10.1016/j.nano.2012.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Weissmann G., Hirschhorn R., Krakauer K. (1969) Effect of melittin upon cellular and lysosomal membranes. Biochem. Pharmacol. 18, 1771–1775 [DOI] [PubMed] [Google Scholar]
- 11. Soman N. R., Baldwin S. L., Hu G., Marsh J. N., Lanza G. M., Heuser J. E., Arbeit J. M., Wickline S. A., Schlesinger P. H. (2009) Molecularly targeted nanocarriers deliver the cytolytic peptide melittin specifically to tumor cells in mice, reducing tumor growth. J. Clin. Invest. 119, 2830–2842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Soman N. R., Lanza G. M., Heuser J. M., Schlesinger P. H., Wickline S. A. (2008) Synthesis and characterization of stable fluorocarbon nanostructures as drug delivery vehicles for cytolytic peptides. Nano Lett. 8, 1131–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pan H., Myerson J. W., Ivashyna O., Soman N. R., Marsh J. N., Hood J. L., Lanza G. M., Schlesinger P. H., Wickline S. A. (2010) Lipid membrane editing with peptide cargo linkers in cells and synthetic nanostructures. FASEB J. 24, 2928–2937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pan H., Ivashyna O., Sinha B., Lanza G. M., Ratner L., Schlesinger P. H., Wickline S. A. (2011) Post-formulation peptide drug loading of nanostructures for metered control of NF-κB signaling. Biomaterials 32, 231–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pan H., Marsh J. N., Christenson E. T., Soman N. R., Ivashyna O., Lanza G. M., Schlesinger P. H., Wickline S. A. (2012) Postformulation peptide drug loading of nanostructures. Methods Enzymol. 508, 17–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Blankenberg S., Barbaux S., Tiret L. (2003) Adhesion molecules and atherosclerosis. Atherosclerosis 170, 191–203 [DOI] [PubMed] [Google Scholar]
- 17. Galkina E., Ley K. (2007) Vascular adhesion molecules in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 27, 2292–2301 [DOI] [PubMed] [Google Scholar]
- 18. Preiss D. J., Sattar N. (2007) Vascular cell adhesion molecule-1: a viable therapeutic target for atherosclerosis? Intl. J. Clin. Pract. 61, 697–701 [DOI] [PubMed] [Google Scholar]
- 19. Libby P. (2002) Inflammation in atherosclerosis. Nature 420, 868–874 [DOI] [PubMed] [Google Scholar]
- 20. Gavina M., Belicchi M., Rossi B., Ottoboni L., Colombo F., Meregalli M., Battistelli M., Forzenigo L., Biondetti P., Pisati F., Parolini D., Farini A., Issekutz A. C., Bresolin N., Rustichelli F., Constantin G., Torrente Y. (2006) VCAM-1 expression on dystrophic muscle vessels has a critical role in the recruitment of human blood-derived CD133+ stem cells after intra-arterial transplantation. Blood 108, 2857–2866 [DOI] [PubMed] [Google Scholar]
- 21. O'Hanlon D. M., Fitzsimons H., Lynch J., Tormey S., Malone C., Given H. F. (2002) Soluble adhesion molecules (E-selectin, ICAM-1, and VCAM-1) in breast carcinoma. Eur. J. Cancer 38, 2252–2257 [DOI] [PubMed] [Google Scholar]
- 22. Regidor P. A., Callies R., Regidor M., Schindler A. E. (1998) Expression of the cell adhesion molecules ICAM-1 and VCAM-1 in the cytosol of breast cancer tissue, benign breast tissue and corresponding sera. Eur. J. Gynaecol. Oncol. 19, 377–383 [PubMed] [Google Scholar]
- 23. Zibara K., Chignier E., Covacho C., Poston R., Canard G., Hardy P., McGregor J. (2000) Modulation of expression of endothelial intercellular adhesion molecule-1, platelet-endothelial cell adhesion molecule-1, and vascular cell adhesion molecule-1 in aortic arch lesions of apolipoprotein E-deficient compared with wild-type mice. Arterioscler. Thromb. Vasc. Biol. 20, 2288–2296 [DOI] [PubMed] [Google Scholar]
- 24. Nakashima Y., Raines E. W., Plump A. S., Breslow J. L., Ross R. (1998) Upregulation of VCAM-1 and ICAM-1 at atherosclerosis-prone sites on the endothelium in the ApoE-deficient mouse. Arterioscler. Thromb. Vasc. Biol. 18, 842–851 [DOI] [PubMed] [Google Scholar]
- 25. Nahrendorf M., Keliher E., Panizzi P., Zhang H., Hembrador S., Figueiredo J. L., Aikawa E., Kelly K., Libby P., Weissleder R. (2009) 18F-4V for PET-CT imaging of VCAM-1 expression in atherosclerosis. JACC Cardiovasc. Imag. 2, 1213–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kelly K. A., Allport J. R., Tsourkas A., Shinde-Patil V. R., Josephson L., Weissleder R. (2005) Detection of vascular adhesion molecule-1 expression using a novel multimodal nanoparticle. Circ. Res. 96, 327–336 [DOI] [PubMed] [Google Scholar]
- 27. Burtea C., Ballet S., Laurent S., Rousseaux O., Dencausse A., Gonzalez W., Port M., Corot C., Elst L. V., Muller R. N. (2012) Development of a magnetic resonance imaging protocol for the characterization of atherosclerotic plaque by using vascular cell adhesion molecule-1 and apoptosis-targeted ultrasmall superparamagnetic iron oxide derivatives. Arterioscler. Thromb. Vasc. Biol. 32, e36–e48 [DOI] [PubMed] [Google Scholar]
- 28. Morawski A. M., Winter P. M., Yu X., Fuhrhop R. W., Scott M. J., Hockett F., Robertson J. D., Gaffney P. J., Lanza G. M., Wickline S. A. (2004) Quantitative “magnetic resonance immunohistochemistry” with ligand-targeted (19)F nanoparticles. Magn. Reson. Med. 52, 1255–1262 [DOI] [PubMed] [Google Scholar]
- 29. Waters E. A., Chen J., Yang X., Zhang H., Neumann R., Santeford A., Arbeit J., Lanza G. M., Wickline S. A. (2008) Detection of targeted perfluorocarbon nanoparticle binding using 19F diffusion weighted MR spectroscopy. Magn. Reson. Med. 60, 1232–1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lakowicz J. R. (1999)) Principles of Fluorescence Spectroscopy, Kluwer Academic/Plenum Press, New York [Google Scholar]
- 31. Mall S., Sharma R. P., East J. M., Lee A. G. (1998) Lipid-protein interactions in the membrane: studies with model peptides. Faraday Discuss. 127–136; discussion 137–157 [DOI] [PubMed] [Google Scholar]
- 32. Powl A. M., East J. M., Lee A. G. (2003) Lipid-protein interactions studied by introduction of a tryptophan residue: the mechanosensitive channel MscL. Biochemistry 42, 14306–14317 [DOI] [PubMed] [Google Scholar]
- 33. Boroujerdi M. (2001) Pharmacokinetics Principles and Applications, McGraw-Hill, New York [Google Scholar]
- 34. Chen J., Lanza G. M., Wickline S. A. (2010) Quantitative magnetic resonance fluorine imaging: today and tomorrow. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2, 431–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kaneda M. M., Caruthers S., Lanza G. M., Wickline S. A. (2009) Perfluorocarbon nanoemulsions for quantitative molecular imaging and targeted therapeutics. Annals Biomed. Eng. 37, 1922–1933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hughes M. S., Marsh J. N., Zhang H., Woodson A. K., Allen J. S., Lacy E. K., Carradine C., Lanza G. M., Wickline S. A. (2006) Characterization of digital waveforms using thermodynamic analogs: detection of contrast-targeted tissue in vivo. IEEE Trans. Ultrason. Ferroelectr. Freq. Control 53, 1609–1616 [DOI] [PubMed] [Google Scholar]
- 37. Hughes M., Marsh J., Lanza G., Wickline S., McCarthy J., Wickerhauser V., Maurizi B., Wallace K. (2011) Improved signal processing to detect cancer by ultrasonic molecular imaging of targeted nanoparticles. J. Acoust. Soc. Am. 129, 3756–3767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Klocek G., Schulthess T., Shai Y., Seelig J. (2009) Thermodynamics of melittin binding to lipid bilayers. Aggregation and pore formation. Biochemistry 48, 2586–2596 [DOI] [PubMed] [Google Scholar]
- 39. Jawien J., Nastalek P., Korbut R. (2004) Mouse models of experimental atherosclerosis. J. Physiol. Pharmacol. 55, 503–517 [PubMed] [Google Scholar]
- 40. Fazio S., Linton M. F. (2001) Mouse models of hyperlipidemia and atherosclerosis. Front. Biosci. 6, D515–D525 [DOI] [PubMed] [Google Scholar]
- 41. Willems Van Dijk K., Hofker M. H., Havekes L. M. (2000) Use of transgenic mice to study the role of apolipoprotein E in lipid metabolism and atherosclerosis. Intl. J. Tiss. Reactions 22, 49–58 [PubMed] [Google Scholar]
- 42. O'Brien K. D., Allen M. D., McDonald T. O., Chait A., Harlan J. M., Fishbein D., McCarty J., Ferguson M., Hudkins K., Benjamin C. D. (1993) Vascular cell adhesion molecule-1 is expressed in human coronary atherosclerotic plaques. Implications for the mode of progression of advanced coronary atherosclerosis. J. Clin. Invest. 92, 945–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. O'Brien K. D., McDonald T. O., Chait A., Allen M. D., Alpers C. E. (1996) Neovascular expression of E-selectin, intercellular adhesion molecule-1, and vascular cell adhesion molecule-1 in human atherosclerosis and their relation to intimal leukocyte content. Circulation 93, 672–682 [DOI] [PubMed] [Google Scholar]
- 44. Davies M. J., Gordon J. L., Gearing A. J., Pigott R., Woolf N., Katz D., Kyriakopoulos A. (1993) The expression of the adhesion molecules ICAM-1, VCAM-1, PECAM, and E-selectin in human atherosclerosis. J. Pathol. 171, 223–229 [DOI] [PubMed] [Google Scholar]
- 45. Cybulsky M. I., Iiyama K., Li H., Zhu S., Chen M., Iiyama M., Davis V., Gutierrez-Ramos J. C., Connelly P. W., Milstone D. S. (2001) A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J. Clin. Invest. 107, 1255–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Matheny H. E., Deem T. L., Cook-Mills J. M. (2000) Lymphocyte migration through monolayers of endothelial cell lines involves VCAM-1 signaling via endothelial cell NADPH oxidase. J. Immunol. 164, 6550–6559 [DOI] [PubMed] [Google Scholar]
- 47. Meraz M. A., White J. M., Sheehan K. C., Bach E. A., Rodig S. J., Dighe A. S., Kaplan D. H., Riley J. K., Greenlund A. C., Campbell D., Carver-Moore K., DuBois R. N., Clark R., Aguet M., Schreiber R. D. (1996) Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell 84, 431–442 [DOI] [PubMed] [Google Scholar]
- 48. Raven J. F., Williams V., Wang S., Tremblay M. L., Muller W. J., Durbin J. E., Koromilas A. E. (2011) Stat1 is a suppressor of ErbB2/Neu-mediated cellular transformation and mouse mammary gland tumor formation. Cell Cycle 10, 794–804 [DOI] [PubMed] [Google Scholar]
- 49. Klover P. J., Muller W. J., Robinson G. W., Pfeiffer R. M., Yamaji D., Hennighausen L. (2010) Loss of STAT1 from mouse mammary epithelium results in an increased Neu-induced tumor burden. Neoplasia 12, 899–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Markowska A., Urasinska E., Domagala W. (2008) In vitro assessment of adhesion molecules expression by human endothelial cells cocultured with c-erbB2-positive and c-erbB2-negative breast carcinoma cell lines. Polish J. Pathol. 59, 49–54 [PubMed] [Google Scholar]
- 51. Chan S. R., Vermi W., Luo J., Lucini L., Rickert C., Fowler A. M., Lonardi S., Arthur C., Young L. J., Levy D. E., Welch M. J., Cardiff R. D., Schreiber R. D. (2012) STAT1-deficient mice spontaneously develop estrogen receptor alpha-positive luminal mammary carcinomas. Breast Cancer Res. 14, R16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Marsh J. N., Partlow K. C., Abendschein D. R., Scott M. J., Lanza G. M., Wickline S. A. (2007) Molecular imaging with targeted perfluorocarbon nanoparticles: quantification of the concentration dependence of contrast enhancement for binding to sparse cellular epitopes. Ultrasound Med. Biol. 33, 950–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jarisch R., Yman L., Boltz A., Sandor I., Janitsch A. (1979) IgE antibodies to bee venom, phospholipase A, melittin and wasp venom. Clin. Allergy 9, 535–541 [DOI] [PubMed] [Google Scholar]
- 54. King T. P., Kochoumian L., Joslyn A. (1984) Melittin-specific monoclonal and polyclonal IgE and IgG1 antibodies from mice. J. Immunol. 133, 2668–2673 [PubMed] [Google Scholar]
- 55. Fehlner P. F., Berg R. H., Tam J. P., King T. P. (1991) Murine T cell responses to melittin and its analogs. J. Immunol. 146, 799–806 [PubMed] [Google Scholar]
- 56. King T. P., Wade D., Coscia M. R., Mitchell S., Kochoumian L., Merrifield B. (1994) Structure-immunogenicity relationship of melittin, its transposed analogues, and D-melittin. J. Immunol. 153, 1124–1131 [PubMed] [Google Scholar]
- 57. Son D. J., Ha S. J., Song H. S., Lim Y., Yun Y. P., Lee J. W., Moon D. C., Park Y. H., Park B. S., Song M. J., Hong J. T. (2006) Melittin inhibits vascular smooth muscle cell proliferation through induction of apoptosis via suppression of nuclear factor-κB and Akt activation and enhancement of apoptotic protein expression. J. Pharmacol. Exp. Ther. 317, 627–634 [DOI] [PubMed] [Google Scholar]
- 58. Moon D. O., Park S. Y., Choi Y. H., Kim N. D., Lee C., Kim G. Y. (2008) Melittin induces Bcl-2 and caspase-3-dependent apoptosis through downregulation of Akt phosphorylation in human leukemic U937 cells. Toxicon 51, 112–120 [DOI] [PubMed] [Google Scholar]
- 59. Wang C., Chen T., Zhang N., Yang M., Li B., Lu X., Cao X., Ling C. (2009) Melittin, a major component of bee venom, sensitizes human hepatocellular carcinoma cells to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis by activating CaMKII-TAK1-JNK/p38 and inhibiting IκBα kinase-NF-κB. J. Biol. Chem. 284, 3804–3813 [DOI] [PubMed] [Google Scholar]