

Abstract
Background
Major histocompatibility complex (MHC) molecules present peptides to T lymphocytes. It is of critical biological and medical importance to elucidate how different MHC alleles bind to a specific set of peptides.
Method
In this study we approach the problem from the algebraic and geometric point of view to analyse MHC-peptide-binding data accumulated over the years. The space of sequence properties (having a particular amino acid at a particular position) of MHC-peptide complexes conveys a geometric structure to these sequence properties in the form of a distance measure, which reveals the peptide binding requirements imposed by the polymorphic sequence characteristics of the MHC molecules.
Results
Comparison of the results of this study with our current knowledge of MHC-peptide binding constraints leads to robust agreement. This study provides the tools to quantitate these binding constraints giving a more detailed account of them and opening the way to make peptide binding predictions for MHC alleles for which there is no peptide elution data. In addition, the geometric representation of MHC-peptide complex sequence data gives a distance measure between amino acids in reference to their ability to meet MHC binding requirements.
Conclusions
The algebraic and geometric view of amino acid sequences provides a theoretical framework to study the function of proteins when there is enough variation in this sequence to account for the variation in their function, as it is the case with MHC molecules in regard to their ability to present peptides.
Background
The cellular immune response depends on antigen presentation to T cells by major histocompatibility complex (MHC) molecules, the antigens presented being small peptides with amino acid sequence limitations determined by the MHC alleles carried by a particular individual. In the study of the immune response it is of critical importance to discover the laws governing which peptides can be presented by which MHC alleles. As peptide receptors, major histocompatibility complex (MHC) molecules are capable of binding different peptides; but each MHC molecule–each allele–exhibits a predilection for a set of peptides with distinct sequence characteristics. From crystallographic studies of MHC-peptide complexes, as well as from sequence analysis of peptides eluted from these complexes, we learn that MHC genetic polymorphism is responsible for the differences in peptide binding between MHC molecules. [1, 2, 3, 4, 5, 6] We claim that peptide selection by a given MHC allele can be presented as a function of the MHC sequence, instead of the current approach to this problem which consists in a function of the allele in question. By setting the amino acids that occupy the different positions in the MHC molecule as the arguments of this function instead of a mere reference to an allele, the door is open to make generalizations from available elution data to include those cases in which no empirical data is yet available.
Over the past decade data on peptide binding to the MHC molecule have been accumulating. [7, 8] Do these data provide by themselves–that is, independently of other data such as crystallographic and chemical-sufficient information to characterise the requirements imposed by the various MHC alleles on what kind of peptide can be presented to T cells? The problem at hand consists in finding the proper theoretical framework to look at and analyse the sequences of these MHC-peptide complexes. We claim geometry provides this theoretical framework. Amino acid sequences can be represented as vectors in a metric space. [9] It is the essence of this metric space that two sequences are at a distance of each other. Our analytical tools are the transformations we can impose on this metric space to manipulate the distances between sequences, that is, between MHC-peptide complexes.
With the duality principle in geometry and the concept of dual spaces we move from a space of distances between amino acid sequences populated by MHC-peptide complexes to a space populated by the sequence properties of these complexes. In this new space we can measure the distance between the properties of the MHC-peptide complexes rather than the distances between the complexes themselves. Looking at the data in this light we can see how the sequence of the MHC molecule affects the sequence restrictions for the peptide allowed to bind to the molecule. This is revealed in the form of proximities between certain sequence properties of the MHC molecule and those of the bound peptide.
In this study we concentrate on the class I human leucocyte antigen (HLA) molecules and nonameric peptides bound to them.
The algebraic and geometric structure of MHC-peptide complexes
Let S be the space of MHC-peptide complexes. In this space each MHC-peptide complex is a point, represented by the vector of their amino acid sequence properties, the values the point takes in the co-ordinates of the space. The co-ordinates (dimensions) of this space correspond to these sequence properties, both peptide sequence properties and MHC sequence properties (only polymorphic positions in the MHC α1 and α2 domains are included). Peptide binding data allows the population of this space with × points. These × points are represented by matrix M of size × n, where n is the dimensionality of the space, that is, the total number of sequence properties, and each row is the vector representing each point. (In this study peptide binding data included 2535 nonameric peptides known to bind alleles HLA-A1, HLA-A11, HLA-A3, HLA-A2, HLA-A24, HLA-A25, HLA-A33, HLA-A31, HLA-A30, HLA-A29, HLA-B45, HLA-B44, HLA-B35, HLA-B53, HLA-B51, HLA-B14, HLA-B8, HLA-B38, HLA-B13, HLA-B27, HLA-B7, HLA-B42, HLA-854, HLA-CW2, and HLA-CW4, obtained from the MHCPEP database.) [8]
Now, let S' be the x-dimensional space of the sequence properties of the MHC-peptide complexes. Each point in this space corresponds to a property of the type 'MHC-peptide complex has amino acid x, at position pi', where pi can be either in the peptide or in the MHC molecule. Each dimension in this space corresponds to a particular MHC-peptide complex. We say S' is the transposed space of S. S is a space of particulars (molecules) and S' is a space of universals (sequence properties). The n points in S' are represented by matrix M' of size n ×, where each row is the vector representing each point. M' is the transposed matrix of M. (See Fig. 1) The points in S become co-ordinate axes in S' and the co-ordinate axes in S become points in S'.
Figure 1.

SPACE S' is the transposed space of SPACE S, the points in the latter become coordinates in the former, the co-ordinates in the latter become points in the former.
To give an example, let us define the sequence properties x, y and z (coordinates or dimensions in space S) as: x ≡ 'having> amino acid D in peptide position 3', y ≡ 'having amino acid E in peptide position 3', and z ≡ 'having amino acid Y in peptide position 9'. Let us also define the MHC-peptide complexes a, b, c and d (points in space S) as; a ≡ [HLA-A*0101-IADMGHLKY], b ≡ [HLA-A*0101-STEPVNILY], c ≡ [HLA-A*2402-TYSAGIVQI], and d ≡ [HLA-A*0207-LLDVPTAAV]. Then, ignoring all other points and dimensions, space S of MHC-peptide complexes is defined as: S = {a(1,0,1), b(0,1,1), c(0,0,0), d(1,0,0)}, where each point is defined by its coordinates and the values of these coordinates indicate that a property holds true ('1') or that it does not ('0'). Space S can then be represented by the matrix:
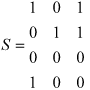
where each row represents a point (an MHC-peptide complex) in the space with its three coordinates. And the transposed space S' of sequence properties is defined as S' = {x (1,0,0,1), y(0,1,0,0,), z(1,1,0,0,)}, and it can be represented by the matrix:
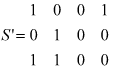
where each row represents a point (a sequence property) in the space with its four coordinates. It can be seen that the matrix of space S' is the transposed matrix of space S. For this reason we call S' the 'transposed space' of S.
This conversion is similar to the conversion of the sentence 'peptide i has amino acid Y at position 2' to the sentence 'having amino acid Y at position 2 is a characteristic of peptide i'. The subject and the predicate in the first sentence become the predicate and the subject in the second sentence respectively. There is a duality principle at work here. As Ramsey indicates, there is no sense in the distinction between 'individual' and 'quality', or between 'particular' and 'universal', the two types being in every way symmetrically related. [10] The conversion of S into S' by transposing M allows the measurement of the distance between the points in S', that is between the sequence properties of MHC-peptide complexes. Particularly, we are interested in the distance between sequence properties specific of the peptide and sequence properties specific of the MHC molecule. The distance between the sequence characteristics of the MHC molecule and those of the peptide has the potential of revealing how peptides bind to MHC molecules and how sequence requirements are imposed by the different MHC alleles on peptides as binding candidates.
Our data unit is the MHC-peptide complex. We define two types of categorical variables of the MHC-peptide complex. First, variables of the following type: amino acid Paa is present at position Pp in the peptide-these variables we call 'p' and the set of them all 'P'. Second, variables of the type: amino acid Haa is present at position Hp in the MHC molecule-these variables we call 'h' and the set of them all 'H'. In other words, a partition of space S' (we call 'K') is made to separate the class of peptide sequence properties (P) from the class of MHC sequence properties (H), so that K = {P, H}, S' = P ∪ H and P ∩ H = Θ. The variables or properties we are talking about- 'having amino acid threonine at position 9 in the peptide', for instance-are bipolar: a peptide either has that amino acid at that position or it has not. In the case of MHC sequence properties, only polymorphic positions in the alpha 1 and 2 domains are considered. Therefore, nonamers are characterised by 180 sequence properties, one for each of the 20 amino acids at each of the nine positions; and MHC alleles are characterised by 207 sequence properties for each of the possible amino acids at each one of the 73 polymorphic positions in the alpha-1 and alpha-2 domains. Only some amino acids are found at each polymorphic site; for instance, at position 65 the amino acids G, Q and R can be found in various alleles; at position 90, only A and D; etc.
A function D of the Cartesian product H × P into R (the line of real numbers) gives the distance between every pair (h, p) of sequence properties of MHC molecules and peptides bound to them. That is, D: H × P → R where D = {(h, p, D(h, p))| h ∈ H, p ∈ P}. (See Fig. 2) In other words, this function takes two arguments: one is an HLA sequence property (e.g. 'having amino acid E at position 46 in the HLA molecule'), and the other argument is a peptide sequence property (e.g. 'having amino acid P at position 2 in the peptide'). The value returned by this function is a distance measure. In measuring the distance between two such properties-points or vectors in S'-there are various alternatives. We use the Ochiai similarity index defined as: [11]
Figure 2.

FUNCTION D, mapping of the cartesian product H × P into R (the real-number line).
![]()
where a is the number of MHC-peptide complexes where both h and p hold, b the number of complexes where h holds but p dos not, c the number of complexes where h does not hold but p does, and d the number of cases where neither h or p hold. In other words, a, b, c and d are the cells in a 2 × 2 contingency table. This similarity index ranges from 0 to 1. In this study the values returned by function D were such that only 1% were greater than 0.4, 0.1% were greater than 0.6, and 0.03% were greater than 0.8. Measures of similarity and measures of dissimilarity- distance-can be linked to each other by means of 'similarity functions'. [11] In this study we may talk about similarity measures, but conceptually it is distances we are dealing with.
To give a concrete example, let us consider the following sequence properties: h1 ≡ 'having amino acid Q at MHC position 65', h2 ≡ 'having amino acid N at MHC position 66', p1 ≡ 'having amino acid P at peptide position 2', p2 ≡ 'having amino acid G at peptide position 5', p3 ≡ 'having amino acid I at peptide position 6'. The values returned by function D for the corresponding members of H × P are as follows:
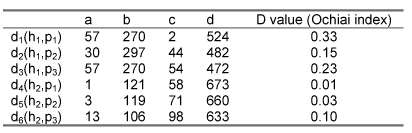
That is, for the first row, there are 57 MHC-peptide complexes in our database where at the MHC position 65 there is a Q and at peptide position 2 there is a P; there are 270 cases where at the MHC position 65 there is a Q but at peptide position 2 there is not a P; there are 2 cases where at the MHC position 65 there is not a Q and at peptide position 2 there is a P; and there are 524 cases where at the MHC position 65 there is not a Q and at peptide position 2 there is not a P. The D value for these two sequence properties is 0.33. Etc.
A relation L(d1, d2) is defined on the function D so that two elements d1(h1, p1) and d1(h1, p2) are related if h1 and h2 are sequence properties characteristic of the same MHC allele, where h1 and h2 are members of H, p1 and p2 are members of P and d1 and d2 are members of D. This relation is not an equivalence relation because although it is reflexive and symmetric, it is not transitive. Therefore the relation L(d1, d2) does not lead to a partition of D, but to a collection Q of overlapping subsets. Each element of Q-subset of D defined by L-represents itself an MHC allele. (See Fig. 3)
Figure 3.

RELATION L, defined on function D so that two elements d1(h1, p1) and d2(h2, p2) are related if h1 and h2 are sequence properties characteristic of the same MHC allele. Collection Q is the set of elements in D related to each other according to L; since L is not an equivalence relation, Q is not a partition.
On each element qi of Q-subset of D corresponding to a particular MHC allele-we define relation N so that two elements of D in qi are related if the distance returned by function D corresponds to the same peptide sequence property p ∈ P. Relation N is an equivalence relation that defines a partition on qi. Let Ui(N) be the partition defined by N on qi, this partition will have at most 180 equivalence classes, one corresponding to each peptide sequence property p ∈ P. In saying that N is defined on qi rather than on Q we are actually considering the product or intersection of two relations: L ∩ N. Since L is not an equivalence relation, nor is L ∩ N, and it does not lead to a partition of Q.
Let function V of Q into R180 (180-dimensional real-number space) return a vector for each MHC allele qi., member of Q, with the maximum value d = D(h, p) in each equivalence class of partition Ui(N). This R180 space we call Y and is the metric space of MHC alleles where each axis corresponds to a peptide sequence property (20 amino acids and 9 peptide positions), and each point in this space is an MHC allele. (See Fig. 4) Function V returns an 180-dimension vector. The co-ordinate value each MHC allele-each point in Y-assumes in each axis is a measure of predilection of the peptides to be bound to that allele for that corresponding peptide sequence property of that axis. If the property is, for instance, having glutamic acid at position 2 in the peptide, alleles that allow peptides with glutamic acid at position 2 will have a high value as their co-ordinate in that axis, and those alleles that do not will have a low value. The vector representing each allele in Y is in fact equivalent to the 20 × 9 'matrix' or table of 'average relative frequencies' or 'binding affinities' that characterise the sequence of peptides that bind to an allele.
Figure 4.

FUNCTION V of Q into Y, returns the 180-dimension vector of peptide binding affinities for each MHC allele.
By giving these data an algebraic and geometric structure we can answer such questions as: 'Which amino acids and in which positions in the MHC molecule are involved in defining peptide binding requirements?', and also 'How different are amino acids in peptides in regard to meeting the binding constraints imposed by the MHC?'
Results
Effect of HLA polymorphism on peptide sequence requirements
The study of function D reveals the binding properties of HLA-peptide complexes. This study consists in the examination of the distribution of the values returned by function D for the various values that the arguments of the function take. For instance, it shows that positions 2 and 9 in the peptide are critical in determining binding, position 2 carrying with it the highest values by far. These positions have been called 'anchor positions'. Next in importance are positions 1 and 3. Positions 4, 5, 6, 7 and 8 appear to play a very secondary role if they play one at all. The only exception to this finding is what takes place with allele HLA-B27. Only this allele carries with it high similarity values with peptide positions between 3 and 9. Whereas other alleles have requirements that affect which amino acids are at positions 2, 9, 1 and 3 (in order of stringency in the requirements), but not at the other positions in the peptide. HLA-B27 imposes requirements that in addition affect which amino acids occupy positions between 3 and 9. This was observed because the complete set of HLA sequence variables with high similarity values at positions between 3 and 9 correspond exactly to the sequence of HLA-B27.
As given by function D, high similarity measures (Ochiai Index > 0.7) for position 2 in the peptide show all but one (position 66) of the polymorphic positions in the HLA molecule described by Chelvanayagam as the 'peptide binding environment' for this position in the peptide: 7, 9, 24, 25, 26, 34, 35, 36, 45, 62, 63, 66, 67, 70, 99, 159, 163,167 (polymorphic positions in bold). [12] Other positions (32, 41, 65, 97, 113 and 158) have high similarity measures suggesting a possible role in the selection of the amino acids that are to occupy this position in the peptide.
Similarity measures for position 9 in the peptide are lower than for position 2, by using a lower cutoff (Ochiai Index > 0.4) function D shows the majority (all except 77, 80, 81 and 143) of the polymorphic positions in Chelvanayagam's 'peptide binding environment' for this position in the peptide: 70, 73, 74, 76, 77, 80, 81, 84, 95, 96, 97, 114,116, 123, 124, 142,143, 146, 147 (polymorphic positions in bold). Other positions (62, 66, 67, 107, 127, 145 and 158) appear to have high similarity measures suggesting a possible role in the selection of the amino acids that are to occupy position 9 in the peptide.
Function D also shows other peculiarities of MHC-peptide binding such as the fact that a basic amino acid (K, R, H) at position 9 in the peptide is associated with the presence of aspartic acid at positions 74, 77 and 116 in the HLA molecule. This had been previously observed by Vasmatzis and co-workers. [13]
Distance between amino acids
Previously we defined a theoretical concept of amino acid distance in terms of the dissimilarity between the chemical properties of amino acids. [9] If we could measure the distance between amino acids in peptides bound to HLA molecules in reference to how they are selected and restricted by the HLA molecule itself to allow binding, then we would have a useful measuring instrument to tell how different peptides themselves are in their ability to bind to HLA molecules.
This can be accomplished by studying the distribution of values returned by function D depending on the amino acid in each position of the peptide (Paa) and ignoring their position (Pp). In other words, we have defined a partition of D in which each equivalence class corresponds to an amino acid. Doing so we obtain a vector for each amino acid with each value in the vector representing a measure of the distribution of the values of function D, such as the average, for each HLA sequence variable (h ∈ H). In this way we have created a new space we call X-Paa where the amino acids that constitute peptides are represented as vectors and the HLA sequence variables as co-ordinate axes.
The distances between amino acids in this space X-Paa show at a high level-small distance- three distinct clusters: 1) tyrosine, phenylalanine and tryptophan; 2) serine, valine and threonine; and 3) isoleucine, methionine and leucine. At a lower level-allowing clusters to form with a greater distance between its members-new clusters appear, like the one formed by glutamine and arginine; but then other clusters start to overlap and become ill-defined, like the supercluster formed by amino acids alanine, serine, valine, threonine, lysine, isoleucine, methionine and leucine. These patterns in space X-Paa show that the distance measure defined in it is coherent. It is difficult to give a theoretical account for these distance measures in terms of known amino acid characteristics, but close to a third of the variability of the distance between amino acids in this space can be explained in terms of their chemical structure and their size, as determined by multiple regression analysis between the empirical distance matrix of X-Paa, as independent variable, and two theoretical distance matrices as dependent variables based on 1) molecular weight, and 2) chemical properties (hydrophobicity, polarity, charge, etc.). This regression analysis gives a coefficient of determination of R2 = 27.5%, which increases to R2 = 29.0% when chemical characteristics are represented as separate variables. Analysis of variance to study the effect of each variable independently of the others shows that, apart from the molecular weight, the chemical characteristics that appear to play a major role are the presence of an aromatic ring or an amide group, or whether the amino acid is a base positively charged; other characteristics, such as presence of an -OH or an -SH group, appear to play a less important role.
In our previous study we defined amino acid distances in terms of their chemical characteristics. In the present study we have been able to measure amino acid distance in empirical terms in regard to the ability of peptide amino acids to meet peptide binding requirements. That this empirical distance measure is coherent is shown by the distinct clusters of amino acids it leads to. In trying to give an account of this empirical distance we find that it can be partially explained in terms of both the theoretical chemical distance scheme we had previously developed and the amino acid molecular weight. As several authors had already pointed out, [14, 15, 16] it is not just the chemical characteristics of amino acids, but their size too, that allows them to fit appropriately in the peptide-binding groove of the MHC molecule. The three distinct amino acid clusters identified ({Y, F, W}, {S, V, T} and {I, M, L}) throw some light into what it is about amino acids in the peptide that MHC molecules recognise as critical in determining binding. Although valine and leucine are very similar in their chemical structure, they are in different clusters. It appears that it is the molecular weight that separates them in spite of their almost identical chemical structure. In fact, the three clusters that stand out in our study could be separated solely based on molecular weight if the rest of the amino acids are not taken into account. The proximity of methionine to leucine and isoleucine indicates that the presence of sulphur in methionine does not make any difference in peptide binding. It is remarkable to find valine (a non-polar amino acid) in the same cluster as serine and threonine (polar amino acids). The same thing with tyrosine (a polar amino acid) that is in the same cluster as phenylalanine and tryptophan (non-polar). The definition of the {Y, F, W} cluster indicates the importance of aromatic rings in determining peptide binding for that is precisely what defines this cluster. One is tempted to conclude that chemical structure- whether an amino acid has an aliphatic chain or an aromatic ring, for instance-is more important than polarity.
The amino-acid distance described in the paper is 'biased' in the sense that it only applies to differences in amino acids in reference to how they determine peptide binding to the HLA molecule. This cannot be extrapolated to the influence of an amino acid in any other protein for any other function.
Peptide-binding profiles for HLA alleles
By representing HLA alleles as vectors in space Y as the mapping created by function V (see above) the door is open to make peptide binding predictions using the traditional algorithms for this task [17, 18] as the vectors in space Y are equivalent to the 'binding-affinity' or 'peptide-side-chain-frequency' matrices [19, 20] used by these algorithms. Space Y is created from data in which the reference to the alleles peptides bind to has been substituted with the amino acid sequence of those alleles. In this way predictions can be made not only for HLA alleles for which there is elution data available, but also for those alleles for which there is no elution data, in so far as the latter share sequence characteristics with the former.
Empirical binding-affinity matrices for each allele can be represented in another space Z with the same dimensions as Y. The difference between Y and Z is that Z contains points only for HLA alleles for which there is peptide binding data, whereas Y has points representing all alleles, even those without peptide binding data available, as long as their sequences have homologies with alleles with peptide binding data. While space Z comes from amino acid frequency counts at various peptide positions for different alleles, space Y comes from distances between sequence properties, by transposing S into S' and looking at sequence properties of HLA -peptide complexes instead of looking at the complexes themselves. For example the vector for HLA-B55 in Y is given in Table 1. This allele was not considered to have enough elution data to be included in the model to create space Y, so its vector is solely based on data from other alleles.
Table 1.
Vector for allele HLA-B55 in space Y.
| Amino | Peptide amino acid position | ||||||||
| Acid | P1 | P2 | P3 | P4 | P5 | P6 | P7 | P8 | P9 |
| A | 0.25 | 0.16 | 0.16 | 0.20 | 0.17 | 0.18 | 0.18 | 0.17 | 0.12 |
| C | 0.10 | 0.05 | 0.14 | 0.11 | 0.04 | 0.11 | 0.06 | 0.06 | 0.03 |
| D | 0.17 | 0.04 | 0.17 | 0.16 | 0.12 | 0.10 | 0.08 | 0.09 | 0.03 |
| E | 0.16 | 0.16 | 0.09 | 0.13 | 0.10 | 0.08 | 0.11 | 0.32 | 0.02 |
| F | 0.27 | 0.12 | 0.24 | 0.13 | 0.13 | 0.14 | 0.14 | 0.08 | 0.22 |
| G | 0.15 | 0.14 | 0.10 | 0.15 | 0.12 | 0.14 | 0.11 | 0.13 | 0.09 |
| H | 0.09 | 0.16 | 0.11 | 0.09 | 0.11 | 0.13 | 0.13 | 0.07 | 0.10 |
| I | 0.16 | 0.14 | 0.20 | 0.12 | 0.12 | 0.20 | 0.13 | 0.19 | 0.35 |
| K | 0.13 | 0.05 | 0.18 | 0.15 | 0.39 | 0.12 | 0.13 | 0.13 | 0.43 |
| L | 0.21 | 0.26 | 0.15 | 0.18 | 0.15 | 0.17 | 0.18 | 0.16 | 0.32 |
| M | 0.13 | 0.09 | 0.11 | 0.03 | 0.08 | 0.05 | 0.21 | 0.08 | 0.15 |
| N | 0.11 | 0.08 | 0.09 | 0.13 | 0.12 | 0.14 | 0.08 | 0.10 | 0.09 |
| P | 0.06 | 0.84 | 0.18 | 0.15 | 0.23 | 0.11 | 0.15 | 0.22 | 0.04 |
| Q | 0.15 | 0.08 | 0.08 | 0.30 | 0.07 | 0.22 | 0.15 | 0.10 | 0.03 |
| R | 0.42 | 0.71 | 0.29 | 0.18 | 0.16 | 0.16 | 0.16 | 0.12 | 0.23 |
| S | 0.14 | 0.14 | 0.12 | 0.13 | 0.19 | 0.26 | 0.10 | 0.16 | 0.04 |
| T | 0.14 | 0.18 | 0.13 | 0.15 | 0.12 | 0.11 | 0.37 | 0.16 | 0.06 |
| V | 0.11 | 0.26 | 0.19 | 0.11 | 0.13 | 0.13 | 0.19 | 0.13 | 0.23 |
| W | 0.05 | 0.07 | 0.08 | 0.13 | 0.06 | 0.08 | 0.13 | 0.07 | 0.10 |
| Y | 0.12 A | 0.25 | 0.42 | 0.10 | 0.08 | 0.14 | 0.16 | 0.08 | 0.25 |
| A | P | R | Q | P | G | L | M | A | |
| A | P | R | T | V | A | L | T | A | |
| A | P | R | Q | P | G | L | M | A | |
| A | P | R | T | V | A | L | T | A | |
| R | P | R | H | Q | G | V | M | V | |
| I | P | Y | H | I | V | N | I | V | |
| A | P | T | G | D | L | P | R | A | |
The top part of the table shows the vector for allele HLA-B55, of size 180, represented as a matrix. (Data for this allele was not used to build the models in this study.) The bottom part of the table shows the sequence of seven nonameric peptides known to bind to HLA-B55. Highlighted in bold are the points of agreement between the model and the sequence of these peptides.
In order to evaluate the prediction made in Table 1 about the sequence of peptides likely to bind to HLA-B55, we have calculated the likelihood ratio (LR) of the prediction made for each position in a nine-amino-acid peptide as derived from 2 × 2 contingency tables. First we determine a cut-off, values of 0.23 or above are considered positive predictions, that is, they indicate that the corresponding amino acid is a likely candidate to appear in a collection of peptides known to bind to HLA-B55; values below 0.23 are negative predictions. At position 1 (P1) 3 amino acids (A, F and R) were predicted to be found in such peptides. In the small collection of peptides presented, 3 amino acids are found at that position (A, R, and I). Therefore we have 2 true positives (TP), A and R; 1 false positive (FP), F; 1 false negative (FN), I; and 16 true negatives (TN), the rest of the amino acids. The likelihood ratio for this position is LR = (2/(2+1))/(1/(1+16)) = 11.33. For position P2 we have TP = 1; FP = 4; FN = 0; TN = 15; and LR = 4.75. For position P3 we have TP = 2; FP = 1; FN = 1;TN = 16; and LR = 11.33. For position P4 we have TP = 1; FP = 0; FN = 3; TN = 17; the LR cannot be calculated because the specificity is 100%, although the sensitivity is only 25%. For position P5 we have TP = 1; FP = 1; FN = 4; TN = 14; and LR = 3. For position P9 we have TP= 1; FP = 6; FN = 1; TN = 12; and LR = 1.52. Considering that by random we get a LR = 1, likelihood ratios above 1, some as high as 11, indicate that the prediction has some substance; especially when the LRs are multiplied with each other as they are to be.
In trying to make peptide binding predictions for alleles without peptide elution data, the question is how good a substitute of Z Y is. Regression analysis was used to answer this question, with the vectors representing HLA-alleles in space Y as the independent variable, and those vectors representing the same alleles in space Z as the dependent variable. In addition, the numerical values of vectors in Y and Z were converted to categorical values and then the pairs of vectors were compared using 2 × 2 contingency tables. (See Table 2.)
Table 2.
Comparison of allele vectors in spaces Y and Z.
| HLA | Numerical | Categorical | ||
| allele | R2 (%) | Sensitivity (%) | Specificity (%) | Likelihood Ratio |
| A1 | 27.1 | 50 | 97 | 15 |
| A2 | 71.9 | 67 | 100 | |
| A3 | 63.7 | 50 | 98 | 22 |
| A11 | 40.5 | 50 | 97 | 18 |
| A24 | 25.8 | 100 | 97 | 36 |
| A29 | 12.7 | 67 | 95 | 13 |
| A31 | 26.8 | 100 | 97 | 30 |
| A33 | 16.0 | 100 | 96 | 25 |
| B7 | 36.6 | 60 | 94 | 10 |
| B8 | 14.4 | 50 | 95 | 10 |
| B14 | 22.9 | 50 | 94 | 9 |
| B27 | 71.8 | 100 | 99 | 86 |
| B35 | 36.9 | 100 | 97 | 30 |
| B38 | 0.2 | 0 | 93 | |
| B44 | 18.3 | 25 | 96 | 6 |
| B51 | 48.6 | 100 | 98 | 45 |
| B53 | 22.4 | 67 | 97 | 20 |
| B54 | 40.9 | 67 | 98 | 30 |
| ALL | 27.7 | 64 | 96 | 18 |
'R2' stands for the coefficient of determination in the regression analysis of numerical values of the vectors in spaces Y and Z (see text) for each allele. (In all cases the p value was 0.000, except for B38, which was 0.51.) Allele vectors values in Y and Z were converted to categorical values (0, 1) using cut-off values and pairs of vectors for each allele were compared using a 2 × 2 contingency table. 'Sensitivity', 'specificity', and 'likelihood ratio' refer to the prediction of a Z vector by the corresponding Y vector, and were calculated from these 2 × 2 tables.
It is clear that the peptide-binding profiles for alleles without experimental elution data cannot be accurate. It is also clear that an approximate prediction can in fact be made for the complete range of class I HLA alleles, the accuracy of the prediction depending on the sequence similarities with alleles for which there is elution data. Table 3 shows tentative predictions of peptide-binding motifs for alleles without sufficient binding data to make direct predictions, based on their respective vectors in space Y.
Table 3.
Predicted peptide-binding motifs for HLA class I alleles without sufficient binding data to make direct predictions.
| Pp | Paa | A23 | A26 | A32 | A34 | A36 | A43 | A66 | A68 | A69 | A74 | B62 | B75 | B72 | B63 | B37 | B61 | B41 | B46 | B48 | B49 | B50 | B52 | B55 | B57 | B58 | 859 | B67 | B73 | B78 | B81 |
| 1 | A | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 8.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | ||||||||||||||||||
| 1 | F | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0-3 | 0.3 | 0.3 | 0.3 | 0,3 | 0,3 | 0,3 | 0,3 | ||||||||
| 1 | R | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.4 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.4 | 0.3 | 0.3 | 0,3 | 04 | 0.3 | 0.3 | 0.4 | 0.4 | 0.5 | 0.3 | 0.4 | |||
| 2 | E | 0.5 | 0.7 | 0.7 | 0.7 | 0.7 | 0.7 | 0.7 | 0.7 | 0.7 | 0.7 | 0.7 | |||||||||||||||||||
| 2 | F | 0.3 | |||||||||||||||||||||||||||||
| 2 | I | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | |||||||||||||||
| 2 | L | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.4 | 0.5 | 0.5 | 0.4 | 0.5 | 0.5 | 0.3 | 0.3 | 0.5 | 0.5 | 0.3 | 0.5 | 0.5 | 0.3 | 0.4 |
| 2 | P | 0.5 | 0.8 | 0.5 | 0.8 | 0.3 | 0.5 | 0.8 | 0.8 | 0.8 | 0.5 | 0.8 | 0.8 | 0.8 | 0.8 | 0.7 | 0.8 | 0.8 | 0.8 | 0.8 | 0.6 | 0.6 | 0.8 | 0.8 | 0.8 | 0,8 | 0.8 | 0.8 | 0.8 | 0.8 | 0.8 |
| 2 | R | 0.4 | 0.4 | 0.4 | 0,4 | 0.4 | 0,4 | 0.4 | 0.5 | 0.5 | 0.7 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.7 | 0.5 | 0.5 | 0.5 | 07 | 0.5 | 0.5 | 0.7 | 0.7 | 0.9 | 0.5 | 07 | |||
| 2 | T | 03 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | |||||||||||||||||||
| 2 | V | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | |||||||||||
| 2 | Y | 0.7 | 0.5 | 0.5 | 0.5 | ||||||||||||||||||||||||||
| 3 | D | 0.3 | 0.4 | 0.3 | 0.4 | 0.4 | 0.4 | ||||||||||||||||||||||||
| 3 | F | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0,3 | 0.3 | |||||||||||||||||||
| 3 | K | 0.3 | 0.3 | ||||||||||||||||||||||||||||
| 3 | R | 0.3 | 0.4 | 0.4 | 0.3 | 0.4 | 0.3 | 0.3 | 0.4 | ||||||||||||||||||||||
| 3 | Y | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 04 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.4 | 0.3 | 0.3 | 0.3 | 0,4 | 0.3 | 0.3 | 0.4 | 0.4 | 0.5 | 0.3 | 0.4 | |||
| 3 | I | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.4 | 0.4 | 0.3 | 0.3 | 0.4 | 0.3 | 0.4 | 0.4 | 0.3 | |||||
| 3 | K | 0.4 | 0.5 | 0.5 | 0.4 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.4 | 0.4 | 0.5 | 0.5 | 04 | 0.4 | 0.4 | 0.4 | |||||||||||
| 9 | L | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 03 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | ||
| 9 | R | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | ||||||||||||||||||
| 8 | V | 0.4 | 0.4 | 0.4 | 0.4 | 0.4 | 0.4 | 0.4 | 0.3 | 0.4 | 0.4 | 0.4 | 0.4 | 0.4 | 0.4 | 0.4 | 0.4 | 0.4 | 0.4 | 0.4 | 0.3 | 0.4 | 0.4 | 0.3 | |||||||
| 9 | Y | 0.3 | 0.6 | 0.4 | 0.3 | 0.6 | 0.6 | 0.4 | 0.4 | 0.4 | 0.4 | 0.6 | 0.3 | 0.3 | 0.6 | 0.6 | 0.3 | 0.3 | 0.3 | 0.3 |
Peptide-binding motifs are based on allele vectors in space Y. Only values for the Ochiai similarity index equal or greater than 0.3 are included. 'Pp' stands for amino acid position in peptide and 'Paa' for the amino acid occupying that position.
MHC pocket classes and peptide-binding motifs
The fact that co-ordinate values in S' are bipolar opens the door to the use of propositional logic. Space S' is not only the space of peptide and HLA sequence properties, which, from the point of view of logic, are considered elementary or atomic propositions; but also the space of all possible propositional functions based on the former elementary propositions. We are interested in two types of functions, those that take their arguments from HLA sequence properties (H), functions of the form f1(h1, h2, ..., hn); and functions that take their arguments from peptide sequence properties (P), functions of the form fII(p1, p2, ..., pm). The latter (fII) are what in the immunology literature are called 'peptide-binding motifs', the former (fI) can be, for instance, the MHC pocket classes Zhang and co-workers describe. [16]
Just as we can measure the distance between two properties h and p in S', we can also measure the distance between two propositional functions fI and fII. For instance, if we define the following elementary sequence properties h1 ≡ (Hp = 45, Haa = 'M'), h2, ≡ (Hp = 45, Haa = 'T'), h3 ≡ (Hp = 67, Haa = 'V'), h4 ≡ (Hp = 67, Haa = 'M'), h5 ≡ (Hp = 66, Haa = 'N'), p1≡ (Pp = 2, Paa = 'V'), p2 ≡ (Pp = 2, Paa = 'S'), and p3 = (Pp = 2, Paa = 'T'), the distance between fI = (h1∨ h2) and (h3∨ h4) and h5 and fII = p1∨ p2 ∨ p3 is 0.48, measured as an Ochiai similarity index, a value reached in less than 1% of the distances measured in space S' indicating unusual proximity. In this case fI is the Zhang MHC pocket class 'P2-IV.2', and fII is the restriction of position 2 in the peptide (Pp = 2) to the {S, V, T} amino acid cluster. See Table 4 for other such distance measurements between fI-type and fII-type propositional functions, that is, between HLA pocket classes and peptide-binding motifs. By the use of propositional logic, and by making a geometric representation of logical propositions, which allows the measurement of the distance between them, we have a convenient tool to study peptide binding in terms of the sequence characteristics of MHC molecules and peptides.
Table 4.
Distance between logical combinations of HLA and peptide sequence properties.
| MHC | HLA sequence variables | Peptide sequence variables | Similarity index |
| pocket class | (distance) | ||
| P2-I.1 | Hp = 9, Haa = Y | Pp = 2, Paa = P | 0.90 |
| Hp = 66, Haa = I | |||
| Hp = 67, Haa = Y or F | |||
| P2-III.1 | Hp = 45, Haa = D or E | Pp = 2, Paa = R or H or K | 0.73 |
| P2-IV.1 | Hp = 45, Haa = M | Pp = 2, Paa = L or I or M | 0.63 |
| Hp = 63, Haa = E | |||
| Hp = 66, Haa = K | |||
| Hp = 67, Haa = V | |||
| P2-IV.2 | Hp = 45, Haa = M or T | Pp = 2, Paa = V or S or T | 0.48 |
| Hp = 66, Haa = N | |||
| Hp = 67, Haa = V or M | |||
| P2.-V.1 | Hp = 9, Haa = S or V | Pp = 2, Paa = Y or F or W | 0.85 |
| Hp = 63, Haa = E or Q | |||
| Hp = 66, Haa = K or R | |||
| PΩ-I | Hp= 116, Haa = Y or F or L | Pp = 9, Paa = I or M or L or V | 0.59 |
| PΩ-II.1 | Hp= 116, Haa = D | Pp = 9, Paa = K or R | 0.73 |
| Hp = 74, Haa = D | |||
| Hp = 77, Haa = D | |||
| PΩ-II.2 | Hp = 116, Haa = D | Pp = 9, Paa = K or R | 0.63 |
| Hp = 74, Haa = D | |||
| PΩ-III | Hp = 116, Haa = D or S | Pp = 9, Paa = Y or F or W or L or I or M | 0.19 |
| Hp = 74, Haa = D or Y | |||
| PΩ-III | Hp = 116, Haa = D or S | Pp = 9, Paa = K or R or Y | 0.63 |
| Hp = 74, Haa = D or Y |
HLA molecule.- Hp: position number; Haa: amino acid. Peptide.- Pp: position number, Paa: amino acid. For the similarity index (Ochiai index), '1' means total similarity and '0' total absence of similarity; in our study only 4% of distance measurements between isolated sequence variables had an index greater than 0.2, 1% had an index greater than 0.4, 0.1% had an index greater than 0.6, and only in 0.03% of measurements the index was greater than 0.8.
The amino acids favoured at position 2 in the peptide for the various HLA pocket classes defined by Zhang et al. provides additional evidence for the amino acid clusters we have identified. The amino acid cluster {S, V, T} is favoured in the Zhang pocket class P2-IV.2, the cluster {I, M, L} in the pocket class P2-IV.1, and the cluster {Y, F, W} in the pocket class P2-V.1.
Conclusions
By looking at the problem of peptide binding to the MHC molecule from an algebraic and geometric perspective, molecules and their properties are seen as vectors in a metric space in which distances can be measured. A range of analytical tools then become available to study these distances, from which important conclusions can be drawn. The positions in the MHC molecule that determine peptide binding requirements are revealed. How different peptide amino acids are in meetings those requirements becomes clear. And we can define allelic peptide-binding profiles in terms of the amino acid sequence of the MHC allele, allowing the prediction of peptide binding for alleles for which there is no binding data as long as they have sequence similarities with alleles for which there is. At this time these predictions are limited by the fact that the correlation of allele vectors in spaces Y and Z is not perfect; but being able to make predictions at all marks the way to use current peptide data to make inferences for alleles for which there is no such data. We should emphasise that all the results presented here were derived automatically, without any input of previous knowledge of the biological question at hand. Our findings come directly and strictly from an automatic data analysis of the MHCPEP data. By means of the analytical methods introduced here knowledge accumulated by many researchers over many years has been reproduced. In addition, these methods have brought to light new facts about peptide binding.
A major limitation in this study is that variables (sequence properties) are assumed to be independent of each other in their effect on peptide binding, when there is good reason to believe they are not. Dropping the assumption of independence, however, comes at a very high cost by increasing the computational complexity of the problem to such a magnitude that makes it apparently intractable. Future progress in elucidating how peptides are selected for their presentation to T cells depends on the development of algorithms to analyse peptide binding data in a combinatorial way that would account for the possibility that the effect of MHC sequence variables is interrelated. Here we have shown that a propositional calculus can be developed to represent any such combination of sequence variables, fI-type propositional functions for MHC sequence variables and fII-type propositional functions for peptide sequence variables. We have also shown that the study of all the possible combinations of variables amounts to evaluating how these two types of functions- fI and fII-hold together in the space S' of empirical data.
Geometry, as the study of abstract spaces, results from the distinction between 'set' and 'space'. A space differs from the mere set of the elements that populate the space by possessing a structure that places the elements of the space in a certain relation with each other. The elements in the space-points, vectors-are close or far from each other, there is a distance between them. By defining a distance measure between the elements of a set we confer a geometric structure to that set converting it into a space. If we can tell how different MHC alleles are from each other– how distant they are–in reference to a particular function, let us say, their ability to present peptides of a specific amino acid sequence, we are actually converting the set of MHC alleles into a metric space where a distance measure has been defined. Now, we can similarly define a distance measure between MHC alleles in terms of their sequence differences. In fact we can define many such distance measures by changing the 'scale' of the co-ordinates, that is, by changing the 'weight' a variable (co-ordinate) has in contributing to the distance measure-each new measure being the result of a transformation imposed on that space. If we find which transformation in the sequence space of MHC molecules results in a distance measure that best parallels-correlates more closely with-the distance measure in the functional space of MHC molecules, we have a geometric model of the function of MHC molecules in terms of their amino acid sequences.
In this paper we talk about the 'transposed space' as the result of transposing the matrix of vectors in the sequence space of MHC-peptide complexes. This matrix operation creates a dual space where the n dimensions in the original space become the points-vectors-in the new space, and the × vectors in the original space become the dimensions-co-ordinates-in the new space. By doing so we convert the distance measure between MHC-peptide complexes into a distance measure between their sequence properties. This last concept-the distance between the sequence properties of MHC-peptide complexes-is a key that opens the door to elucidate the peptide binding requirements imposed by the MHC amino acid sequence.
We conclude that algebra and geometry provide a convenient theoretical framework to study the function of proteins as amino acid sequences when there is enough variation in this sequence to account for the variation in their function, as we have seen to be the case with MHC molecules in regard to their ability to present peptides. The algebraic and geometric concepts presented here are the foundation for the design of an information model to create a database and the algorithms to manipulate it. They are not presented for the sake of advancing a unique and peculiar theory, but with an entirely practical intent. Although databases and computer programmes are typically presented as implementations, it is preferable to present them formally in mathematical terms so that their true nature comes to light. The theoretical concepts presented here allowed us to manipulate MHC-peptide-binding data in a successful manner.
Although in this paper we have centred our attention on the conceptualisation of the problem and on the methodological aspects of data analysis, we are aware that the results presented here depend on the quality of empirical data used in the analysis. A critical review of the data sets currently available [7,8,21] indicate that careful auditing of the data, as well as continuous compilation of new empirical data are necessary.
Contributor Information
Pedro Cano, Email: pcano@mdanderson.org.
Bo Fan, Email: BFan1@JANUS.JNJ.com.
References
- Bjorkman PJ, Saper MA, Samraoui B, Bennett WS, Strominger JL, Wiley DC. Structure of the human class I histocompatibility antigen, HLA-A2. Nature. 1987;329:506–512. doi: 10.1038/329506a0. [DOI] [PubMed] [Google Scholar]
- Bjorkman PJ, Saper MA, Samraoui B, Bennett WS, Strominger JL, Wiley DC. The foreign antigen binding site and T cell recognition regions of class I histocompatibility antigens. Nature. 1987;329:512–518. doi: 10.1038/329512a0. [DOI] [PubMed] [Google Scholar]
- Falk K, Rotzschke 0, Stevanovic S, Jung G, Rammensee HG. Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules. Nature. 1991;351:290–296. doi: 10.1038/351290a0. [DOI] [PubMed] [Google Scholar]
- Rammensee HG, Falk K, Rotzschke O. MHC molecules as peptide receptors. Curr Opin Immunol. 1993;5:35–44. doi: 10.1016/0952-7915(93)90078-7. [DOI] [PubMed] [Google Scholar]
- Rammensee HG. Chemistry of peptides associated with MHC class I and class II molecules. Curr Opin Immunol. 1995;7:85–96. doi: 10.1016/0952-7915(95)80033-6. [DOI] [PubMed] [Google Scholar]
- Van Bleek GM, Nathenson SG. Isolation of an endogenously processed immunodominant viral peptide from the class I H-2Kb molecule. Nature. 1990;348:213–216. doi: 10.1038/348213a0. [DOI] [PubMed] [Google Scholar]
- Rammensee HG, Friede T, Stevanoviic S. MHC ligands and peptide motifs: first listing. Immunogenetics. 1995;41:178–228. doi: 10.1007/BF00172063. [DOI] [PubMed] [Google Scholar]
- Brusic V, Rudy G, Harrison LC. MHCPEP, a database of MHC-binding peptides: update 1997. Nucleic Acids Res. 1998;26:368–371. doi: 10.1093/nar/26.1.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano P, Fan B, Stass S. A geometric study of the amino acid sequence of class I HLA molecules. Immunogenetics. 1998;48:324–334. doi: 10.1007/s002510050439. [DOI] [PubMed] [Google Scholar]
- Ramsey FP. Universals. In Philosophical Papers Edited by Mellor DH Cambridge University Press Cambridge, 1990.
- Joly S, Le Calve G. Similarity functions. In Classification and dissimilarity analysis Edited by Van Cutsen B Springer-Verlag, New York, 1994.
- Chelvanayagam G. A roadmap for HLA-A, HLA-B, and HLA-C peptide binding specificities. Immunogenetics. 1996;45:15–26. doi: 10.1007/s002510050162. [DOI] [PubMed] [Google Scholar]
- Vasmatzis G, Zhang C, Cornette JL, DeLisi C. Computational determination of side chain specificity for pockets in class I MHC molecules. Mol Immunol. 1996;33:1231–1239. doi: 10.1016/S0161-5890(96)00090-9. [DOI] [PubMed] [Google Scholar]
- Smith KJ, Reid SW, Stuart Dl, McMichael AJ, Jones EY, Bell Jl. An altered position of the alpha 2 helix of MHC class I is revealed by the crystal structure of HLA-B*3501. Immunity. 1996;4:203–213. doi: 10.1016/s1074-7613(00)80429-x. [DOI] [PubMed] [Google Scholar]
- Smith KJ, Reid SW, Harlos K, McMichael AJ, Stuart Dl, Bell Jl, Jones EY. Bound water structure and polymorphic amino acids act together to allow the binding of different peptides to MHC class I HLA-B53. Immunity. 1996;4:215–228. doi: 10.1016/s1074-7613(00)80430-6. [DOI] [PubMed] [Google Scholar]
- Zhang C, Anderson A, DeLisi C. Structural principles that govern the peptide-binding motifs of class I MHC molecules. J Mol Biol. 1998;281:929–947. doi: 10.1006/jmbi.1998.1982. [DOI] [PubMed] [Google Scholar]
- D'Amaro J, Houbiers JG, Drijfhout JW, Brandt RM, Schipper R, Bavinck JN, Melief CJ, Kast WM. A computer program for predicting possible cytotoxic T lymphocyte epitopes based on HLA class I peptide-binding motifs. Hum Immunol. 1995;43:13–18. doi: 10.1016/0198-8859(94)00153-H. [DOI] [PubMed] [Google Scholar]
- Parker KC, Bednarek MA, Coligan JE. Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains. Immunol. 1994;152:163–175. [PubMed] [Google Scholar]
- Barber LD, Gillece-Castro B, Percival L, Li X, Clayberger C, Parham P. Overlap in the repertoires of peptides bound in vivo by a group of related class I HLA-B allotypes. Curr Biol. 1995;5:179–190. doi: 10.1016/s0960-9822(95)00039-x. [DOI] [PubMed] [Google Scholar]
- Kubo RT, Sette A, Grey HM, Appella E, Sakaguchi K, Zhu NZ, Arnott D, Sherman N, Shabanowitz J, Michel H, et al. Definition of specific peptide motifs for four major HLA-A alleles. J Immunol. 1994;152:3913–3924. [PubMed] [Google Scholar]
- Rammensee HG, Bachmann J, Stevanovic S. MHC ligands and Peptide Motifs Chapman & Hall, New York, 1997.