

Abstract
Replication origin licensing builds a fundamental basis for DNA replication in all eukaryotes. This occurs during the late M to early G1 phases in which chromatin is licensed by loading of the MCM2-7 complex, an essential component of the replicative helicase. In the following S phase, only a minor fraction of chromatin-bound MCM2-7 complexes are activated to unwind the DNA. Therefore, it is proposed that the vast majority of MCM2-7 complexes license dormant origins that can be used as backups. Consistent with this idea, it has been repeatedly demonstrated that a reduction (~60%) in chromatin-bound MCM2-7 complexes has little effect on the density of active origins. In this study, however, we describe the first exception to this observation. A reduction of licensed origins due to Mcm4chaos3 homozygosity reduces active origin density in primary embryonic fibroblasts (MEFs) in a C57BL/6J (B6) background. We found that this is associated with an intrinsically lower level of active origins in this background compared to others. B6 Mcm4chaos3/chaos3 cells proliferate slowly due to p53-dependent upregulation of p21. In fact, the development of B6 Mcm4chaos3/chaos3 mice is impaired and a significant fraction of them die at birth. While inactivation of p53 restores proliferation in B6 Mcm4chaos3/chaos3 MEFs, it paradoxically does not rescue animal lethality. These findings indicate that a reduction of licensed origins may cause a more profound effect on cell types with lower densities of active origins. Moreover, p53 is required for the development of mice that suffer from intrinsic replication stress.
Introduction
Replication origin licensing is a prerequisite for eukaryotic DNA replication. Between the late M and early G1 phases, replication origins are licensed by the chromatin loading of heterohexamers of the minichromosome maintenance proteins (MCM2-7), essential components of the pre-replication complex (reviewed in Lei 2005; Sclafani et al. 2002). In the following S phase, licensed origins fire when MCM2-7 complexes form active replicative helicases along with cofactors CDC45 and GINS (Ilves et al. 2010; Moyer et al. 2006). During S phase, DNA synthesis occurs exclusively from these licensed origins. After origins fire, the departure of active MCM2-7 complexes from once-fired origins and the displacement of inactive MCM2-7 complexes from replicated DNA return chromatin to the unlicensed state (Todorov et al. 1995; Yan et al. 1993). The expression of geminin, a licensing inhibitor, also prohibits re-licensing of once-fired origins during S phase (McGarry and Kirschner 1998; Wohlschlegel et al. 2000). Therefore, origin licensing primes origins for replication only prior to S phase entry, thereby preventing re-replication of DNA.
Deregulation of origin licensing is closely linked with cancer development (Blow and Gillespie 2008; Ha et al. 2004; Ishimi et al. 2003). Upregulation of the MCM2-7 proteins has been especially investigated as a cancer marker (Lei 2005). However, this probably occurs at a later stage of cancer, as inactivation of p53 increases MCM2-7 expression (Scian et al. 2008). Previously, our studies as well as others have shown that a reduced level of licensed origins causes spontaneous tumors in mice, suggesting a causative role of deregulated origin licensing in cancer development (Chuang et al. 2010; Kunnev et al. 2010; Pruitt et al. 2007; Shima et al. 2007). As only a minor fraction of licensed origins are used in unperturbed S phase, it has been demonstrated that a reduction of licensed origins has little effect on the density of active origins (Ge et al. 2007; Ibarra et al. 2008; Kunnev et al. 2010). Rather, it seems only to result in a reduction in the number of dormant origins (Ge et al. 2007; Ibarra et al. 2008; Woodward et al. 2006). Dormant origins are defined as those which remain mostly unused in unperturbed S phase but can be activated to survive perturbed S phase, possibly to compensate for slow fork progression (Ge et al. 2007; Woodward et al. 2006). However, our study using Mcm4chaos3 mice revealed that such dormant origins also play a critical role in rescuing stalled replication forks in unperturbed S phase (Kawabata et al. 2011). Therefore, we concluded that this reduction in the number of dormant origins results in the accumulation of stalled forks, leading to intrinsic replication stress and spontaneous tumorigenesis. Taken together, we propose that dormant origins exist in such vast excess because of their role in the rescue of stalled forks in unperturbed S phase.
Mcm4 is an essential gene that encodes a component of the MCM2-7 complex. We have previously shown that homozygosity for a Mcm4 null allele (Mcm4Gt(RRE056)Byg, hereafter Mcm4− for the sake of simplicity) causes early embryonic lethality before implantation (Shima et al. 2007). Mcm4chaos3 was originally isolated from a mouse mutagenesis screen, causing only a single amino acid change (Phe345Ile), therefore Mcm4chaos3 homozygous mice are viable (Shima et al. 2007). Compound heterozygotes carrying both Mcm4chaos3 and Mcm4− alleles have extended viability compared to Mcm4−/− embryos but their survival is greatly affected by their genetic background (Shima et al. 2007). While Mcm4chaos3/− embryos in a C57BL/6J (B6) background die around mid-gestation, those in a mixed background between B6 and C3HeB/FeJ (C3H) are viable for a few days after birth (Shima et al. 2007). Therefore, the development of Mcm4chaos3/− embryos is severely impaired in the B6 background. However, the underlying mechanism for this phenomenon has not been investigated.
In this study, we report that the viability of Mcm4chaos3/chaos3 mice is also reduced in the B6 background. By comparative analyses of B6, C3H and their F1 embryonic fibroblasts for DNA replication kinetics and cell proliferation, we found that this reduced viability can most likely be attributed to an intrinsically lower level of active replication origins in B6 cells. Unlike other genetic backgrounds, a reduced number of dormant origins due to Mcm4chaos3 homozygosity substantially lowered the levels of active origins in the B6 background, causing upregulation of p21WAF1/Cip1, a CDK inhibitor (Xiong et al. 1993), in a p53-dependent manner. This study also describes a complex interaction between Trp53 and Mcm4, suggesting that normal p53 function is required to support viability and tumor suppression in mice that suffer from intrinsic replication stress. Moreover, our data suggest the interesting possibility that fundamental DNA replication kinetics, such as active origin density, may be genetically controlled.
Materials and Methods
Animals
Trp53 mice (B6.129S2-Trp53tm1Tyj/J) and p21 mice (B6;129S2-Cdkn1atm1Tyj/J) were purchased from the Jackson Laboratory. Mcm4chaos3 was introduced into the C57BL/6J and C3HeB/FeJ backgrounds by backcrossing 7 times (N8). F1 mice were produced by intercrossing the B6 and C3H Mcm4chaos3 lines. All mice were genotyped by PCR on tail DNA. Primer sequences are available upon request. All experiments involving mice were approved by the Institutional Animal Care and Use Committee (IACUC).
Generation of MEFs
MEFs were obtained from 12.5–14.5 dpc embryos using a standard procedure described previously and cultured in standard conditions (Shima et al. 2007). A homogenized embryo was plated onto a 10 cm dish (p0), replated onto a 15 cm dish (p1), and frozen in three vials. For experiments, one vial was thawed and the cells were cultured on a 10 cm dish for a few days (p2). The resulting 6–9 × 106 cells (p3) were then replated for experimental use.
Antibodies
We used Abcam anti-MCM antibodies (ab3159, ab4460, ab4459, ab17967, ab4458 and ab2360 for MCM2, MCM3, MCM4, MCM5, MCM6 and MCM7, respectively), anti-CDC45 (Abcam; ab56476), anti-Pol delta (Abcam; ab10362), anti-pan actin (Thermo Scientific; MS-1295-P1ABX), anti-p21 (BD Pharmingen; 556431), anti-phospho-p53, anti-phospho-RAD17, anti-γH2AX, anti-RPA32, anti-phospho-CHK1 (Cell Signaling; #9284, #3421, #2577, #2208 and #2341 respectively), and anti-RAD51 (Calbiochem, PC130).
Western blot
Cell extracts were harvested with Laemmli’s sample buffer, run on 8 or 12% SDS-polyacrylamide gel, and transferred to polyvinylidene membrane. Membranes were incubated with primary antibodies, followed by incubation with the appropriate secondary antibodies. Proteins were visualized using the Immobilon Western Chemiluminescent HRP substrate (Millipore, WBKLS0500). Cell fractionation was performed using the Qproteome Nuclear Protein Kit (Qiagen).
DNA fiber
We used the DNA fiber protocol described by Sugimura et al. (Sugimura et al. 2007). Cells were sequentially labeled with modified dUTPs for DNA fiber analysis. Cells were first cultured in the presence of digoxigenin-dUTPs for 20 min to label ongoing forks red. After washing with PBS, cells were then cultured in the presence of biotin-dUTPs for 30 min. To allow efficient incorporation of the dUTPs, a treatment with hypotonic buffer (10mM HEPES, 30mM KCl, pH7.4) precedes each dUTP labeling. To visualize the labeled DNA fibers, cells were mixed with a 10-fold excess of unlabeled cells, fixed, and dropped onto slides. After cell lysis, the DNA fibers were released and extended by tilting the slides. Incorporated dUTPs were visualized by immunofluorescent detection using anti digoxigenin-Rhodamine (Roche) and streptavidin-Alexa-Fluor-488 (Invitrogen). Images were captured using the Axio Imager A1 microscope (Zeiss). The average origin-to-origin distances as well as fork velocities were analyzed by t-test between wildtype and Mcm4chaos3/chaos3 cells.
Cell cycle analysis
A modified version of the Click-iT™ Assay from Invitrogen was used (Hamelik and Krishan 2009). This modified assay uses a hypotonic lysis solution to isolate individual nuclei, thereby avoiding unnecessary fixation and permeabilization steps. Cells were cultured for two days and then treated with a dose of 20μM 5-ethynyl-2′-deoxyuridine (EdU) for 45 minutes to label replicating cells. Cells were then harvested via trypsinization and incubated with Nonidet P-40 solution, vortexed, and washed with 1% BSA in PBS. Then, the normal protocol for the Click-iT™ reaction to detect EdU incorporation was followed. Finally, cells were treated with Rnase A (200 μg/ml) and stained with propidium iodide (4 μg/ml) prior to analysis by flow cytometry. The FACSCalibur cytometer (BD biosciences) was used to collect all data and to specifically gate the diploid population. The FL2 and FL4 channels were used to observe DNA content and EdU incorporation, respectively.
Immunofluorescence microscopy
To observe discrete foci of RPA32 and phospho-RAD17, and chromatin-bound MCM2, cells were pretreated with 0.5% Triton X-100 in PBS for 1 minute before fixation with paraformaldehyde (PFA) for 15 minutes. After permeabilization in 0.1% Triton X-100 in PBS for 15 min, cells were subjected to treatment with the Image-iT signal enhancer (Invitrogen) before being incubated with primary antibodies. Staining with the appropriate secondary antibodies and fluorescence microscopy (Zeiss) were used to visualize staining. All procedures were performed at room temperature.
Results
The viability of Mcm4chaos3/chaos3 mice is significantly decreased in a C57BL/6J background
We have generated a B6 Mcm4chaos3 line (N8) in which more than 99% of the genome is of B6 origin. In the process of breeding, we found that a fraction of B6 Mcm4chaos3/chaos3 pups die a few days after birth. Among 48 offspring generated from Mcm4chaos3/+ intercrosses, only 3 Mcm4chaos3/chaos3 mice were alive at weaning age, significantly fewer than the expected number of 12 (p<0.003, χ2-test). By timed mating, we verified that Mcm4chaos3/chaos3 embryos were present at the expected ratios throughout gestation (data not shown). In contrast, 14 Mcm4chaos3/chaos3 mice were alive among 54 offspring in the C3HeB/FeJ (C3H) background (p=0.875, χ2-test). As Mcm4chaos3/chaos3 mice were also born at the expected ratios in the F1 background (data not shown), reduced viability was unique to the B6 background. Moreover, B6 Mcm4chaos3/chaos3 embryos were about 15% smaller in size (p<0.005, t-test) than their Mcm4chaos3/+ littermates (Figs. 1A and 1B). We also found that B6 Mcm4chaos3/chaos3 mice were prone to microphthalmia (Fig. 1C), which was observed at a higher rate (18%) than wildtype B6 mice (3.2%) (Houghtaling et al. 2003; Kalter 1968). As retina size is primarily determined by cell number (Green et al. 2003), these findings suggest that the rate of cell proliferation is significantly reduced in B6 Mcm4chaos3/chaos3 embryos. In fact, B6 Mcm4chaos3/chaos3 embryonic fibroblasts (MEFs) ceased to proliferate earlier in vitro compared to wildtype cells (Fig. 1D). Although this reduced rate of cell proliferation is likely to contribute to the semi-lethality of B6 Mcm4chaos3/chaos3 pups, the direct cause of death remains to be determined.
Figure 1.
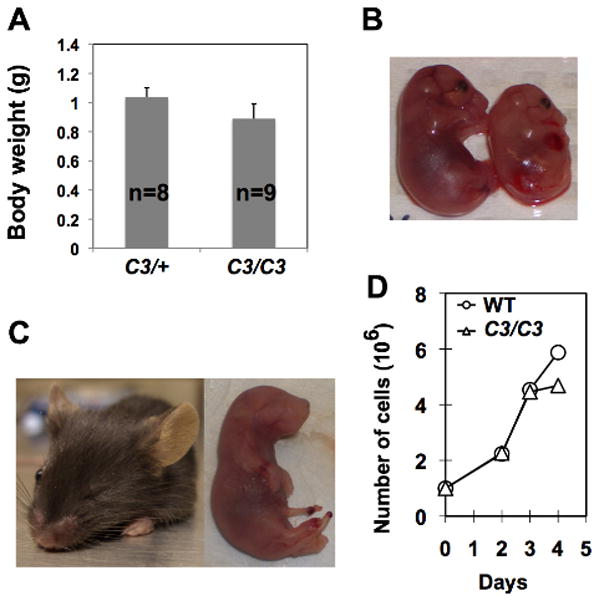
Developmental defects in B6 Mcm4chaos3/chaos3 mice are associated with a decreased rate of cell proliferation. A) Average body weights for Mcm4chaos3/+ (C3/+) and Mcm4chaos3/chaos3 (C3/C3) embryos (17.5–18.5 days post-coitum, dpc). Bars indicate standard deviation. B) Representative images of 16.5-dpc B6 Mcm4chaos3/+ (left) and Mcm4chaos3/chaos3 (right) embryos. C) Microphthalmia in adult (left) and newborn (right) B6 Mcm4chaos3/chaos3 mice. D) Proliferation of B6 wildtype (WT) and Mcm4chaos3/chaos3 (C3/C3) MEFs in vitro.
The density of active origins is significantly decreased in Mcm4Chaos3/Chaos3 MEFs in the B6 background in which active origin density is intrinsically lower
We have shown that Mcm4chaos3 homozygosity decreases the amount of the MCM2-7 proteins (Shima et al. 2007), leading to a reduced number of dormant origins (Kawabata et al. 2011). It has been previously reported that a reduction of licensed origins has little effect on the density of active origins (Ge et al. 2007; Ibarra et al. 2008; Kunnev et al. 2010). Indeed, we also reported no significant difference in the average density of active origins between wildtype and Mcm4chaos3/chaos3 cells in the F1 background (Kawabata et al. 2011). However, the smaller size of B6 Mcm4chaos3/chaos3 embryos and their reduced level of cell proliferation prompted us to investigate a potential strain difference in DNA replication kinetics in response to a reduction of licensed origins using the DNA fiber technique (Sugimura et al. 2007). In this method, newly synthesized DNA is sequentially labeled with two kinds of modified nucleotides, which are detected on stretched DNA fibers using immunofluorescence (Fig. 2A). Surprisingly, we found that B6 Mcm4chaos3/chaos3 cells had a significantly larger origin-to-origin (ori-to-ori) distance (156±14 kb) than wildtype cells (112±5.0 kb)(Fig. 2B left, also see Fig. S1), indicating a decreased density of active origins. However, C3H Mcm4chaos3/chaos3 cells showed no difference from wildtype cells of the same background, much like data obtained previously in the F1 background (Fig. 2B left, also see Kawabata et al. 2011). These data suggest that, upon a reduction in the number of licensed origins, a decreased density of active origins occurs uniquely in the B6 background. As described elsewhere (Kawabata et al. 2011), we also repeatedly found that Mcm4chaos3/chaos3 cells have a faster fork velocity compared to wildtype cells regardless of genetic background (Fig. 2B right). We think that this faster fork velocity could be attributed to the biochemical properties of the mutant helicase, whose activity is somewhat more efficient than the wildtype helicase (Kawabata et al. 2011). It should also be noted that the average ori-to-ori distance in B6 wildtype cells (112±5.0 kb) was significantly larger than that of C3H or F1 wildtype cells (95.0±5.0 kb and 49.1±2.6 kb, respectively), suggesting that B6 cells have a lower level of active origins than C3H and F1 cells. Moreover, the average fork velocity in wildtype B6 cells (1.7±0.05 kb/min) was more than twice as fast as wildtype C3H and F1 cells (0.84±0.02 kb/min and 0.61±0.01 kb/min, respectively) (Fig. 2B right). These data collectively suggest that the fundamental kinetics of DNA replication, such as origin density and fork velocity, may be under the control of genetic factors.
Figure 2.
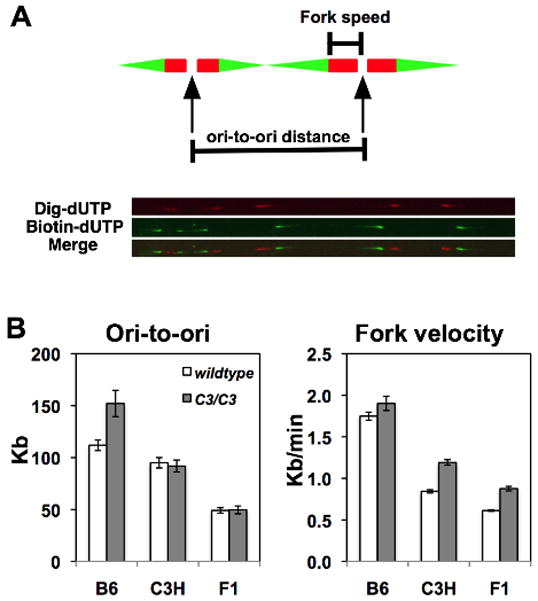
The DNA fiber assay reveals active origin density and fork velocity. A) A schematic presentation of DNA fiber measurements. Two kinds of modified nucleotides (digoxigenin-dUTPs and biotin-dUTPs) were sequentially incorporated into newly synthesized DNA during S phase, revealing the direction of fork movement (Red to Green). Fork velocity was determined by measuring from the start of the red track to the start of the green track. To estimate the density of active origins, the distances between adjacent origins (ori-to-ori) were measured. A merged image of digoxigenin- and biotin-labeled DNA is shown at the bottom. B) Genetic background has a strong effect on active origin density and fork velocity. Unlike C3H and F1 Mcm4chaos3/chaos3 cells (C3/C3), B6 Mcm4chaos3/chaos3 cells have a significantly longer average inter-origin distance than wildtype (WT) cells (left). Forks are also faster in Mcm4chaos3/chaos3 cells regardless of genetic background (right). These data were obtained from three independent experiments using MEFs from different embryos. Bars represent standard errors. Note that the F1 data have been previously published (Kawabata et al. 2011) but are shown here for a better comparison, with permission from Elsevier (License number 2665400529110).
B6 Mcm4chaos3/chaos3 MEFs display a reduced level of chromatin-bound PCNA and upregulated p21 expression
To understand what leads to a decreased density of active origins in B6 Mcm4chaos3/chaos3 cells, we investigated the levels of replication proteins in B6 wildtype and Mcm4chaos3/chaos3 MEFs by Western blot. Consistent with our previous findings, all MCM proteins were significantly reduced in B6 Mcm4chaos3/chaos3 cells (Fig. 3)(Chuang et al. 2010; Kawabata et al. 2011; Shima et al. 2007). Semi-quantitative analysis allowed us to estimate that ~60% of all chromatin-bound MCM2-7 proteins are lost in B6 Mcm4chaos3/chaos3 cells (Fig. 3 left). This reduction rate was very similar to that was found in Mcm4Chaos3/Chaos3 cells in other backgrounds (Chuang et al. 2010; Kawabata et al. 2011). However, we found that B6 Mcm4chaos3/chaos3 cells also exhibited highly upregulated expression of p21WAF1/Cip1 (hereafter p21) and its increased chromatin binding (Fig. 3, right). Chromatin-bound PCNA and Polδ levels were also appreciably reduced (>30% loss, see Fig. 3, left), while a normal level of chromatin-bound CDC45, a co-factor of the MCM2-7 helicase (Ilves et al. 2010; Moyer et al. 2006), was retained in B6 Mcm4chaos3/chaos3 cells. The latter observation suggests that PCNA may dissociate from chromatin after CDC45 loading for the formation of the active helicase. It has been reported that the dissociation of PCNA (and other proteins involved in lagging strand synthesis such as Polδ) from chromatin occurs after partial inhibition of DNA replication (Shimura et al. 2008). We therefore confirmed increased levels of markers for stalled replication forks in B6 Mcm4chaos3/chaos3 cells (Fig. S2A) as previously observed in F1 Mcm4Chaos3/Chaos3 cells (Kawabata et al. 2011). We also found that B6 Mcm4chaos3/chaos3 cells consistently exhibit a higher basal level of phosphorylated CHK1 (Fig. S2B), the downstream effector of the major checkpoint kinase ATR (Zhao and Piwnica-Worms 2001). This was not the case in the F1 background (Kawabata et al. 2011). Thus, B6 cells might be hypersensitive to an accumulation of stalled forks, leading to upregulation of p21 and the removal of PCNA from chromatin.
Figure 3.
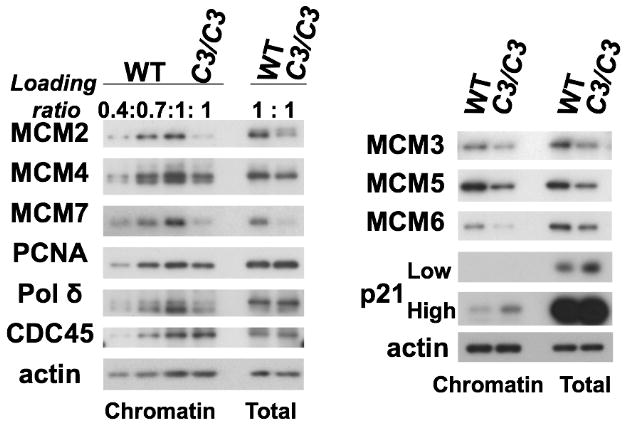
Levels of chromatin-bound PCNA, Pol δ and the MCM2-7 proteins are significantly reduced in B6 Mcm4chaos3/chaos3 (C3/C3) MEFs. Reduction rates for chromatin-bound MCM2/4/7 and the other replication proteins in Mcm4chaos3/chaos3 cells were estimated by referencing wildtype (WT) proteins loaded in different amounts (left). In Mcm4chaos3/chaos3 cells, the MCM3/5/6 proteins were also reduced in both the chromatin fraction and the total cell lysate (right). Note that upregulation of p21 also occurs in Mcm4chaos3/chaos3 cells. Actin was used as a loading control. Samples were harvested at Day 2 (Fig. 1D) when there was no difference in proliferation between B6 wildtype and Mcm4chaos3/chaos3 cells.
C3H and F1 Mcm4chaos3/chaos3 MEFs proliferate normally with wildtype levels of chromatin-bound PCNA
Next, we examined whether a reduction in the number of licensed origins causes universal p21 upregulation and/or a reduced level of chromatin-bound PCNA in primary MEFs. We tested this using Mcm4chaos3/chaos3 cells in the C3H and F1 backgrounds, as Mcm4chaos3/chaos3 pups are fully viable and normal in size in these backgrounds (data not shown). Despite a similarly reduced amount of MCM7 in both total cell lysates and chromatin fractions (Fig. 4A), C3H and F1 Mcm4chaos3/chaos3 cells proliferated normally in vitro (Fig. 4B) and retained wildtype levels of chromatin-bound PCNA levels (Fig. 4A, right). As B6 wildtype cells have an apparently lower amount of MCM7 in both total cell lysates and chromatin fractions compared to other backgrounds (Fig. 4A), we tested if this is reproducibly observed. Indeed, we confirmed that B6 wildtype cells display lower levels of not only MCM7 but also MCM2 and MCM4 compared to F1 and C3H wildtype cells (Fig. S3A). This suggests that B6 MEFs have a relatively lower level of licensed origins. Moderate p21 upregulation was also seen in F1 and C3H Mcm4chaos3/chaos3 cells (Fig. 4C). Interestingly, however, we found that B6 wildtype cells express p21 at an intrinsically lower level compared to C3H and F1 wildtype cells (Figs. 4C and S3B). Given this lower basal level of p21 expression, it appears that p21 upregulation occurs most drastically in the B6 background upon a reduction in the number of licensed origins. As B6 cells have a relatively lower amount of MCM proteins (Fig. S3A), we then tested the possibility that further reduction of MCMs by Mcm4chaos3 homozygosity might activate the licensing checkpoint, leading to p21 upregulation. The licensing checkpoint is typically triggered by severe levels (>90%) of MCM depletion (Shreeram et al. 2002), and its activation is associated with the co-upregulation of p21 and p27 (Machida et al. 2005). This in turn inhibits CDK2, thereby blocking the S-phase entry of cells with an extremely low number of licensed origins. However, p27 was not upregulated in B6 Mcm4chaos3/chaos3 cells (Fig. S3B). B6 Mcm4chaos3/chaos3 cells also did not show an accumulation of G1 phase cells and their cell cycle profiles were very similar to those reported for other genetic backgrounds (Fig. S3C). These data collectively suggest that p21 upregulation occurs independently from the licensing checkpoint.
Figure 4.
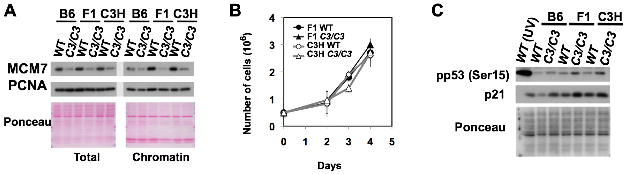
C3H and F1 Mcm4chaos3/chaos3 cells proliferate normally despite reduced levels of the MCM 2-7 proteins, but activate p53. A) MCM7 protein levels were significantly reduced both in total cell lysates and chromatin fractions in B6, C3H and F1 Mcm4chaos3/chaos3 (C3/C3) cells compared to their respective wildtype (WT) cells. However, the reduction of chromatin-bound PCNA was most severe in B6 Mcm4chaos3/chaos3 cells. B) F1 and C3H Mcm4chaos3/chaos3 MEFs proliferate at the same rates as their respective wildtype control cells in vitro. Bars show standard deviations. C) Regardless of p21 expression levels, strong p53 activation is observed in F1 and C3H Mcm4chaos3/chaos3 cells. Note that the level of p21 expression appears to be intrinsically lower in B6 wildtype cells.
Transcriptional upregulation of p21, a result of p53 activation, is elevated in B6 Mcm4chaos3/chaos3 MEFs
p53 plays a major role in the transcriptional regulation of p21 (Bunz et al. 1998) and is constitutively activated in Mcm4chaos3/chaos3 cells in all backgrounds through its phosphorylation at serine 15 (Figs. 4C and S3B)(Shieh et al. 1997). To test the role of p53 in p21 upregulation in B6 Mcm4chaos3/chaos3 cells, we introduced Trp53 nullizygosity (Trp53−/−) into B6 wildtype and Mcm4chaos3/chaos3 MEFs (Jacks et al. 1994). In Trp53−/−;Mcm4chaos3/chaos3 cells, p21 was barely detectable (Fig. 5A), and chromatin-bound PCNA was restored to a wildtype level (Fig. 5B). Moreover, inactivation of p53 improved the rate of proliferation of B6 Mcm4chaos3/chaos3 cells (Fig. 5C) by increasing the number of S phase cells from 19.0% to 33.5% (Figs. S3C and 5D). Therefore, we conclude that activation of the p53/p21 axis is a major cause of the decreased rate of proliferation of B6 Mcm4chaos3/chaos3 cells.
Figure 5.

Loss of p53 restores chromatin-bound PCNA levels and allows for the proliferation of B6 Mcm4chaos3/chaos3 MEFs with an increased level of DNA damage. A) p21 upregulation in B6 Mcm4chaos3/chaos3 cells is p53 dependent. Loss of p53 also results in increased levels of pRAD17. B) Chromatin-bound PCNA levels in Trp53−/−;Mcm4chaos3/chaos3 cells are comparable to wildtype levels. Loss of p53 does not change the expression of PCNA (left) but promotes its chromatin-binding (right). C) Loss of p53 improves the proliferation of B6 Mcm4chaos3/chaos3 and wildtype cells in vitro. D) p53 inactivation drastically increases the percentage of S phase cells. The diploid population was gated and analyzed for DNA content and EdU incorporation by flow cytometry. The averages of three independent measurements are shown with standard errors. E) Loss of p53 increases the levels of chromatin-bound MCM2. Intensity of MCM2 staining within individual cell was measured and its distribution was plotted. At least 90 cells per genotype were analyzed (left). Representative images of MCM2 and dUTP staining are shown for each genotype (right). F) Loss of p53 results in the upregulation of RAD51 as well as a drastic increase in γH2AX. This occurs regardless of genotype. UV-irradiated (30 J/m2) wildtype cells were used as a positive control for the phosphorylated form of p53 and RAD17 (A) and γH2AX (F).
Inactivation of p53 results in an increase in chromatin-bound MCM levels, and DNA damage and RAD51 upregulation
Loss of p53 resulted in increased levels of multiple MCM proteins as well as their increased chromatin binding both in wildtype and Mcm4chaos3/chaos3 cells (Fig. 5B). However, this could simply be a result of a drastically increased rate of cell proliferation caused by p53 inactivation (Fig. 5C–D). Therefore, we analyzed MCM2 levels per cell by immunocytostaining, which revealed significantly increased levels of chromatin-bound MCM2 both in wildtype and B6 Mcm4chaos3/chaos3 cells lacking p53 (Fig. 5E, left). These data are consistent with a previous finding that the p53/p21 axis negatively regulates the transcription of the Mcm genes (Scian et al. 2008). It should be noted that the highest level of MCM2 staining was observed in non-S phase cells, most likely those in G1 phase (see the lack of dUTP incorporation in Fig. 5E, right). After S phase entry, MCM2 staining appears to drop immediately, as cells with higher levels of dUTP incorporation have very little MCM2 staining (Fig. 5E. right). Furthermore, we found that loss of p53 resulted in: 1) an increase in the phosphorylated form of RAD17, a checkpoint protein (Bao et al. 2001) (Fig. 5A), 2) upregulation of RAD51 recombinase (Fig. 5F) and 3) an increase in phosphorylated histone H2AX (γH2AX), a marker of DNA damage such as DNA double strand breaks (Rogakou et al. 1998) (Fig. 5F), suggesting an increase in homologous recombination events possibly due to replication fork collapse. These data are consistent with p53’s role in genome stability in response to replication stress (Gatz and Wiesmuller 2006; Kumari et al. 2004).
Mcm4 and Trp53 genetically interact in a complex manner during development and tumorigenesis
Since Trp53 nullizygosity improved the proliferation of B6 Mcm4chaos3/chaos3 MEFs (Fig. 5C), we next asked if the newborn lethality found in B6 Mcm4chaos3/chaos3 mice could also be rescued by Trp53 nullizygosity. We intercrossed B6 Trp53+/−;Mcm4chaos3/chaos3 mice to generate Trp53−/−;Mcm4chaos3/chaos3 mice. Surprisingly, we recovered only two Trp53−/−;Mcm4chaos3/chaos3 mice out of a total of 49 mice at weaning age. This is significantly lower than the expected number of 12.25 (p=0.0026), suggesting that loss of p53 actually promoted the newborn lethality of B6 Mcm4chaos3/chaos3 mice. This indicates that Trp53−;Mcm4chaos3 double homozygosity supports cell proliferation in vitro but not necessarily normal development. After an extended period of breeding, we were able to generate a total of five Trp53−/−;Mcm4chaos3/chaos3 mice. They all died with thymic lymphomas between 14–19 weeks of age, significantly faster (p < 0.03) than the 21.7-week latency for thymic lymphomas for Trp53−/− mice in our colony (Fig. 6A). To further test the collaboration between Trp53− and Mcm4chaos3 in tumorigenesis, we next investigated the effect of Trp53− heterozygosity on B6 Mcm4chaos3 homozygotes. Approximately 30% of Trp53+/− mice reportedly develop osteosarcomas by 17 months of age (Jacks et al. 1994). Consistent with this observation, only three out of 12 Trp53+/− mice had osteosarcomas by 16 months of age. In contrast, all Trp53+/−;Mcm4chaos3/chaos3 mice succumbed to tumors with a mean latency of 8.3 months (Fig. 6B). We found that a loss of the wildtype Trp53 allele occurred in a few such tumors (Fig. S4). Osteosarcomas were the most prominent tumor type (Fig. 6C), while one histiocytic sarcoma and one lymphoma were also observed which were typically seen in B6 Mcm4chaos3/chaos3 mice (Kawabata et al. 2011). These data suggest that Trp53 dictates the tumor spectrum in Trp53;Mcm4chaos3 double mutants. Finally, we tested the role of p21 in Mcm4chaos3 tumorigenesis. The average tumor-free survival of p21+/−;Mcm4chaos3/chaos3 mice was 12.7 months (Fig. 6D), very similar to what has been seen in Mcm4chaos3/chaos3 mice in different backgrounds (Kawabata et al. 2011; Shima et al. 2007). Therefore, it seems unlikely that p21 upregulation contributes to tumor suppression in Mcm4chaos3/chaos3 mice.
Figure 6.

Inactivation of p53 promotes spontaneous tumorigenesis in Mcm4chaos3/chaos3 mice, but a loss of p21 does not. A) Tumor-free survival curves of Trp53−/− and Trp53−/−;Mcm4chaos3/chaos3 mice. B) Tumor-free survival curves of Trp53+/− and Trp53+/−;Mcm4chaos3/chaos3 mice. C) An X-ray photo of an osteosarcoma observed in a Trp53+/−;Mcm4chaos3/chaos3 mouse. D) Tumor-free survival curves of p21+/−;Mcm4chaos3/chaos3 and p21−/−;Mcm4chaos3/chaos3 mice. All Trp53;Mcm4chaos3 experiments were performed in a B6 congenic background, whereas the p21;Mcm4chaos3 cancer study was conducted in a mixed background between B6 and C3H.
Discussion
In this study, we investigated the mechanisms underlying the reduced viability of B6 Mcm4chaos3/chaos3 mice. As this reduced viability is associated with a decreased rate of cell proliferation in B6 Mcm4chaos3/chaos3 embryos (Fig. 1), we focused on the molecular mechanisms involving DNA replication. DNA fiber data show that the basic kinetics of DNA replication such as active origin density and fork velocity are considerably different in wildtype MEFs among the three genetic backgrounds we tested, revealing that B6 MEFs possess the lowest density of active origins. Moreover, Mcm4chaos3 homozygosity results in a further reduction of the density of active origins in this genetic background (Fig. 2B). Notably, this was the first observation that a reduction in the number of licensed origins significantly reduces the density of active origins. Therefore, we went on to investigate what leads to this phenomenon. We found that B6 Mcm4chaos3/chaos3 cells have reduced levels of chromatin-bound PCNA and polδ (Fig 3, left) and upregulation of p21 (Fig. 3, right). The interaction between p21 and PCNA is well established, and is known to inhibit the loading of replicative polymerases such as polδ (Cazzalini et al. 2003; Oku et al. 1998; Waga et al. 1994). Therefore, the lower level of chromatin-bound polδ in B6 Mcm4chaos3/chaos3 cells can be explained by this p21-PCNA interaction. However, p21 does not inhibit the loading of PCNA onto chromatin (Cazzalini et al. 2003; Waga et al. 1994). Thus, there must be an additional mechanism responsible for the removal of PCNA from chromatin. Previous studies reported that replication inhibitions trigger the dispersal of PCNA from replication factories (Rossi et al. 2006; Shimura et al. 2008; Stokes and Michael 2003). In agreement with these findings, B6 Mcm4chaos3/chaos3 cells consistently exhibit a higher basal level of pCHK1 compared to wildtype cells (Fig. S2B), indicative of inherently increased levels of replication stress. Compared to other genetic backgrounds, B6 cells have a relatively lower number of licensed origins (Fig. S3A), and thus may be more sensitive to an increased level of stalled forks upon a further reduction of licensed origins due to Mcm4chaos3 homozygosity. In sum, we propose that the dispersal of PCNA from chromatin in B6 Mcm4chaos3/chaos3 cells mainly occurs as a response to intrinsic replication stress, contributing to the reduced rate of cell proliferation in B6 Mcm4chaos3/chaos3 embryos. However, we do not entirely exclude the possibility that other factors also contribute to this unique phenomenon.
An alternative explanation for the reduced rate of cell proliferation in B6 Mcm4chaos3/chaos3 embryos would be an increased level of licensing checkpoint activation. However, we do not think this is likely, as the licensing checkpoint is typically induced following a high level (>90%) of licensing factor depletion (Liu et al. 2009; Machida et al. 2005; Nevis et al. 2009; Shreeram et al. 2002). Therefore, we do not think that the reduction level of MCM proteins (~60% compared to wildtype) in B6 Mcm4chaos3/chaos3 cells is sufficient to activate this checkpoint (see Fig. 3). Furthermore, it has been proposed that this checkpoint blocks the S phase entry of cells with an extremely low number of licensed origins, as these cells would most likely experience severe genome instability during DNA replication (Ge and Blow 2009). B6 Mcm4chaos3/chaos3 cells, however, exhibit no signs of G1 accumulation or a drastic decrease in S phase cells (Fig. S3C). Their cell cycle profiles are similar to those reported for other genetic backgrounds (Chuang et al. 2010; Shima et al. 2007). In this context, the reduced level of chromatin-bound PCNA specifically seen in B6 Mcm4chaos3/chaos3 cells cannot be explained by the activation of the licensing checkpoint. Finally, B6 Mcm4chaos3/chaos3 cells are proficient in the ATR-CHK1 pathway (Fig. S2B), which is mainly activated in response to the presence of stalled forks during S phase (Cimprich and Cortez 2008).
It is known that p53 is activated upon replication stress (Gottifredi et al. 2001; Marusyk et al. 2007; Nayak and Das 2002). Although there has been much controversy as to whether p53 transactivates p21 upon replication stress, it apparently depends on cell type (Gottifredi et al. 2001; Nayak and Das 2002). In our study, transactivation of p21 by p53 occurred profoundly in B6 Mcm4chaos3/chaos3 MEFs. Our results show that this is responsible for the slower rate of proliferation and the lower level of chromatin-bound PCNA observed in B6 Mcm4chaos3/chaos3 cells (Fig. 5B–C). In fact, loss of the p53/p21 axis not only corrected these unique phenotypes but also drastically increased the expression levels of the MCM proteins as well as their chromatin binding in B6 Mcm4chaos3/chaos3 cells. The latter observation suggests the possibility that the activation of the p53/p21 axis, which is also observed in Mcm4chaos3/chaos3 cells in other backgrounds to some extent, may also cause repression of Mcm gene expression, thereby contributing to lower MCM protein levels. Supporting this idea, it has been reported that transcript levels of Mcm genes are 30–50% lower in Mcm4chaos3/chaos3 cells than wildtype cells (Chuang et al. 2010).
It turns out that p53 activation in B6 Mcm4chaos3/chaos3 cells is crucial for both development and tumor suppression. Generally, a loss of p53 supports or extends the survival of embryos mutant for an essential DNA repair gene, which otherwise die earlier in development due to increased apoptosis (Lim and Hasty 1996; Shu et al. 1999; Xu et al. 2001). However, the interaction between Mcm4 and Trp53 appears to be much more complex. While loss of p53 improved the proliferation of B6 Mcm4chaos3/chaos3 MEFs in vitro, it also promoted the lethality of B6 Mcm4chaos3/chaos3 mice. Notably, the same observation has been made for Mcm2 and Trp53 in a recent study (Kunnev et al. 2010). Together, it can be interpreted that the concomitant impairment of the MCM2-7 complex and p53 is largely incompatible with normal development. A major cause for this impaired development is an enhanced level of genome instability due to loss of p53 (see Figs. 5A and 5F), which, however, promotes spontaneous tumorigenesis in rare surviving double mutants (Fig. 6 and Kunnev et al. 2010). Importantly, this synthetic lethality and collaboration in tumorigenesis for the Trp53 and Mcm genes does not appear to be strain-specific, as this was observed in a mixed background (Kunnev et al. 2010). In contrast, we did not observe an acceleration of spontaneous tumorigenesis in p21−/−;Mcm4chaos3/chaos3 mice (Fig. 6D). Therefore, whatever p21’s role in Mcm4chaos3/chaos3 cells is, it is unlikely to have a strong effect on the suppression of Mcm4chaos3 tumors.
Recent studies showed that Atr, an essential gene for checkpoint activation in response to replication stress, also exhibits synthetic lethality with Trp53 in mice (Murga et al. 2009; Ruzankina et al. 2009). Because of the established role of ATR in stabilizing replication forks, Atr deficient cells suffer from intrinsic replication stress. Under such conditions, p53’s role in genome stability would become more critical to sustain animal viability. As surviving double mutants for Atr and Trp53 exhibit premature aging but are not tumor prone, it appears that Atr is required for tumor formation even in the absence of p53 (Murga et al. 2009). We have previously shown that Mcm4chaos3/chaos3 cells suffer from intrinsic replication stress due to the accumulation of stalled replication forks (Kawabata et al. 2011). In this context, Mcm4chaos3/chaos3 embryos require functional p53 to support animal development. However, having an intact ATR pathway, surviving Trp53−/−:Mcm4chaos3/chaos3 mice exhibit accelerated tumorigenesis as a result of enhanced genome instability.
Microphthalmia occurs relatively frequently in the B6 strain (Kalter 1968), but B6 Mcm4chaos3/chaos3 mice exhibit this condition at an even higher rate. Interestingly, the same microphthalmia phenotype was also observed in Fanconi anemia mouse models for Fanca, Fancc and Fancd2 when maintained in a B6 background (Carreau 2004; Houghtaling et al. 2003; Wong et al. 2003). Given the role of the Fanconi anemia proteins in replication fork stability (Gari et al. 2008; Howlett et al. 2005; Wang et al. 2008), B6 cells might be intrinsically hypersensitive to replication stress. The inter-strain difference in DNA replication kinetics found in this study supports this idea. Among the three genetic backgrounds investigated in this study and our previous study (Kawabata et al. 2011), B6 cells had the lowest density of active origins (Fig. 2B). Comparing average ori-to-ori distances, F1 cells had much higher densities of active origins (49.1±2.6 kb, see also Kawabata et al. 2011) than either parental inbred strains (B6 and C3H wildtype cells, 112±5.0 kb and 95.0±5.0 kb, respectively). As F1 Mcm4chaos3/chaos3 mice are fully viable, this higher density of active origins could be a factor in supporting viability. The density of active origins in cells in another mixed background (129x Balb/c) (Kunnev et al. 2010) is also very similar to what was seen in our F1 cells in Fig. 2 (see also Kawabata et al. 2011). These observations suggest that a function of hybrid vigor may be to increase active origin density.
A positive correlation between replicon size and fork velocity has already been reported (Conti et al. 2007; Courbet et al. 2008). Larger replicons are typically replicated by faster forks, while slower forks are often found in smaller replicons. Consistent with these observations, we also found an inverse relationship between active origin density and fork velocity in the three genetic backgrounds we analyzed. Reflecting a relatively lower density of active origins, the average fork velocity was significantly faster in wildtype B6 cells (1.7±0.05 kb/min) than in wildtype C3H (0.84±0.02 kb/min) or F1 cells (0.61 ±0.01 kb/min, from Kawabata et al. 2011). A recent study demonstrated that fork velocity determines replicon size by modulating chromatin loop length (Courbet et al. 2008). For example, when fork velocity is slow, inefficient (dormant) origins fire more frequently, causing chromatin looping to become smaller in the following G1 phase (Courbet et al. 2008). Our findings suggest the intriguing possibility that fork velocity could be genetically regulated, thereby determining chromatin looping and thus origin density.
Supplementary Material
Acknowledgments
We thank Dr. Kazuto Sugimura for the DNA fiber protocol and Dr. David Largaespada for his critical reading of the manuscript. This study was supported by grants (to N.S.) from Susan G Komen for the Cure (BCTR0707864) and the NCI (R01-CA148806). The final publication is available at www.springerlink.com doi: 10.1007/s00335-011-9333-7.
References
- Bao S, Tibbetts RS, Brumbaugh KM, Fang Y, Richardson DA, Ali A, Chen SM, Abraham RT, Wang XF. ATR/ATM-mediated phosphorylation of human Rad17 is required for genotoxic stress responses. Nature. 2001;411:969–974. doi: 10.1038/35082110. [DOI] [PubMed] [Google Scholar]
- Blow JJ, Gillespie PJ. Replication licensing and cancer--a fatal entanglement? Nat Rev Cancer. 2008;8:799–806. doi: 10.1038/nrc2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–1501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- Carreau M. Not-so-novel phenotypes in the Fanconi anemia group D2 mouse model. Blood. 2004;103:2430. doi: 10.1182/blood-2003-11-3946. [DOI] [PubMed] [Google Scholar]
- Cazzalini O, Perucca P, Riva F, Stivala LA, Bianchi L, Vannini V, Ducommun B, Prosperi E. p21CDKN1A does not interfere with loading of PCNA at DNA replication sites, but inhibits subsequent binding of DNA polymerase delta at the G1/S phase transition. Cell Cycle. 2003;2:596–603. [PubMed] [Google Scholar]
- Chuang CH, Wallace MD, Abratte C, Southard T, Schimenti JC. Incremental Genetic Perturbations to MCM2-7 Expression and Subcellular Distribution Reveal Exquisite Sensitivity of Mice to DNA Replication Stress. PLoS Genet. 2010;6 doi: 10.1371/journal.pgen.1001110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9:616–627. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti C, Sacca B, Herrick J, Lalou C, Pommier Y, Bensimon A. Replication fork velocities at adjacent replication origins are coordinately modified during DNA replication in human cells. Mol Biol Cell. 2007;18:3059–3067. doi: 10.1091/mbc.E06-08-0689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courbet S, Gay S, Arnoult N, Wronka G, Anglana M, Brison O, Debatisse M. Replication fork movement sets chromatin loop size and origin choice in mammalian cells. Nature. 2008;455:557–560. doi: 10.1038/nature07233. [DOI] [PubMed] [Google Scholar]
- Gari K, Decaillet C, Stasiak AZ, Stasiak A, Constantinou A. The Fanconi anemia protein FANCM can promote branch migration of Holliday junctions and replication forks. Mol Cell. 2008;29:141–148. doi: 10.1016/j.molcel.2007.11.032. [DOI] [PubMed] [Google Scholar]
- Gatz SA, Wiesmuller L. p53 in recombination and repair. Cell Death Differ. 2006;13:1003–1016. doi: 10.1038/sj.cdd.4401903. [DOI] [PubMed] [Google Scholar]
- Ge XQ, Blow JJ. The licensing checkpoint opens up. Cell Cycle. 2009;8:2320–2322. [PMC free article] [PubMed] [Google Scholar]
- Ge XQ, Jackson DA, Blow JJ. Dormant origins licensed by excess Mcm2-7 are required for human cells to survive replicative stress. Genes Dev. 2007;21:3331–3341. doi: 10.1101/gad.457807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottifredi V, Shieh S, Taya Y, Prives C. p53 accumulates but is functionally impaired when DNA synthesis is blocked. Proc Natl Acad Sci U S A. 2001;98:1036–1041. doi: 10.1073/pnas.021282898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green ES, Stubbs JL, Levine EM. Genetic rescue of cell number in a mouse model of microphthalmia: interactions between Chx10 and G1-phase cell cycle regulators. Development. 2003;130:539–552. doi: 10.1242/dev.00275. [DOI] [PubMed] [Google Scholar]
- Ha SA, Shin SM, Namkoong H, Lee H, Cho GW, Hur SY, Kim TE, Kim JW. Cancer-associated expression of minichromosome maintenance 3 gene in several human cancers and its involvement in tumorigenesis. Clin Cancer Res. 2004;10:8386–8395. doi: 10.1158/1078-0432.CCR-04-1029. [DOI] [PubMed] [Google Scholar]
- Hamelik RM, Krishan A. Click-iT assay with improved DNA distribution histograms. Cytometry A. 2009;75:862–865. doi: 10.1002/cyto.a.20780. [DOI] [PubMed] [Google Scholar]
- Houghtaling S, Timmers C, Noll M, Finegold MJ, Jones SN, Meyn MS, Grompe M. Epithelial cancer in Fanconi anemia complementation group D2 (Fancd2) knockout mice. Genes Dev. 2003;17:2021–2035. doi: 10.1101/gad.1103403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett NG, Taniguchi T, Durkin SG, D’Andrea AD, Glover TW. The Fanconi anemia pathway is required for the DNA replication stress response and for the regulation of common fragile site stability. Hum Mol Genet. 2005;14:693–701. doi: 10.1093/hmg/ddi065. [DOI] [PubMed] [Google Scholar]
- Ibarra A, Schwob E, Mendez J. Excess MCM proteins protect human cells from replicative stress by licensing backup origins of replication. Proc Natl Acad Sci U S A. 2008;105:8956–8961. doi: 10.1073/pnas.0803978105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilves I, Petojevic T, Pesavento JJ, Botchan MR. Activation of the MCM2-7 Helicase by Association with Cdc45 and GINS Proteins. Mol Cell. 2010;37:247–258. doi: 10.1016/j.molcel.2009.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimi Y, Okayasu I, Kato C, Kwon HJ, Kimura H, Yamada K, Song SY. Enhanced expression of Mcm proteins in cancer cells derived from uterine cervix. Eur J Biochem. 2003;270:1089–1101. doi: 10.1046/j.1432-1033.2003.03440.x. [DOI] [PubMed] [Google Scholar]
- Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- Kalter H. Sporadic congenital malformations of newborn inbred mice. Teratology. 1968;1:193–199. doi: 10.1002/tera.1420010208. [DOI] [PubMed] [Google Scholar]
- Kawabata T, Luebbe SW, Yamaguchi S, Ilves I, Matise I, Buske T, Botchan MR, Shima N. Stalled fork rescue via dormant replication origins in unchallenged S phase promotes proper chromosome segregation and tumor suppression. Molecular Cell. 2011;41:543–553. doi: 10.1016/j.molcel.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari A, Schultz N, Helleday T. p53 protects from replication-associated DNA double-strand breaks in mammalian cells. Oncogene. 2004;23:2324–2329. doi: 10.1038/sj.onc.1207379. [DOI] [PubMed] [Google Scholar]
- Kunnev D, Rusiniak ME, Kudla A, Freeland A, Cady GK, Pruitt SC. DNA damage response and tumorigenesis in Mcm2-deficient mice. Oncogene. 2010;29:3630–3638. doi: 10.1038/onc.2010.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei M. The MCM complex: its role in DNA replication and implications for cancer therapy. Curr Cancer Drug Targets. 2005;5:365–380. doi: 10.2174/1568009054629654. [DOI] [PubMed] [Google Scholar]
- Lim DS, Hasty P. A mutation in mouse rad51 results in an early embryonic lethal that is suppressed by a mutation in p53. Mol Cell Biol. 1996;16:7133–7143. doi: 10.1128/mcb.16.12.7133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Slater DM, Lenburg M, Nevis K, Cook JG, Vaziri C. Replication licensing promotes cyclin D1 expression and G1 progression in untransformed human cells. Cell Cycle. 2009;8:125–136. doi: 10.4161/cc.8.1.7528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machida YJ, Teer JK, Dutta A. Acute reduction of an origin recognition complex (ORC) subunit in human cells reveals a requirement of ORC for Cdk2 activation. J Biol Chem. 2005;280:27624–27630. doi: 10.1074/jbc.M502615200. [DOI] [PubMed] [Google Scholar]
- Marusyk A, Wheeler LJ, Mathews CK, DeGregori J. p53 mediates senescence-like arrest induced by chronic replicational stress. Mol Cell Biol. 2007;27:5336–5351. doi: 10.1128/MCB.01316-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGarry TJ, Kirschner MW. Geminin, an inhibitor of DNA replication, is degraded during mitosis. Cell. 1998;93:1043–1053. doi: 10.1016/s0092-8674(00)81209-x. [DOI] [PubMed] [Google Scholar]
- Moyer SE, Lewis PW, Botchan MR. Isolation of the Cdc45/Mcm2-7/GINS (CMG) complex, a candidate for the eukaryotic DNA replication fork helicase. Proc Natl Acad Sci U S A. 2006;103:10236–10241. doi: 10.1073/pnas.0602400103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murga M, Bunting S, Montana MF, Soria R, Mulero F, Canamero M, Lee Y, McKinnon PJ, Nussenzweig A, Fernandez-Capetillo O. A mouse model of ATR-Seckel shows embryonic replicative stress and accelerated aging. Nat Genet. 2009;41:891–898. doi: 10.1038/ng.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak BK, Das GM. Stabilization of p53 and transactivation of its target genes in response to replication blockade. Oncogene. 2002;21:7226–7229. doi: 10.1038/sj.onc.1205889. [DOI] [PubMed] [Google Scholar]
- Nevis KR, Cordeiro-Stone M, Cook JG. Origin licensing and p53 status regulate Cdk2 activity during G(1) Cell Cycle. 2009;8:1952–1963. doi: 10.4161/cc.8.12.8811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oku T, Ikeda S, Sasaki H, Fukuda K, Morioka H, Ohtsuka E, Yoshikawa H, Tsurimoto T. Functional sites of human PCNA which interact with p21 (Cip1/Waf1), DNA polymerase delta and replication factor C. Genes Cells. 1998;3:357–369. doi: 10.1046/j.1365-2443.1998.00199.x. [DOI] [PubMed] [Google Scholar]
- Pruitt SC, Bailey KJ, Freeland A. Reduced Mcm2 Expression Results in Severe Stem/Progenitor Cell Deficiency and Cancer. Stem Cells. 2007;25:3121–3121. doi: 10.1634/stemcells.2007-0483. [DOI] [PubMed] [Google Scholar]
- Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- Rossi R, Lidonnici MR, Soza S, Biamonti G, Montecucco A. The dispersal of replication proteins after Etoposide treatment requires the cooperation of Nbs1 with the ataxia telangiectasia Rad3-related/Chk1 pathway. Cancer Res. 2006;66:1675–1683. doi: 10.1158/0008-5472.CAN-05-2741. [DOI] [PubMed] [Google Scholar]
- Ruzankina Y, Schoppy DW, Asare A, Clark CE, Vonderheide RH, Brown EJ. Tissue regenerative delays and synthetic lethality in adult mice after combined deletion of Atr and Trp53. Nat Genet. 2009;41:1144–1149. doi: 10.1038/ng.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scian MJ, Carchman EH, Mohanraj L, Stagliano KE, Anderson MA, Deb D, Crane BM, Kiyono T, Windle B, Deb SP, Deb S. Wild-type p53 and p73 negatively regulate expression of proliferation related genes. Oncogene. 2008;27:2583–2593. doi: 10.1038/sj.onc.1210898. [DOI] [PubMed] [Google Scholar]
- Sclafani RA, Tecklenburg M, Pierce A. The mcm5-bob1 bypass of Cdc7p/Dbf4p in DNA replication depends on both Cdk1-independent and Cdk1-dependent steps in Saccharomyces cerevisiae. Genetics. 2002;161:47–57. doi: 10.1093/genetics/161.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh SY, Ikeda M, Taya Y, Prives C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell. 1997;91:325–334. doi: 10.1016/s0092-8674(00)80416-x. [DOI] [PubMed] [Google Scholar]
- Shima N, Alcaraz A, Liachko I, Buske TR, Andrews CA, Munroe RJ, Hartford SA, Tye BK, Schimenti JC. A viable allele of Mcm4 causes chromosome instability and mammary adenocarcinomas in mice. Nat Genet. 2007;39:93–98. doi: 10.1038/ng1936. [DOI] [PubMed] [Google Scholar]
- Shimura T, Torres MJ, Martin MM, Rao VA, Pommier Y, Katsura M, Miyagawa K, Aladjem MI. Bloom’s syndrome helicase and Mus81 are required to induce transient double-strand DNA breaks in response to DNA replication stress. J Mol Biol. 2008;375:1152–1164. doi: 10.1016/j.jmb.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shreeram S, Sparks A, Lane DP, Blow JJ. Cell type-specific responses of human cells to inhibition of replication licensing. Oncogene. 2002;21:6624–6632. doi: 10.1038/sj.onc.1205910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Z, Smith S, Wang L, Rice MC, Kmiec EB. Disruption of muREC2/RAD51L1 in mice results in early embryonic lethality which can Be partially rescued in a p53(−/−) background. Mol Cell Biol. 1999;19:8686–8693. doi: 10.1128/mcb.19.12.8686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes MP, Michael WM. DNA damage-induced replication arrest in Xenopus egg extracts. J Cell Biol. 2003;163:245–255. doi: 10.1083/jcb.200306006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimura K, Takebayashi S, Ogata S, Taguchi H, Okumura K. Non-denaturing fluorescence in situ hybridization to find replication origins in a specific genome region on the DNA fiber. Biosci Biotechnol Biochem. 2007;71:627–632. doi: 10.1271/bbb.60662. [DOI] [PubMed] [Google Scholar]
- Todorov IT, Attaran A, Kearsey SE. BM28, a human member of the MCM2-3-5 family, is displaced from chromatin during DNA replication. J Cell Biol. 1995;129:1433–1445. doi: 10.1083/jcb.129.6.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waga S, Hannon GJ, Beach D, Stillman B. The p21 inhibitor of cyclin-dependent kinases controls DNA replication by interaction with PCNA. Nature. 1994;369:574–578. doi: 10.1038/369574a0. [DOI] [PubMed] [Google Scholar]
- Wang LC, Stone S, Hoatlin ME, Gautier J. Fanconi anemia proteins stabilize replication forks. DNA Repair (Amst) 2008;7:1973–1981. doi: 10.1016/j.dnarep.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlschlegel JA, Dwyer BT, Dhar SK, Cvetic C, Walter JC, Dutta A. Inhibition of eukaryotic DNA replication by geminin binding to Cdt1. Science. 2000;290:2309–2312. doi: 10.1126/science.290.5500.2309. [DOI] [PubMed] [Google Scholar]
- Wong JC, Alon N, McKerlie C, Huang JR, Meyn MS, Buchwald M. Targeted disruption of exons 1 to 6 of the Fanconi Anemia group A gene leads to growth retardation, strain-specific microphthalmia, meiotic defects and primordial germ cell hypoplasia. Hum Mol Genet. 2003;12:2063–2076. doi: 10.1093/hmg/ddg219. [DOI] [PubMed] [Google Scholar]
- Woodward AM, Gohler T, Luciani MG, Oehlmann M, Ge X, Gartner A, Jackson DA, Blow JJ. Excess Mcm2-7 license dormant origins of replication that can be used under conditions of replicative stress. J Cell Biol. 2006;173:673–683. doi: 10.1083/jcb.200602108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D. p21 is a universal inhibitor of cyclin kinases. Nature. 1993;366:701–704. doi: 10.1038/366701a0. [DOI] [PubMed] [Google Scholar]
- Xu X, Qiao W, Linke SP, Cao L, Li WM, Furth PA, Harris CC, Deng CX. Genetic interactions between tumor suppressors Brca1 and p53 in apoptosis, cell cycle and tumorigenesis. Nat Genet. 2001;28:266–271. doi: 10.1038/90108. [DOI] [PubMed] [Google Scholar]
- Yan H, Merchant AM, Tye BK. Cell cycle-regulated nuclear localization of MCM2 and MCM3, which are required for the initiation of DNA synthesis at chromosomal replication origins in yeast. Genes Dev. 1993;7:2149–2160. doi: 10.1101/gad.7.11.2149. [DOI] [PubMed] [Google Scholar]
- Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol. 2001;21:4129–4139. doi: 10.1128/MCB.21.13.4129-4139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.