

Abstract
MicroRNAs are important post-transcriptional regulators in immune cells, but how viral infection regulates microRNA expression to shape dendritic cell responses has not been well characterized. We identified 20 miRNAs that were differentially expressed in primary murine dendritic cells in response to the double-stranded RNA agonist poly(I:C), a subset of which were modestly regulated by influenza infection. miR-451 was unique because it was induced more strongly in primary splenic and lung dendritic cells by live viral infection than by purified agonists of pattern recognition receptors. We determined that miR-451 regulates a subset of pro-inflammatory cytokine responses. Three types of primary dendritic cells treated with anti-sense RNA antagomirs directed against miR-451 secreted elevated levels of IL-6, TNF, CCL5/RANTES, and CCL3/MIP1α, and these results were confirmed using miR-451null cells. miR-451 negatively regulates YWHAZ/14-3-3ζ protein levels in various cell types, and we measured a similar inhibition of YWHAZ levels in dendritic cells. It is known that YWHAZ can control the activity of two negative regulators of cytokine production: FOXO3, which is an inhibitory transcription factor, and ZFP36/Tristetraprolin, which binds to AU-rich elements within 3′-UTRs to destabilize cytokine mRNAs. Inhibition of miR-451 expression correlated with increased YWHAZ protein expression and decreased ZFP36 expression, providing a possible mechanism for the elevated secretion of IL-6, TNF, CCL5/RANTES, and CCL3/MIP1α. miR-451 levels are themselves increased by IL-6 and type I interferon, potentially forming a regulatory loop. These data suggest that viral infection specifically induces a miRNA that directs a negative regulatory cascade to tune dendritic cell cytokine production.
Introduction
Dendritic cell recognition of microbes is instrumental for instructing innate and adaptive immunity to clear pathogens and establish immunological memory. The use of purified agonists has revealed the critical role of pattern recognition receptors in orchestrating dendritic cell responses, which has important implications for vaccine adjuvant design (1, 2). Influenza is an enveloped negative sense single-stranded RNA virus that is thought to be detected primarily by RNA-specific pattern recognition receptors. This concept is supported by impaired cytokine responses by influenza-infected cells lacking the RNA sensors TLR3, TLR7, and MAVS and no clear demonstration of host recognition of the 11 influenza-encoded proteins (3–7). Viral entry, assembly, and budding perturb normal cell biology, but it is unclear how sentinel cells integrate these potential signals from live viral infection with foreign RNA detection.
This study identifies a microRNA that is induced more strongly by influenza infection than stimulation with the purified double-stranded RNA agonist poly(I:C), a viral mimetic which is known to be detected by endosomal TLR3 and cytosolic RIG-I (8–10). MicroRNAs (miRNA) are important post-transcriptional regulators in immune cells but their roles within dendritic cells have not been well characterized (11). These small non-coding RNAs negatively regulate protein levels by interacting with target mRNAs by partial base pair complementarity, which blocks translation or triggers mRNA degradation (12). miRNAs can act as fine-tuners, to titrate the levels of translatable mRNA, as well as switches, to repress protein production by maintaining mRNA levels below a threshold (13). Fine-tuning of protein levels by miRNAs has been shown to regulate developmental programs, cellular responses to infection, and to provide a restraint on inflammation (14–17). Expression of miRNAs and target mRNAs can be cell type-restricted, resulting in a cell lineage-specific role for this class of negative regulators in many systems (18).
The identification of negative regulatory networks is particularly relevant to understanding and manipulating dendritic cell biology. These cells rapidly secrete high levels of cytokines that orchestrate inflammatory responses aimed at controlling replicating pathogens. Cytokines drive a number of positive feedback loops and therefore their production must therefore be tightly controlled to limit chronic inflammatory sequelae. Pro-inflammatory cytokine production can be inhibited at multiple regulatory points: signaling, transcription, RNA stability, translation, and secretion (19). The transcription factor FOXO3 negatively regulates pro-inflammatory cytokine gene expression and FOXO3null dendritic cells secrete increased IL-6, TNF, and CCL2/MCP-1 following viral infection (20). The protein ZFP36/Tristetraprolin post-transcriptionally represses the expression of numerous pro-inflammatory cytokines, including TNF, IL-6, CCL2/MCP-1, CCL3/MIP1α, CCL4/MIP-1β, CXCL2/MIP-2 by binding to AU-rich elements in their mRNA 3′-UTRs and promoting mRNA decay (21–24). Both FOXO3 and ZFP36 are inhibited by YWHAZ/14-3-3ζ, an adaptor protein that modulates the activity of binding partners by controlling subcellular localization or kinase activity (25). YWHAZ binds FOXO3 and ZFP36 via phosphoserine-dependent interactions to inhibit the activity of these negative regulators by sequestering them from nucleic acid interactions (26–30). Regulation of YWHAZ itself is less well-characterized. YWHAZ function is altered by its phosphorylation state and its levels are stoichiometrically limiting in cells, rendering YWHAZ activity sensitive to the regulation of its expression level (25). Here, we demonstrate that YWHAZ levels are inhibited by a miRNA specifically induced by viral infection. Reduced YWHAZ levels can relieve repression of ZFP36, resulting in negative regulation of pro-inflammatory cytokine expression by dendritic cells.
Materials and Methods
Cell culture and mice
C57Bl/6 mice (Charles River) or MyD88null mice (Institute for Systems Biology, Seattle) were injected subcutaneously between the shoulder blades with 6x106 B6-melanoma cells expressing Flt3L to expand the dendritic cell compartment. After 2 weeks, spleens and lungs were isolated, enzymatically dissociated using Liberase Blendzyme III (Roche) and dendritic cells isolated by autoMACS magnetic bead purification using pan-DC beads (specific for CD11c and PDCA1; Miltenyi Biotec). Cell purity was measured by flow cytometry of CD11c expression and was >95% in each experiment. This purification scheme yielded a mixture of conventional CD11chi dendritic cells as well as PDCA1+ plasmacytoid cells, and the ratio of these two populations was consistent between experiments. These splenic dendritic cells were used for all experiments, unless otherwise noted. To isolate specific dendritic cell populations, Flt3L-expanded spleens were enzymatically dissociated, red blood cells removed using ACK lysis solution (Sigma), B cells depleted using CD19+ autoMACS microbeads, stained using CD11c-FITC (BD) and PDCA1-APC (Miltenyi Biotec) and sorted for CD11c+PDCA1− (myeloid DC) and PDCA1+ (plasmacytoid DC) expression using a FACS Aria. Bone marrow was isolated from the femurs and tibia of C57Bl/6 mice, red blood cells removed by ACK lysis, and cells cultured at a density of 5x106 cells per mL in RPMI-1640 containing 10% heat-inactivated FBS, 2 mM L-glutamine, penicillin, streptomycin, 100 μM β-mercaptoethanol, and 100 ng/mL Flt3L (Peprotech) for 7-8 days without medium change. The JAWS II dendritic cell line was obtained from ATCC and cultured as recommended. The JC4 red blood cell line was used to compare miRNA expression between erythoid and dendritic cells. CD8+ T cells were isolated from the spleens of OT-1 mice (Jackson Laboratories) by mechanical dissociation and positive selection using CD8-specific microbeads and autoMACS purification. Cells were stained using 1 μM CFSE in PBS containing 5% FBS for 10 min at room temperature, followed by 3 washes with PBS. miR-144/miR-451null mice generated by the deletion of a 388 bp segment of genomic DNA containing both the pre-miR-144 and pre-miR-451 sequences by homologous recombination in 129 embryonic stem cells (26). Mice were backcrossed 5 times onto the C57Bl/6 background (verified to be >95% C57Bl/6), and wild type littermates used for infection experiments. These mice are deficient for both miR-451 and miR-144, but miR-144 expression is undetectable in dendritic cells. Experiments using mice were approved by an IACUC committee and performed in compliance with the U.S. Department of Health and Human Services Guide for the Care and Use of Laboratory Animals.
Nucleofection
Antagomirs were introduced by nucleofection using the Amaxa mouse dendritic cell nucleofection kit and program Y-01 according to the manufacturer’s protocol (Lonza). 2x106 cells were combined with 24 μL of 5 nM antagomir in 100 μL. Media was gently added to cells without centrifugation, and cells were then cultured with the indicated stimuli.
Retroviral transduction of dendritic cells
1x107 Phoenix cells plated in a 10 cm petri dish in 5 mls of complete media without antibiotics were transfected with 12 μg of DNA and 36 μl of Lipofectamine 2000 in 3 mls of OptiMEM (Life Technologies). After 18 hours, the transfection mix was replaced with 10 mls of complete media and the virus allowed to grow for 3 days. 7-8x106 bone marrow cells from C57Bl/6 mice femurs were cultured in 10 mls of complete media with 10ng/ml IL-3, 20 ng/ml IL-6 and 25 ng/ml SCF for 3 days. After 3 days of culture, 1x106 bone marrow cells were transduced by centrifuging at 1800 rcf at 32 oC for 2 hours with 5 mls of retroviral supernatant and 4 μg/ml polybrene and then incubated for 3 more hours at 37 oC followed by a media change containing IL-3, IL-6 and SCF. After 3 days, media was replaced with fresh media containing 100 ng/ml FLT3L and then incubated for 5 days before selection with 5 μg/ml puromycin for 3 days. An expression construct expressing murine miR-451 alone with flanking sequences 100 bp upstream and 198 bp downstream was cloned into a retroviral MSCV-GFP vector. Packaged virus was generated using Phoenix ecotropic cells, transduced onto JAWS II cells, and stably selected using 15 μg/mL puromycin. Retroviral constructs expressing shRNAs specific for Ywhaz or luciferase (control) or overexpressing Ywhaz have been described previously (Yu et al., 2010).
Cell stimulation
Cells were infected with H1N1 influenza A/Puerto Rico/8/34 (PR8, Charles River) (or H3N2 influenza X31 where indicated, Paul Thomas, St. Jude’s, Memphis, TN) at an MOI of 10 virions per dendritic cell and EMCV at an MOI of 2. Cells were treated with 30 μM poly(I:C) (Invitrogen), 30 μM R848, 1 μg/mL LPS (List Biological), 500 U/mL IFN-β (PBL InterferonSource), IL-6 (Peprotech), or 1 μg/mL SIINFEKL ova peptide. Virus was inactivated by UV and used at an MOI equivalent to live virus. UV-inactivated virus was assessed to be non-infectious using the TC-1 mouse lung epithelial cell line and flow cytometry using an antibody specific for viral NP protein (Argene). Splenic dendritic cells were infected with influenza or stimulated with poly(I:C), pulsed with ova peptide SIINFEKL and cultured with CFSE-labeled CD8+ OT-1 T cells for 3 days. CFSE dilution in live (7-AAD−) cells is shown for cultures containing dendritic cells treated with miR-451 antagomir or control antagomir. DC:T cell co-cultures were cultured at a ratio of 1:2, treated as described, and supernatants collected after 24 h or 72 h.
Flow cytometry
Cells were blocked using 1 ug/mL anti-FcR antibody (2.4G2) in PBS containing 2% FBS and 2 mM EDTA and stained using fluorescently-labeled CD11c, MHCII or CD80-specific antibodies (BD Biosciences) or PDCA1-specific antibody (Miltenyi Biotec), and fixed in 10% formalin. Fluorescence of antibody-stained cells or cells nucleofected with FITC-conjugated antagomirs was measured using a FACSCalibur.
MicroRNA array profiling
miRNA profiling was performed using a miRNA array (Exiqon) on primary Flt3L in vivo-expanded dendritic cells infected with influenza PR8 (MOI=10), stimulated with poly(I:C) (30 μg/mL poly(I:C)) or cultured in media alone for 8 h. The quality of the total RNA was verified by an Agilent 2100 Bioanalyzer profile. One μg total RNA from sample and reference were labeled with Hy3™ and Hy5™ fluorescent label, respectively, using the miRCURY™ LNA Array power labeling kit (Exiqon, Denmark) following the procedure described by the manufacturer. The Hy3™-labeled samples and a Hy5™-labeled reference RNA sample were mixed pair-wise and hybridized to the miRCURY™ LNA array version 10.0 (Exiqon, Denmark), which contains capture probes targeting all miRNAs for all species registered in the miRBASE version 11.0 at the Sanger Institute. The hybridization was performed according to the miRCURY™ LNA array manual using a Tecan HS4800 hybridization station (Tecan, Austria). After hybridization the microarray slides were scanned and stored in an ozone free environment (ozone level below 2.0 ppb) in order to prevent potential bleaching of the fluorescent dyes. The miRCURY™ LNA array microarray slides were scanned using the Agilent G2565BA Microarray Scanner System (Agilent Technologies, Inc., USA) and the image analysis was carried out using the ImaGene 8.0 software (BioDiscovery, Inc., USA). The quantified signals were normalized using the global Lowess (LOcally WEighted Scatterplot Smoothing) regression algorithm. miRNAs were considered to be expressed if measured above the detection limit in all 3 independent biological replicates of at least one experimental condition. miRNAs were considered to be differentially expressed if the ratio relative to mock was ≥1.5 in either influenza-infected or poly(I:C)-treated samples and a p value <0.001. Student’s t test was performed in stimulated cells relative to mock. The full data set has been deposited in GEO (Accession # GSE36316; http://www.ncbi.nlm.nih.gov/geo/).
Molecular biology
For quantitative RT-PCR measurements, RNA was isolated using Trizol, cDNA was synthesized from DNase-treated RNA using random primers or miR-specific primers, and qRT-PCR performed using gene- or miRNA-specific primers and probes (ABI). Expression was normalized to Ef-1 (Eif1a) (mRNA) or sno202 (miRNA). Viral RNA was quantified by RT-PCR using primers specific for influenza M gene (Forward 5′ CAT GGA ATG GCT AAA GAC AAG ACC, Reverse 5′ CCA TTA AGG GCA TTT TGG ACA, Probe FAM-5′ TTT GTG TTC ACG CTC ACC GTG CCC A –TAMRA) and normalized to the level of mouse EF-1. Western blot analysis of total protein was performed on cellular lysates prepared using RIPA buffer containing protease and phosphatase inhibitors and nuclear extracts prepared by hypotonic lysis. Equal quantities were run on 4-12% gradient SDS-PAGE gels, transferred to PVDF, and serial incubations of membranes were performed with the indicated antibodies [14-3-3ζ/YWHAZ (Santa Cruz sc-1019), FOXO3a (Cell Signaling #2497), phospho-FOXO3a (Ser294, Cell Signaling #5538), ZFP36/Tristetraprolin (Santa Cruz sc8458), LAMINB1 (Invitrogen) or β-ACTIN-HRP (ab20271, Abcam)]. Species-specific secondary antibodies conjugated to HRP were visualized by enhanced chemiluminescent detection and the signal was quantified by densitometry. Cytokine levels in cell supernatants were measured using a 16-32-plex Luminex panel (Millipore).
Statistical analysis
Means ± SEM for independent biological replicates are shown unless stated otherwise in the text. P values were determined using an unpaired two-tailed Student’s t test, assuming equal variances on all experimental data sets.
Results
Viral infection regulates microRNA expression
To explore how viral infection modulates dendritic cell gene expression post transcriptionally, we profiled 578 miRNAs in primary murine splenic dendritic cells infected in vitro with influenza A virus or stimulated with poly(I:C) for 8 hours. Influenza is a negative-sense single-stranded RNA virus which activates dendritic cells through RIG-I, TLR3, and TLR7 (3, 7, 31), while poly(I:C) is a double-stranded RNA agonist that stimulates cells via TLR3, MDA5, and RIG-I-dependent signaling. The expression of 246 miRNAs was detectable in at least one experimental condition, and the concentrations of the majority of these miRNAs were not altered by these stimuli. However, 21 miRNAs exhibited expression changes ≥ 1.5-fold (p <0.001) following poly(I:C) treatment or viral infection relative to mock-treated cells. 18 of these differentially expressed miRNAs were up-regulated and 3 were down-regulated (Fig. 1A-B). Regulated expression of these microRNAs within dendritic cells has not been described previously, except for miR-155, which is known to be induced in dendritic cells and macrophages by inflammatory stimuli (32, 33). Poly(I:C) stimulation affected miRNA expression more potently than influenza infection for nearly all miRNAs, which is consistent with the overall weaker effect of influenza infection on global gene expression in dendritic cells (data not shown). Double-stranded RNA, either from viruses or polyI:C, is a well-characterized agonist of dendritic cell responses. However, it is unclear how additional aspects of viral infection, including expression of viral proteins or disruption of cellular processes by the viral life cycle, regulate host transcription. The induction of miR-451 expression by influenza infection and not poly(I:C) stimulation was therefore notable (Fig. 1A). miR-451 regulates erythroid lineage differentiation (26, 34–36) and has not been characterized within dendritic cells.
Figure 1. Viral stimulation regulates microRNA expression in dendritic cells.
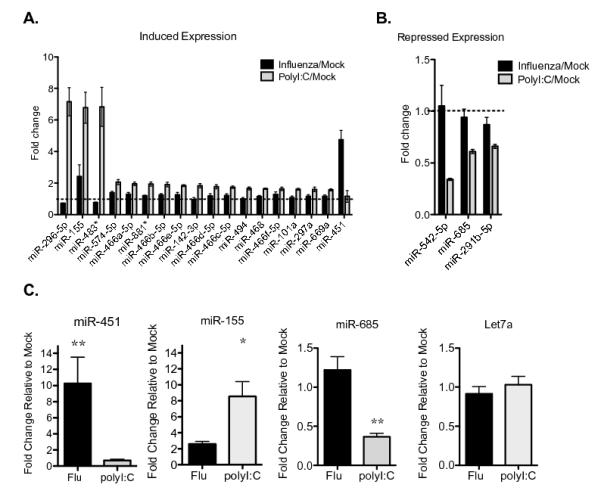
(A) The expression of 578 miRNAs was measured in splenic dendritic cells infected with influenza or stimulated with poly(I:C) for 8 hours in vitro and compared with unstimulated cells cultured in parallel (mock). The means ± SEM for three independent biological replicates were calculated, and the fold change relative to unstimulated cells is shown. The dotted line indicates where expression is equivalent to unstimulated cells. miRNAs with a ≥1.5-fold increase in expression in stimulated relative to unstimulated cells and p<0.001 are shown; n=3. (B) The miRNAs with a ≥1.5 mean fold decrease in expression and p<0.001 following stimulation are shown, as described in A; n=3. (C) Quantitative RT-PCR was performed on miRNA isolated from splenic dendritic cells infected with influenza, stimulated with poly(I:C), or cultured in parallel without stimulation for 8 h. Expression of the indicated miRNAs was normalized to sno202 expression, and the data are displayed as fold change in stimulated cells relative to mock; means ± SEM for n=3-8 independent biological experiments are shown. *p<0.05, **p<0.01.
We used quantitative RT-PCR to confirm that miR-451 is increased following influenza infection for 8 hours (Fig. 1C). We also validated the increased expression of miR-155, which is more strongly induced by poly(I:C) when compared with influenza, the decreased expression of miR-685 by poly(I:C) more than by influenza infection, and the stable expression of Let-7a, which was not altered by stimulation in the miRNA array profiling dataset (Fig 1C). We explored the range of stimuli that increase miR-451 expression in primary dendritic cells, using doses that trigger robust transcriptional responses in dendritic cells. There was not a requirement for live virus, as both live and UV-inactivated influenza induced miR-451 (Fig 2A). miR-451 expression was increased most strongly by the H1N1 influenza strain PR8, the cytokines type I interferon (IFN-β) or IL-6, but not by a variety of TLR agonists (poly(I:C), LPS, R848) relative to mock-treated cells (Fig. 2A). Induction of miR-451 was partially dependent on type I interferon signaling: IFNARnull dendritic cells, which cannot respond to the type I interferons that are produced following influenza infection, exhibited lower miR-451 induction than wild-type cells (Fig 2B). To test whether elevated interferon production by influenza-infected dendritic cells could provide a possible mechanism for the high expression level of miR-451 in virally-infected cells compared with cells treated with poly(I:C) or R848, we measured type I and type III interferon expression. Influenza infection did not induce significantly more IFN-α, IFN-β, or IFN-λ compared with poly(I:C) or R848 stimulation (Fig. 2C and data not shown). While R848 is a well-established inducer of type I IFN expression via TLR7-MyD88-dependent signaling cascades, miR-451 is up-regulated in the absence MyD88 (Fig. 2D). miR-451 expression also increased in influenza-infected primary murine lung dendritic cells (Fig. 2E). The dendritic cells isolated from spleens and lungs are comprised of both myeloid and plasmacytoid dendritic cells. To determine which type of dendritic cell up-regulates miR-451 following influenza infection, we isolated myeloid (CD11c+PDCA1-) and plasmacytoid (PDCA1+) cells by FACS and observed that miR-451 was up-regulated in infected myeloid DCs (Fig. 2F). Dendritic cells express approximately 10-fold less miR-451 than the high levels measured in erythroid cells (Fig. 2G) (36). In erythroid cells, miR-451 and miR-144, encoded in the same bicistronic locus, are both expressed. In contrast, miR-144 expression was not detectable by microarray (GEO # GSE36316) or quantitative RT-PCR (data not shown) in dendritic cells.
Figure 2. miR-451 expression is more strongly induced by virus, IL-6, and type I IFN compared with purified pattern recognition receptor agonists.
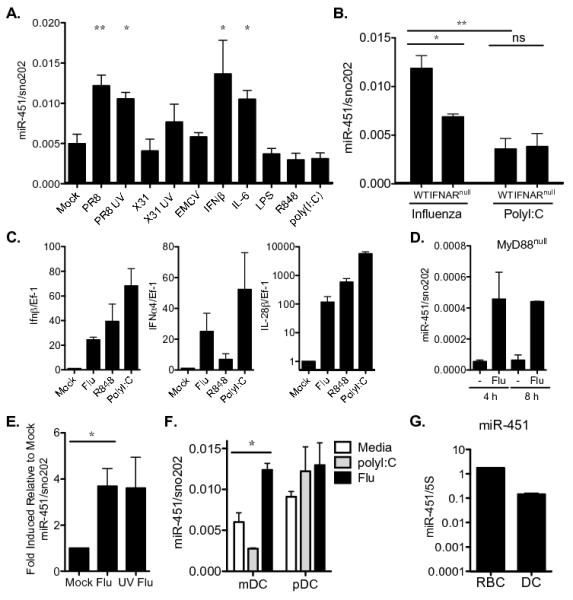
(A) Quantitative RT-PCR was performed on splenic dendritic cells stimulated with the indicated agonists for 6 h, and miR451 expression was normalized to sno202 expression. n=2 and are representative of ≥2 independent experiments. (B) Quantitative RT-PCR of miR-451 expression was performed as described in A on WT or IFNARnull splenic dendritic cells stimulated with the indicated agonists for 6 h; n=2 and are representative of ≥2 independent experiments. (C) Quantitative RT-PCR was performed on splenic dendritic cells stimulated with the indicated agonists for 4 h. Type I IFN (IFNβ1 and IFNα4) and type III IFN (IL-28β/IFN-λ) gene expression was normalized to Ef-1 and fold induction relative to mock-cultured cells are shown; n=3 independent experiments. (D) MyD88null splenic dendritic cells were cultured for 4 h or 8 h with or without influenza PR8 infection, and miR-451 expression quantified by qRT-PCR as described in A; n=2. (E) Quantitative RT-PCR was performed as described in A on primary lung dendritic cells treated with equivalent numbers of live or UV-inactivated influenza PR8 virions (MOI=10) for 8 h; means ± SEM for n=3-6 are shown. (F) Conventional myeloid (mDC, CD11c+PDCA1−) and plasmacytoid (pDC, PDCA1+) dendritic cells were isolated by FACS and infected with influenza, stimulated with poly(I:C), or cultured in media alone for 18 h. miR-451 expression was measured by qRT-PCR and normalized to sno202 expression; means ± SEM for n=3 are shown, p=0.01. (G) miR-451 expression was measured by qRT-PCR in splenic dendritic cells infected with influenza PR8 for 6 h and compared with JC4 red blood cells, and expression plotted relative to 5S rRNA expression. *p<0.05, **p<0.01.
Antagomir knock-down of miR-451 expression does not alter dendritic cell co-stimulatory activity
To determine the functional consequence of miR-451 expression by dendritic cells infected with influenza, we introduced an LNA-stabilized RNA oligonucleotide that is antisense to miR-451 (miR-451 antagomir) into primary splenic dendritic cells by nucleofection and compared responses to cells identically treated with a scrambled oligonucleotide control (control antagomir). We observed uniform uptake of the FITC-conjugated antagomirs (Fig. 3A) and functional inhibition of miR-451 expression following treatment with the miR-451 antagomir by quantitative RT-PCR after 21 h (Fig. 3B). miR-451 levels were decreased by >99% based on the detection limit of the assay. miR-451 antagomir treatment did not decrease expression levels of other miRNAs compared with control antagomir treatment (data not shown) and did not decrease miR-451 levels when RNA was isolated immediately after nucleofection and before the antagomir could lead to degradation of its target (Fig. 3B). These controls confirm the specificity of the RT-PCR assay and show that the presence of the antagomir does not simply inhibit the qRT-PCR reaction. Cells responded to the process of nucleofection of RNA antagomirs, in which RNA is physically delivered to the cytosol, by inducing miR-451 expression (Fig. 3C). This induction of miR-451 expression is less than that produced by influenza infection and more than that produced by poly(I:C) treatment (Fig. 1C).
Figure 3. Antagomir knock-down of miR-451 expression does not alter dendritic cell antigen presentation.

(A) FITC-labeled microRNA antagomirs were delivered to splenic dendritic cells by nucleofection and their uptake was assessed by flow cytometry after 24 h of culture (filled histogram) relative to mock-treated cells (unfilled histogram). Data are representative of 3 independent experiments. (B) miR-451 antagomir or control antagomir were delivered to splenic dendritic cells by nucleofection. RNA was isolated immediately following nucleofection, allowing insufficient time for the antagomir to interact specifically with target miRNA in order to assess whether the presence of antagomirs inhibit the qRT-PCR reaction (0 h). Cells were also cultured for 21 h prior to RNA isolation, which allowed for time for specific miRNA-antagomir interactions (21 h). Quantitative RT-PCR was performed and miR-451 expression levels are shown relative to sno202 expression levels. ND = not detected as below the detection limit of the assay. Data are representative of ≥ 3 independent experiments. (C) Cells were nucleofected with either control antagomir, antagomir specific for miR-685, or untreated, then cultured for 21 h. miR-451 expression levels were quantified by qRT-PCR as described in B, normalized to sno202 expression levels and shown relative to cells that did not undergo nucleofection. Means ± SEM are shown for 3 independent experiments. (D) Splenic dendritic cells from wild type or miR-451null mice or wild type cells treated with antagomirs were infected with influenza for 24 h. Cell activation was assessed by staining for surface expression of MHCII or CD80 and overlay plots are shown for cells with normal (dark line) or reduced (light line) miR-451 expression. Data are representative of 3 independent experiments. (E) Splenic dendritic cells were isolated from WT or miR-451null mice and wild type cells treated with miR-451 antagomir or control antagomir where indicated. Dendritic cells were infected with influenza, pulsed with ova peptide SIINFEKL, and cultured with CD8+ OT-1 T cells for 3 days. IFN-γ was measured in the supernatants of three biological replicates and the means ± SEM are shown. *p<0.05.
Antigen presentation is a principal function of dendritic cells. We observed that wild type dendritic cells treated with miR-451 antagomir or control antagomir or cells derived from wild type and miR-451null mice were similar in their ability to activate T cells. The surface expression of two molecules necessary for antigen presentation to CD4+ T cells, MHCII and the co-stimulatory molecule CD80, was independent of miR-451 expression (Fig. 3D). Loss of miR-451, by either antagomir treatment or genetic deletion, did not alter dendritic cell survival (data not shown). Inhibition of miR-451 expression did not alter the T cell stimulatory capacity of influenza-infected dendritic cells, as assessed by IFN-γ production (Fig. 3E) or CFSE dilution by antigen-specific CD8+ T lymphocytes (data not shown), and similar results were obtained following poly(I:C) stimulation (data not shown).
miR-451 regulates the production of a distinct set of cytokines by influenza-infected dendritic cells
Inhibition of miR-451 expression had a striking effect on cytokine production by primary splenic dendritic cells following influenza infection in vitro. Levels of secreted IL-6, TNF, CCL5/RANTES, and CCL3/MIP-1α were all consistently increased in cells treated with miR-451 antagomir relative to cells treated with control antagomir (Fig. 4A). Additionally, in some experiments, secretion of CXCL2/MIP-2, CCL4/MIP-1β, CXCL1/KC, and CCL2/MCP-1 by cells treated with miR-451 antagomir was also elevated relative to control-treated cells. In contrast, there were no significant differences in the secretion of CXCL10/IP-10, IL-1β, CXCL9/MIG, GM-CSF, IL-10, CCL11/Eotaxin, IL-9, IL-13, IL-7, IL-15, or G-CSF (Supplemental Fig. 1). Cytokine secretion by dendritic cells treated with miR-451 antagomir and control antagomir was equivalent following stimulation with poly(I:C), an agonist that does not induce miR-451 expression (Fig. 4A).
Figure 4. miR-451 regulates the production of a distinct set of cytokines by influenza-infected dendritic cells.
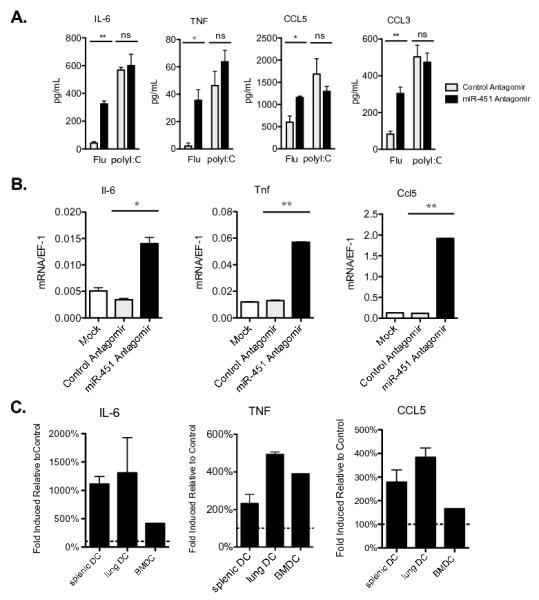
(A) Primary splenic dendritic cells treated with miR-451 or control antagomirs were infected with influenza or treated with poly(I:C) for 24 h and secreted cytokine levels were quantified by ELISA. Means ± SEM for n≥3 experiments are shown. (B) Splenic dendritic cells were treated with antagomir and infected with influenza as described in A, RNA isolated after 24 h, qRT-PCR performed, and gene expression was plotted relative to Ef-1 expression. Means ± SEM for n=3 experiments are shown. (C) Primary splenic and lung dendritic cells and Flt3L-derived bone marrow dendritic cells were treated with miR-451 antagomir or control antagomir prior to infection with influenza for 24 h. Cytokines were measured in culture supernatants by ELISA. The data are displayed as the fold-increase in secreted cytokine levels in cells treated with miR-451 antagomir relative to control antagomir for each type of dendritic cell. Means ± SEM for n=3 experiments are shown. *p<0.05, **p<0.01.
miRNAs can negatively regulate protein levels by blocking translation or by targeting mRNAs for degradation. To explore the mechanism of action of miR-451, quantitative RT-PCR was performed and increased steady-state mRNA levels of Il-6, Tnf, and Ccl5/Rantes mRNA were measured in cells treated with antagomir specific for miR-451 (Fig. 4B). To establish whether miR-451 negatively regulates expression of this group of cytokines in multiple types of dendritic cells, we performed ELISAs on supernatants collected from murine splenic, lung, and Flt3L-derived bone marrow dendritic cells 24 h after influenza infection and measured increased levels of IL-6, TNF, and CCL5/RANTES secreted by all three types of dendritic cells (Fig. 4C). We also detected increased levels of IFN-β mRNA and protein secretion by miR-451 antagomir-treated dendritic cells in response to influenza infection (Fig. 5A-B). We measured the expression of a panel of type I IFN-regulated genes and identified an increased type I IFN signature in miR-451 antagomir treated cells (Fig. 5A). This did not correlate with improved antiviral capacity to limit viral transcription in miR-451 antagomir-treated cells, as viral RNA loads were equivalent to those in control cells (Fig. 5C).
Figure 5. miR-451 knockdown increases type I interferon production and interferon-regulated antiviral gene expression but does not impact dendritic cell antiviral capacity.
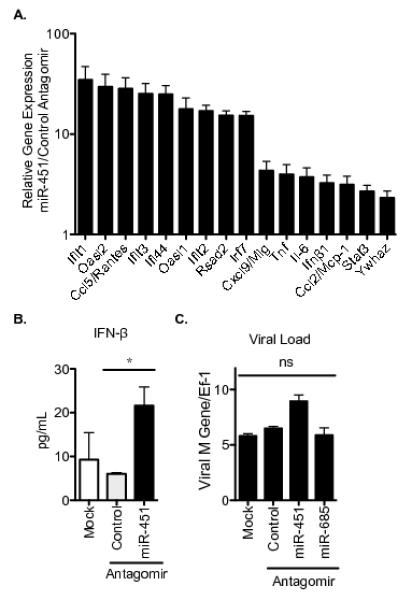
(A) Cells were treated with miR-451 antagomir or control antagomir and infected with influenza. RNA was isolated after 21 h and qRT-PCR performed using gene specific primers and normalized to Ef-1 expression. Data is plotted as the mean expression level in miR-451 antagomir-treated cells relative to control antagomir. n=2 and representative of ≥2 experiments. (B) Cells were treated as described in A for 21 h and levels of secreted IFN-β were quantified by ELISA. Means ± SEM for 3 independent experiments are shown. **p<0.01. (C) Cells were treated as described in A or treated with the miR-685 antagomir as a negative control. RNA was isolated after 2 h (confirming equivalent initial viral load) and after 24 h and qRT-PCR performed. Viral M gene relative to host Ef-1 was quantified and means ± SEM are shown; n=2, and representative of 3 experiments; ns = not significant, p>0.05.
The cytokine phenotype observed after miR-451 suppression by antagomir was confirmed in primary dendritic cells by comparing cytokine production in wild type and miR-451null dendritic cells, although the differences observed were more subtle. When we measured a panel of 26 cytokines and chemokines (measured in Figure 1 and Supplemental Figure 1), we observed increased secretion of IL-6, TNF, CXCL2/MIP-2, and CXCL1/KC by miR-451null dendritic cells following influenza infection (Fig. 6A). We measured analogous increases in Il6, Tnf, and Ifnβ at the mRNA level (Fig. 6B). To test whether antagomir treatment modulated dendritic cell cytokine production independently of the decrease in miR-451 expression, miR-451null splenic dendritic cells were treated with miR-451 antagomir or control antagomir and compared with wild type cells. IL-6 and CCL3/MIP-1α production in response to influenza infection was equivalent in miR-451null and wild type dendritic cells treated with miR-451 antagomir. Furthermore, the increase in secretion of these two cytokines was similar in miR-451null dendritic cells and wild-type dendritic cells treated with miR-451 antagomir (Supplemental Fig. 2A).
Figure 6. Confirmation of the miR-451 phenotype using miR-451null dendritic cells and gain of function studies.
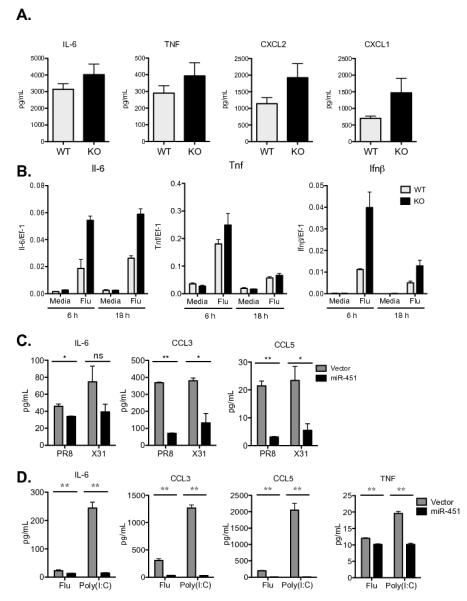
(A) Primary splenic dendritic cells isolated from wild type or miR-451null mice were infected with influenza for 24 hours and secreted cytokine levels were quantified by ELISA. Means ± SEM for n≥3 are shown. (B) Splenic dendritic cells were treated as described in A, RNA isolated after 6 h or 18 h, qRT-PCR performed, and the expression of Il-6, Tnf, and Ifn-β was plotted relative to EF-1 expression. Means ± SEM for n=3 are shown. (C) JAWS II dendritic cells over-expressing miR-451 or vector alone were infected with the influenza A strains PR8 or X31 for 24 h and secreted cytokines were quantified by ELISA. Means ± SEM for n=3 are shown. (D) Bone marrow-derived dendritic cells over-expressing miR-451 or vector alone were infected with the influenza A strain PR8 or stimulated with poly(I:C) for 21 h and secreted cytokines quantified by ELISA. Means ± SEM for n=3 are shown; **p<0.01.
Over-expression of miR-451 led to a reciprocal decrease in cytokine secretion following infection with two different influenza A viral strains. Stable lines of the JAWS II dendritic cells over-expressing miR-451 or vector alone were generated by retroviral transduction and drug selection. miR-451 over-expression impaired the secreted levels of IL-6, CCL3/MIP-1α and CCL5/RANTES (Fig. 6C), while TNF production by these cells was below the assay detection limit. Decreased levels of CXCL2/MIP2 and CCL4/MIP1β were also measured along with normal levels of all other assayed cytokines (data not shown). These data were corroborated using bone marrow-derived dendritic cells transduced to over-express miR-451, which resulted in decreased levels of IL-6, CCL3/MIP-1α, CCL5/RANTES, and TNF (Fig. 6D). While poly(I:C) does not induce miR-451 expression or result in differential cytokine production in miR-451 antagomir-treated cells (Fig. 4A), poly(I:C) stimulation of cells engineered to over-express miR-451 results in increased cytokine secretion compared with control cells (Fig. 6D).
miR-451 regulates cytokine expression by targeting Ywhaz
We and others have demonstrated that miR-451 targets the 3′-UTR of Ywhaz/14-3-3ζ mRNA in multiple cell types (26, 35, 37, 38). In agreement with these studies, YWHAZ protein levels were significantly increased in miR-451null dendritic cells compared with wild type cells (Fig. 7A). Moreover, Ywhaz mRNA was increased by miR-451 antagomir treatment in dendritic cells (Fig. 5). Thus, expression of miR-451 correlates with suppression of Ywhaz mRNA and protein expression in dendritic cells, similar to what we reported for erythroid cells (39). YWHAZ belongs to the 14-3-3 family of phosphoserine binding proteins that interact with functionally diverse signaling proteins to modulate their activities. YWHAZ can bind and sequester ZFP36 and FOXO3 from interacting with nucleic acids, thereby inhibiting the actions of these two negative regulators of IL-6 and TNF expression (20, 28). YWHAZ sequesters FOXO3 in the cytoplasm, preventing it from repressing transcription in the nucleus (28, 29). ZFP36 promotes mRNA destabilization by binding to AU-rich elements in the 3′-UTRs in some inflammatory cytokine mRNAs, including TNF, IL-6, and CCL3, and is negatively regulated by interactions with YWHAZ (27, 30). ZFP36 protein levels were reduced in miR-451null cells, which have increased YWHAZ expression (Fig. 7A). We measured increased phosphorylation of FOXO3 on Ser294, which targets this protein for degradation, and decreased nuclear translocation of FOXO3, both of which reflect decreased FOXO3 activity in cells with increased YWHAZ levels (Fig. 7A).
Figure 7. miR-451 negatively regulates Ywhaz expression in dendritic cells.
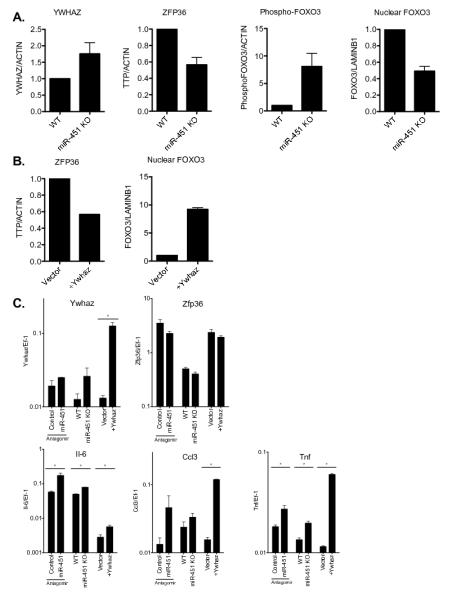
(A) Whole cell lysates were prepared from splenic dendritic cells isolated from wild type or miR-451null mice and infected in vitro with influenza PR8 for 6 hours. Western blots were performed using antibodies specific for YWHAZ, ZFP36, or phosphorylated FOXO3, signals quantified using densitometry, normalized to the expression level of actin, and displayed relative to the levels in wild type cells. Nuclear extracts were prepared from cells treated under identical conditions, probed for FOXO3 expression, normalized to the level of LaminB1, and displayed relative to the levels in wild type cells. Means ± SEM are shown for 2-7 samples. (B) Whole cell lysates or nuclear extracts were prepared from splenic dendritic cells stably expressing YWHAZ or vector alone, infected in vitro with influenza PR8 for 6 hours, and Western blotting performed as described in A. Means ± SEM; n=2. (C) Splenic dendritic cells were prepared from wild type or miR-451null mice, wild type cells were treated with control antagomir or miR-451 antagomir, or transduced to over-express YWHAZ or vector alone. Following infection with influenza PR8 for 24 hours, RNA was isolated and quantitative RT-PCR was performed. Gene expression was calculated relative to Ef-1 and means ± SEM are shown for 2-3 samples. * denotes p<0.05.
We have shown that decreased expression of miR-451 correlates with increased YWHAZ expression, decreased nuclear FOXO3, reduced ZFP36 expression, and increased levels of IL-6, TNF, CCL3/MIP1α and CCL5/RANTES. While causal relationships between these observations have been established in other systems, YWHAZ has not been directly connected to FOXO3 or ZFP36 activities or cytokine expression in dendritic cells. We were unable to investigate these mechanisms through YWHAZ knockdown experiments due to observed nonspecific effects of shRNAs on IL-6 secretion in dendritic cells (Supplemental Fig. 2C). However, overexpression of Ywhaz by retroviral transduction in Flt3L-expanded bone marrow-derived dendritic cells decreased ZFP36 protein, similar to what we observed in miR-451null cells where Ywhaz is de-repressed. In contrast, FOXO3 nuclear localization, was increased in cells overexpressing YWHAZ (Fig. 7B), which is not concordant with our observation that reduced miR-451 levels correlate with increased YWHAZ expression and decreased nuclear localization of FOXO3 (Fig. 7A). Figure 7C shows comparable regulation of Ywhaz, Zfp36, IL-6, Ccl3/Mip1a, and Tnf in cells with decreased miR-451 levels using antagomirs or genetic ablation, or cells exogenously over-expressing Ywhaz.
These data can be synthesized into a working model shown in Figure 8. Influenza infection increases miR-451 expression, which dampens pro-inflammatory cytokine expression. Our data supports a model whereby miR-451 targets the 3′-UTR of YWHAZ to decrease protein levels. This relieves a brake on ZFP36 activity, which is a negative regulator of pro-inflammatory cytokine expression. Poly(I:C) stimulation of cells, which does not increase miR-451 levels, yields no miR-451-dependent cytokine phenotype in antagomir-treated cells. In cells with low miR-451 expression, increased YWHAZ protein and mRNA levels correlate with decreased ZFP36 levels, and increased cytokine expression. Increasing YWHAZ expression by transduction resulted in an analogous decrease in ZFP36 and increase in IL-6 and TNF expression. These IL-6 and IFN-β levels can positively regulate miR-451 expression, which together serves to buffer cytokine expression.
Figure 8. miR-451 regulates a negative cascade to modulate pro-inflammatory cytokine expression.
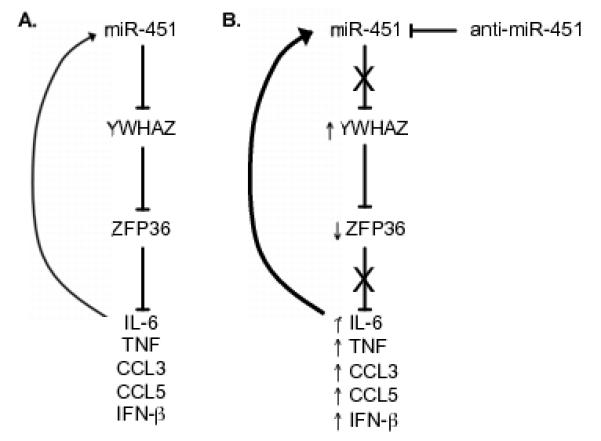
(A) miR-451 targets the 3′-UTR of YWHAZ to decrease protein levels. YWHAZ binds ZFP36 to decrease binding to AU-rich elements in the 3′-UTR of the proinflammatory cytokines TNF, IFN-β, and IL-6 and the chemokines CCL3 and CCL5. IL-6 and IFN-β positively regulate miR-451 expression in dendritic cells. (B) Antagomir data supports the following model for the observed increased expression of IL-6, TNF, CCL3, CCL5, and IFN-β in primary dendritic cells treated with miR-451 antagomir. Antagomir treatment increases YWHAZ protein and mRNA levels, which correlates with decreased ZFP36 levels. Increasing YWHAZ expression by transduction resulted in an analogous decrease in ZFP36 and increase in IL-6, TNF, and chemokine expression. Increased IL-6 and IFN-β levels can positively regulate miR-451 expression while miR-451 expression culminates in decreased IL-6 expression, which together serves to buffer IL-6 expression.
Discussion
miRNAs provide a layer of post-transcriptional regulation of dendritic cell responses to infection. Global profiling of differential miRNA expression in primary dendritic cells responding to live viral infection or a purified RNA agonist of the TLR and RLR detection pathways resulted in the novel description of 16 up-regulated and 3 down-regulated miRNAs and confirmed the known induction of miR-155 expression (32, 33)(Fig. 1). The consequence of regulated expression of these miRNAs remains to be assessed using gain-of-function, loss-of-function, and mechanistic studies, as utilized here to characterize the relevance of miR-451 expression. It was also noteworthy that the expression of the vast majority of miRNAs was unaffected in response to these stimuli. Induction of miR-451 expression was unique to influenza-infected cells, and a transcriptional response specific to influenza infection that cannot be recapitulated by purified agonists is unusual. While we observed that this virus-induced response was dampened in IFNARnull cells and addition of exogenous IL-6, IFN-β or UV-inactivated virus could also induce miR-451 expression (Fig. 2), future studies are required to characterize both how dendritic cells recognize influenza infection and the signaling leading to up-regulated miR-451 expression. Interestingly, nucleofection of any short RNA antagomir also increased miR-451 levels (Fig. 3), raising the possibility that cells recognize and respond to live viral infection and physical delivery of RNA into the cytosol using similar mechanisms. Dendritic cells respond to fusion between viral and cellular membranes during infection by inducing expression of interferon-stimulated genes, and this fusion-dependent response was dependent on the adaptor STING (40), providing a potential mechanism underlying our observation that live and UV-killed influenza induces miR-451 expression.
It is known that antigen presentation and cytokine production are two features of DC maturation that can be uncoupled, highlighting the existence of distinct control points of DC activation. We observed increased pro-inflammatory cytokine production by miR-451null cells or cells treated with miR-451 antagomirs compared with their wild-type counterparts, but saw no effect on MHCII or co-stimulatory molecule expression nor differences in the ability of these cells to activate antigen-specific CD8+ T cells (Fig. 3). While the studies comparing antagomirs and miR451 knockout cells were concordant in affecting the same subset of cytokines (Fig. 7C), the miR-451-regulated cytokine phenotype was consistently larger in the antagomir studies. This is reminiscent of the larger effect of antagomir-mediated miR-451 knockdown on erythropoiesis observed when compared with genetic knockout (41). Many miRNAs regulate developmental pathways, so dendritic cells that develop in the absence of miR-451may activate compensatory pathways, leading to differences in response magnitude when compared with normally differentiated wild type dendritic cells that have antagomir-mediated knockdown of miR-451 levels. The cytokine levels that are tuned by miR-451 play critical roles in the immune system. IL-6 and TNF drive acute inflammatory responses during sepsis as well as serve a progressive role in chronic inflammatory diseases such as rheumatoid arthritis, and TNF-blocking antibodies are a standard of care for treating rheumatoid arthritis. Type I interferons activate expression of a module of antiviral effectors during acute viral infections as well as during underlying chronic inflammatory diseases such as lupus and type I diabetes (42, 43). CCL5/RANTES is a potent chemoattractant for T cells along with eosinophils and basophils and CCL3/MIP1α chemoattracts neutrophils and monocytes. miR-451 knockdown had inconsistent effects on increasing secretion of other chemokines (CCL4/MIP-1β, CXCL2/MIP2, CXCL9/MIG, and CXCL1/KC), with differences reaching statistical significance in some experiments but not all. We observed specificity in the cytokines affected by miR-451 expression, which did not alter secretion of IL-10, eotaxin, and IL-1β. Together, the pro-inflammatory cytokines and chemokines that are more highly expressed in the absence of miR-451 activate a broad range of inflammatory cells, highlighting the importance of stringent regulatory mechanisms.
miR-451 has been shown in multiple systems to inhibit YWHAZ protein expression (35, 37–39). Increased levels of YWHAZ in cells with reduced miR-451 levels following antagomir treatment or genetic ablation can be linked to increased levels of inflammatory cytokine expression by two possible mechanisms. YWHAZ binds two inducible attenuators of dendritic cell activation: ZFP36 and FOXO3. Zfp36 expression is highly induced by stimulation with TLR agonists or cytokines such as TNF or type I IFN (21). ZFP36 is an RNA binding protein that targets AU-rich mRNAs such as TNF, IL-6, and CCL3/MIP1α for degradation. YWHAZ has been shown in multiple cell types to sequester ZFP36 from binding target mRNAs. Increasing YWHAZ levels by genetic over-expression or miR-451 antagomir treatment correlated with decreased ZFP36 levels (Fig. 7A-C). YWHAZ is also a known negative regulator of FOXO3, a transcription factor that interacts with the transcriptional machinery to negatively regulate transcription of cytokines such as IL-6 and TNF (20, 44). FOXO3 is controlled by four mechanisms: transcription, post-translational modifications, cellular localization, and degradation (45). FOXO3 expression is induced following TLR stimulation and phosphorylation of FOXO3 can lead to degradation or binding to YWHAZ, which excludes FOXO3 from the nucleus to prevent transcriptional repression (28). We have previously demonstrated that miR-451 expression increased FOXO3 activity (39). In the current study, we detected a similar positive correlation between miR-451 and FOXO3 nuclear localization (and thereby activity). However, over-expression of YWHAZ did not lend mechanistic support to this observed correlation. This could result from a technical limitation, due to the sensitivity of dendritic cells to perturbation by the experimental manipulation of gene expression, or result from a unique pattern of regulation of FOXO3 by YWHAZ in dendritic cells. Our data proposes one miR-451 target that could explain the observed cytokine phenotype, but does not exclude a role for other YWHAZ binding partners or other miR-451 target mRNAs that could contribute to our observed miR-451-dependent cytokine phenotype.
miR-451 plays an important role in erythroid lineage differentiation and miR-451 expression had not previously been described in dendritic cells. miR-144 is encoded on the same pri-miRNA transcript as miR-451 and the two mature miRNAs are co-expressed in erythroid; in contrast, while pri-miR-451 and pri-miR-144 could both be measured in dendritic cells, mature miR-144 expression was undetectable in dendritic cells (data not shown). miR-451 is the only miRNA that can be processed by Ago2 in the absence of Dicer (46)(47)(48), and differences in miRNA processing between myeloid and erythroid cells provide a possible mechanism for the unique expression pattern of processed mature miR-451 and miR-144. Alternatively, miR-451 is expressed at much higher levels than miR-144 in erythroid cells (36, 39). It could be that the locus is expressed and processed similarly in both erythroid and dendritic cells but that mir-144 is found at sub-threshold levels in the latter because of lower overall expression of the locus. miR-451 targeting of Ywhaz is conserved between erythroid and dendritic cells yet the consequences, erythroid differentiation and resistance to oxidant stress and dendritic cell pro-inflammatory cytokine production, are distinct. The cell lineage-specific expression of YWHAZ binding partners (ZFP36 is highly expressed in activated dendritic cells) and distinct FOXO3-dependent transcriptional programs (erythroid cells lack IL-6 and TNF expression) could explain the different phenotypic effects of miR-451 expression between cell types.
miR-451, ZFP36, and FOXO3 are all transcriptionally induced by viral stimuli, and the phenotypes of Zfp36null and Foxo3null mice show that their contribution to negative feedback is more important than providing a homeostatic restraint on inflammation (20, 22). miRNAs have modest effects on the protein levels of targets, decreasing concentrations by an average of 2-fold (49, 50). The phenotypic effects of miRNAs can be larger than their effects on steady-state protein levels when target expression is at a threshold, when they target multiple members of the same pathway, or when multiple miRNAs bind to a given mRNA target (13). Signaling molecules and transcription factors are both particularly sensitive to changes in concentration (11). miRNAs can therefore more potently inhibit cytokine transcripts by targeting the regulators of cytokine gene expression. Here, we characterize a miRNA that targets a multifunctional protein, YWHAZ, that is present in limiting quantity (25), providing another mechanism for amplifying the effect of small modulations in protein levels.
Cells most efficiently process information using biochemical networks with high sensitivity to changes in input signals and low sensitivity to stochastic fluctuations. Regulatory circuits enable cells to buffer propagated noise while maintaining sensitivity to signals (51). While positive regulatory circuits are instrumental in cellular differentiation programs, negative regulatory circuits generate homeostasis or oscillatory behavior. Negative feedback can serve as a buffer by attenuating noise as well as providing stability by limiting the range over which the concentrations of network components can fluctuate (52, 53). Our data adds a third negative regulator in a negative feedback cascade that buffers pro-inflammatory cytokine secretion by influenza-infected dendritic cells. It will be interesting to determine whether induction of miR-451 by influenza infection and the correlating restraint on cytokine secretion offers a net benefit to the host or pathogen.
Supplementary Material
Acknowledgements
We thank A. Diercks, V. Litvak, K. Kennedy and A. Lampano for advice and S. Fallen and R. Suen for technical assistance.
Footnotes
This work was funded by NIH grants #HHSN272200800058C Systems Influenza Contract and HHSN272200700038C Systems Immunology Contract.
The authors have no conflicting financial interests.
References
- 1.Amit I, Garber M, Chevrier N, Leite AP, Donner Y, Eisenhaure T, Guttman M, Grenier JK, Li W, Zuk O, Schubert LA, Birditt B, Shay T, Goren A, Zhang X, Smith Z, Deering R, McDonald RC, Cabili M, Bernstein BE, Rinn JL, Meissner A, Root DE, Hacohen N, Regev A. Unbiased reconstruction of a mammalian transcriptional network mediating pathogen responses. Science. 2009;326:257–263. doi: 10.1126/science.1179050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chevrier N, Mertins P, Artyomov MN, Shalek AK, Iannacone M, Ciaccio MF, Gat-Viks I, Tonti E, DeGrace MM, Clauser KR, Garber M, Eisenhaure TM, Yosef N, Robinson J, Sutton A, Andersen MS, Root DE, von Andrian U, Jones RB, Park H, Carr SA, Regev A, Amit I, Hacohen N. Systematic discovery of TLR signaling components delineates viral-sensing circuits. Cell. 2011;147:853–867. doi: 10.1016/j.cell.2011.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pichlmair A, Schulz O, Tan CP, Näslund TI, Liljeström P, Weber F, Reis e Sousa C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science. 2006;314:997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- 4.Le Goffic R, Pothlichet J, Vitour D, Fujita T, Meurs E, Chignard M, Si-Tahar M. Cutting Edge: Influenza A virus activates TLR3-dependent inflammatory and RIG-Idependent antiviral responses in human lung epithelial cells. J. Immunol. 2007;178:3368–3372. doi: 10.4049/jimmunol.178.6.3368. [DOI] [PubMed] [Google Scholar]
- 5.Loo Y-M, Fornek J, Crochet N, Bajwa G, Perwitasari O, Martinez-Sobrido L, Akira S, Gill MA, García-Sastre A, Katze MG, Gale M., Jr Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J. Virol. 2008;82:335–345. doi: 10.1128/JVI.01080-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh C-S, Reis e Sousa C, Matsuura Y, Fujita T, Akira S. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 7.Guillot L, Le Goffic R, Bloch S, Escriou N, Akira S, Chignard M, Si-Tahar M. Involvement of toll-like receptor 3 in the immune response of lung epithelial cells to double-stranded RNA and influenza A virus. J. Biol. Chem. 2005;280:5571–5580. doi: 10.1074/jbc.M410592200. [DOI] [PubMed] [Google Scholar]
- 8.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of doublestranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 9.Sun Q, Sun L, Liu H-H, Chen X, Seth RB, Forman J, Chen ZJ. The specific and essential role of MAVS in antiviral innate immune responses. Immunity. 2006;24:633–642. doi: 10.1016/j.immuni.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 10.Kumar H, Kawai T, Kato H, Sato S, Takahashi K, Coban C, Yamamoto M, Uematsu S, Ishii KJ, Takeuchi O, Akira S. Essential role of IPS-1 in innate immune responses against RNA viruses. J. Exp. Med. 2006;203:1795–1803. doi: 10.1084/jem.20060792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leung AKL, Sharp PA. MicroRNA functions in stress responses. Mol. Cell. 2010;40:205–215. doi: 10.1016/j.molcel.2010.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mukherji S, Ebert MS, Zheng GXY, Tsang JS, Sharp PA, van Oudenaarden A. MicroRNAs can generate thresholds in target gene expression. Nat. Genet. 2011;43:854–859. doi: 10.1038/ng.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xiao C, Rajewsky K. MicroRNA control in the immune system: basic principles. Cell. 2009;136:26–36. doi: 10.1016/j.cell.2008.12.027. [DOI] [PubMed] [Google Scholar]
- 15.Skalsky RL, Cullen BR. Viruses, microRNAs, and host interactions. Annu. Rev. Microbiol. 2010;64:123–141. doi: 10.1146/annurev.micro.112408.134243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boldin MP, Taganov KD, Rao DS, Yang L, Zhao JL, Kalwani M, Garcia-Flores Y, Luong M, Devrekanli A, Xu J, Sun G, Tay J, Linsley PS, Baltimore D. miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J. Exp. Med. 2011;208:1189–1201. doi: 10.1084/jem.20101823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chassin C, Kocur M, Pott J, Duerr CU, Gütle D, Lotz M, Hornef MW. miR-146a mediates protective innate immune tolerance in the neonate intestine. Cell Host Microbe. 2010;8:358–368. doi: 10.1016/j.chom.2010.09.005. [DOI] [PubMed] [Google Scholar]
- 18.Jansen BJH, Sama IE, Eleveld-Trancikova D, van Hout-Kuijer MA, Jansen JH, Huynen MA, Adema GJ. MicroRNA genes preferentially expressed in dendritic cells contain sites for conserved transcription factor binding motifs in their promoters. BMC Genomics. 2011;12:330. doi: 10.1186/1471-2164-12-330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Naka T, Fujimoto M, Tsutsui H, Yoshimura A. Negative regulation of cytokine and TLR signalings by SOCS and others. Adv. Immunol. 2005;87:61–122. doi: 10.1016/S0065-2776(05)87003-8. [DOI] [PubMed] [Google Scholar]
- 20.Dejean AS, Beisner DR, Ch’en IL, Kerdiles YM, Babour A, Arden KC, Castrillon DH, DePinho RA, Hedrick SM. Transcription factor Foxo3 controls the magnitude of T cell immune responses by modulating the function of dendritic cells. Nat. Immunol. 2009;10:504–513. doi: 10.1038/ni.1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sauer I, Schaljo B, Vogl C, Gattermeier I, Kolbe T, Müller M, Blackshear PJ, Kovarik P. Interferons limit inflammatory responses by induction of tristetraprolin. Blood. 2006;107:4790–4797. doi: 10.1182/blood-2005-07-3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kratochvill F, Machacek C, Vogl C, Ebner F, Sedlyarov V, Gruber AR, Hartweger H, Vielnascher R, Karaghiosoff M, Rülicke T, Müller M, Hofacker I, Lang R, Kovarik P. Tristetraprolin-driven regulatory circuit controls quality and timing of mRNA decay in inflammation. Mol. Syst. Biol. 2011;7:560. doi: 10.1038/msb.2011.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blackshear PJ. Tristetraprolin and other CCCH tandem zinc-finger proteins in the regulation of mRNA turnover. Biochem. Soc. Trans. 2002;30:945–952. doi: 10.1042/bst0300945. [DOI] [PubMed] [Google Scholar]
- 24.Kang J-G, Amar MJ, Remaley AT, Kwon J, Blackshear PJ, Wang P, Hwang PM. Zinc finger protein tristetraprolin interacts with CCL3 mRNA and regulates tissue inflammation. J. Immunol. 2011;187:2696–2701. doi: 10.4049/jimmunol.1101149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fu H, Subramanian RR, Masters SC. 14-3-3 proteins: structure, function, and regulation. Annu. Rev. Pharmacol. Toxicol. 2000;40:617–647. doi: 10.1146/annurev.pharmtox.40.1.617. [DOI] [PubMed] [Google Scholar]
- 26.Yu D, dos Santos CO, Zhao G, Jiang J, Amigo JD, Khandros E, Dore LC, Yao Y, D’Souza J, Zhang Z, Ghaffari S, Choi J, Friend S, Tong W, Orange JS, Paw BH, Weiss MJ. miR-451 protects against erythroid oxidant stress by repressing 14-3-3zeta. Genes Dev. 2010;24:1620–1633. doi: 10.1101/gad.1942110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stoecklin G, Stubbs T, Kedersha N, Wax S, Rigby WFC, Blackwell TK, Anderson P. MK2-induced tristetraprolin:14-3-3 complexes prevent stress granule association and ARE-mRNA decay. EMBO J. 2004;23:1313–1324. doi: 10.1038/sj.emboj.7600163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tzivion G, Dobson M, Ramakrishnan G. FoxO transcription factors; Regulation by AKT and 14-3-3 proteins. Biochim. Biophys. Acta. 2011;1813:1938–1945. doi: 10.1016/j.bbamcr.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 29.Dobson M, Ramakrishnan G, Ma S, Kaplun L, Balan V, Fridman R, Tzivion G. Bimodal regulation of FoxO3 by AKT and 14-3-3. Biochim. Biophys. Acta. 2011;1813:1453–1464. doi: 10.1016/j.bbamcr.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun L, Stoecklin G, Van Way S, Hinkovska-Galcheva V, Guo R-F, Anderson P, Shanley TP. Tristetraprolin (TTP)-14-3-3 complex formation protects TTP from dephosphorylation by protein phosphatase 2a and stabilizes tumor necrosis factor-alpha mRNA. J. Biol. Chem. 2007;282:3766–3777. doi: 10.1074/jbc.M607347200. [DOI] [PubMed] [Google Scholar]
- 31.Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, Iwasaki A, Flavell RA. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc. Natl. Acad. Sci. U.S.A. 2004;101:5598–5603. doi: 10.1073/pnas.0400937101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ceppi M, Pereira PM, Dunand-Sauthier I, Barras E, Reith W, Santos MA, Pierre P. MicroRNA-155 modulates the interleukin-1 signaling pathway in activated human monocyte-derived dendritic cells. Proc. Natl. Acad. Sci. U.S.A. 2009;106:2735–2740. doi: 10.1073/pnas.0811073106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martinez-Nunez RT, Louafi F, Friedmann PS, Sanchez-Elsner T. MicroRNA-155 modulates the pathogen binding ability of dendritic cells (DCs) by downregulation of DC-specific intercellular adhesion molecule-3 grabbing non-integrin (DCSIGN) J. Biol. Chem. 2009;284:16334–16342. doi: 10.1074/jbc.M109.011601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dore LC, Amigo JD, Dos Santos CO, Zhang Z, Gai X, Tobias JW, Yu D, Klein AM, Dorman C, Wu W, Hardison RC, Paw BH, Weiss MJ. A GATA-1-regulated microRNA locus essential for erythropoiesis. Proc. Natl. Acad. Sci. U.S.A. 2008;105:3333–3338. doi: 10.1073/pnas.0712312105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Patrick DM, Zhang CC, Tao Y, Yao H, Qi X, Schwartz RJ, Jun-Shen Huang L, Olson EN. Defective erythroid differentiation in miR-451 mutant mice mediated by 14-3-3zeta. Genes Dev. 2010;24:1614–1619. doi: 10.1101/gad.1942810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rasmussen KD, Simmini S, Abreu-Goodger C, Bartonicek N, Di Giacomo M, Bilbao-Cortes D, Horos R, Von Lindern M, Enright AJ, O’Carroll D. The miR-144/451 locus is required for erythroid homeostasis. J. Exp. Med. 2010;207:1351–1358. doi: 10.1084/jem.20100458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Z, Luo X, Ding S, Chen J, Chen T, Chen X, Zha H, Yao L, He X, Peng H. MicroRNA-451 regulates p38 MAPK signaling by targeting of Ywhaz and suppresses the mesangial hypertrophy in early diabetic nephropathy. FEBS Lett. 2012;586:20–26. doi: 10.1016/j.febslet.2011.07.042. [DOI] [PubMed] [Google Scholar]
- 38.Bergamaschi A, Katzenellenbogen BS. Tamoxifen downregulation of miR-451 increases 14-3-3ζ and promotes breast cancer cell survival and endocrine resistance. Oncogene. 2012;31:39–47. doi: 10.1038/onc.2011.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu D, dos Santos CO, Zhao G, Jiang J, Amigo JD, Khandros E, Dore LC, Yao Y, D’Souza J, Zhang Z, Ghaffari S, Choi J, Friend S, Tong W, Orange JS, Paw BH, Weiss MJ. miR-451 protects against erythroid oxidant stress by repressing 14-3-3zeta. Genes Dev. 2010;24:1620–1633. doi: 10.1101/gad.1942110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holm CK, Jensen SB, Jakobsen MR, Cheshenko N, Horan KA, Moeller HB, Gonzalez-Dosal R, Rasmussen SB, Christensen MH, Yarovinsky TO, Rixon FJ, Herold BC, Fitzgerald KA, Paludan SR. Virus-cell fusion as a trigger of innate immunity dependent on the adaptor STING. Nat. Immunol. 2012;13:737–743. doi: 10.1038/ni.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Papapetrou EP, Korkola JE, Sadelain M. A genetic strategy for single and combinatorial analysis of miRNA function in mammalian hematopoietic stem cells. Stem Cells. 2010;28:287–296. doi: 10.1002/stem.257. [DOI] [PubMed] [Google Scholar]
- 42.Baechler EC, Gregersen PK, Behrens TW. The emerging role of interferon in human systemic lupus erythematosus. Curr. Opin. Immunol. 2004;16:801–807. doi: 10.1016/j.coi.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 43.Heinig M, Petretto E, Wallace C, Bottolo L, Rotival M, Lu H, Li Y, Sarwar R, Langley SR, Bauerfeind A, Hummel O, Lee Y-A, Paskas S, Rintisch C, Saar K, Cooper J, Buchan R, Gray EE, Cyster JG, Erdmann J, Hengstenberg C, Maouche S, Ouwehand WH, Rice CM, Samani NJ, Schunkert H, Goodall AH, Schulz H, Roider HG, Vingron M, Blankenberg S, Münzel T, Zeller T, Szymczak S, Ziegler A, Tiret L, Smyth DJ, Pravenec M, Aitman TJ, Cambien F, Clayton D, Todd JA, Hubner N, Cook SA. A transacting locus regulates an anti-viral expression network and type 1 diabetes risk. Nature. 2010;467:460–464. doi: 10.1038/nature09386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang S-T, Chang C-C, Yen M-C, Tu C-F, Chu C-L, Peng Y-T, Chen D-Y, Lan J-L, Lin C-C. RNA interference-mediated silencing of Foxo3 in antigen-presenting cells as a strategy for the enhancement of DNA vaccine potency. Gene Ther. 2011;18:372–383. doi: 10.1038/gt.2010.146. [DOI] [PubMed] [Google Scholar]
- 45.Peng SL. Foxo in the immune system. Oncogene. 2008;27:2337–2344. doi: 10.1038/onc.2008.26. [DOI] [PubMed] [Google Scholar]
- 46.Cheloufi S, Dos Santos CO, Chong MMW, Hannon GJ. A dicerindependent miRNA biogenesis pathway that requires Ago catalysis. Nature. 2010;465:584–589. doi: 10.1038/nature09092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cifuentes D, Xue H, Taylor DW, Patnode H, Mishima Y, Cheloufi S, Ma E, Mane S, Hannon GJ, Lawson ND, Wolfe SA, Giraldez AJ. A novel miRNA processing pathway independent of Dicer requires Argonaute2 catalytic activity. Science. 2010;328:1694–1698. doi: 10.1126/science.1190809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang J-S, Maurin T, Robine N, Rasmussen KD, Jeffrey KL, Chandwani R, Papapetrou EP, Sadelain M, O’Carroll D, Lai EC. Conserved vertebrate mir-451 provides a platform for Dicer-independent, Ago2-mediated microRNA biogenesis. Proc. Natl. Acad. Sci. U.S.A. 2010;107:15163–15168. doi: 10.1073/pnas.1006432107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Selbach M, Schwanhäusser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008;455:58–63. doi: 10.1038/nature07228. [DOI] [PubMed] [Google Scholar]
- 50.Baek D, Villén J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of microRNAs on protein output. Nature. 2008;455:64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dublanche Y, Michalodimitrakis K, Kümmerer N, Foglierini M, Serrano L. Noise in transcription negative feedback loops: simulation and experimental analysis. Mol. Syst. Biol. 2006;2:41. doi: 10.1038/msb4100081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Becskei A, Serrano L. Engineering stability in gene networks by autoregulation. Nature. 2000;405:590–593. doi: 10.1038/35014651. [DOI] [PubMed] [Google Scholar]
- 53.Paulsson J. Summing up the noise in gene networks. Nature. 2004;427:415–418. doi: 10.1038/nature02257. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.