

Abstract
Formin proteins and their associated factors cooperate to assemble unbranched actin filaments in diverse cellular structures. The Saccharomyces cerevisiae formin Bni1 and its associated nucleation-promoting factor (NPF) Bud6 generate actin cables and mediate polarized cell growth. Bud6 binds to both the tail of the formin and G-actin, thereby recruiting monomeric actin to the formin to create a nucleation seed. Here, we structurally and functionally dissect the nucleation-promoting C-terminal region of Bud6 into a Bni1-binding “core” domain and a G-actin binding “flank” domain. The ∼2-Å resolution crystal structure of the Bud6 core domain reveals an elongated dimeric rod with a unique fold resembling a triple-helical coiled-coil. Binding and actin-assembly assays show that conserved residues on the surface of this domain mediate binding to Bni1 and are required for NPF activity. We find that the Bni1 dimer binds two Bud6 dimers and that the Bud6 flank binds a single G-actin molecule. These findings suggest a model in which a Bni1/Bud6 complex with a 2:4 subunit stoichiometry assembles a nucleation seed with Bud6 coordinating up to four actin subunits.
The assembly of diverse filamentous actin arrays in cells is dependent on machinery that catalyzes the otherwise inefficient step of actin nucleation. A variety of actin nucleators and nucleation-promoting factors (NPFs) have now been identified, including Arp2/3 complex, WASp/WAVE family members, formins, Spire, Cobl, Lmod, JMY, adenomatous polyposis coli (APC), and Bud6 (reviewed in refs. 1–4). Although each is unique in its detailed mechanism of actin assembly, many of these factors are surprisingly related in their overall strategy for promoting polymerization from a pool of free actin monomers. Additionally, efficient actin assembly often requires cooperation between an actin nucleator and one or more NPFs. A majority of these nucleators or NPFs contain multiple WASP homology-2 (WH2) domains, a short (17–27 aa) motif that binds actin monomers (2). The need for nucleators to recruit multiple actin monomers stems from the fact that actin dimers and trimers are extremely unstable, short-lived species. The smallest stable actin species is a tetramer, which has a Kd of 0.14 μM (5). The classic Arp2/3 complex exploits interactions with WH2-containing WASP-family NPFs in its mechanism of actin filament nucleation. Together, the Arp2 and Arp3 subunits in the complex resemble a short-pitched actin dimer and bind to two NPF molecules (6–8), each of which brings in at least one actin monomer. This is thought to generate a four-actin cross-filament seed for a daughter filament that rapidly polymerizes at an angle of 70° to the mother filament (9). Other WH2-containing proteins can act independently to nucleate actin filaments, likely by arraying multiple actin subunits into a nucleation seed. For example, Cobl has three WH2 domains and an unusually long linker sequence separating its second and third WH2 domains, which is critical for nucleation (10). This has led to the proposal that Cobl arranges monomers into a cross-filament trimer that serves as a seed for polymerization.
Formin-family nucleators generally lack WH2 domains, and they also differ from the Arp2/3 complex in that they nucleate unbranched actin filaments, such as those found in cytokinetic rings, filopodia, stress fibers, and yeast actin cables (4, 11). Formins vary in their domain structure, reflecting their diverse cellular roles and mechanisms of regulation, but all contain the highly conserved formin homology-2 (FH2) domain. The FH2 domain consists of two rod-shaped subdomains tied together in a head-to-tail arrangement by flexible linkers to form a closed ring. Each side of the dimer contains two actin-binding surfaces, allowing the FH2 dimer to organize two or three actin subunits into a filament-like orientation that can function as a nucleus for filament polymerization (12, 13). Formins associate with the “barbed” end of a nascent filament, and the flexible nature of the FH2 dimer affords it the surprising ability to “stair-step” on the elongating barbed end as new actin subunits are incorporated (4, 14). Elongation is also facilitated by the formin homology-1 (FH1) domain, a segment immediately adjacent to the FH2 domain that contains multiple proline-rich motifs and can accelerate elongation by recruiting profilin-bound actin subunits to the site of incorporation at the barbed end of the growing filament (15, 16).
Formins were initially proposed to nucleate actin filaments solely using their FH2 domains, perhaps by stabilizing transiently formed dimers or trimers (17) because the isolated FH2 domain lacks significant affinity for actin monomers (12). However, recent work has revealed that an adjacent C-terminal tail region, often containing the diaphanous autoregulatory domain (DAD), directly binds actin monomers and works together with the FH2 domain to stimulate nucleation (18). Other formins may use a WH2-like element distinct from the DAD domain for this purpose (19, 20). Furthermore, a growing number of formins have been shown to bind directly to actin monomer-recruiting NPFs. For example, the Drosophila formin Cappuccino and its mammalian counterparts Fmn1 and Fmn2 bind to Spire, an actin nucleator that contains an array of four WH2 domains (21). The mammalian formin mDia1 interacts with APC protein, an actin nucleator that binds monomers but does not have identifiable WH2 domains (22). In budding yeast, the formin Bni1 binds Bud6, which also binds actin monomers but does not have a clearly recognizable WH2 domain (23).
Bud6 localizes to the bud tip and neck, and it was first identified in a yeast two-hybrid screen for actin-interacting proteins (24). In Schizosaccharomyces pombe, the microtubule plus end-associated protein Tea1, formin For3, and Bud6 form a large polarity complex that resides at the cell tips and promotes localized actin cable assembly, and the triple knockout of these three genes leads to severe defects in cell polarity (25). Bud6 also contributes to the maintenance of septin-dependent diffusion barriers in the endoplasmic reticulum and nuclear membranes, which limit membrane protein diffusion between the mother and daughter cell compartments (26, 27). The N-terminal half of Bud6 is required for its in vivo localization and for its function in cortical capture of astral microtubule ends (28, 29). It has also been shown to bind microtubules directly (28, 29). The C-terminal half (residues 489–788) directly facilitates actin filament assembly by the formin Bni1 (23). Bud6 enhances the nucleation phase, rather than the elongation phase, of Bni1-mediated actin filament assembly, and this NPF effect requires separable interactions of Bud6 with Bni1 and with actin monomers (30).
To understand better how Bud6 functions together with Bni1 in actin assembly, we dissected the structural and functional properties of a C-terminal fragment of Bud6 that contains NPF activity (c-Bud6, residues 550–788). We find that c-Bud6 can functionally be divided into two parts: a trypsin stable core (residues 550–688) that contains the Bni1 binding site and a flank (residues 699–788) that binds to actin monomers. Although the core domain retains the ability to bind Bni1, it inhibits, rather than stimulates, actin nucleation by Bni1, likely because it obstructs the actin monomer recruitment activity of the Bni1 tail region. The crystal structure of the trypsin stable Bud6 core reveals a unique rod-shaped dimeric fold. Conserved surfaces at either end of the core domain and at its center are critical for Bni1 binding and NPF activity. Through a series of native gel shift, size exclusion chromatography (SEC) multiangle light scattering (MALS), and isothermal titration calorimetry (ITC) experiments, we determined the stoichiometry of association of Bud6 with Bni1 and with actin monomers. These structural and functional data inform an emerging model for the mechanism of actin nucleation by this nucleator/NPF pair.
Results
Functional Dissection of c-Bud6.
The C-terminal half of Bud6 (residues 489–788) binds both Bni1 and monomeric actin, and it strongly enhances the rate of Bni1-mediated actin nucleation (23, 30, 31). We sought to determine whether the binding sites for Bni1 and for actin lie in distinct structural domains of Bud6. We prepared a slightly shorter C-terminal fragment of Bud6 (c-Bud6, residues 550–788; Fig. 1A) that exhibited essentially identical enhancement of Bni1 activity as previously described for the construct using residues 489–788 and subjected it to limited proteolysis to identify proteolytically stable domains. Digestion with trypsin removed ∼11 kDa from c-Bud6, leaving a stable 17-kDa core (Fig. 1B). N-terminal sequencing and intact mass determination by MS revealed that this core domain contains residues 552–688. Based on these results, we designed, expressed, and purified two additional Bud6 constructs for further study: Bud6core (residues 550–688) and Bud6flank (residues 699–788).
Fig. 1.

Dissection of c-Bud6 functional domains. (A) Domain structures of Bud6 and Bni1. The residue boundaries of the constructs studied here are highlighted. (B) Trypsin digest of c-Bud6. Aliquots of c-Bud6 alone (uncut) or c-Bud6 treated with trypsin for the indicated time (in hours) were analyzed by Coomassie-stained SDS/PAGE. (C) Native-PAGE analysis of Bni1/Bud6 complexes. Bni1-FH2C was mixed with either Bud6core or Bud6flank and resolved on an 8–25% gradient native gel. Note that Bni1-FH2C does not migrate as a single species but that it completely shifts the well-defined Bud6core band to a much slower migrating position. (D) Native-PAGE analysis of the Bni1tail and its interactions with Bud6core and Bud6flank. The proteins alone (lanes 1–3) or mixtures of an excess of Bni1tail with Bud6core (lane 4) or Bud6flank (lane 5) were resolved on 20% native gel. Note that Bni1tail completely shifts the Bud6core band. (E) SEC of the Bni1 tail region alone (red trace) or mixed in excess with the Bud6core protein (blue trace). SDS/PAGE analysis of the elution fractions from the Bni1/Bud6 mixture (Lower) shows that a fraction of the Bni1 tail protein elutes together with the Bud6core, indicating that the complex is stable on gel filtration. (F) Native-PAGE analysis of Bud6core, Bud6flank, and G-actin. The Bud6 proteins alone or mixed with G-actin in G-buffer, as indicated, were resolved on 20% native gel. Note that Bud6flank shifts the G-actin band to a more slowly migrating position, indicating formation of a complex.
We also prepared a Bni1 construct encompassing its FH2 domain and C-terminal tail region (Bni1-FH2C, residues 1348–1953; Fig. 1A) and tested its binding to Bud6core or Bud6flank using a native-PAGE gel shift assay (Fig. 1C). Native-PAGE analysis of Bud6core and Bud6flank in isolation revealed a predominant, rapidly migrating band for both constructs (Fig. 1C, lanes 2 and 3). Although Bni1-FH2C alone does not migrate as a well-defined species (lane 1), addition to Bud6core gives rise to a new, slowly migrating band corresponding to the complex and depletes the band corresponding to free Bud6core (Fig. 1C, lane 4). In contrast, Bni1-FH2C does not alter the migration of Bud6flank (Fig. 1C, lane 5). The binding site of Bud6 on Bni1 has been mapped to a region spanning residues 1750–1824 (31). We identified a smaller fragment of Bni1 (Bni1tail, residues 1794–1837) that partially overlaps with the previously identified region and is sufficient to mediate binding to Bud6core. As we observed with the longer Bni1-FH2C construct, Bni1tail completely shifted Bud6core but did not affect migration of Bud6flank in a native-PAGE gel shift assay (Fig. 1D). Bni1tail also comigrates with Bud6core by gel filtration (Fig. 1E). We conclude that the regions represented by the Bud6core and Bniltail constructs are sufficient to mediate the interaction between these proteins.
We next tested binding of Bud6core or Bud6flank to monomeric actin using the native gel shift assay (Fig. 1F). In isolation, G-actin, Bud6core, and Bud6flank all migrated at approximately the same position on a 20% native-PAGE gel. When Bud6flank and G-actin were mixed, a slower migrating band was observed and the band at the position of the isolated proteins was depleted (Fig. 1F, lane 4). In contrast, no shifted band was apparent when Bud6core and G-actin were mixed (Fig. 1F, lane 5). Taken together with the previous results, these data show c-Bud6 has a modular structure composed of the Bud6core domain, which is sufficient to bind Bni1tail, and the Bud6flank domain, which is sufficient to mediate interaction with G-actin.
Structure of Bud6core.
Although we were unable to grow crystals of intact c-Bud6, the trypsin-stable core fragment crystallized readily. Bud6core was highly expressed in Escherichia coli and yielded two crystal forms. We determined the structure by single-wavelength anomalous dispersion (SAD) phasing to dmin = 2.9 Å using selenomethionine (SeMet)-substituted protein. The SeMet crystals were of space group P21 and contained a dimer in the asymmetrical unit. Density modification resulted in a readily interpretable map (Fig. S1A). Iterative model building and refinement produced a model with good statistics and geometry (Table 1). A much higher resolution dataset to 2.04 Å was collected on a native crystal in space group C2221 (Table 1). We determined the second structure by molecular replacement using the refined P21 structure as a search model. The asymmetrical unit of the C2221 crystals contains a single Bud6core subunit, and the dimer is formed by crystallographic symmetry. The final C2221 model includes Bud6 residues 553–677 and 127 water molecules, and it was refined to an R value of 0.192 (Rfree = 0.227) to 2.04 Å. Ramachandran analysis reveals excellent geometry, with 100.0% of the residues in preferred regions. A representative 2Fo − Fc σA-weighted electron density map is shown in Fig. S1B. Apart from some deviation from perfect twofold symmetry in the P21 structure, the two models are essentially the same, and we describe the higher resolution C2221 structure in the following analysis.
Table 1.
X-ray data collection and refinement
| Native | SeMet | |
| Data collection | ||
| Wavelength, Å | 0.97949 | 0.97934 |
| Space group | C2221 | P21 |
| a, b, c, Å/α, Å/β, Å/γ; ° | 58.9, 95.2, 70.8 | 53.3, 67.4, 58.8, 90.0, 94.5, 90.0 |
| Resolution, Å | 2.04 | 2.90 |
| Unique reflections | 12,868 | 17,136 |
| Redundancy | 3.5 | 1.9 |
| Completeness, % | 99.4 (97.2) | 95.3 (71.7) |
| I/σ | 17.6 (4.1) | 15.8 (2.0) |
| Rsym, %* | 6.4 (27.6) | 4.4 (28.2) |
| Phasing | ||
| Phasing power | 1.65 | |
| Figure of merit (acentric/centric) | 0.31/0.10 | |
| Refinement | ||
| Resolution range, Å | 35.4–2.04 | 35.0–2.90 |
| R-factor/Rfree† | 0.192/0.227 | 0.235/0.278 |
| Bond length deviation, Å | 0.007 | 0.010 |
| Bond angle deviation, ° | 0.9 | 1.2 |
| Average B factor, Å2 | 29.8 | 91.0 |
| Minimum B factor, Å2 | 16.4 | 57.7 |
| Maximum B factor, Å2 | 65.0 | 193.3 |
| Ramachandran plot, % | ||
| Preferred region (outlier) | 100.0 (0) | 92.3 (0) |
If not indicated otherwise, values in parentheses are for a high-resolution bin of 1/10 of total volume.
*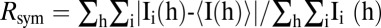 , where Ii(h) is the ith measurement and 〈I(h)〉 is the mean of all measurements of I(h) for Miller indices h.
, where Ii(h) is the ith measurement and 〈I(h)〉 is the mean of all measurements of I(h) for Miller indices h.
†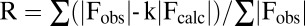 . Rfree is obtained for a test set of reflections (7.5% and10% for native and SAD, respectively).
. Rfree is obtained for a test set of reflections (7.5% and10% for native and SAD, respectively).
Bud6core is an elongated, rod-shaped dimer, with dimensions of ∼120 × 20 × 20 Å (Fig. 2A). The N-terminal portion of each polypeptide chain forms an initial helix α1 that runs approximately half the length of the rod. After a short loop of five residues, a very long second helix (α2) runs the entire length of the domain. The second helix is continuous but kinked sharply at a proline residue, Pro-629 (Fig. 2A). The two subunits interlock to form an overall triple-helical topology. The N termini of the subunits are close together (10 Å apart) at the middle of the rod. The C termini are at the opposite ends of the rod, 116 Å away from each other. Consistent with the dimer we observe in the crystal structure, we find that both c-Bud6 and Bud6core are stable dimers in solution, as measured by SEC-MALS (Fig. 2B).
Fig. 2.

Structure of Bud6core. (A) Overall structure of Bud6core dimer. Two chains are colored green and blue. The proline residue, Pro-629, that causes sharp bend of the second long helix is indicated by a stick representation of its side chain, in magenta. (B) SEC-MALS analysis of Bud6core and c-Bud6 reveals that both are dimers in solution. Purified Bud6core (residues 550–688) and c-Bud6 (residues 550–788) were analyzed on a Superdex 200 gel filtration column coupled to a MALS detector. The elution profile as measured by refractive index is shown for Bud6core as a thin red trace and for c-Bud6 as a thin black trace. The thicker horizontal traces indicate the measured molar mass, ∼32.7 kDa for Bud6core (expected molar mass for a Bud6core dimer is 32.7 kDa) and ∼54.4 kDa for c-Bud6 (theoretical molar mass is 55.6 kDa for a c-Bud6 dimer). (C) Intersubunit hydrophobic interactions of the Bud6core. Chain A is shown in green, and chain B is shown in blue. Designations of α1, α2_N, and α2_C are the same as in Fig. S4B. Side chains are shown in sphere presentation with the Van der Waals radius. (D) Number of polar interactions also stabilizes the dimer; hydrogen bonds are indicated by dashed lines.
The fold of the Bud6core dimer is unique, because a distance matrix alignment (DALI) search of structurally related proteins returned no match of any obvious biological relevance or evolutionary relatedness (32). However, Bud6core does share some structural similarity with a domain-swapped form of a spectrin repeat (Fig. S2), whose last helix is replaced by the same element of another spectrin repeat. In addition, a similar overall topology has been observed in the heterodimeric Vps27/Hse1 complex, in which two helices from one subunit and a third helix from another subunit form a three-helix bundle at each end of a barbell-like structure (33) (Fig. S3). Bud6core superimposes on the Vps27/Hse1 complex with an rmsd of 3.0 Å for 87 of 91 Vps27 residues and an rmsd of 4.2 Å for 85 of 88 Hse1 residues superimposed.
The interface between the two subunits of Bud6core is quite extensive, burying a total surface area of 5,196 Å2 (2,598 Å2 per subunit). The hydrophobic packing of Bud6core closely resembles that of a triple-helical coiled-coil. A coiled-coil is a ubiquitous protein motif that typically exhibits a “heptad repeat” in its primary sequence. The seven positions in the heptad repeat are labeled a through g, and interactions between the helices in the coiled-coil are mediated by hydrophobic residues at positions a and d in the repeat, which exhibit knobs-into-holes side-chain packing. Oppositely charged residues are sometimes present at positions e and g of interacting helices (34). We used the program SOCKET (35) to examine the heptad register of Bud6core (Fig. S4A). The hydrophobic and polar interactions of Bud6core are plotted in a helical wheel representation in Fig. S4B. The dominant feature stabilizing the dimeric structure is the formation of eight layers of three interacting hydrophobic residues, one contributed by each helix (Fig. 2C). These eight sets of interacting residues include four symmetrically equivalent layers on each half of the rod. The interacting residues in the four unique layers are Ile-A584a, Val-A606d, and Phe-B670a (layer 1); Leu-A580d, Leu-A610a, and Ala-B666a (layer 2); Val-A577a, Ala-A613d, and Leu-B663a (layer 3); and Leu-A573d, Val-A617a, and Leu-B659d (layer 4). (Here, we use the capital letter A or B preceding the residue number to indicate chain A or B in the dimer and the italicized letter after the residue number to indicate the heptad repeat position). The corresponding interactions are found on the opposite end of the rod-shaped dimer but with participating residues of chains A and B exchanged.
In addition to the hydrophobic packing, a number of polar interactions stabilize the dimer. These include intersubunit salt bridges Arg-556/Glu-645 and Lys-628/Glu653 as well as hydrogen bonds Gln-621/Lys-660 and Trp-635/Glu-645 (Fig. 2D).
Identification of Functional Surfaces on the Bud6core Domain.
We aligned the sequences of Bud6 homologs from 46 fungi using CLUSTAL W (36) and analyzed the rate of evolutionary variation at each position using the ConSurf server (37). The aligned sequences are presented in Dataset S1. Mapping of conservation onto the surface of the Bud6core domain reveals three conserved patches (Fig. 3A). All three of these patches lie on one face of the Bud6core domain; two of the patches, which are identical due to symmetry, are located near the ends of the rod, and we refer to them as the “distal” patches. The third “central” patch is at the center of the domain and consists of two contiguous areas that are identical due to symmetry.
Fig. 3.

Identification of conserved functional surface residues on Bud6core. (A) Surface conservation of Bud6core. Each residue is labeled with a color ranging from the most conserved (magenta) to the most variable (cyan) as analyzed with ConSurf (37). The eight residues that were mutated for biochemical analysis are indicated on the surface in the zoomed-in view. The two identical central patches are contiguous; residues contributing to these patches are distinguished by the use of prime symbols in their labels. (B) Concentration-dependent effects of WT and mutant c-Bud6 polypeptides on Bni1-mediated actin assembly. Monomeric actin (2 μM, 2.5% pyrene-labeled) was polymerized in the presence of 10 nM Bni1 (FH1-FH2-C) and variable concentrations of c-Bud6 as indicated. Each data point is an average of at least two trials in which the maximum rate of actin assembly over the course of the reaction was determined. All values were normalized to the rate of actin assembly occurring in the presence of 10 nM Bni1 alone. The Kapp value for each mutant was calculated by determining the concentration of mutant c-Bud6 required to increase the rate of Bni1-mediated assembly to half of the maximal rate observed for WT c-Bud6.
We designed a series of point mutants in the central and distal patches to test the role of these surfaces in enhancing the actin assembly activity of Bni1. To accomplish this, we prepared c-Bud6 constructs (residues 550–788) containing individual mutations in the central (K632A, E636A, L639A, and Q646A) or distal (Q581A, D582A, E585S, and D662A) patch and tested their abilities to stimulate Bni1-mediated actin assembly using bulk pyrene-actin assembly assays. For WT c-Bud6, we observed a sixfold increase in the rate of Bni1-mediated actin assembly, with a half-maximal stimulatory effect at 10 nM c-Bud6 (Fig. 3B). Two of the four mutants in the central conserved patch (K632A and L639A) were markedly defective in stimulating Bni1. We note that the K632A was able to induce a WT level of stimulation, but at much higher concentrations (more than 10-fold greater than that required with WT c-Bud6). In contrast, the L639A mutant showed much less stimulation of Bni1 activity than WT, even at the highest concentration tested. Only one of the mutations in the distal conserved patch (E585S) showed a significant defect in promoting Bni1-mediated actin assembly; however, it was still able to induce a WT-like level of stimulation at the highest concentration tested. The remaining three distal patch mutants were only mildly impaired in this assay (Q581A, D582A, and D662A; Fig. 3B). To assess whether the decreased actin assembly observed with the c-Bud6 mutants stemmed from a decrease in nucleation, we calculated the concentration of filaments present at 50% polymerization in reactions containing 200 nM of each of the c-Bud6 fragments. The three c-Bud6 mutants strongly impaired in Bni1 stimulation (E585S, K632A, and L639A) showed a significant reduction in the concentration of filaments produced compared with reactions containing WT c-Bud6, confirming that the mutants specifically disrupt the nucleation-promoting activity of c-Bud6 (Fig. S5 A and B).
Mutations in the Bud6 Core Domain Specifically Impair Binding to Bni1.
Our bulk pyrene-actin assembly assays show that the core domain of Bud6 is directly involved in stimulating actin assembly via Bni1. We considered that the defects associated with the mutations might arise from (i) weakening of Bud6’s affinity for Bni1; (ii) a neomorphic effect of the mutations; or (iii) interference with some other, as yet undefined, aspect of its nucleation-promoting mechanism. To explore this issue further, we used bulk actin assembly assays to measure the ability of Bud6core (either WT or that containing one of the eight point mutations described above) to compete with c-Bud6 for binding to Bni1 FH1-COOH. Because Bud6core does not bind G-actin (Fig. 1F), we reasoned that Bud6core binding to Bni1 FH1-COOH should block c-Bud6–mediated delivery of G-actin to Bni1. As expected, WT Bud6core displayed a concentration-dependent ability to attenuate the stimulatory effect of c-Bud6 on Bni1 (Fig. 4A). Of the eight Bud6 point mutants we tested, the three that were defective in stimulating Bni1 activity when introduced into c-Bud6 (E585S, K632A, and L639A) also showed defects, when introduced into Bud6core, in competing with c-Bud6 for binding to Bni1 (Fig. 4B). Because none of the c-Bud6 mutants showed obvious defects in interacting with G-actin (Fig. S6 A and B), we conclude that the conserved patches within Bud6core specifically mediate the interaction of Bud6 with Bni1 to promote actin assembly. In addition, a previous mutagenesis study of Bud6 based on primary sequence conservation identified triple-alanine mutants that disrupted binding to Bni1; these mutations also map to the central conserved patch on Bud6core (30). Collectively, these mutational data show that both the distal and central conserved patches mediate binding to Bni1.
Fig. 4.

Competition of Bud6core and c-Bud6 polypeptides for binding to Bni1 (FH1-FH2-C). (A) Monomeric actin (2 μM) was assembled in the presence of 10 nM Bni1 (FH1-FH2-C), 100 nM WT c-Bud6, and indicated concentrations of WT Bud6core. (B) Reactions were performed as in A, except using the indicated mutant Bud6core polypeptides. Each data point represents a single trial, where the slope of the raw curve was measured at 50% polymerization. Red datasets indicate mutations that impair the NPF activity of c-Bud6 (Fig. 3B); blue datasets indicate mutations in c-Bud6 that were pseudo-WT for stimulating Bni1.
Bud6core Inhibits Bni1-Mediated Actin Assembly.
The ability to bind both Bni1 and G-actin is critical for the function of Bud6, and Bud6 enhances nucleation by recruiting actin monomers to the Bni1/Bud6 complex (30). As in many formins, the isolated FH2 domain of Bni1 is competent to nucleate filaments, but its nucleation activity is enhanced strongly by inclusion of additional C-terminal tail sequences that encompass the autoregulatory DAD domain (18). Because the Bud6 binding site has been mapped to sequences overlapping with the DAD domain, we asked what effect Bud6core would have on nucleation by Bni1 FH1-FH2-C. We found that increasing concentrations of Bud6core inhibited actin assembly but that inhibition plateaued at ∼40% of the activity of Bni1 alone (Fig. 5 A and B). The residual activity was comparable to that expected for the isolated FH2 domain (18); thus, the observed inhibition by Bud6core likely stems from blocking of the actin monomer binding site in the Bni1 C terminus. The apparent inhibition constant Kapp is 29 nM (Fig. 5B), similar to the apparent affinity constant for stimulation of assembly by c-Bud6, confirming that Bud6core mediates binding to Bni1 with little or no contribution from the flank region.
Fig. 5.

Bud6core domain inhibits Bni1-mediated actin assembly. (A) Monomeric actin (2 μM, 5% pyrene-labeled) was assembled in the presence of 10 nM C-Bni1 (FH1-FH2-C) and a range of concentrations of Bud6core domain. AU, arbitrary units; These are the raw fluorescence readings. (B) Concentration-dependent effects of Bud6core domain on the rate of C-Bni1–mediated actin assembly. The percentage of activity was determined from the slopes of the raw curves as in A. The slope for C-Bni1 in the absence of Bud6 was defined as 100% activity. Each data point is an average of two independent trials.
Bni1 Dimer Binds Two c-Bud6 Dimers, and Each Bud6flank Binds One Actin.
The dimeric structure of c-Bud6 raises the obvious question of the stoichiometry of the binding interaction with Bni1, which is also a dimer. To determine the stoichiometry of the complex, we titrated a fixed amount of Bud6core with increasing ratios of Bni1-FH2C and analyzed the mixtures by SEC (Fig. 6A). A molar ratio of 0.5 was sufficient to eliminate the peak corresponding to free Bud6core, suggesting that one Bni1 dimer binds two Bud6core dimers. Consistent with this hypothesis, analysis of the c-Bud6/Bni1-FH2C complex by SEC-MALS revealed a molar mass of ∼240 kDa (Fig. 6B), corresponding to a 2:1 complex of c-Bud6 and Bni1-FH2C dimers. Similarly, SEC-MALS analysis of the slightly smaller Bud6core/Bni1-FH2C complex revealed a molar mass consistent with that expected for a 2:1 complex (expected mass of ∼208 kDa, measured molar mass = 195 kDa; Fig. S7A). Finally, titration of Bni1-FH2C with c-Bud6 in a native gel shift assay revealed a two-step migration shift indicative of two independent binding sites for c-Bud6 on the Bni1 dimer and a 2:1 complex at saturation (Fig. S7 B and C).
Fig. 6.

Stoichiometry of Bud6 interaction with Bni1 and G-actin. (A) Analysis of Bni1-FH2C/Bud6core mixtures by SEC. Bud6core alone, or with addition of a 0.25 or 0.5 molar ratio of Bni1-FH2C as indicated, was analyzed by SEC on a Superdex 200 column. Note that a 0.5 ratio of Bni1-FH2C was sufficient to ablate the free Bud6core peak (blue trace). Quantitation of Bud6core and Bni1-FH2C in the peak eluting at ∼11 mL by densitometry of the elution fractions resolved by SDS/PAGE confirmed a 2:1 ratio of Bud6core to Bni1-FH2C. (B) SEC-MALS analysis of c-Bud6/Bni1-FH2C indicated a molar mass of ∼240 kDa. Molar mass (black) and refractive index (red) are plotted vs. elution volume from a Superdex 200 size-exclusion column. A 2:1 complex of c-Bud6/Bni1 dimers has an expected mass of ∼250 kDa. (C) Analysis of Bud6 binding to Bni1tail by ITC. (Left) Titration of Bud6core with Bni1tail yielded a stoichiometry of n = 0.48 and Kd = 0.75 μM. (Right) Titration of c-Bud6 with Bni1tail produced similar results (n = 0.49 and Kd = 1.1 μM). (D) Native-PAGE analysis indicating that Bud6flank forms a 1:1 complex with G-actin. The amount of G-actin was fixed, and the amount of Bud6flank was gradually increased by the molar ratio indicated. (E) SDS/PAGE analysis of bands from a Bud6flank/G-actin native-PAGE gel shift assay. The upper bands from Bud6flank/G-actin molar ratios of 1 and 2 in D were excised, combined, and resolved on a 20% SDS/PAGE denaturing gel. (F) Native-PAGE analysis indicates that c-Bud6 forms a 1:1 complex with G-actin. A fixed amount of c-Bud6 was combined with an increasing molar ratio of G-actin as indicated and analyzed on 20% native gel. All protein concentrations were determined by amino acid analysis.
We further characterized the Bni1/Bud6 interaction using ITC. Titration of c-Bud6 or Bud6core with the Bni1tail protein revealed a binding stoichiometry of ∼0.5 for both Bud6 constructs (Fig. 6C), indicating that the Bud6 dimer engages a single Bni1 tail. We attempted to analyze binding of Bud6 to Bni1-FH2C with ITC, but we were unable to achieve concentrations of the Bni1-FH2C protein sufficient to yield a reliable calorimetric signal on titration with either c-Bud6 or Bud6core.
We also used a native gel shift approach to study binding of the Bud6flank domain to G-actin. Titration of a fixed concentration of G-actin with increasing ratios of Bud6flank indicated a binding stoichiometry of 1:1 (Fig. 6D). As noted above, G-actin and Bud6flank alone migrate in a similar position on native-PAGE gel shift assay, but the complex shifts to a slower migrating position. This shift was complete at a stoichiometry of 1:1, and SDS/PAGE analysis of the excised shifted band confirmed that it contains both species (Fig. 6E). Because each Bud6 dimer has two flank domains and each flank binds a single G-actin molecule, we expect that each Bud6 dimer binds two molecules of G-actin. Indeed, native gel shift analysis shows that when G-actin and c-Bud6 reach a molar ratio of 1:1, the faster migrating c-Bud6 band is completely shifted to a slower migrating position (Fig. 6F, lanes 3–5).
Bud6 and p160rock Do Not Share a Conserved Formin-Binding Domain.
Although there are no established homologs of Bud6 in higher eukaryotes, we and others have previously noted a region of sequence similarity between c-Bud6 and the Rho kinase p160rock (23, 38). This region corresponds to the Bud6core domain and a portion of the flank (residues 517–753) that is 22% identical and ∼46% similar to a 234-residue region of p160rock. Because of the unique sequence patterns associated with coiled-coil type proteins, they can exhibit considerable sequence similarity, irrespective of evolutionary or functional relatedness (34). Nevertheless, given the extended length of this region of similarity and the fact that p160rock was recently shown to bind the formin Fhod1 (39), we sought to determine whether this region represented a structural and functional domain conserved between Bud6 in fungi and p160rock in higher eukaryotes. We expressed, crystallized, and determined the structure of a portion of human p160rock that aligns in primary sequence to the Bud6core domain (40). The crystallized regions are 17% identical over 166 residues. The p160rock structure is shown side by side with Bud6core (Fig. S8). Unlike Bud6core, the p160rock domain is a simple, parallel, dimeric coiled-coil. We conclude that there is no apparent structural or functional relationship between these domains, despite their considerable similarity in primary amino acid sequence.
Discussion
Structural studies are crucial for elucidating in detail the mechanisms by which formin proteins and their binding partners assemble actin structures. The yeast formin Bni1 has been a centrally important model for investigation of formin structure and function; studies of Bni1 yielded the discovery of the actin assembly activity of formins (41, 42), the crystal structure of the FH2 domain dimer (12), and the structure of an FH2/actin complex (13). In this study, we provide structural information for Bud6, which serves as an NPF for Bni1 (30). We functionally dissected the C-terminal half of Bud6, in which this NPF activity is vested, into two defined regions: a Bni1-binding core domain (Bud6core, residues 550–688) and an actin-binding flank region (Bud6flank, residues 689–788). The high-resolution crystal structure of the Bud6core domain revealed an elongated (116 Å) rod-shaped dimer with an overall architecture reminiscent of a triple-helical coiled-coil. The Bud6 domain is twofold symmetrical, and we find that conserved residues on the surface of the domain map to three regions: two identical distal patches near each end of the rod and a symmetrical central patch. We identified residues in these conserved surfaces that are important for Bud6 enhancement of actin nucleation by Bni1: two from the central patch (K632A and L637A) and one from the distal patch (E585S). Further, our data indicate that these surfaces on the Bud6core mediate direct binding to Bni1, although they do not exclude the possibility that they also have other mechanistic roles in promoting nucleation.
Bud6 is known to bind to a region partially overlapping the DAD domain at the C terminus of the Bni1 FH2 domain (31). To aid in exploring the relative dimensions and spatial relationships of Bni1, actin, and Bud6, we created a schematic model of their interactions using available structural information (Fig. 7). In addition to the present structure of the Bud6 core domain, we considered the Bni1 FH2 domain dimer and the Bni1 FH2 in complex with actin (13), which defines the interaction of the formin with actin subunits. The latter also reveals a filament-like orientation of actin subunits, which represents a plausible filament nucleus. These components are drawn approximately to scale and with sites of interaction closely representing those revealed in crystal structures. At present, no structural information is available for Bni1tail and its interaction with Bud6core or for the Bud6flank in complex with actin. Thus, these contacts are drawn based on biochemical data. As illustrated, dimeric Bni1, in association with a nascent actin filament, would be expected to present two widely spaced binding sites for Bud6 oriented toward the pointed end of the nucleating filament. The Bud6-binding sites in Bni1 (labeled Bni1tail) lie at the end of the long αT helix, apparently allowing binding to Bud6 without steric interference. Our binding studies establish a 1:2 stoichiometry of binding; that is, the Bni1 dimer binds two Bud6 dimers (Fig. 6), with each Bni1 tail engaging one Bud6 dimer. Simultaneous engagement of both Bni1 tails by a single Bud6 dimer would position the Bud6core domain across the FH2 ring and would be expected to block elongation and interfere with actin binding to the FH2 domain. This is clearly inconsistent with our observed enhancement of Bni1 nucleation by Bud6. Additionally, the Bud6 dimer does not span between Bni1 dimers, because SEC-MALS of the Bni1/Bud6 complex reveals a homogeneous peak with a molar mass consistent with the proposed 1:2 stoichiometry (Fig. 6B). To date, we have been unable to obtain crystals of a Bni1/Bud6 complex.
Fig. 7.

Structurally informed model for collaboration between Bud6 and Bni1 in actin filament nucleation. The figure is drawn based on the structural and binding data presented here, as well as structures of Bni1 and its complex with actin (12, 13). The two sides of the formin dimer are shown in green and purple; actin subunits are colored yellow, orange, gray, and cyan; and the Bud6 dimer is shown in yellow and red. The Bni1 FH2 dimer binds two Bud6 dimers, which, in turn, bind a total of four actin subunits to assemble a nucleation seed with Bni1. It is important to note that the number of actin subunits required for productive nucleation by Bni1 and Bud6 remains to be determined and the depiction of which actin subunits are coordinated by the Bud6 flank domains is hypothetical. It is possible that the Bud6flank dissociates from actin on formation of a stable nucleus, because c-Bud6 binds to G-actin but not F-actin. Additionally, it is unclear whether Bud6 and Bni1 can simultaneously engage the same actin subunit, because the Bud6flank binding site on actin remains to be elucidated.
How might Bud6 present actin monomers to Bni1 to facilitate nucleation? The 116-Å length of the Bud6core domain easily spans the length of two actin subunits in the longitudinal direction of an actin filament, and the actin-binding Bud6flank segments extend from either end of the core domain. Therefore, it is tempting to propose that four actin monomers could be brought into register to form a cross-filament seed by the action of two Bud6 dimers in association with the Bni1 dimer (Fig. 7). It is interesting to note that Bud6 is expected to be positioned toward the pointed end of the filament; this organization may allow it to facilitate nucleation while not interfering with elongation at the barbed end, which depends on recruitment of profilin-actin by the FH1 domain. Further study will be required to establish whether the Bni1/Bud6 complex remains intact during elongation or whether it must dissociate to allow processive capping by Bni1.
All known actin nucleators or their NPFs can recruit multiple actin monomers, either because they harbor tandem actin monomer-binding motifs or via oligomerization, which brings several single actin monomer-binding sites together (43). The Arp2/3 complex is a unique example in having two actin-related subunits that are believed to mimic two actin subunits and binding two molecules of WASp, each of which brings in at least one conventional actin subunit to spark formation of a nucleus (7, 44). Another unusual example is the bacterial type III-secreted effector protein TRAP, expressed in Chlamydia trachomatis, which contains only a single WH2 domain but oligomerizes to promote actin nucleation in vitro (45). More typical are the NPFs with multiple actin monomer-binding motifs, such as Spire, Lmod, Cobl, JMY, and possibly APC. Among these NPFs, Spire is perhaps the most studied as an actin assembly factor. In addition to a series of four WH2 motifs, Spire contains an N-terminal KIND domain that mediates binding to the C-terminal tail of its cognate formin Cappuccino (21). The four WH2 domains in Spire are closely spaced and can create a linear arrangement of monomers in one strand but may also promote lateral actin subunit interactions (46). Irrespective of whether lateral interactions arise in association with a single Spire molecule, two Spire molecules can bind to one Cappuccino dimer, which could catalyze formation of a two-stranded seed. Indeed, the formin-dimerized Spire is a much more potent nucleator.
Despite their many differences and a lack of detectable structural or evolutionary relatedness, Spire, Bud6, and APC share functional and mechanistic similarities. Each appears to cooperate with its respective formin in assembling specific structures; Spire and Cappuccino are both required for proper assembly of an actin mesh in the course of oocyte development (47), whereas Bud6 augments actin cable assembly by Bni1 and APC interacts with Diaphanous to promote pseudocleavage furrow formation and extension in Drosophila embryos (48). Spire, Bud6, and APC each bind just C-terminal to the FH2 domain of their respective formin, likely blocking the actin-monomer binding ability of this portion of the formin but apparently superseding it functionally by contributing the ability to bind multiple actin monomers. Dimeric Bud6 is expected to recruit a total of four actin monomers in association with the Bni1 dimer, whereas Spire could array as many as eight actin subunits with its multiple WH2 domains in association with Cappuccino dimer. APC dimerizes, like Bud6, and then binds at least four actin monomers with high affinity (49, 50). There is no known homolog of Bud6 in higher eukaryotes, but the mechanistic similarities among Bud6, Spire, and APC suggest that potential formin NPFs need not share similar sequences or structures. The combination of a formin tail-binding domain with the ability to bind at least two actin monomers may suffice. It will be interesting to see if additional formin NPFs that satisfy these two criteria can be identified.
Materials and Methods
Protein Preparation.
Bud6 (residues 550–788, 550–688, and 699–788) and Bni1 (residues 1794–1837) were all expressed as N-terminal GST-tobacco etched virus (TEV)–tagged fusion proteins using a modified pET-30 vector. To express Bud6 (residues 550–688), the plasmid was transformed into Rosetta2(DE3) cells (Novagen) and grown at 37 °C to OD600 = 0.5. The temperature of the culture was then shifted to 20 °C, and cells were induced with 0.25 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for 15 h. Cells were lysed by sonication in lysis buffer (1× PBS, 5 mM DTT, 2 mM EDTA, 1 mM PMSF) and cleared by high-speed centrifugation. The supernatant was incubated with glutathione Sepharose resin (GE Healthcare) for 3 h at 4 °C and washed, and the protein was eluted with elution buffer [50 mM Tris (pH 8.0), 150 mM NaCl, 5 mM DTT, 10 mM glutathione]. TEV protease was added to remove the GST tag at 4 °C overnight. Bud6 (residues 550–688) was depleted of TEV and further purified by anion-exchange chromatography at pH 7.5. The purified protein was dialyzed against protein buffer [20 mM Hepes (pH 7.5), 100 mM NaCl, 5 mM DTT], concentrated to 10 mg/mL, and stored at −80 °C. SeMet protein was obtained as above, except for the following: SeMet was incorporated by metabolic inhibition (51), and 10 mM DTT was added in all buffers.
Bud6 (residues 550–788) E. coli cells were grown at 30 °C after induction and induced with 0.5 mM IPTG for 5 h. Bni1 (residues 1348–1953) was expressed as an N-terminal 6× His-TEV–tagged fusion protein. Its expression condition was similar to that of Bud6 (residues 550–788).
Rabbit skeletal muscle actin was purified (52) and labeled with pyrenyliodoacetamide as described (53, 54). Bni1 (FH1-FH2-C) was expressed in Saccharomyces cerevisiae and purified as described (55).
Crystallization, Data Collection, and Processing.
Initial screens of crystallization conditions were carried out using 96-well format Index and Crystal Screen HT kits (Hampton Research) on a Phoenix robot (Art Robbins Instruments). After optimization, crystals of Bud6 (residues 550–688) were grown by using hanging-drop vapor diffusion at 20 °C. SeMet Bud6 protein was crystallized by mixing 2 μL of 10 mg/mL protein with an equal volume of mother liquid consisting of 9% (wt/vol) PEG 3350, 85 mM sodium malonate (pH 7.0), 10 mM DTT, and 25% (vol/vol) glycerol, and it was equilibrated against 0.5 mL of mother liquor. Crystals grew to full size in 2 d and were directly frozen out of mother liquor into liquid nitrogen. All datasets were collected at the Northeastern Collaborative Access Team 24ID-C beamline at Argonne National Laboratory. A SAD dataset was collected at the peak wavelength in a single 180° rotation. A much higher resolution dataset was collected on a native protein crystal grown under conditions consisting of 12% PEG 3350, 175 mM sodium malonate (pH 7.0), 5 mM DTT, and 20% glycerol. Crystals were rods instead of plates, as were those of the SeMet protein, and took 2 wk to grow. Integration, scaling, and merging of the diffraction data were performed with HKL2000 (56). A summary of the data collection statistics is given in Table 1.
Structure Determination.
The substructure determination, phasing, and density modification were performed using autoSHARP (57). For the SAD dataset, five (of six) selenium sites were identified, and the resulting density modified map had clear electron density for most residues. An almost complete structure was manually built into the map using Coot (58) and was initially refined to an Rfree value of 0.29 with crystallography and NMR system (59). The refinement consisted of alternating rounds of torsion angle molecular dynamics simulated annealing, individual restrained thermal factor refinement, and model building in Coot. The resulting coordinates were used as a search model for molecular replacement to solve crystal form 2 using Phaser (60). Of the two Bud6 dimer chains, chain A gave a much better solution than chain B (Z-score: 19.4 vs. 13.1), and the resulting model was rebuilt using ARP/wARP (61). All final refinements were performed using PHENIX (62). The final model of the SAD dataset contains residues 555–679 for each chain of the dimer. Model statistics are shown in Table 1. The atomic coordinates and diffraction data have been deposited in the Protein Data Bank [PDB ID codes 2OKQ (native) and 2ONX (SAD)].
Native Gel Analysis.
Bni1-FH2C (residues 1348–1953), c-Bud6 (residues 550–788), Bud6flank (residues 699–788), and G-actin were subjected to amino acid analysis to determine their concentrations. Bni1-FH2C at a concentration of 12 μM (all concentrations are final) was mixed with different concentrations of c-Bud6 at 3.3 μM, 6.6 μM, 13.1 μM, and 26.2 μM. The reactions were incubated on ice for 10 min and then fractionated on either 7.5% or 8–25% gradient native gel (PhastGel; GE Healthcare). For titration of Bud6 flank with G-actin, Bud6 flank was buffer-exchanged into G-buffer first. Then, 30 μM G-actin was mixed with Bud6 flank at 7.4 μM, 14.9 μM, 30 μM, and 59.3 μM. Twenty percent homogeneous native gel (PhastGel) was used.
Trypsin Digest of Bud6.
Purified c-Bud6 (residues 550–788) was diluted to 2 mg/mL, to which trypsin was added at a mass ratio of 1:2,500 at 4 °C. Samples were removed for SDS/PAGE analysis every hour. c-Bud6 was reduced to Bud6core (residues 552–688) in 1 h and was stable up to 9 h.
MALS Analysis of Bud6 and Bud6/Bni1 Complex.
Purified c-Bud6 (residues 550–788) or Bud6core was separated on a Superdex 200 column (GE Healthcare) preequilibrated with 20 mM Hepes (pH 7.5), 150 mM KCl, and 2 mM Tris(2-carboxyethyl)phosphine (TCEP), and connected to a Wyatt miniDAWN TREOS three-angle light scattering detector and Optilab-rEX refractive index detector. MALS analysis of c-Bud6/Bni1-FH2C complex was done in a slightly different buffer: 20 mM Tris (pH 8.0), 100 mM NaCl, and 4 mM DTT.
Titration of Bud6core with Bni1-FH2C on SEC.
A Superdex 200 column was preequilibrated with 20 mM Tris⋅HCl, 100 mM NaCl, and 2 mM TCEP, and the elution profiles of a 6-nmol (100 μg) aliquot of Bud6core alone or with different molar ratios of Bni1-FH2C were measured by SEC. To prepare the complexes, 6 nmol (100 μg) of Bud6 core protein was mixed with 1.5 nmol (103.9 μg, 0.25-fold) or 3 nmol (207.8 μg, 0.5-fold) of Bni1-FH2C protein. After incubation at 4 °C for 30 min, the mixtures were loaded onto a Superdex 200 GL 10/300 (GE Healthcare).
ITC.
ITC experiments were performed on an ITC200 (GE Healthcare). Bni1 tail (residues 1794–1837), c-Bud6 (residues 550–788), and Bud6core (residues 550–688) were dialyzed extensively against a buffer containing 50 mM Tris⋅HCl, 150 mM NaCl, and 2 mM TCEP. The reference power was set to 6 μCal/s, and the cell contents were stirred continuously at 250 rpm throughout the titrations. Heat changes were monitored as Bni1tail was titrated into c-Bud6 (67 µM) and Bud6core (64 µM) proteins. Bni1tail at a concentration of 1.2 mM was titrated into the Bud6 protein in a series of 24 injections of 17.75 µL each injection at 25 °C, with a 3-min delay between each injection. A binding isotherm was generated by plotting the heat change evolved per injection against the molar ratio of Bni1tail to c-Bud6 and Bud6 core. Titration curves were fit by a single binding site model using Origin software (version 7; ORIGINLab).
In Vitro Actin Assembly Assays.
Gel-filtered monomeric actin (2 µM final, 2.5% pyrene-labeled) in G-Buffer [10 mM Tris (pH 8.0), 0.2 mM ATP, 0.2 mM CaCl2, 0.2 mM DTT] was converted to Mg-ATP-actin 2 min immediately before use in reactions. A total of 47 μL of actin was added to 10 μL of control buffer or proteins in the same buffer and 3 μL of 20× initiation mix (40 mM MgCl2, 10 mM ATP, 1 M KCl). Pyrene fluorescence was monitored at an excitation of 365 nm and emission of 407 nm at 25 °C in an Infinite M200 plate reader (Tecan). Rates of assembly were calculated from slopes of the curves at ∼50% polymerization. In cases in which the density of filaments produced was determined, previously described methods were used to calculate filament concentrations (63), where the rate of Bni1-mediated elongation was assumed to be 5.3 subunits per second (15). For suppression of spontaneous nucleation assays, 4 μM monomeric actin (2.5% pyrene-labeled) was used.
Supplementary Material
Acknowledgments
We thank the beamline staff at the Northeastern Collaborative Access Team at the Advanced Photon Source, Argonne National Laboratory, for assistance with data collection. This work was supported, in part, by National Institutes of Health (NIH) Grants GM071834 (to M.J.E.) and GM083137 (to B.L.G.). The Northeastern Collaborative Access Team beamlines are supported by Award RR-15301 from the National Center for Research Resources at the NIH.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org [PDB ID codes 3OKQ (native) and 3ONX (SAD)].
See Author Summary on page 20178 (volume 109, number 50).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1203035109/-/DCSupplemental.
References
- 1.Chesarone MA, Goode BL. Actin nucleation and elongation factors: Mechanisms and interplay. Curr Opin Cell Biol. 2009;21(1):28–37. doi: 10.1016/j.ceb.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dominguez R. Actin filament nucleation and elongation factors—Structure-function relationships. Crit Rev Biochem Mol Biol. 2009;44(6):351–366. doi: 10.3109/10409230903277340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Campellone KG, Welch MD. A nucleator arms race: Cellular control of actin assembly. Nat Rev Mol Cell Biol. 2010;11(4):237–251. doi: 10.1038/nrm2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goode BL, Eck MJ. Mechanism and function of formins in the control of actin assembly. Annu Rev Biochem. 2007;76:593–627. doi: 10.1146/annurev.biochem.75.103004.142647. [DOI] [PubMed] [Google Scholar]
- 5.Sept D, McCammon JA. Thermodynamics and kinetics of actin filament nucleation. Biophys J. 2001;81(2):667–674. doi: 10.1016/S0006-3495(01)75731-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Robinson RC, et al. Crystal structure of Arp2/3 complex. Science. 2001;294(5547):1679–1684. doi: 10.1126/science.1066333. [DOI] [PubMed] [Google Scholar]
- 7.Padrick SB, Doolittle LK, Brautigam CA, King DS, Rosen MK. Arp2/3 complex is bound and activated by two WASP proteins. Proc Natl Acad Sci USA. 2011;108(33):E472–E479. doi: 10.1073/pnas.1100236108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ti SC, Jurgenson CT, Nolen BJ, Pollard TD. Structural and biochemical characterization of two binding sites for nucleation-promoting factor WASp-VCA on Arp2/3 complex. Proc Natl Acad Sci USA. 2011;108(33):E463–E471. doi: 10.1073/pnas.1100125108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boczkowska M, et al. X-ray scattering study of activated Arp2/3 complex with bound actin-WCA. Structure. 2008;16(5):695–704. doi: 10.1016/j.str.2008.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ahuja R, et al. Cordon-bleu is an actin nucleation factor and controls neuronal morphology. Cell. 2007;131(2):337–350. doi: 10.1016/j.cell.2007.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chesarone MA, DuPage AG, Goode BL. Unleashing formins to remodel the actin and microtubule cytoskeletons. Nat Rev Mol Cell Biol. 2010;11(1):62–74. doi: 10.1038/nrm2816. [DOI] [PubMed] [Google Scholar]
- 12.Xu Y, et al. Crystal structures of a Formin Homology-2 domain reveal a tethered dimer architecture. Cell. 2004;116(5):711–723. doi: 10.1016/s0092-8674(04)00210-7. [DOI] [PubMed] [Google Scholar]
- 13.Otomo T, et al. Structural basis of actin filament nucleation and processive capping by a formin homology 2 domain. Nature. 2005;433(7025):488–494. doi: 10.1038/nature03251. [DOI] [PubMed] [Google Scholar]
- 14.Mizuno H, et al. Rotational movement of the formin mDia1 along the double helical strand of an actin filament. Science. 2011;331(6013):80–83. doi: 10.1126/science.1197692. [DOI] [PubMed] [Google Scholar]
- 15.Kovar DR, Harris ES, Mahaffy R, Higgs HN, Pollard TD. Control of the assembly of ATP- and ADP-actin by formins and profilin. Cell. 2006;124(2):423–435. doi: 10.1016/j.cell.2005.11.038. [DOI] [PubMed] [Google Scholar]
- 16.Vavylonis D, Kovar DR, O’Shaughnessy B, Pollard TD. Model of formin-associated actin filament elongation. Mol Cell. 2006;21(4):455–466. doi: 10.1016/j.molcel.2006.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pring M, Evangelista M, Boone C, Yang C, Zigmond SH. Mechanism of formin-induced nucleation of actin filaments. Biochemistry. 2003;42(2):486–496. doi: 10.1021/bi026520j. [DOI] [PubMed] [Google Scholar]
- 18.Gould CJ, et al. The formin DAD domain plays dual roles in autoinhibition and actin nucleation. Curr Biol. 2011;21(5):384–390. doi: 10.1016/j.cub.2011.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chhabra ES, Higgs HN. INF2 Is a WASP homology 2 motif-containing formin that severs actin filaments and accelerates both polymerization and depolymerization. J Biol Chem. 2006;9(36):26754–26767. doi: 10.1074/jbc.M604666200. [DOI] [PubMed] [Google Scholar]
- 20.Heimsath EG, Jr, Higgs HN. The C terminus of formin FMNL3 accelerates actin polymerization and contains a WH2 domain-like sequence that binds both monomers and filament barbed ends. J Biol Chem. 2012;287(5):3087–3098. doi: 10.1074/jbc.M111.312207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quinlan ME, Hilgert S, Bedrossian A, Mullins RD, Kerkhoff E. Regulatory interactions between two actin nucleators, Spire and Cappuccino. J Cell Biol. 2007;179(1):117–128. doi: 10.1083/jcb.200706196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okada K, et al. Adenomatous polyposis coli protein nucleates actin assembly and synergizes with the formin mDia1. J Cell Biol. 2010;189(7):1087–1096. doi: 10.1083/jcb.201001016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moseley JB, et al. A conserved mechanism for Bni1- and mDia1-induced actin assembly and dual regulation of Bni1 by Bud6 and profilin. Mol Biol Cell. 2004;15(2):896–907. doi: 10.1091/mbc.E03-08-0621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Amberg DC, Zahner JE, Mulholland JW, Pringle JR, Botstein D. Aip3p/Bud6p, a yeast actin-interacting protein that is involved in morphogenesis and the selection of bipolar budding sites. Mol Biol Cell. 1997;8(4):729–753. doi: 10.1091/mbc.8.4.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feierbach B, Verde F, Chang F. Regulation of a formin complex by the microtubule plus end protein tea1p. J Cell Biol. 2004;165(5):697–707. doi: 10.1083/jcb.200403090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luedeke C, et al. Septin-dependent compartmentalization of the endoplasmic reticulum during yeast polarized growth. J Cell Biol. 2005;169(6):897–908. doi: 10.1083/jcb.200412143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shcheprova Z, Baldi S, Frei SB, Gonnet G, Barral Y. A mechanism for asymmetric segregation of age during yeast budding. Nature. 2008;454(7205):728–734. doi: 10.1038/nature07212. [DOI] [PubMed] [Google Scholar]
- 28.Jin H, Amberg DC. The secretory pathway mediates localization of the cell polarity regulator Aip3p/Bud6p. Mol Biol Cell. 2000;11(2):647–661. doi: 10.1091/mbc.11.2.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Delgehyr N, Lopes CS, Moir CA, Huisman SM, Segal M. Dissecting the involvement of formins in Bud6p-mediated cortical capture of microtubules in S. cerevisiae. J Cell Sci. 2008;121(Pt 22):3803–3814. doi: 10.1242/jcs.036269. [DOI] [PubMed] [Google Scholar]
- 30.Graziano BR, et al. Mechanism and cellular function of Bud6 as an actin nucleation-promoting factor. Mol Biol Cell. 2011;22(21):4016–4028. doi: 10.1091/mbc.E11-05-0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moseley JB, Goode BL. Differential activities and regulation of Saccharomyces cerevisiae formin proteins Bni1 and Bnr1 by Bud6. J Biol Chem. 2005;280(30):28023–28033. doi: 10.1074/jbc.M503094200. [DOI] [PubMed] [Google Scholar]
- 32.Holm L, Sander C. Protein structure comparison by alignment of distance matrices. J Mol Biol. 1993;233(1):123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- 33.Prag G, et al. The Vps27/Hse1 complex is a GAT domain-based scaffold for ubiquitin-dependent sorting. Dev Cell. 2007;12(6):973–986. doi: 10.1016/j.devcel.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Burkhard P, Stetefeld J, Strelkov SV. Coiled coils: A highly versatile protein folding motif. Trends Cell Biol. 2001;11(2):82–88. doi: 10.1016/s0962-8924(00)01898-5. [DOI] [PubMed] [Google Scholar]
- 35.Walshaw J, Woolfson DN. Socket: A program for identifying and analysing coiled-coil motifs within protein structures. J Mol Biol. 2001;307(5):1427–1450. doi: 10.1006/jmbi.2001.4545. [DOI] [PubMed] [Google Scholar]
- 36.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ashkenazy H, Erez E, Martz E, Pupko T, Ben-Tal N. ConSurf 2010: Calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010;38(Web Server issue):W529–W533. doi: 10.1093/nar/gkq399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martin SG, Rincón SA, Basu R, Pérez P, Chang F. Regulation of the formin for3p by cdc42p and bud6p. Mol Biol Cell. 2007;18(10):4155–4167. doi: 10.1091/mbc.E07-02-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hannemann S, et al. The Diaphanous-related Formin FHOD1 associates with ROCK1 and promotes Src-dependent plasma membrane blebbing. J Biol Chem. 2008;283(41):27891–27903. doi: 10.1074/jbc.M801800200. [DOI] [PubMed] [Google Scholar]
- 40.Tu D, et al. Crystal structure of a coiled-coil domain from human ROCK I. PLoS ONE. 2011;6(3):e18080. doi: 10.1371/journal.pone.0018080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sagot I, Rodal AA, Moseley J, Goode BL, Pellman D. An actin nucleation mechanism mediated by Bni1 and profilin. Nat Cell Biol. 2002;4(8):626–631. doi: 10.1038/ncb834. [DOI] [PubMed] [Google Scholar]
- 42.Evangelista M, Pruyne D, Amberg DC, Boone C, Bretscher A. Formins direct Arp2/3-independent actin filament assembly to polarize cell growth in yeast. Nat Cell Biol. 2002;4(1):32–41. doi: 10.1038/ncb718. [DOI] [PubMed] [Google Scholar]
- 43.Qualmann B, Kessels MM. New players in actin polymerization—WH2-domain-containing actin nucleators. Trends Cell Biol. 2009;19(6):276–285. doi: 10.1016/j.tcb.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 44.Padrick SB, et al. Hierarchical regulation of WASP/WAVE proteins. Mol Cell. 2008;32(3):426–438. doi: 10.1016/j.molcel.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jewett TJ, Fischer ER, Mead DJ, Hackstadt T. Chlamydial TARP is a bacterial nucleator of actin. Proc Natl Acad Sci USA. 2006;103(42):15599–15604. doi: 10.1073/pnas.0603044103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen CK, Sawaya MR, Phillips ML, Reisler E, Quinlan ME. Multiple forms of Spire-actin complexes and their functional consequences. J Biol Chem. 2012;287(13):10684–10692. doi: 10.1074/jbc.M111.317792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dahlgaard K, Raposo AA, Niccoli T, St Johnston D. Capu and Spire assemble a cytoplasmic actin mesh that maintains microtubule organization in the Drosophila oocyte. Dev Cell. 2007;13(4):539–553. doi: 10.1016/j.devcel.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Webb RL, Zhou MN, McCartney BM. A novel role for an APC2-Diaphanous complex in regulating actin organization in Drosophila. Development. 2009;136(8):1283–1293. doi: 10.1242/dev.026963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Okada I, Fujiki S, Iwase S, Abe H. Stabilization of actin filaments prevents germinal vesicle breakdown and affects microtubule organization in Xenopus oocytes. Cytoskeleton (Hoboken) 2012;69(5):312–323. doi: 10.1002/cm.21028. [DOI] [PubMed] [Google Scholar]
- 50.Breitsprecher D, et al. Rocket launcher mechanism of collaborative actin assembly defined by single-molecule imaging. Science. 2012;336(6085):1164–1168. doi: 10.1126/science.1218062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Van Duyne GD, Standaert RF, Karplus PA, Schreiber SL, Clardy J. Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J Mol Biol. 1993;229(1):105–124. doi: 10.1006/jmbi.1993.1012. [DOI] [PubMed] [Google Scholar]
- 52.Spudich JA, Watt S. The regulation of rabbit skeletal muscle contraction. I. Biochemical studies of the interaction of the tropomyosin-troponin complex with actin and the proteolytic fragments of myosin. J Biol Chem. 1971;246(15):4866–4871. [PubMed] [Google Scholar]
- 53.Higgs HN, Blanchoin L, Pollard TD. Influence of the C terminus of Wiskott-Aldrich syndrome protein (WASp) and the Arp2/3 complex on actin polymerization. Biochemistry. 1999;38(46):15212–15222. doi: 10.1021/bi991843+. [DOI] [PubMed] [Google Scholar]
- 54.Pollard TD, Cooper JA. Methods to characterize actin filament networks. Methods Enzymol. 1982;85 Pt B:211–233. doi: 10.1016/0076-6879(82)85022-2. [DOI] [PubMed] [Google Scholar]
- 55.Moseley JB, Maiti S, Goode BL. Formin proteins: purification and measurement of effects on actin assembly. Methods Enzymol. 2006;406:215–234. doi: 10.1016/S0076-6879(06)06016-2. [DOI] [PubMed] [Google Scholar]
- 56.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 57.Vonrhein C, Blanc E, Roversi P, Bricogne G. Automated structure solution with autoSHARP. Methods Mol Biol. 2007;364:215–230. doi: 10.1385/1-59745-266-1:215. [DOI] [PubMed] [Google Scholar]
- 58.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 59.Brunger AT. Version 1.2 of the Crystallography and NMR system. Nat Protoc. 2007;2(11):2728–2733. doi: 10.1038/nprot.2007.406. [DOI] [PubMed] [Google Scholar]
- 60.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Crystallogr D Biol Crystallogr. 2005;61(Pt 4):458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 61.Morris RJ, Perrakis A, Lamzin VS. ARP/wARP and automatic interpretation of protein electron density maps. Methods Enzymol. 2003;374:229–244. doi: 10.1016/S0076-6879(03)74011-7. [DOI] [PubMed] [Google Scholar]
- 62.Adams PD, et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Harris ES, Higgs HN. Biochemical analysis of mammalian formin effects on actin dynamics. Methods Enzymol. 2006;406:190–214. doi: 10.1016/S0076-6879(06)06015-0. [DOI] [PubMed] [Google Scholar]
