

Abstract
Background
Mitochondrial dysfunction (MtD) has been observed in approximately five percent of children with autism spectrum disorders (ASD). MtD could impair highly energy-dependent processes such as neurodevelopment, thereby contributing to autism. Most of the previous studies of MtD in autism have been restricted to the biomarkers of energy metabolism, while most of the genetic studies have been based on mutations in the mitochondrial DNA (mtDNA). Despite the mtDNA, most of the proteins essential for mitochondrial replication and function are encoded by the genomic DNA; so far, there have been very few studies of those genes. Therefore, we carried out a detailed study involving gene expression and genetic association studies of genes related to diverse mitochondrial functions.
Methods
For gene expression analysis, postmortem brain tissues (anterior cingulate gyrus (ACG), motor cortex (MC) and thalamus (THL)) from autism patients (n=8) and controls (n=10) were obtained from the Autism Tissue Program (Princeton, NJ, USA). Quantitative real-time PCR arrays were used to quantify the expression of 84 genes related to diverse functions of mitochondria, including biogenesis, transport, translocation and apoptosis. We used the delta delta Ct (∆∆Ct) method for quantification of gene expression. DNA samples from 841 Caucasian and 188 Japanese families were used in the association study of genes selected from the gene expression analysis. FBAT was used to examine genetic association with autism.
Results
Several genes showed brain region-specific expression alterations in autism patients compared to controls. Metaxin 2 (MTX2), neurofilament, light polypeptide (NEFL) and solute carrier family 25, member 27 (SLC25A27) showed consistently reduced expression in the ACG, MC and THL of autism patients. NEFL (P = 0.038; Z-score 2.066) and SLC25A27 (P = 0.046; Z-score 1.990) showed genetic association with autism in Caucasian and Japanese samples, respectively. The expression of DNAJC19, DNM1L, LRPPRC, SLC25A12, SLC25A14, SLC25A24 and TOMM20 were reduced in at least two of the brain regions of autism patients.
Conclusions
Our study, though preliminary, brings to light some new genes associated with MtD in autism. If MtD is detected in early stages, treatment strategies aimed at reducing its impact may be adopted.
Keywords: Autism, Mitochondria, Postmortem brain, NEFL, Uncoupling protein, Metaxin
Background
Autism is a complex neurodevelopmental disorder characterized by deficiencies in social interaction and communication, and repetitive and stereotyped behaviors. Autistic disorder, Asperger syndrome, and pervasive developmental disorder-not otherwise specified (PDD-NOS) comprise a heterogeneous group of neurodevelopmental disorders known as autism spectrum disorders (ASD). The abnormalities are usually identified in the early years of childhood and often coexist with impairments in cognitive functioning, learning, attention and sensory processing. According to a recent report, the prevalence of this disorder has risen to 1 in 110, with a male to female ratio of 4.5:1 [1].
A growing body of evidence from biochemical and neuroimaging studies has suggested that a disturbed brain bioenergetic metabolism underlies the pathophysiology of autism in some cases. Magnetic resonance spectroscopy studies have shown, in the brain of autism patients, abnormal levels of metabolites relating to brain bioenergetics, such as decreased levels of phosphocreatine and N-acetyl-aspartate, and elevated lactate [2,3].
Mitochondria serve as the energy powerhouses of eukaryotic cells, since they generate most of the adenosine triphosphate (ATP), the source of chemical energy in cells. The findings of abnormal brain bioenergetics, therefore, support an involvement of mitochondrial dysfunction (MtD) in the pathogenesis of ASD [4]. Diminished levels of ATP have been observed in autism brain [2]. Rats induced for MtD have been found to exhibit certain brain, behavioral and metabolic changes consistent with ASD, including microglial activation, reduced levels of glutathione, repetitive behaviors, social interaction deficits, hyperactivity and oxidative stress (OS) [5-8].
In a systematic review and meta-analysis, MtD was observed in approximately five percent of children with ASD; developmental regression, seizures, motor delay and gastrointestinal abnormalities were found to be significantly more prevalent in children with ASD/MtD as compared with the general ASD population [9]. Defective lymphocytic mitochondria [10] and ultrastructural abnormalities of mitochondria [11,12] have been reported in autism. Nutritional supplements (for example, carnitine, vitamin B) and/or antioxidants (for example, co-enzyme Q10) have been found to be beneficial in the treatment of some children with ASD/MtD [13-15].
Recent studies have reported brain region-specific deficits of mitochondrial electron transport chain complexes in autism [16,17]. Upregulated expression of the mitochondrial aspartate/glutamate carrier (SLC25A12) [18,19], and evidence of hypoxia, as measured by a reduction in the anti-apoptotic protein Bcl-2 and an increase in the pro-apoptotic protein p53 [20,21], has also been reported in autism brain.
Several of the previous studies of MtD in autism were restricted to the biomarkers of energy metabolism, while most of the genetic studies were based on mutations in the mitochondrial DNA (mtDNA). Despite the mtDNA, most of the proteins essential for mitochondrial replication and function are encoded by the genomic DNA; so far, there have been very few studies of those genes. We aimed at elucidating the role of MtD in the pathogenesis of autism. Using the postmortem brains of autism patients and healthy controls, we compared the expression of 84 genes involved in diverse functions of the mitochondria such as, biogenesis, transport, translocation and apoptosis. Furthermore, we analyzed the genetic association of three of these genes with autism, in two independent studies involving family-based samples of different origins.
Methods
Gene expression studies of human postmortem brains
Postmortem brain tissues
Postmortem brain samples of autism patients and healthy controls were provided by the Autism Tissue Program (http://www.autismtissueprogram.org), NICHD Brain and Tissue Bank for Developmental Disorders (NICHD BTB; Baltimore, MD, USA; http://medschool.umaryland.edu/btbank/) and Harvard Brain Tissue Resource Center (HBTRC; Belmont, MA; http://www.brainbank.mclean.org/). Frozen tissue samples from anterior cingulate gyrus (ACG), motor cortex (MC) and thalamus (THL) were used in the study. Demographic characteristics of the samples (ACG: eight autism, ten controls; MC: seven autism, eight controls; THL: eight autism, nine controls) are described in Table 1.
Table 1.
Postmortem brain tissue information
| Sample IDa | Diagnosis | Age (years) | Gender | PMI (hours) | Race | Cause of death | Brain regionsb |
|---|---|---|---|---|---|---|---|
| 1065 |
Control |
15 |
M |
12 |
Caucasian |
Multiple injuries |
ACG, THL |
| 1297 |
Control |
15 |
M |
16 |
African American |
Multiple injuries |
ACG, MC, THL |
| 1407 |
Control |
9 |
F |
20 |
African American |
Asthma |
ACG, MC, THL |
| 1541 |
Control |
20 |
F |
19 |
Caucasian |
Head injuries |
ACG, MC, THL |
| 1649 |
Control |
20 |
M |
22 |
Hispanic |
Multiple injuries |
ACG, MC, THL |
| 1708 |
Control |
8 |
F |
20 |
African American |
Asphyxia, multiple injuries |
ACG, MC, THL |
| 1790 |
Control |
13 |
M |
18 |
Caucasian |
Multiple injuries |
ACG |
| 1793 |
Control |
11 |
M |
19 |
African American |
Drowning |
ACG, MC, THL |
| 1860 |
Control |
8 |
M |
5 |
Caucasian |
Cardiac Arrhythmia |
ACG |
| 4543 |
Control |
28 |
M |
13 |
Caucasian |
Multiple injuries |
MC, THL |
| 4638 |
Control |
15 |
F |
5 |
Caucasian |
Chest injuries |
ACG |
| 4722 |
Control |
14 |
M |
16 |
Caucasian |
Multiple injuries |
MC, THL |
| 797 |
Autism |
9 |
M |
13 |
Caucasian |
Drowning |
ACG, THL |
| 1638 |
Autism |
20 |
F |
50 |
Caucasian |
Seizure |
ACG, MC, THL |
| 4231 |
Autism |
8 |
M |
12 |
African American |
Drowning |
ACG, MC, THL |
| 4721 |
Autism |
8 |
M |
16 |
African American |
Drowning |
ACG, MC, THL |
| 4899 |
Autism |
14 |
M |
9 |
Caucasian |
Drowning |
ACG, MC, THL |
| 5000 |
Autism |
27 |
M |
8.3 |
NA |
NA |
ACG, MC, THL |
| 6294 |
Autism |
16 |
M |
NA |
NA |
NA |
ACG, MC, THL |
| 6640 | Autism | 29 | F | 17.83 | NA | NA | ACG, MC, THL |
aAutism Tissue Program (ATP) identifier; bBrain regions for which, each sample was available; ACG, anterior cingulate gyrus; F, female; M, male; MC, motor cortex; NA, not available; PMI, postmortem interval; THL, thalamus.
The difference in age and postmortem interval (PMI) between the autism and control groups was examined by t-test. Fisher’s Exact test was used to examine the difference in sex distribution between the two groups (see Additional file 1).
RNA extraction
The brain tissues (approximately 75 mg obtained by macrodissection) were homogenized by ultrasonication, and total RNA was extracted using TRIzol™ Reagent (Invitrogen, Carlsbad, CA, USA) in accordance with the manufacturer’s protocol. The RNA samples were further purified using an RNeasy™ Micro Kit (QIAGEN GmbH, Hilden, Germany) following the manufacturer’s instructions; this protocol includes a DNase treatment step. The quantity (absorbance at 260 nm) and quality (ratio of absorbance at 260 nm and 280 nm) of RNA were estimated with a NanoDrop ND-1000 Spectrophotometer (Scrum, Tokyo, Japan). As per the requirements for the subsequent array experiment, the following criteria were met for all of the RNA samples: 1) A260:A230 ratio, >1.7; 2) A260:A280 ratio, between 1.8 and 2.0 (the A260:A280 ratio of all our RNA samples were in the range of 2.0 to 2.1), and 3) concentration of total RNA, >40 ng/μl.
First strand cDNA synthesis
First-strand cDNA was synthesized from 500 ng of total RNA using the RT2 First Strand Kit (SABiosciences, Frederick, MD, USA) following the manufacturer’s protocol. The kit contains a genomic DNA elimination step and a built-in external RNA control that helps monitor reverse transcription efficiency and tests for contaminating inhibitors during quantitative PCR (qPCR).
qPCR
We used the RT2 ProfilerTM PCR Array Human Mitochondria (SABiosciences) for quantifying the expression of 84 genes related to the biogenesis and functions of mitochondria. The array also has five reference genes (beta-2-microglobulin (B2M), hypoxanthine phosphoribosyltransferase 1 (HPRT1), ribosomal protein L13a (RPL13A), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and actin beta (ACTB)), three reverse transcription controls (RTCs), three positive PCR controls (PPCs), and one genomic DNA control (GDC), making up to a total of 96 assays. The details of the genes are provided in Additional files 2 and 3. The 384-well format of the array includes four replicates of each of the 96 assays. It makes use of the SYBRTM Green method of qPCR analysis. The qPCR reactions were carried out according to the manufacturer’s protocol, in ABI PRISM 7900HT SDS (Applied Biosystems (ABI), Foster City, CA, USA).
Data analysis
The threshold cycle (Ct) values obtained from qPCR were analyzed by the ∆∆Ct method using RT2 Profiler PCR Array Data Analysis (Microsoft Excel-based program of SABiosciences). It calculates:
1) ∆Ct of each gene = Ct of gene of interest - average Ct of chosen reference genes
2) ∆∆Ct for each gene across two groups; ∆∆Ct = ∆Ct (autism group) - ∆Ct (control group)
3) fold-change for each gene from control group to autism group as 2 ^ (−∆∆Ct)
Based on the Kolmogorov-Smirnov test, the expression of all genes was found to follow a normal distribution. Therefore, a t-test, which was also the default option in our data analysis program, was used to examine any significant difference in gene expression between the control and autism groups.
The statistical program also performs the following functions: 1) interprets all Ct values ≥35 as a negative call; 2) examines genomic DNA contamination in the samples based on the Ct of GDC (Ct <35 will indicate genomic DNA contamination); 3) examines the presence of impurities in RNA samples based on the Ct value of PPC (Ct should be 20 ± 2 on each array and should not vary by more than two cycles between the arrays being compared); and 4) interprets any inhibition of reverse transcription based on the Ct values of RTC and PPC (values <5 for Ct RTC - Ct PPC is indicative of no apparent inhibition).
Genetic association study
An association study, rather than deep sequencing, is considered as a cost-effective approach for studying complex traits like autism.
AGRE subjects
841 pedigree samples (3211 individuals in total) were obtained from Autism Genetic Resource Exchange (AGRE; http://www.agre.org; Los Angeles, CA, USA) [22]. This includes 1467 patients (1178 males; 289 females) with autism. Pedigree information for each individual, along with the diagnoses based on Autism Diagnostic Interview-Revised (ADI-R) [23], are available in the AGRE website. Families with a non-idiopathic autism flag (for example, fragile-X, abnormal brain imaging results, dysmorphic features, birth trauma) recorded for any of its members were excluded from the study.
Japanese subjects
The aforementioned AGRE samples were predominantly of Caucasian origin. Therefore, Japanese samples were used in an effort to replicate the AGRE genetic association results in samples of a different ethnicity. The Japanese trio samples included 188 children with ASD [gender: 155 males, 33 females; age: 10.49 ± 4.75 years (mean ± SD); IQ: 82.06 ± 26.6] and both parents for each child. All of the subjects were Japanese, born and living in the areas of central Japan including Chukyo, Tokai and Kanto. The purpose of the study was fully explained to the participants, and written informed consent was obtained. This study was approved by the Ethics Committee of Hamamatsu University School of Medicine. The diagnosis of autism was based on ADI-R [23] and DSM-IV-TR [24]. We are licensed to privately use a Japanese version of ADI-R by Professor C. Lord. ADI-R scores were available for 100 patients. All of the autistic individuals underwent screening to exclude comorbid psychiatric illnesses (for example, schizophrenia, affective disorders, mental retardation, and personality or behavioral disorders) by means of the Structured Clinical Interview for DSM-IV (SCID) [25]. Individuals with a history of neurological disorders (for example, epilepsy, head injury) or genetic disorders (for example, fragile X syndrome, tuberous sclerosis) were excluded.
Single nucleotide polymorphism (SNP) selection
The genomic structures of metaxin 2 (MTX2; 2q31.1), neurofilament, light polypeptide 68 kDa (NEFL; 8p21.2), and solute carrier family 25, member 27 (SLC25A27; 6p12.3) were based on the University of California, Santacruz (UCSC; http://genome.ucsc.edu/) February 2009 draft assembly of the human genome. MTX2 (68.626 kb) consists of 11 exons, NEFL (5.664 kb) of 4 exons, and SLC25A27 (25.238 kb) of 9 exons. SNPs (minor allele frequency >0.1) for the association study were selected from the International HapMap Project (http://www.hapmap.org) database on Caucasian and Japanese populations. Additional file 4 gives the list of SNPs chosen for the three genes using the pairwise tagging option of Haploview v4.1 (http://www.broad.mit.edu/mpg/haploview).
Genotyping
Assay-on-Demand/Assay-by-design SNP genotyping products (ABI) were used to score genotypes, based on the TaqMan™ assay method [26]. Genotypes were determined in ABI PRISM 7900HT SDS (ABI), and analyzed using SDS v2.0 software (ABI).
Statistical analysis
FBAT v2.0.3 (http://biosun1.harvard.edu/~fbat/fbat.htm) was used to examine the associations of SNPs with autism. FBAT-MM option was used for multimarker test. FBAT provides valid tests of association in the presence of linkage even when using multiple affected siblings from families of variable structure. In addition to performing tests of association for individual markers, FBAT allows for tests of association with haplotypes that may be phase ambiguous. Linkage disequilibrium (LD) plots based on D′ values were constructed using Haploview. Haplotype association was also examined using this software. Power analysis was done using the Genetic Power Calculator (http://pngu.mgh.harvard.edu/~purcell/gpc/dtdt.html).
Results
Gene expression analysis using human postmortem brain samples
There was no significant difference in age, postmortem interval (PMI) or sex distribution between the autism and control groups in any of the brain regions analyzed (see Additional file 1).
In the qPCR experiment, genomic DNA contamination was not observed for any of the RNA samples; Ct GDC was >35 for all the samples, indicating that genomic DNA contamination, if present, was too low to affect the gene expression results. Ct PPC was 20 ± 2 for all the arrays compared, showing the apparent absence of impurities in the RNA samples. Further, there was no indication of any inhibition of reverse transcription for any of the samples, since Ct RTC - Ct PPC was <5 for all the samples.
For normalization of gene expression, the following reference genes were selected for the various brain regions: 1) ACG: RPL13A, GAPDH and ACTB; 2) MC: B2M, RPL13A, GAPDH and ACTB; 3) THL: B2M, HPRT1 and GAPDH. The chosen reference genes for each brain region did not show any significant difference in expression between the control and autism groups.
We observed brain region-specific alterations in the expression of several genes in the autism group compared to the control group (Table 2). A total of 22 genes in ACG, 15 genes in MC and 12 genes in THL showed aberrant expression in autism patients compared to controls. These genes belong to the following functional groups: 1) membrane polarization and potential, 2) mitochondrial transport, 3) small molecule transport, 4) targeting proteins to mitochondria, 5) mitochondrion protein import, 6) outer membrane translocation, 7) inner membrane translocation, 8) mitochondrial fission and fusion, 9) mitochondrial localization, and 10) apoptosis (Table 2). A majority of the genes showed a reduced expression in autism as compared to the controls. Eleven genes each belonging to the SLC25A mitochondrial transporter family and TIMM/TOMM family of translocases, showed altered expression in autism.
Table 2.
Genes with altered expressions in autism postmortem brains
|
Gene |
Anterior Cingulate Gyrus |
Motor Cortex |
Thalamus |
|||
|---|---|---|---|---|---|---|
| Fold change | Pvalue* | Fold change | Pvalue* | Fold change | Pvalue* | |
|
AIPb,d,e |
|
|
−1.473 |
0.048 |
|
|
|
BCL2a,b,j |
1.356 |
0.045 |
|
|
|
|
|
DNAJC19b,d,e |
|
|
−1.868 |
0.037 |
−1.520 |
0.030 |
|
DNM1Li,j |
−1.658 |
0.020 |
−1.603 |
0.045 |
|
|
|
HSP90AA1b |
|
|
|
|
1.662 |
0.044 |
|
LRPPRCi |
−1.486 |
0.025 |
−1.530 |
0.018 |
|
|
|
MFN2b,d,h,i |
−1.446 |
0.021 |
|
|
|
|
|
MIPEPb,d,e |
|
|
−2.003 |
0.012 |
|
|
|
MTX2b |
−1.795 |
0.044 |
−2.055 |
0.017 |
−2.511 |
0.002 |
|
NEFLi |
−4.208 |
0.014 |
−2.935 |
0.025 |
−6.006 |
0.012 |
|
RHOT2i |
−1.338 |
0.019 |
|
|
|
|
|
SLC25A12c |
−2.112 |
0.013 |
−1.913 |
0.008 |
|
|
|
SLC25A14c |
−1.876 |
0.034 |
−1.924 |
0.010 |
|
|
|
SLC25A15c |
−1.402 |
0.036 |
|
|
|
|
|
SLC25A22c |
−2.061 |
0.007 |
|
|
|
|
|
SLC25A24c |
|
|
−1.690 |
0.008 |
−1.625 |
0.019 |
|
SLC25A25c |
|
|
|
|
1.830 |
0.044 |
|
SLC25A27c |
−2.132 |
0.025 |
−2.167 |
0.042 |
−2.644 |
0.026 |
|
SLC25A3c |
|
|
−1.668 |
0.020 |
|
|
|
SLC25A37c |
|
|
|
|
1.973 |
0.046 |
|
SLC25A4c |
−1.614 |
0.034 |
|
|
|
|
|
SLC25A5c |
|
|
−1.548 |
0.027 |
|
|
|
SOD2j |
|
|
|
|
3.919 |
0.036 |
|
TIMM17Ag |
−1.809 |
0.011 |
|
|
|
|
|
TIMM17Bg |
−1.601 |
0.006 |
|
|
|
|
|
TIMM23g |
|
|
−1.734 |
0.022 |
|
|
|
TIMM44g |
|
|
|
|
1.769 |
0.034 |
|
TIMM50g |
−1.513 |
0.045 |
|
|
|
|
|
TIMM8Ag |
−1.612 |
0.021 |
|
|
|
|
|
TIMM9g |
|
|
|
|
−1.940 |
0.010 |
|
TOMM20f |
−1.812 |
0.025 |
−1.745 |
0.017 |
|
|
|
TOMM22f |
−1.450 |
0.024 |
|
|
|
|
|
TOMM34f |
|
|
|
|
−1.995 |
0.002 |
|
TOMM70Af |
−1.525 |
0.041 |
|
|
|
|
|
TP53a,b,j |
1.769 |
0.021 |
|
|
|
|
| TSPOb,d | 1.720 | 0.037 | ||||
aMembrane polarization and potential; bMitochondrial transport; cSmall molecule transport; dTargeting proteins to mitochondria; eMitochondrion protein import; fOuter membrane translocation; gInner membrane translocation; hMitochondrial fission and fusion; iMitochondrial localization; jApoptotic genes; *P values were calculated by t-test.
The genes MTX2, NEFL and SLC25A27showed consistently reduced expression in all the three brain regions (ACG, MC and THL) of autism patients (Figure 1). The most pronounced reduction, in autism brains, was observed for NEFL [−4.208 fold (P = 0.014) in ACG; -2.935 fold (P = 0.025) in MC; -6.006 fold (P = 0.012) in THL].
Figure 1.
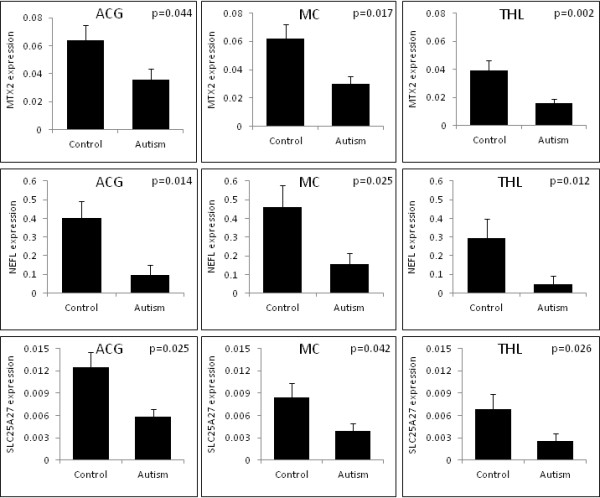
Reduced expression of MTX2, NEFL and SLC25A27 in autism brain. Comparison of the expression of MTX2, NEFL and SLC25A27 in the various brain regions of autism patients and healthy controls. MTX2, NEFL and SLC25A27showed significantly reduced expression in all the three brain regions of autism patients. The P values (t-test) are given at the top right corner of each graph. The y-axis of each graph represents the relative expression of the respective gene normalized to the reference genes. The gene expression is normalized against the average Ct of the chosen reference genes for each brain region. The following reference genes were selected for the various brain regions: a) anterior cingulate gyrus (ACG): RPL13A, GAPDH and ACTB b) motor cortex (MC): B2M, RPL13A, GAPDH and ACTB c) thalamus (THL): B2M, HPRT1 and GAPDH.
The expression of DNAJC19, DNM1L, LRPPRC, SLC25A12, SLC25A14, SLC25A24 and TOMM20 were consistently reduced in at least two of the brain regions of autism patients compared to controls (Table 2).
None of the P values of altered expression of genes withstand multimarker testing (conventional Bonferroni approach).
Genetic association study
Power analysis showed that the AGRE sample size of 841 families provided 37.3% and 95.5% power to detect an odds ratio of 1.2 and 1.5 respectively, for an allele frequency of 0.156 at an α of 0.05. However, the sample size of Japanese trios (188 trios) was underpowered to detect an association.
NEFL showed a nominal association with autism in the AGRE samples (Table 3; Figure 2). The SNP rs2979704 in the untranslated region (UTR) of exon 4 showed a significant association with autism (P = 0.038; Z-score 2.066). After multimarker testing, there was a tendency for association (P = 0.083). No significant haplotype association was observed. There was no association of NEFL with autism in the Japanese samples (see Additional file 5.1).
Table 3.
FBAT analysis ofNEFLin AGRE family samples
| Marker | Location | Allelea | Familiesb | Frequency | Z-scorec | Pvalued |
|---|---|---|---|---|---|---|
| rs2979704 |
Exon 4 UTR |
T |
356 |
0.844 |
2.066 |
0.038 |
| |
|
C |
|
0.156 |
−2.066 |
|
| rs3761 |
Exon 4 UTR |
G |
291 |
0.894 |
−1.655 |
0.097 |
| |
|
A |
|
0.106 |
1.655 |
|
| rs2979687 |
5’ |
C |
583 |
0.654 |
0.859 |
0.390 |
| |
|
T |
|
0.346 |
−0.859 |
|
| P value after multimarker testing | 0.083 | |||||
aMajor allele is listed first; bNumber of informative families used by FBAT; cPositive score indicates the risk allele, negative score indicates the protective allele; dP <0.05, additive model, significant P values are indicated in bold italic; UTR, Untranslated region.
Figure 2.
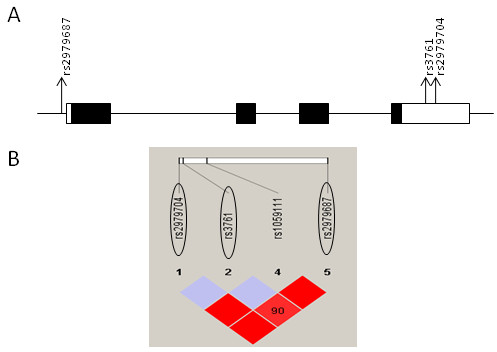
Linkage disequilibrium (LD) plot of NEFL. (A) Genomic structure of NEFL showing the locations of SNPs (denoted by arrows) used in the association study of AGRE samples. Exons are indicated by boxes, with translated regions in closed boxes and untranslated regions in open boxes. (B) LD structure of NEFL in Caucasian samples, based on D′ values. Tag single nucleotide polymorphisms (SNPs) are encircled.
SLC25A27 showed a nominal association with autism in Japanese samples (Table 4). The SNP rs6901178 in intron 4 showed an association with autism (P = 0.046; Z-score 1.990. After multimarker testing, there was a tendency for association (P = 0.073). No significant haplotype association was observed. There was no association of SLC25A27 with autism in the AGRE samples (see Additional file 5.2).
Table 4.
FBAT analysis ofSLC25A27in Japanese family samples
| Marker | Location | Allelea | Familiesb | Frequency | Z-scorec | Pvalued |
|---|---|---|---|---|---|---|
| rs12192544 |
5’ |
C |
68 |
0.888 |
0.000 |
1.000 |
| |
|
G |
|
0.112 |
0.000 |
|
| rs9381469 |
Intron 3 |
G |
138 |
0.532 |
0.147 |
0.883 |
| |
|
A |
|
0.468 |
−0.147 |
|
| rs6901132 |
Intron 4 |
A |
132 |
0.573 |
0.899 |
0.368 |
| |
|
G |
|
0.427 |
−0.899 |
|
| rs6901178 |
Intron 4 |
G |
61 |
0.896 |
−1.990 |
0.046 |
| |
|
A |
|
0.104 |
1.990 |
|
| rs2270450 |
Exon 9 UTR |
C |
109 |
0.771 |
0.877 |
0.380 |
| |
|
T |
|
0.229 |
−0.877 |
|
| P value after multimarker testing | 0.073 | |||||
aMajor allele is listed first; bNumber of informative families used by FBAT; cPositive score indicates the risk allele, negative score indicates the protective allele; dP <0.05, additive model, significant P values are indicated in bold italics; UTR, Untranslated region.
MTX2 did not show any significant association with autism in the AGRE or Japanese samples (see Additional files 5.3 and 5.4).
Discussion
Our study of MtD in autism involves a wide array of genes related to diverse mitochondrial functions. We report brain region-specific alterations in the expression of these genes in autism. MTX2, NEFL and SLC25A27 showed consistently reduced expression in the ACG, MC and THL of autism patients. We also observed nominal genetic association of NEFL and SLC25A27 with autism.
Gene expression was analyzed in three brain regions: ACG, MC and THL. ACG has been found to be involved in emotion formation and processing, learning and memory [27,28]; MC in planning, control and execution of voluntary motor functions [29]; and THL in the processing and relaying of sensory information [30]. In autistic individuals, abnormalities of anterior cingulate have been found to be linked with impairments in cognitive control [31], social orientation [32], social target detection [33], and response monitoring [34]. Increased white matter volume of MC has been reported to be associated with motor impairments in autistic children [35]. Impairments in auditory, tactile, and visual sensory stimuli processing, found in autistic individuals, have been attributed to THL abnormalities [36]. Reduced thalamic volume has also been observed in autism [37].
Brain samples from the cerebellum and cortices had been used in previous studies of MtD in autism. The brain regions (ACG, MC and THL) used in our study have not been reported elsewhere. The differences in the results of our study and other whole genome transcriptomic analyses of autism brain [17,38,39] might be due to the differences in the regions of brain that were analyzed. The differences in metabolic demands or brain region-specific pathophysiology could affect the expression of mitochondrial genes. The etiological heterogeneity and criteria for sample selection might also have influenced the results since MtD is observed in only a subset of autistic individuals.
NEFL, SLC25A27 and MTX2 showed reduced expression in all the three brain regions of autism patients. NEFL is located in 8p21.2, which has been suggested as a susceptible region for autism in a genome-wide association study [40]. Moreover, 8p is known as a potential hub for developmental neuropsychiatric disorders [41]. Being a major constituent of neurofilaments, NEFL plays a pivotal function in the assembly and maintenance of axonal cytoskeleton [42]. Knocking out of Nefl has been found to reduce axonal caliber and conduction velocity in mice [43]. Sensorimotor impairments and reversal learning deficits have been observed in Nefl transgenic mice [44]. NEFL has also been found to have a vital role in regulating mitochondrial morphology, fusion, and motility in neurons [45,46]. Reduced NEFL expression may thus restrict mitochondrial translocation to areas of the cell requiring energy. We observed a nominal association of an NEFL SNP with autism in the AGRE samples. However, this SNP is located in the UTR of exon 4 and might not have a functional significance.
SLC25A27, also known as uncoupling protein 4 (UCP4), belongs to the large family of mitochondrial anion carrier proteins that are located on the inner mitochondrial membrane. It is expressed predominantly in the central nervous system (CNS) [47]. It has also been suggested to have roles in the reduction of reactive oxygen species [48], neuroprotection against OS and ATP deficiency [49], inhibition of apoptosis [50], neuronal cell differentiation [51], mitochondrial biogenesis [52], and mitochondrial calcium homeostasis [53]. Downregulation of SLC25A27 could thus have detrimental effects on these processes. Pharmacological targeting of neuronal uncoupling proteins (UCPs) represents an important avenue to combat MtD. Fatty acids have been reported to activate UCPs [54,55]. Consequently, a ketogenic diet has been found to increase the protein levels and activities of UCPs, including that of SLC25A27 [56]. We observed a nominal association of SLC25A27 with autism in Japanese samples. However, rs6901178, the SNP that showed association, is located in intron 4 and might not have a functional significance.
MTX2, located on the cytosolic face of the outer mitochondrial membrane, has been suggested to function as an import receptor for mitochondrial preproteins, a crucial process for cell survival [57,58]. It also plays a major role in the regulation of apoptosis [59]. In this study, we observed a downregulation of MTX2 in the ACG, MC and THL of autism patients; however, we did not observe an association of this gene with autism.
We also observed, in autism brains, region-specific alterations in the expression of several other mitochondria-related genes (Table 2). These genes fall into the ten functional groups as described in Additional file 3 and presented below:
1) Membrane polarization and potential (MPP): MPP plays a crucial role in energy production, maintenance of calcium homeostasis, protein import and cell survival [60,61]. We observed, in the ACG of autism patients, an elevated expression of BCL2 and TP53, which are involved in the maintenance of MPP.
2) Mitochondrial transport: In brain, the proper localization of mitochondria in the neurons is necessary for the generation of synaptic and action potentials, regulation of intracellular calcium dynamics and ATP synthesis [62,63]. In various regions of autism brains, we observed alterations in the expression of several genes related to mitochondrial transport, such as, AIP, BCL2, DNAJC19, HSP90AA1, MFN2, MIPEP, TP53 and TSPO. The expression of DNAJC19 was downregulated in the MC and THL of autism patients.
3) Small molecule transport, SLC25A family: The expression of several members of SLC25A solute carrier family was altered, with most of them being downregulated, in autism. Mitochondrial solute carriers transport a variety of solutes (di- and tri-carboxylates, keto acids, amino acids, nucleotides and coenzymes/cofactors) across the inner mitochondrial membrane [64]. We observed a reduced expression of SLC25A12 and SLC25A14 in the ACG and MC of autism patients. However, upregulated expression of SLC25A12 has been observed in some prior studies [18,19]. The brain regions used in this study were different from those in the aforementioned studies. The variation in metabolic demands of different brain regions could consequently affect the expression of mitochondrial genes. There are also conflicting reports about the association of SLC25A12 with autism [65-67]. The expression of SLC25A24 was reduced in the MC and THL of autism patients.
4) Targeting proteins to mitochondria.
5) Mitochondria protein import: Of the hundreds of proteins that are found within the mitochondria, the mitochondrial genome encodes only 13, and the rest must be imported from the cytosol [68]. The nuclear-encoded, cytoplasmically synthesized proteins should be precisely targeted and imported to the mitochondria. In this study, the expression of several genes involved in protein targeting and import were found to be altered, with the majority of them being downregulated, in autism brains. Among these, DNAJC19 was downregulated in the MC and THL of autism patients.
6) Outer membrane translocation.
7) Inner membrane translocation: The TIMM/TOMM translocases are involved in the translocation of nuclear DNA-encoded mitochondrial proteins across the outer and inner mitochondrial membranes [69]. Several genes belonging to the TIMM/TOMM family showed altered expression in autism brain. TOMM20 showed a reduced expression in the ACG and MC of autism patients.
8) Mitochondrial fission and fusion: The expression of MFN2, one of the genes involved in the regulation of mitochondrial fission and fusion was found to be downregulated in the ACG of autism patients. Mitochondrial fission and fusion are crucial in maintaining the integrity of mitochondria, electrical and biochemical connectivity, turnover of mitochondria, segregation and protection of mtDNA, and programmed cell death [70]. In the neurons, this is involved in the formation and function of synapses in the dendritic spines and axons [71,72].
9) Mitochondrial localization: We observed reduced expression of DNM1L, LRPPRC, MFN2 and RHOT2, localized predominantly in the mitochondria. These genes are involved in the biogenesis, maintenance of morphology and integrity, trafficking, and homeostasis of mitochondria [73-75]. The expression of DNM1L and LRPPRC were reduced in the ACG and MC of autism patients.
10) Apoptosis: The expression of apoptotic genes were altered, with most of them being upregulated, in the brain of autism patients. Recent studies have demonstrated a possible association between neural cell death and autism [76,77].
A two-way ANOVA showed that the expression of all the genes that were differentially expressed in two or more brain regions of autism were dependent on the disease status rather than being region-specific (data not shown).
It is not yet clear if MtD is the cause or effect of autism. ASD patients have often been found to manifest biochemical or neuropathological traits linked with altered mitochondrial function. Since mitochondrial abnormalities often result in CNS dysfunction, leading to developmental regression, learning disability, and various behavioral disturbances, ASD could be an important clinical presentation of MtD [78]. However, the clinical features, and the biochemical and genetic abnormalities in ASD patients with an underlying MtD have been found to be heterogeneous. In addition, several of the biochemical abnormalities indicative of MtD may occur in the absence of any relevant genetic alterations [79]. On the contrary, mitochondrial abnormalities might also manifest as a secondary to certain pathophysiological processes involved in autism, such as immune dysregulation, OS and altered calcium homeostasis [79]. Even though it is possible that a greater proportion of individuals with ASD might have MtD at the genetic level, it may not be manifested clinically.
We observed only nominal association of NEFL and SLC25A27 with autism. Recent studies have indicated that only a subset of autism may be associated with the biochemical endophenotype of mitochondrial energy deficiency [80]. Therefore, related genes might not show a strong association with the disorder. Considering the highly heterogeneous nature of autism, nominal associations of genes with subtle effects on the disease phenotype should not be ignored. The small sample size of the Japanese trios is, however, a serious limitation of this study. MTX2, NEFL and SLC25A27 were selected for genetic association studies since their expression was reduced in all of the three brain regions analyzed. Nevertheless, there would have been other important genes directly impacting mitochondrial functions, albeit differential expression in just one or two brain regions of autism patients. However, a detailed study involving several genes was not possible due to financial constraints.
Factors inherent in postmortem brain studies, and beyond the investigator’s control, might have influenced our results. We did not have sufficient data regarding brain pH. However, large-scale gene analysis have shown that brain pH or PMI has no significant correlation with RNA integrity [81,82]. The pH could be lower in the postmortem brains of individuals who suffered prolonged agonal states, such as in respiratory arrest, multi-organ failure and coma [83]. However, the cause of death was sudden for most of the subjects included in our study. So, we assume that brain pH might not have affected the gene expression. The other concern is the effect of medication; antidepressants, antipsychotics and selective serotonin re-uptake inhibitors are known to inhibit mitochondrial activities [84,85]. In this study, medication status was available for only three autism patients, two of whom had received more than two classes of drugs (drug doses unknown). Therefore, it was difficult to examine the effects of medication on gene expression. Another matter of concern is that the cause of death for a majority of the autism patients was seizure or drowning, where the latter could also have been due to seizures. Seizure activity has been known to impair mitochondrial energy production by altering the activity of mitochondrial enzymes involved in ATP production [86,87]. In this study, we have not examined the expression of any genes directly involved in mitochondrial energy production. Therefore, we assume that the cause of death might not have influenced our results. Moreover, it is not yet clear if MtD is the cause or effect of seizures.
Conclusions
Our study, though preliminary, brings to light some new genes associated with MtD in autism. Dysfunction of these genes could lead to defects in mitochondrial activities, including energy metabolism, thus augmenting and disseminating several brain abnormalities related to autism.
Abbreviations
ACG: anterior cingulate gyrus; ADI-R: autism diagnostic interview-revised; AGRE: Autism Genetic Resource Exchange; ASD: autism spectrum disorders; CNS: central nervous system; Ct: threshold cycle; GDC: genomic DNA control; LD: Linkage disequilibrium; MC: motor cortex; MPP: membrane polarization and potential; MtD: mitochondrial dysfunction; OS: oxidative stress; PDD-NOS: pervasive developmental disorder-not otherwise specified; PMI: postmortem interval; PPCs: positive PCR controls; qPCR: real-time PCR; RTCs: reverse transcription controls; SCID: Structured Clinical Interview for DSM-IV; SNP: single nucleotide polymorphism; THL: thalamus; UCPs: uncoupling proteins; UCSC: University of California Santa Cruz; UTR: untranslated region.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AA was involved in conception, design, conducting experiments, data analysis and drafting of article. IT, KY, YI and TT were involved in analysis and interpretation of data. HM, TM, SY, MT, KJT, KM, YIwata, KS and HI were involved in drafting the article. KN, TS, TY and NM were involved in revising the article critically. All authors read and approved the final manuscript.
Supplementary Material
Demographic characteristics of postmortem brain samples.
Human Mitochondria RT2ProfilerTMPCR Array: Gene Table. Description of genes included in the array.
Human Mitochondria RT2ProfilerTMPCR Array: Functional Gene Groupings. Classification of genes according to their functions.
SNPs selected for the genetic association study.
FBAT analysis. Results of FBAT analysis of genes without significant association.
Contributor Information
Ayyappan Anitha, Email: anitha@hama-med.ac.jp.
Kazuhiko Nakamura, Email: nakamura@hama-med.ac.jp.
Ismail Thanseem, Email: thanseem@hama-med.ac.jp.
Kazuo Yamada, Email: k_yamada@brain.riken.jp.
Yoshimi Iwayama, Email: yoshimi@brain.riken.jp.
Tomoko Toyota, Email: t_toyota@brain.riken.jp.
Hideo Matsuzaki, Email: matsu@hama-med.ac.jp.
Taishi Miyachi, Email: jupiter3@hama-med.ac.jp.
Satoru Yamada, Email: Satoru_Yamada@member.metro.tokyo.jp.
Masatsugu Tsujii, Email: mtsujii@sass.chukyo-u.ac.jp.
Kenji J Tsuchiya, Email: tsuchiya@hama-med.ac.jp.
Kaori Matsumoto, Email: km@hama-med.ac.jp.
Yasuhide Iwata, Email: iwata@hama-med.ac.jp.
Katsuaki Suzuki, Email: k-suzuki@hama-med.ac.jp.
Hironobu Ichikawa, Email: hironobu_ichikawa@tmhp.jp.
Toshiro Sugiyama, Email: eptsugi@hama-med.ac.jp.
Takeo Yoshikawa, Email: takeo@brain.riken.go.jp.
Norio Mori, Email: morin@hama-med.ac.jp.
Acknowledgements
We thank Dr. Jane Pickett, Director of Brain Resources and Data, Autism Tissue Program, for facilitating brain tissue collection. Human tissue was obtained from the NICHD Brain and Tissue Bank for Developmental Disorders at the University of Maryland, Baltimore, Maryland. Tissue samples were also provided by the Harvard Brain Tissue Resource Center, which is supported in part by PHS grant number R 24 MH 068855. This work was supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (23591700 to AA and 23390288 to KN). A part of this study is the result of "Integrated research on neuropsychiatric disorders" carried out under the Strategic Research Program for Brain Sciences by the Ministry of Education, Culture, Sports, Science and Technology of Japan. We thank Tae Takahashi and Mika Oyaizu for technical assistance.
References
- Rice C. Prevalence of autism spectrum disorders-Autism and Developmental Disabilities Monitoring Network, United States 2006. MMWR Surveill Summ. 2009;58:1–20. [PubMed] [Google Scholar]
- Minshew NJ, Goldstein G, Dombrowski SM, Panchalingam K, Pettegrew JW. A preliminary 31P MRS study of autism: evidence for undersynthesis and increased degradation of brain membranes. Biol Psychiatry. 1993;33:762–773. doi: 10.1016/0006-3223(93)90017-8. [DOI] [PubMed] [Google Scholar]
- Chugani DC, Sundram BS, Behen M, Lee ML, Moore GJ. Evidence of altered energy metabolism in autistic children. Prog Neuropsychopharmacol Biol Psychiatry. 1999;23:635–641. doi: 10.1016/S0278-5846(99)00022-6. [DOI] [PubMed] [Google Scholar]
- Lombard J. Autism: a mitochondrial disorder? Med Hypotheses. 1998;50:497–500. doi: 10.1016/S0306-9877(98)90270-5. [DOI] [PubMed] [Google Scholar]
- Schwab MA, Sauer SW, Okun JG, Nijtmans LG, Rodenburg RJ, van den Heuvel LP, Drose S, Brandt U, Hoffmann GF, Ter Laak H, Kolker S, Smeitink JA. Secondary mitochondrial dysfunction in propionic aciduria: a pathogenic role for endogenous mitochondrial toxins. Biochem J. 2006;398:107–112. doi: 10.1042/BJ20060221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacFabe DF, Cain DP, Rodriguez-Capote K, Franklin AE, Hoffman JE, Boon F, Taylor AR, Kavaliers M, Ossenkopp KP. Neurobiological effects of intraventricular propionic acid in rats: possible role of short chain fatty acids on the pathogenesis and characteristics of autism spectrum disorders. Behav Brain Res. 2007;176:149–169. doi: 10.1016/j.bbr.2006.07.025. [DOI] [PubMed] [Google Scholar]
- MacFabe DF, Rodriguez-Captoe K, Hoffman JE, Franklin AE, Mohammed-Asef Y, Taylor AR, Boon F, Cain DP, Kavaliers M, Possmayer F, Ossenkopp K. A novel rodent model of autism: intraventricular infusions of propionic acid increase locomotor activity and induce neuroinflammation and oxidative stress in discrete regions of adult rat brain. Am J Biochem Biotech. 2008;4:146–166. [Google Scholar]
- Shultz SR, MacFabe DF, Ossenkopp KP, Scratch S, Whelan J, Taylor R, Cain DP. Intracerebroventricular injection of propionic acid, an enteric bacterial metabolic end-product, impairs social behavior in the rat: implications for an animal model of autism. Neuropharmacology. 2008;54:901–911. doi: 10.1016/j.neuropharm.2008.01.013. [DOI] [PubMed] [Google Scholar]
- Rossignol DA, Frye RE. Mitochondrial dysfunction in autism spectrum disorders: a systematic review and meta-analysis. Mol Psychiatry. 2012;17:290–314. doi: 10.1038/mp.2010.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giulivi C, Zhang YF, Omanska-Klusek A, Ross-Inta C, Wong S, Hertz-Picciotto I, Tassone F, Pessah IN. Mitochondrial dysfunction in autism. JAMA. 2010;304:2389–2396. doi: 10.1001/jama.2010.1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fillano JJ, Goldenthal MJ, Rhodes CH, Marin-Garcia J. Mitochondrial dysfunction in patients with hypotonia, epilepsy, autism, and developmental delay: HEADD syndrome. J Child Neurol. 2002;17:435–439. doi: 10.1177/088307380201700607. [DOI] [PubMed] [Google Scholar]
- Gargus JJ, Imtiaz F. Mitochondrial energy-deficient endophenotype in autism. Am J Biochem Biotech. 2008;4:198–207. [Google Scholar]
- Filipek PA, Juranek J, Smith M, Mays LZ, Ramos ER, Bocian M, Masser-Frye D, Laulhere TM, Modahl C, Spence MA, Gargus JJ. Mitochondrial dysfunction in autistic patients with 15q inverted duplication. Ann Neurol. 2003;53:801–804. doi: 10.1002/ana.10596. [DOI] [PubMed] [Google Scholar]
- Tsao CY, Mendell JR. Autistic disorder in 2 children with mitochondrial disorders. J Child Neurol. 2007;22:1121–1123. doi: 10.1177/0883073807306266. [DOI] [PubMed] [Google Scholar]
- Ezugha H, Goldenthal M, Valencia I, Anderson CE, Legido A, Marks H. 5q14.3 deletion manifesting as mitochondrial disease and autism: case report. J Child Neurol. 2010;25:1232–1235. doi: 10.1177/0883073809361165. [DOI] [PubMed] [Google Scholar]
- Chauhan A, Gu F, Essa MM, Wegiel J, Kaur K, Brown WT, Chauhan V. Brain region-specific deficit in mitochondrial electron transport chain complexes in children with autism. J Neurochem. 2011;117:209–220. doi: 10.1111/j.1471-4159.2011.07189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg MR, Rubin RA, Falcone T, Ting AH, Natowicz MR. Brain transcriptional and epigenetic associations with autism. PLoS One. 2012;7:e44736. doi: 10.1371/journal.pone.0044736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepagnol-Bestel AM, Maussion G, Boda B, Cardona A, Iwayama Y, Delezoide AL, Moalic JM, Muller D, Dean B, Yoshikawa T, Gorwood P, Buxbaum JD, Ramoz N, Simonneau M. SLC25A12 expression is associated with neurite outgrowth and is upregulated in the prefrontal cortex of autistic subjects. Mol Psychiatry. 2008;13:385–397. doi: 10.1038/sj.mp.4002120. [DOI] [PubMed] [Google Scholar]
- Palmieri L, Papaleo V, Porcelli V, Scarcia P, Gaita L, Sacco R, Hager J, Rousseau F, Curatolo P, Manzi B, Militerni R, Bravaccio C, Trillo S, Schneider C, Melmed R, Elia M, Lenti C, Saccani M, Pascucci T, Puglisi-Allegra S, Reichelt KL, Persico AM. Altered calcium homeostasis in autism-spectrum disorders: evidence from biochemical and genetic studies of the mitochondrial aspartate/glutamate carrier AGC1. Mol Psychiatry. 2010;15:38–52. doi: 10.1038/mp.2008.63. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Halt AR. Altered levels of Bcl2 and p53 proteins in parietal cortex reflect deranged apoptotic regulation in autism. Synapse. 2001;42:281–284. doi: 10.1002/syn.10002. [DOI] [PubMed] [Google Scholar]
- Araghi-Niknam M, Fatemi SH. Levels of Bcl-2 and P53 are altered in superior frontal and cerebellar cortices of autistic subjects. Cell Mol Neurobiol. 2003;23:945–952. doi: 10.1023/B:CEMN.0000005322.27203.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geschwind DH, Sowinski J, Lord C, Iversen P, Shestack J, Jones P, Ducat L, Spence SJ. The autism genetic resource exchange: a resource for the study of autism and related neuropsychiatric conditions. Am J Hum Genet. 2001;69:463–466. doi: 10.1086/321292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord C, Rutter M, Le Couteur A. Autism Diagnostic Interview-Revised: a revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J Autism Dev Disord. 1994;24:659–685. doi: 10.1007/BF02172145. [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fourth edition, Text Revision. Washington, DC: American Psychiatric Association; 2000. [Google Scholar]
- American Psychiatric Association. User's Guide for the Structured Clinical Interview for DSM-IV Axis I Disorders SCID-1: Clinician Version. Washington, DC: American Psychiatric Association; 1997. [Google Scholar]
- Ranade K, Chang MS, Ting CT, Pei D, Hsiao CF, Olivier M, Pesich R, Hebert J, Chen YD, Dzau VJ, Curb D, Olshen R, Risch N, Cox DR, Botstein D. High-throughput genotyping with single nucleotide polymorphisms. Genome Res. 2001;11:1262–1268. doi: 10.1101/gr.157801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takenouchi K, Nishijo H, Uwano T, Tamura R, Takigawa M, Ono T. Emotional and behavioral correlates of the anterior cingulate cortex during associative learning in rats. Neuroscience. 1999;93:1271–1287. doi: 10.1016/S0306-4522(99)00216-X. [DOI] [PubMed] [Google Scholar]
- Bush G, Luu P, Posner MI. Cognitive and emotional influences in anterior cingulate cortex. Trends Cogn Sci. 2000;4:215–222. doi: 10.1016/S1364-6613(00)01483-2. [DOI] [PubMed] [Google Scholar]
- Evarts EV. In: Handbook of Physiology-The Nervous System, II. Brookhart JM, Mountcastle VB, editor. Maryland: American Physiological Society; 1981. Role of motor cortex in voluntary movements in primates; pp. 1083–1120. [Google Scholar]
- McCormick DA, Bal T. Sensory gating mechanisms of the thalamus. Curr Opin Neurobiol. 1994;4:550–556. doi: 10.1016/0959-4388(94)90056-6. [DOI] [PubMed] [Google Scholar]
- Agam Y, Joseph RM, Barton JJ, Manoach DS. Reduced cognitive control of response inhibition by the anterior cingulate cortex in autism spectrum disorders. NeuroImage. 2010;52:336–347. doi: 10.1016/j.neuroimage.2010.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundy P. Annotation: the neural basis of social impairments in autism: the role of the dorsal medial-frontal cortex and anterior cingulate system. J Child Psychol Psychiatry. 2003;44:793–809. doi: 10.1111/1469-7610.00165. [DOI] [PubMed] [Google Scholar]
- Dichter GS, Felder JN, Bodfish JW. Autism is characterized by dorsal anterior cingulate hyperactivation during social target detection. Soc Cogn Affect Neurosci. 2009;4:215–226. doi: 10.1093/scan/nsp017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakkar KN, Polli FE, Joseph RM, Tuch DS, Hadjikhani N, Barton JJ, Manoach DS. Response monitoring, repetitive behaviour and anterior cingulate abnormalities in autism spectrum disorders (ASD) Brain. 2008;131:2464–2478. doi: 10.1093/brain/awn099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostofsky SH, Burgess MP, Gidley Larson JC. Increased motor cortex white matter volume predicts motor impairment in autism. Brain. 2007;130:2117–2122. doi: 10.1093/brain/awm129. [DOI] [PubMed] [Google Scholar]
- Minshew NJ, Goldstein G, Siegel DJ. Neuropsychologic functioning in autism: profile of a complex information processing disorder. J Int Neuropsychol Soc. 1997;3:303–316. [PubMed] [Google Scholar]
- Tsatsanis KD, Rourke BP, Klin A, Volkmar FR, Cicchetti D, Schultz RT. Reduced thalamic volume in high-functioning individuals with autism. Biol Psychiatry. 2003;53:121–129. doi: 10.1016/S0006-3223(02)01530-5. [DOI] [PubMed] [Google Scholar]
- Garbett K, Ebert PJ, Mitchell A, Lintas C, Manzi B, Mirnics K, Persico AM. Immune transcriptome alterations in the temporal cortex of subjects with autism. Neurobiol Dis. 2008;30:303–311. doi: 10.1016/j.nbd.2008.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, Mill J, Cantor RM, Blencowe BJ, Geschwind DH. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474:380–384. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauritsen MB, Als TD, Dahl HA, Flint TJ, Wang AG, Vang M, Kruse TA, Ewald H, Mors O. A genome-wide search for alleles and haplotypes associated with autism and related pervasive developmental disorders on the Faroe Islands. Mol Psychiatry. 2006;11:37–46. doi: 10.1038/sj.mp.4001754. [DOI] [PubMed] [Google Scholar]
- Tabares-Seisdedos R, Rubenstein JL. Chromosome 8p as a potential hub for developmental neuropsychiatric disorders: implications for schizophrenia, autism and cancer. Mol Psychiatry. 2009;14:563–589. doi: 10.1038/mp.2009.2. [DOI] [PubMed] [Google Scholar]
- Muma NA, Hoffman PN. Neurofilaments are intrinsic determinants of axonal caliber. Micron. 1993;24:677–683. doi: 10.1016/0968-4328(93)90044-2. [DOI] [Google Scholar]
- Elder GA, Friedrich VL Jr, Bosco P, Kang C, Gourov A, Tu PH, Lee VM, Lazzarini RA. Absence of the mid-sized neurofilament subunit decreases axonal calibers, levels of light neurofilament (NF-L), and neurofilament content. J Cell Biol. 1998;141:727–739. doi: 10.1083/jcb.141.3.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filali M, Dequen F, Lalonde R, Julien JP. Sensorimotor and cognitive function of a NEFL(P22S) mutant model of Charcot-Marie-Tooth disease type 2E. Behav Brain Res. 2011;219:175–180. doi: 10.1016/j.bbr.2010.12.022. [DOI] [PubMed] [Google Scholar]
- Wagner OI, Lifshitz J, Janmey PA, Linden M, McIntosh TK, Leterrier JF. Mechanisms of mitochondria-neurofilament interactions. J Neurosci. 2003;23:9046–9058. doi: 10.1523/JNEUROSCI.23-27-09046.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentil BJ, Minotti S, Beange M, Baloh RH, Julien JP, Durham HD. Normal role of the low-molecular-weight neurofilament protein in mitochondrial dynamics and disruption in Charcot-Marie-Tooth disease. FASEB J. 2012;26:1194–1203. doi: 10.1096/fj.11-196345. [DOI] [PubMed] [Google Scholar]
- Mao W, Yu XX, Zhong A, Li W, Brush J, Sherwood SW, Adams SH, Pan G. UCP4, a novel brain-specific mitochondrial protein that reduces membrane potential in mammalian cells. FEBS Lett. 1999;443:326–330. doi: 10.1016/S0014-5793(98)01713-X. [DOI] [PubMed] [Google Scholar]
- Liu D, Chan SL, de Souza-Pinto NC, Slevin JR, Wersto RP, Zhan M, Mustafa K, de Cabo R, Mattson MP. Mitochondrial UCP4 mediates an adaptive shift in energy metabolism and increases the resistance of neurons to metabolic and oxidative stress. Neuromolecular Med. 2006;8:389–414. doi: 10.1385/NMM:8:3:389. [DOI] [PubMed] [Google Scholar]
- Wei Z, Chigurupati S, Bagsiyao P, Henriquez A, Chan SL. The brain uncoupling protein UCP4 attenuates mitochondrial toxin-induced cell death: role of extracellular signal-regulated kinases in bioenergetics adaptation and cell survival. Neurotox Res. 2009;16:14–29. doi: 10.1007/s12640-009-9039-8. [DOI] [PubMed] [Google Scholar]
- Zhang M, Wang B, Ni YH, Liu F, Fei L, Pan XQ, Guo M, Chen RH, Guo XR. Overexpression of uncoupling protein 4 promotes proliferation and inhibits apoptosis and differentiation of preadipocytes. Life Sci. 2006;79:1428–1435. doi: 10.1016/j.lfs.2006.04.012. [DOI] [PubMed] [Google Scholar]
- Smorodchenko A, Rupprecht A, Sarilova I, Ninnemann O, Brauer AU, Franke K, Schumacher S, Techritz S, Nitsch R, Schuelke M, Pohl EE. Comparative analysis of uncoupling protein 4 distribution in various tissues under physiological conditions and during development. Biochim Biophys Acta. 2009;1788:2309–2319. doi: 10.1016/j.bbamem.2009.07.018. [DOI] [PubMed] [Google Scholar]
- Andrews ZB, Diano S, Horvath TL. Mitochondrial uncoupling proteins in the CNS: in support of function and survival. Nat Rev Neurosci. 2005;6:829–840. doi: 10.1038/nrn1767. [DOI] [PubMed] [Google Scholar]
- Chan SL, Liu D, Kyriazis GA, Bagsiyao P, Ouyang X, Mattson MP. Mitochondrial uncoupling protein-4 regulates calcium homeostasis and sensitivity to store depletion-induced apoptosis in neural cells. J Biol Chem. 2006;281:37391–37403. doi: 10.1074/jbc.M605552200. [DOI] [PubMed] [Google Scholar]
- Jezek P. Fatty acid interaction with mitochondrial uncoupling proteins. J Bioenerg Biomembr. 1999;31:457–466. doi: 10.1023/A:1005496306893. [DOI] [PubMed] [Google Scholar]
- Jimenez-Jimenez J, Ledesma A, Zaragoza P, Gonzalez-Barroso MM, Rial E. Fatty acid activation of the uncoupling proteins requires the presence of the central matrix loop from UCP1. Biochim Biophys Acta. 2006;1757:1292–1296. doi: 10.1016/j.bbabio.2006.05.027. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Rippy NA, Dorenbos K, Concepcion RC, Agarwal AK, Rho JM. The ketogenic diet increases mitochondrial uncoupling protein levels and activity. Ann Neurol. 2004;55:576–580. doi: 10.1002/ana.20062. [DOI] [PubMed] [Google Scholar]
- Armstrong LC, Komiya T, Bergman BE, Mihara K, Bornstein P. Metaxin is a component of a preprotein import complex in the outer membrane of the mammalian mitochondrion. J Biol Chem. 1997;272:6510–6518. doi: 10.1074/jbc.272.10.6510. [DOI] [PubMed] [Google Scholar]
- Armstrong LC, Saenz AJ, Bornstein P. Metaxin 1 interacts with metaxin 2, a novel related protein associated with the mammalian mitochondrial outer membrane. J Cell Biochem. 1999;74:11–22. doi: 10.1002/(SICI)1097-4644(19990701)74:1<11::AID-JCB2>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Ono K, Wang X, Kim SO, Armstrong LC, Bornstein P, Han J. Metaxin deficiency alters mitochondrial membrane permeability and leads to resistance to TNF-induced cell killing. Protein Cell. 2010;1:161–173. doi: 10.1007/s13238-010-0017-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimroth P, Kaim G, Matthey U. Crucial role of the membrane potential for ATP synthesis by F(1)F(o) ATP synthases. J Exp Biol. 2000;203:51–59. doi: 10.1242/jeb.203.1.51. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Ward MW. Mitochondrial membrane potential and neuronal glutamate excitotoxicity: mortality and millivolts. Trends Neurosci. 2000;23:166–174. doi: 10.1016/S0166-2236(99)01534-9. [DOI] [PubMed] [Google Scholar]
- Hollenbeck PJ, Saxton WM. The axonal transport of mitochondria. J Cell Sci. 2005;118:5411–5419. doi: 10.1242/jcs.02745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAskill AF, Kittler JT. Control of mitochondrial transport and localization in neurons. Trends Cell Biol. 2010;20:102–112. doi: 10.1016/j.tcb.2009.11.002. [DOI] [PubMed] [Google Scholar]
- Palmieri F. The mitochondrial transporter family (SLC25): physiological and pathological implications. Pflugers Arch. 2004;447:689–709. doi: 10.1007/s00424-003-1099-7. [DOI] [PubMed] [Google Scholar]
- Turunen JA, Rehnstrom K, Kilpinen H, Kuokkanen M, Kempas E, Ylisaukko-Oja T. Mitochondrial aspartate/glutamate carrier SLC25A12 gene is associated with autism. Autism Res. 2008;1:189–192. doi: 10.1002/aur.25. [DOI] [PubMed] [Google Scholar]
- Correia C, Coutinho AM, Diogo L, Grazina M, Marques C, Miguel T, Ataide A, Almeida J, Borges L, Oliveira C, Oliveira G, Vicente AM. Brief report: High frequency of biochemical markers for mitochondrial dysfunction in autism: no association with the mitochondrial aspartate/glutamate carrier SLC25A12 gene. J Autism Dev Disord. 2006;36:1137–1140. doi: 10.1007/s10803-006-0138-6. [DOI] [PubMed] [Google Scholar]
- Ramoz N, Reichert JG, Smith CJ, Silverman JM, Bespalova IN, Davis KL, Buxbaum JD. Linkage and association of the mitochondrial aspartate/glutamate carrier SLC25A12 gene with autism. Am J Psychiatry. 2004;161:662–669. doi: 10.1176/appi.ajp.161.4.662. [DOI] [PubMed] [Google Scholar]
- Douglas MG, McCammon MT, Vassarotti A. Targeting proteins into mitochondria. Microbiol Rev. 1986;50:166–178. doi: 10.1128/mr.50.2.166-178.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedemann N, Frazier AE, Pfanner N. The protein import machinery of mitochondria. J Biol Chem. 2004;279:14473–14476. doi: 10.1074/jbc.R400003200. [DOI] [PubMed] [Google Scholar]
- Berman SB, Pineda FJ, Hardwick JM. Mitochondrial fission and fusion dynamics: the long and short of it. Cell Death Differ. 2008;15:1147–1152. doi: 10.1038/cdd.2008.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Okamoto K, Hayashi Y, Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119:873–887. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Li H, Chen Y, Jones AF, Sanger RH, Collis LP, Flannery R, McNay EC, Yu T, Schwarzenbacher R, Bossy B, Bossy-Wetzel E, Bennett MV, Pypaert M, Hickman JA, Smith PJ, Hardwick JM, Jonas EA. Bcl-xL induces Drp1-dependent synapse formation in cultured hippocampal neurons. Proc Natl Acad Sci USA. 2008;105:2169–2174. doi: 10.1073/pnas.0711647105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova E, Shurland DL, Ryazantsev SN, van der Bliek AM. A human dynamin-related protein controls the distribution of mitochondria. J Cell Biol. 1998;143:351–358. doi: 10.1083/jcb.143.2.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santel A, Fuller MT. Control of mitochondrial morphology by a human mitofusin. J Cell Sci. 2001;114:867–874. doi: 10.1242/jcs.114.5.867. [DOI] [PubMed] [Google Scholar]
- Fransson A, Ruusala A, Aspenstrom P. Atypical Rho GTPases have roles in mitochondrial homeostasis and apoptosis. J Biol Chem. 2003;278:6495–6502. doi: 10.1074/jbc.M208609200. [DOI] [PubMed] [Google Scholar]
- Fujita E, Dai H, Tanabe Y, Zhiling Y, Yamagata T, Miyakawa T, Tanokura M, Momoi MY, Momoi T. Autism spectrum disorder is related to endoplasmic reticulum stress induced by mutations in the synaptic cell adhesion molecule, CADM1. Cell Death Dis. 2010;1:e47. doi: 10.1038/cddis.2010.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou H, Yu Y, Sheikh AM, Malik M, Yang K, Wen G, Chadman KK, Brown WT, Li X. Association of upregulated Ras/Raf/ERK1/2 signaling with autism. Genes Brain Behav. 2011;10:615–624. doi: 10.1111/j.1601-183X.2011.00702.x. [DOI] [PubMed] [Google Scholar]
- Nissenkorn A, Zeharia A, Lev D, Watemberg N, Fattal-Valevski A, Barash V, Gutman A, Harel S, Lerman-Sagie T. Neurologic presentations of mitochondrial disorders. J Child Neurol. 2000;15:44–48. doi: 10.1177/088307380001500110. [DOI] [PubMed] [Google Scholar]
- Palmieri L, Persico AM. Mitochondrial dysfunction in autism spectrum disorders: cause or effect? Biochim Biophys Acta. 2010;1797:1130–1137. doi: 10.1016/j.bbabio.2010.04.018. [DOI] [PubMed] [Google Scholar]
- Haas RH. Autism and mitochondrial disease. Dev Disabil Res Rev. 2010;16:144–153. doi: 10.1002/ddrr.112. [DOI] [PubMed] [Google Scholar]
- Barton AJ, Pearson RC, Najlerahim A, Harrison PJ. Pre- and postmortem influences on brain RNA. J Neurochem. 1993;61:1–11. doi: 10.1111/j.1471-4159.1993.tb03532.x. [DOI] [PubMed] [Google Scholar]
- Popova T, Mennerich D, Weith A, Quast K. Effect of RNA quality on transcript intensity levels in microarray analysis of human post-mortem brain tissues. BMC Genomics. 2008;9:91. doi: 10.1186/1471-2164-9-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JZ, Vawter MP, Walsh DM, Tomita H, Evans SJ, Choudary PV, Lopez JF, Avelar A, Shokoohi V, Chung T, Mesarwi O, Jones EG, Watson SJ, Akil H, Bunney WE Jr. Myers RM: Systematic changes in gene expression in postmortem human brains associated with tissue pH and terminal medical conditions. Hum Mol Genet. 2004;13:609–616. doi: 10.1093/hmg/ddh065. [DOI] [PubMed] [Google Scholar]
- Rumbach L, Warter JM, Rendon A, Marescaux C, Micheletti G, Waksman A. Inhibition of oxidative phosphorylation in hepatic and cerebral mitochondria of sodium valproate-treated rats. J Neurol Sci. 1983;61:417–423. doi: 10.1016/0022-510X(83)90174-0. [DOI] [PubMed] [Google Scholar]
- Weinbach EC, Costa JL, Nelson BD, Claggett CE, Hundal T, Bradley D, Morris SJ. Effects of tricyclic antidepressant drugs on energy-linked reactions in mitochondria. Biochem Pharmacol. 1986;35:1445–1451. doi: 10.1016/0006-2952(86)90108-5. [DOI] [PubMed] [Google Scholar]
- Liang LP, Patel M. Seizure-induced changes in mitochondrial redox status. Free Radic Biol Med. 2006;40:316–322. doi: 10.1016/j.freeradbiomed.2005.08.026. [DOI] [PubMed] [Google Scholar]
- Kovac S, Domijan AM, Walker MC, Abramov AY. Prolonged seizure activity impairs mitochondrial bioenergetics and induces cell death. J Cell Sci. 2012;125:1796–1806. doi: 10.1242/jcs.099176. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Demographic characteristics of postmortem brain samples.
Human Mitochondria RT2ProfilerTMPCR Array: Gene Table. Description of genes included in the array.
Human Mitochondria RT2ProfilerTMPCR Array: Functional Gene Groupings. Classification of genes according to their functions.
SNPs selected for the genetic association study.
FBAT analysis. Results of FBAT analysis of genes without significant association.