

Abstract
The steroid cholesterol is an essential component of eukaryotic membranes, and it functionally modulates membrane proteins, including G protein-coupled receptors. To reveal insight into how cholesterol modulates G protein-coupled receptors, we have used dynamic single-molecule force spectroscopy to quantify the mechanical strength and flexibility, conformational variability, and kinetic and energetic stability of structural segments stabilizing the human β2-adrenergic receptor (β2AR) in the absence and presence of the cholesterol analog cholesteryl hemisuccinate (CHS). CHS considerably increased the kinetic, energetic, and mechanical stability of almost every structural segment at sufficient magnitude to alter the structure and functional relationship of β2AR. One exception was the structural core segment of β2AR, which establishes multiple ligand binding sites, and its properties were not significantly influenced by CHS.
Keywords: atomic force microscopy, energy landscape, intermolecular and intramolecular interactions, proteoliposomes
The human β2-adrenergic receptor (β2AR) is one of the best-characterized G protein-coupled receptors (GPCRs). β2AR belongs to the class A GPCRs and is expressed in pulmonary and cardiac myocyte tissue (1, 2). Together with its close relative β1AR, β2AR senses adrenalin in bronchial vasculature and noradrenalin in cardiac muscle. The implication in a broad spectrum of diseases, like asthma or heart failure, makes β2AR a potential therapeutic target (3–7). Several crystal structures of β2AR have been determined during the last years (8–12), providing unique insights into the structure–function relationship of GPCRs.
Cellular membranes functionally modulate a large number of membrane proteins (13–18). Such functional modulation is facilitated by chemical and physical interactions between membrane proteins and phospholipids, sphingolipids, cholesterol, and other components of the cell membrane. Similar to many other membrane proteins, GPCRs are regulated by their hetero- and homooligomeric assembly and the membrane composition. Because the heterogeneous composition of cellular membranes changes dynamically, the functional state of GPCRs depends on the location in the cell membrane and the state of the cell (19, 20). The steroid cholesterol modulates chemical and physical properties of cellular membranes and plays a role in the dynamic formation of sphingolipid-enriched assemblies of lipids and membrane proteins. These metastable assemblies, or lipid rafts, can functionally regulate membrane proteins by different mechanisms (19). Indirect regulation of membrane proteins, including GPCRs, can be observed through the ability of cholesterol to modulate biophysical properties of a lipid bilayer (21), whereas the direct regulation of membrane proteins can occur through specific interactions (22–25). Although it is not completely understood how cells control the distribution of cholesterol and by which mechanisms cholesterol functionally regulates GPCRs, insights into these processes are of cell biological and pharmacological importance (26, 27).
Similar to other GPCRs, cholesterol modulates the physiological function of β2AR (26, 28). Furthermore, cholesterol and the more water-soluble cholesterol analog cholesteryl hemisuccinate (CHS) enhance the thermal stability of β2AR (26, 29). Additionally, cholesterol facilitates interactions between GPCRs and seems to be helpful in crystallizing β2AR (9). A recently published X-ray crystallography model of human β2AR showed cholesterol to fit into a shallow surface groove formed by transmembrane α-helices H1, H2, H3, and H4 (30). This structural model unraveled possible interactions between cholesterol and β2AR. However, understanding the extent to which the interactions established by cholesterol change kinetic, energetic, and mechanical properties of structural regions in the receptor requires additional insight.
Intra- and intermolecular interactions stabilizing membrane proteins can be quantified and localized using atomic force microscopy (AFM) -based single-molecule force spectroscopy (SMFS) (31, 32). SMFS enables the assignment of these interactions to individual secondary structure elements, such as transmembrane α-helices, β-strands, or polypeptide loops of membrane proteins embedded in lipid membranes. Measuring the kinetic response of the individual structural elements by dynamic SMFS (DFS) allows quantification of their unfolding free energy, kinetic stability, conformational variability, and mechanical flexibility (33). Both SMFS and DFS have been applied to characterize these parameters of various membrane proteins and follow how they change on changing their functional state or physiological environment (34–39).
In this work, we have reconstituted human β2AR into phospholipid bilayers in the presence and absence of the cholesterol analog CHS and used SMFS and DFS to quantify and localize the inter- and intramolecular interactions established in β2AR. CHS is a more water-soluble cholesterol ester, and it is much easier to handle for in vitro studies than cholesterol, which is one main reason that CHS is widely used in structural biology and biophysical studies as the cholesterol analog. Because CHS and cholesterol are structurally quite similar, they are supposed to show similar properties in interacting and functionally modulating membrane proteins, such as GPCRs (29, 40, 41). Accordingly, GPCRs are frequently characterized in the presence of CHS, and the structural and functional results are interpreted as being obtained in the presence of cholesterol (40–44). To correlate our SMFS measurements with complementary structural and functional insights revealed of β2AR reconstituted into liposomes containing CHS (29, 30, 40), we characterized β2AR reconstituted into lipid membranes in the presence and absence of the cholesterol analog CHS. This approach allows a precise correlation of the CHS-induced interactions quantified by SMFS and provides unique insight into how cholesterol modulates kinetic, energetic, and mechanical properties of structural regions of β2AR.
Results
SMFS of Human β2AR in the Presence and Absence of Cholesterol.
To characterize the influence of cholesterol on the interactions stabilizing human β2AR, we reconstituted the receptor into liposomes containing phospholipids [1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC)] or phospholipids and CHS (DOPC/CHS) (Materials and Methods). For SMFS, the proteoliposomes were densely adsorbed onto freshly cleaved mica (Fig. S1). The AFM stylus was approached and pushed onto the proteoliposomes with a force of ∼700 pN for 0.5 s (Fig. 1A). Subsequently, the cantilever was retracted, and its deflection was recorded as a force-distance (F-D) curve. In ∼0.5% of all approach-and-retract cycles (n = 300,000), a single β2AR attached to the AFM stylus and the F-D curve recorded while retracting the cantilever showed a sawtooth-like pattern of force peaks. These sawtooth-like patterns were similar for β2AR reconstituted in DOPC (Fig. 1B, Upper) and DOPC/CHS (Fig. 1C, Upper) liposomes. Each force peak of an F-D curve reflected the unfolding of a structural segment of β2AR. The magnitude of the force peak revealed the strength of the interaction that stabilized a structural segment against unfolding. These interactions were composed of inter- and intramolecular interactions. For analysis, we selected only F-D curves with an overall length of ∼70–90 nm, because they describe the complete unfolding of the receptor from its terminus (Materials and Methods). To highlight common features among the F-D curves, they were superimposed and displayed as density plots (Fig. 1 B and C, Lower). The superimpositions of F-D curves recorded of β2AR reconstituted in DOPC (Fig. 1B) and DOPC/CHS (Fig. 1C) showed a characteristic pattern of eight force peaks. The presence of the cholesterol analog CHS did not change the positions of the force peaks. However, the magnitude of the force peaks increased in presence of CHS (Fig. 1 and Fig. S2). This difference implies that the cholesterol analog increases the strength of interactions stabilizing β2AR.
Fig. 1.

SMFS of β2AR reconstituted into liposomes composed of either phospholipids (DOPC) or phospholipids and cholesterol (DOPC/CHS). (A) Pressing the AFM stylus onto the proteoliposomes promotes the unspecific attachment of a single β2AR polypeptide to the stylus. Withdrawal of the AFM cantilever stretches the polypeptide and induces the sequential unfolding of β2AR. (B and C) Selection of F-D curves recorded on N-terminal unfolding of β2AR reconstituted into DOPC (B, Upper) and DOPC/CHS (C, Upper) liposomes. Density plots of superimposed F-D curves (B, Lower and C, Lower) highlight their common features. Number of superimposed F-D curves: n = 100 (B) and n = 100 (C). Red numbers on top of each WLC curve (red dashed lines) indicate the average contour lengths (in amino acids) revealed from fitting each force peak of each superimposed F-D curve. Gray scale bars allow evaluation of how frequently individual force peaks were populated.
Mapping Interactions That Stabilize Structural Segments of β2AR.
After having identified that the superimposed F-D spectra (Fig. 1 B and C, Lower) correspond to the unfolding of β2AR from the N-terminal end (SI Text and Figs. S3 and S4), we mapped the interactions to the β2AR structure. When exerting sufficient force to the N-terminal end, β2AR unfolds in a sequence of steps. Every unfolding step is reported by a force peak of the F-D curve (Fig. 1B). An unfolding step, in which a stable structural segment unfolds, describes the transfer of one unfolding intermediate to the next intermediate (31). To assign the unfolding steps and stable structural segments, every force peak of an F-D curve was fitted using the worm-like chain (WLC) model (Materials and Methods). Each WLC fit revealed the contour length of the unfolded polypeptide that connected the AFM stylus and the unfolding intermediate of the receptor. The contour lengths of all force peaks allowed determination of all unfolding steps of β2AR (Fig. 1 B and C and Table S1). In the first unfolding step, the N terminus and the N-terminal transmembrane α-helix of β2AR unfolded. Next, the unfolded polypeptide linking the AFM stylus and the stable structural segments that remained folded and anchored in the membrane was elongated and stretched. As soon as the stretching force exceeded the stability of the next structural segment, this structural segment unfolded as well. This sequential unfolding of one stable structural segment after the other continued until the entire β2AR has been unfolded. In summary, the eight force peaks detected eight unfolding steps, each reflecting the unfolding of a stable structural segment. Mapped onto the secondary structure, these stable structural segments show where inter- and intramolecular interactions stabilized β2AR (Fig. 2). Because the common unfolding peaks detected for β2AR in the presence and absence of CHS showed no differences in their position (Fig. 1), we can conclude that CHS did not stabilize different structural segments (Fig. 2). However, apparently, the strength of the interactions stabilizing β2AR depended on the presence of CHS (Fig. S2). In the next section, we investigate the nature of these interactions.
Fig. 2.

Structural segments stabilizing human β2AR. Secondary (A) and tertiary (B) structure models of β2AR. Each color represents a structural segment that is stabilized by inter- and intramolecular interactions. (A) Black amino acids highlight the end of the previous and the beginning of the next stable structural segment. This position corresponds to the mean contour length (given in parentheses) revealed from WLC curves fitting the force peaks common in every F-D curve. Amino acids colored at less intensity give the SD of locating the average force peak (Table S1). Membrane compensation (Materials and Methods) was applied for the boundaries of stable structural segments that had to be assumed to be within the membrane or at the membrane surface opposite to the puling AFM stylus. All seven transmembrane α-helices of β2AR are labeled with bold numerals (H1–H7). Cytoplasmic and extracellular loops are indicated C1, C2, and C3 and E1, E2, and E3, respectively. H8 denotes the short C-terminal α-helix 8 at the cytoplasmic side. The secondary structure model (A) of C-terminally truncated β2AR carrying an N-terminal FLAG epitope (blue) followed by a tobacco etch virus (TEV) protease cleavage site (green) was taken from ref. 8. The tertiary structure model (B) was taken from Protein Data Bank ID code 3D4S.
Cholesterol Changes the Free-Energy Landscape of β2AR.
The most probable force required to unfold a stable structural segment of a protein depends on the loading rate (pulling force applied vs. time) (45). Thus, the unfolding force is only a relative measure of the stability of a structural segment exposed to mechanical stress. However, the kinetic, energetic, and mechanical properties of a folded structure that, at equilibrium, resides in an energy valley can be described by a free-energy landscape (Fig. S5). To approximate these parameters (33, 45), we recorded F-D curves at seven pulling velocities (100, 300, 600, 900, 1,200, 2,500, and 5,000 nm/s) (Figs. S6 and S7). To investigate the effect of cholesterol on the energy landscape of β2AR, DFS was carried out in the absence and presence of CHS. Then, we determined the most probable unfolding force F* (Figs. S8 and S9) and loading rate rf* (Figs. S10 and S11) of every force peak representing a stable structural segment at different pulling velocities. We plotted the most probable unfolding force vs. the most probable loading rate for every stable structural segment in a so-called DFS plot (Fig. 3).
Fig. 3.

Loading rate-dependent interactions stabilizing structural segments of β2AR depend on cholesterol. DFS plots of each stable structural segment of β2AR reconstituted into DOPC (red) and DOPC/CHS (black) liposomes. Shown is the most probable unfolding force (Figs. S8 and S9) against the most probable loading rate (Figs. S10 and S11). Solid lines show the DFS fits from which xu and k0 were obtained (Table 1). Error bars indicate SE of the most probable unfolding force and loading rate.
As theoretically predicted (46, 47) and experimentally verified using membrane proteins (35, 36, 48–51), increasing the loading rate increases the unfolding forces. We observed a linear relationship between the most probable unfolding force and the logarithm of the loading rate for every interaction. This linearity suggests that, for every stable structural segment, a single energy barrier separated the folded state from the unfolded state (Fig. S5) (46). The DFS data were fitted using Eq. 2 (Fig. 3) to reveal the ground-to-transition state distance xu and transition rate k0, and Eqs. 3 and 4 were used to estimate the unfolding free-energy ΔG‡ and the mechanical spring constant κ for every stable structural segment of β2AR (Materials and Methods). The statistical significance of these differences was estimated using a nonlinear sum-of-squares F test (Table S2) (52, 53). Several stable structural segments showed statistically significant differences in the presence of CHS (Table 1), suggesting that the energy barriers stabilizing the individual structural regions of β2AR changed. In the next section, we describe these cholesterol-induced changes.
Table 1.
Parameters characterizing the free-energy barrier (xu, k0, and ΔG‡) and spring constant (κ) of structural segments stabilizing β2AR reconstituted into DOPC and DOPC/CHS liposomes
|
xu ± SD (nm) |
k0 ± SD (s–1) |
|||
| Stable structural segment | DOPC | DOPC/CHS | DOPC | DOPC/CHS |
| [H1.1] | 0.33 ± 0.02 | 0.44 ± 0.04 | 3.935 ± 1.210 | 0.077 ± 0.073 |
| [H1.2-C1] | 0.26 ± 0.02 | 0.29 ± 0.02 | 1.418 ± 0.429 | 0.248 ± 0.178 |
| [H2.1] | 0.32 ± 0.02 | 0.33 ± 0.02 | 1.250 ± 0.496 | 0.290 ± 0.174 |
| [H2.2-E1] | 0.37 ± 0.02 | 0.45 ± 0.05 | 2.174 ± 0.592 | 0.166 ± 0.140 |
| [H3-C2-H4-E2-H5.1] | 0.55 ± 0.04 | 0.55 ± 0.03 | 0.055 ± 0.041 | 0.038 ± 0.022 |
| [H5.2-C3-H6.1] | 0.26 ± 0.01 | 0.29 ± 0.02 | 0.443 ± 0.162 | 0.018 ± 0.014 |
| [H6.2-E3-H7-H8] | 0.45 ± 0.03 | 0.49 ± 0.03 | 1.376 ± 0.384 | 0.320 ± 0.118 |
| [CT] | 0.54 ± 0.03 | 0.59 ± 0.05 | 0.126 ± 0.054 | 0.011 ± 0.008 |
| Stable structural segment | ΔG‡ ± SD (kBT) | κ ± SD (N/m) | ||
| DOPC | DOPC/CHS | DOPC | DOPC/CHS | |
| [H1.1] | 17.1 ± 0.3 | 21.0 ± 0.9 | 1.26 ± 0.11 | 0.88 ± 0.13 |
| [H1.2-C1] | 18.1 ± 0.3 | 19.8 ± 0.7 | 2.39 ± 0.17 | 2.02 ± 0.26 |
| [H2.1] | 18.2 ± 0.4 | 19.7 ± 0.6 | 1.51 ± 0.17 | 1.49 ± 0.18 |
| [H2.2-E1] | 17.6 ± 0.3 | 20.2 ± 0.8 | 1.07 ± 0.10 | 0.84 ± 0.15 |
| [H3-C2-H4-E2-H5.1] | 21.3 ± 0.7 | 21.7 ± 0.6 | 0.58 ± 0.07 | 0.59 ± 0.06 |
| [H5.2-C3-H6.1] | 19.2 ± 0.4 | 22.4 ± 0.8 | 2.43 ± 0.21 | 2.23 ± 0.25 |
| [H6.2-E3-H7-H8] | 18.1 ± 0.3 | 19.6 ± 0.4 | 0.73 ± 0.08 | 0.69 ± 0.08 |
| [CT] | 20.5 ± 0.4 | 23.0 ± 0.7 | 0.59 ± 0.06 | 0.54 ± 0.06 |
Errors represent SDs. Differences were considered significant and highlighted bold when P values approached P < 0.001 from F tests (Table S2) and values did not overlap within their SD.
Unfolding of β2AR Starts in the Membrane.
The distance xu separating the folded state of a stable structural segment from the transition state characterizes how far a structural segment can be stretched until it unfolds (31, 33). We determined these transition state distances to be <1 nm for β2AR in the absence and presence of CHS (Table 1). Because the thickness of the proteoliposome membrane is ∼7.7 nm (Fig. S1) and the transition state distance is <1 nm, we can follow that a stable structural segment that transverses the membrane unfolds in the membrane. After this unfolding event, the AFM tip mechanically extracts the unfolded polypeptide from the membrane until the unfolded polypeptide is fully stretched. This finding is in agreement with results obtained on other transmembrane proteins, whereas stable structural segments first unfold in the membrane before being extracted from the membrane by the pulling AFM tip (31, 33, 54). Furthermore, our results suggest that the mechanically induced unfolding of β2AR in the lipid membrane is not altered by the presence of the cholesterol analog.
Cholesterol Increases Kinetic and Energetic Stability of β2AR.
Most prominent among the changes induced by CHS is that almost every stable structural segment of β2AR increased kinetic and energetic stability (Table 1). The free-energy barriers that stabilize each structural segment against unfolding increased height in the presence of CHS. Particularly, the energy barriers stabilizing the structural segments [H1.1], [H1.2-C1], [H2.1], [H2.2-E1], [H5.2-C3-H6.1], [H6.2-E3-H7-H8], and [CT] were significantly higher in the presence of CHS. For individual structural segments, the free-energy increase caused by CHS ranged between 1.5 ([H2.1] and [H6.2-E3-H7-H8]) and 3.9 kBT ([H1.1]). The exception was the largest structural segment [H3-C2-H4-E2-H5.1]; its small increase of the energy barrier (0.4 kBT) was not significant. Synchronous to the free-energy barrier heights, the structural segments significantly increased kinetic stability (reciprocal of the transition rate k0) in the presence of CHS (Table 1). Some stable structural segments increased their kinetic stability considerably. For example, in the presence of CHS, stable structural segment [H1.1] increased kinetic stability by a factor of 50, [H1.2-C1] increased by a factor of 6, [H2.1] and [H6.2-E3-H7-H8] increased by a factor of 4, [H5.2-C3-H6.1] increased by a factor of 25, and [CT] increased by a factor of 11. However, the transition rate of the structural core segment [H3-C2-H4-E2-H5.1] remained unchanged in the presence of CHS.
Cholesterol Increases Conformational Variability and Decreases Mechanical Rigidity of Transmembrane α-Helices H1 and H2.
The conformational variability of a structure can be approximated by the distance between ground state and transition state xu (55, 56). With increasing transition state distance, the energy valley stabilizing a structural segment becomes wider. Consequently, the structural segment can adopt more conformational substates and therefore, enhances conformational variability. On the contrary, if an energy valley stabilizing a structural segment narrows, the conformational variability of the structural segment decreases (Fig. S5). The only stable structural segments that significantly increased transition state distance xu by ∼20–30% and increased conformational variability were [H1.1] and [H2.2-E1] (Table 1). Although the other stable structural segments of β2AR showed similar trends in the presence of CHS, these differences were insignificant.
The spring constant κ quantifies the mechanical rigidity of a stable structural segment (49, 57). Although all stable structural segments slightly decreased their mechanical rigidity (spring constant) in the presence of CHS, this decrease was statistically significant only for structural segment [H1.1], which decreased the spring constant by ∼30% to 0.88 N/m.
Discussion
Cholesterol Strengthens Interactions of Almost Every Structural Region of β2AR.
Using SMFS, we characterized interactions that stabilize the human β2AR reconstituted in DOPC liposomes. F-D spectra recorded during mechanical unfolding of β2AR showed a reproducible pattern of force peaks (Fig. 1B). This pattern suggests that a characteristic interaction network stabilized structural segments within β2AR. The presence of CHS did not alter the position of the force peaks (Fig. 1C and Figs. S6 and S7). Hence, we conclude that interactions established in the presence of CHS stabilized the same structural segments of β2AR as detected in the absence of CHS. However, CHS significantly increased the magnitude of individual force peaks and thus, increased the strengths of the interactions stabilizing the structural segments within β2AR (Fig. 3). Because this trend was observed for all stable structural segments (except for [H3-C2-H4-E2-H5.1]) and all pulling velocities, we conclude that CHS increased the mechanical stability of β2AR. This increased mechanical stability may result from direct interactions between CHS and β2AR. However, because cholesterol also affects the properties of the lipid membrane (21, 58, 59) and because cholesterol establishes direct interactions with some but not all structural regions of β2AR (30), we assume that CHS also affected the stability of β2AR indirectly.
Cholesterol Increases the Free-Energy Barrier and Kinetic Stability of Every Stable Structural Segment, Except for the Structural Core Segment.
The strengths of the interactions stabilizing the structural segments of β2AR depended on the loading rate (Fig. 3). This dependency was used to characterize the energy valley and barrier stabilizing every structural segment (Table 1). Except for the structural core segment [H3-C2-H4-E2-H5.1], CHS significantly affected the energy landscape of every stable structural segment of β2AR. Generally, the free-energy barriers stabilizing these structural segments increased height in the presence of CHS. Consequently, the stable structural segments reduced transition rate and increased kinetic stability. Thus, CHS increased the kinetic and energetic stability of β2AR. However, the energy landscape of individual structural segments stabilizing β2AR and thus, their properties changed quite individually. These changes, which are structurally mapped in Fig. 4, will be discussed in the next sections. To verify that our findings were not introduced by the Bell–Evans model (Eq. 2), we have analyzed the DFS plots using the alternative Dudko–Szabo–Hummer model (Fig. S12) (60). This alternative analysis showed that the properties of every stable structural segment of β2AR (except for the structural core segment) changed in the same manner as revealed from the Bell–Evans model.
Fig. 4.

Mapping the kinetic, energetic, and mechanical properties of β2AR in the absence (A) and presence (B) of cholesterol. Structural segments stabilizing β2AR (Protein Data Bank ID code 3D4S) are mapped on the left. Transition state distance xu, transition rate k0, free-energy barrier height ΔG‡, and spring constant κ of stable structural segments in the absence of cholesterol (A) and the presence of cholesterol (B). The color of the β2AR backbone roughly indicates the value for each parameter as indicated by the scale bars. A, Upper and B, Upper show β2AR from the side view; A, Lower and B, Lower show β2AR from the extracellular view. Values were taken from Table 1.
[H1.1].
The presence of CHS affected kinetic, energetic, and mechanical stability of structural segment [H1.1]. The cholesterol analog increased the transition state distance by 33% to 0.44 nm, which enhanced the conformational variability of the extracellular region of transmembrane α-helix H1. Furthermore, CHS significantly decreased the transition rate and thus, increased the kinetic stability of [H1.1] 50-fold. Moreover, CHS increased the free-energy barrier ΔG‡ stabilizing [H1.1] by ∼4 kBT (∼23%). Finally, [H1.1] was the only structural segment of β2AR that significantly lowered the spring constant in the presence of CHS from 1.26 to 0.88 N/m (∼30%). This finding implies that CHS increases mechanical flexibility of this stable structural segment.
The X-ray structure suggests that α-helix H1 establishes direct interactions with cholesterol (30). The DFS data show that CHS kinetically and energetically stabilizes the extracellular part of α-helix H1 and enhances its mechanical flexibility. It is thought that GPCRs, including β2AR, can assemble into dimers in the plasma membrane (61, 62). Although the functional importance of this dimerization and the exact location of the dimer interface in GPCRs are not fully understood (63), it may be speculated that cholesterol promotes β2AR dimerization, because ordered cholesterol molecules were found in the interface between α-helices H1 and H8 of two symmetry-related β2ARs (9). It has been proposed for several other class A GPCRs that α-helix H1 is involved in receptor oligomerization (40, 64, 65). Therefore, it may be speculated that increasing the kinetic and energetic stability and increasing the mechanical flexibility of [H1.1] may favor oligomerization of β2AR.
[H1.2-C1].
In the presence of CHS, the energy landscape stabilizing structural segment [H1.2-C1] showed minor but statistically significant changes. [H1.2-C1] decreased transition rate and thus, increased kinetic stability sixfold. The free energy of [H1.2-C1] increased by 1.7 kBT (∼9%), indicating that this structural segment stabilizes in the presence of cholesterol. Structural models show cholesterol binding to the C-terminal part of transmembrane α-helix H1 (30), which could directly change the properties of structural segments [H1.1] and [H1.2-C1] as detected by DFS. Additional changes may be caused by direct interactions between CHS and residue Y70 at the interface between structural segments [H1.2-C1] and [H2.1]. The minor changes detected for [H1.2-C1] correlate with the finding that Y70 seems to be the least important residue for cholesterol binding and establish only van der Waals interactions with ring A of cholesterol (30). In the absence of additional information, it may be speculated that electrostatic interactions between hydrophilic residues of intracellular loop C1 (e.g., K60 and R63) and the cholesterol hydroxyl group could also contribute to the cholesterol-induced changes of [H1.2-C1].
[H2.1].
CHS reduced the transition rate of [H2.1] sixfold and increased the height of the free-energy barrier by 1.5 kBT (∼8%). The ground-to-transition state distance xu and the spring constant κ of [H2.1] remained unchanged. Thus, in the presence of the cholesterol analog, the kinetic and energetic stability of [H2.1] enhanced significantly. These changes may result from direct interactions between β2AR and CHS, which interact with [H2.1] through residues T73, S74 and C77 (30). The work by Hanson et al. (30) showed that CHS increases the packing value for transmembrane α-helix H2, which correlates with an increased thermal stability of β2AR. Our DFS data show that the increased packing value induced by CHS increases the kinetic and energetic stability of α-helix H2.
[H2.2-E1].
CHS slightly increased the ground-to-transition state distance by 0.08 nm and thus, the conformational variability of [H2.2-E1]. Furthermore, the cholesterol analog increased the kinetic stability of this structural segment 13-fold and increased the height of the free-energy barrier by 2.6 kBT (∼15%). CHS increases the packing value of α-helix H2 and interacts with [H2.2-E1] through an interaction with V81 (30). The changes in [H2.2-E1] might also be indirectly induced by electrostatic interactions between the cholesterol hydroxyl group and positively charged residues (e.g., K97) of extracellular loop E1.
[H3-C2-H4-E2-H5.1].
Although transmembrane α-helices H3 and H4 are supposed to establish direct interactions with cholesterol (30), the energy landscape of the core segment remained unchanged by the presence of CHS. This finding was surprising, because we recently detected that [H3-C2-H4-E2-H5.1] changes the energy landscape in the presence of agonists and inverse agonists (66). However, the core segment [H3-C2-H4-E2-H5.1] exposed a relatively high conformational variability (high xu) and high mechanical elasticity (low κ) compared with the other stable structural segments of β2AR in the presence of CHS. Interestingly, the structural core segment contains multiple ligand binding sites (10, 11). It has been also shown that cholesterol is required for efficient ligand binding of β2AR (67). Thus, our finding that the core segment of β2AR retains its relatively high conformational variability and mechanical flexibility in the presence of the cholesterol analog may be of functional importance for ligand binding.
[H5.2-C3-H6.1].
CHS significantly increased the kinetic stability of [H5.2-C3-H6.1] 25-fold and increased the height of the energy barrier by 3.2 kBT (∼17%). The distance between ground and transition state as well as the spring constant of [H5.2-C3-H6.1] remained unchanged. These changes are surprising, because CHS does not directly interact with the stable structural segment [H5.2-C3-H6.1]. Thus, it can be assumed that CHS induced these changes by modulation of the biophysical properties of the lipid membrane (21, 58, 59) and/or indirect interactions in β2AR.
As assessed by proteolysis and split receptor studies, cotransfection of a plasmid encoding the N terminus to α-helix H5 and a plasmid encoding α-helix H6 to the C terminus generates a functional split receptor (68). The interface between both receptor fragments locates at structural segment [H5.2-C3-H6.1]. Furthermore, the N- and C-terminal regions of cytoplasmic loop C3 are involved in G-protein activation and crucial for the formation of interactions between GPCR and G protein (69). These GPCR and G-protein interactions require cholesterol (70). Thus, it may be speculated that the kinetic and energetic stability of [H5.2-C3-H6.1] increased by CHS may play a role in regulating the interactions between β2AR and G proteins.
[H6.2-E3-H7-H8].
In the presence of CHS, structural segment [H6.2-E3-H7-H8] significantly increased kinetic stability fourfold and increased free energy by 1.5 kBT (∼8%). The oligomerization of class A GPCRs involves the interface between α-helices H1 and H8 (64, 65). Similar to stable structural segments [H1.1] and [H1.2-C1], it may be speculated that CHS affects oligomerization of β2AR by modulating the kinetic and energetic stability of [H6.2-E3-H7-H8]. Furthermore, α-helix H8, which is parallel to the membrane, might be affected by the composition of the membrane (e.g., through electrostatic interactions with polar lipid head groups).
[CT].
CHS induced significant changes in [CT], which increased kinetic stability 10-fold and free energy by 2.5 kBT (∼12%). The C-terminal end of β2AR is not known to contribute to cholesterol binding to the receptor (30). Thus, we speculate that the changing properties of segment [CT] could be indirectly caused by CHS modulating the biophysical properties of the lipid bilayer. However, [CT] contains charged amino acid residues, which might establish electrostatic interactions with the interfacial region of the membrane to which the cholesterol hydroxyl group contributes. [CT] is functionally important, because it interacts with GPCR kinases, arrestin, and other signaling molecules (71). For instance, the C-terminal binding domain is required to direct the trafficking of β2AR to cholesterol-rich caveolae (72). Therefore, we assume that the properties of [CT] changed by CHS, in turn, influence the way that signaling molecules interact with β2AR.
Conclusions
Cholesterol considerably increases the strength of interactions stabilizing structural segments of β2AR. These interactions introduced by the cholesterol analog CHS were sufficient to increase the kinetic, energetic, and mechanical stability of all structural segments stabilizing β2AR, except for the structural core segment [H3-C2-H4-E2-H5.1]; its properties were not significantly influenced by CHS. Because the core segment of β2AR is involved in ligand binding, this finding indicates that cholesterol may not necessarily influence the binding of a ligand to the structural core segment. In our experiments, we could not distinguish the extent to which the changes introduced to all other stable structural segments were caused by the binding of cholesterol to the receptor, the cholesterol-induced stabilization through receptor oligomerization, or the ability of cholesterol to modulate the properties of the lipid bilayer indirectly. At least, the stable structural segments of β2AR that do not expose cholesterol binding sites must have changed properties through indirect interactions mediated by cholesterol.
Our experiments show that cholesterol kinetically, energetically, and mechanically stabilizes the human β2AR. It is assumed that GPCRs being stabilized in such ways restrict their numbers of energetically favorable conformations (33, 73). Because cholesterol significantly modulates the structural properties of β2AR and β2AR shows a functionally different behavior in the presence of cholesterol (26, 28), it can be concluded that the observed kinetic, energetic, and mechanical stabilization of β2AR is of functional relevance. Such findings raise questions, such as whether cholesterol-dependent stabilization is the case for every class A GPCR to which β2AR belongs. Furthermore, it remains to be shown whether the stable structural segments of other GPCRs change similarly as observed for β2AR in the presence of cholesterol. Interestingly, the fact that CHS increases the mechanical and kinetic stability of the receptor supports the hypothesis that cholesterol is an essential component to crystallize a stable conformation of β2AR for structural determination (9).
Taken together, the unchanged structural core segment containing multiple ligand binding sites and the changed properties of all other stable structural segments may represent a mechanism of how cholesterol modulates β2AR. Previous SMFS measurements have shown that the magnitudes of the kinetic, energetic, and mechanical properties that change the functional state of β2AR on ligand binding (66) are similar to the magnitudes changing by the presence of cholesterol. Thus, one may speculate that the altered structural properties of the GPCR in the presence of cholesterol may cause the GPCR to respond differently to environmental changes, such as ligand binding. However, additional experiments that could quantify such an altered response of GPCRs in the presence of cholesterol must be performed.
Materials and Methods
Preparation of β2AR Proteoliposomes.
Spodoptera frugiperda (Sf9) insect cells were grown at 27 °C in suspension cultures in ESF-921 medium (Expression Systems) supplemented with 0.5 mg/mL gentamicin. To generate baculovirus for the β2AR, a BestBac baculovirus expression system (Expression Systems) was used. We used human β2AR with a truncated C-terminal end (48 aa) and an N-terminal FLAG epitope followed by a tobacco etch virus (TEV) protease cleavage site (Fig. 2A). β2AR was expressed using Sf9 cells at a density of ∼3 × 106 cells/mL for ∼48 h. Cells expressing β2AR were assessed by immunofluorescence and harvested by centrifugation (15 min at 5,000 × g). Before purification, cell pellets were stored at −80 °C. A three-step purification procedure was used to purify β2AR from Sf9 cells as described (40). To form a lipid stock solution at 10 and 20 mg/mL, DOPC (Avanti Polar Lipids) and the cholesterol analog CHS (Steraloids) were mixed and dissolved in chloroform. Next, DOPC and CHS were added to a glass vial, with DOPC at 10-fold (vol) excess. Chloroform was evaporated under a fine stream of argon gas. To prepare DOPC lipids without cholesterol, CHS was excluded from all preparation steps. After that point, the lipids were dried for 1 h in vacuum. Then, the lipids were resuspended in 100 mM NaCl, 1% (vol/vol) octyl glucoside, and 20 mM Hepes (pH 7.5), vortexed, and sonicated in an ice-water bath for 1 h. The lipid mixtures were stored at −80 °C.
β2AR was reconstituted as described (40). Briefly, a 300-µL sample was prepared containing a lipid-to-β2AR ratio of 1,000:1 (mol:mol). The lipid/β2AR mixture was mixed with reconstitution buffer until reaching a volume of 300 µL and placed on ice for 2 h. Vesicles were formed removing detergent using a 25 × 0.8 cm Sephadex G-50 (fine) column and reconstitution buffer (100 mM NaCl, 20 mM Hepes, pH 7.5).
SMFS and DFS.
SMFS was carried out using an automated AFM-based SMFS apparatus (ForceRobot 300; JPK Instruments) (74). SMFS data at 5,000 nm/s were recorded using 16-bit data acquisition hardware (NI PCI-6251; National Instruments). The 60-μm-long silicon nitride cantilevers (A-BioLever, BL-RC150 VB; Olympus) had nominal resonance frequencies of ∼8 kHz in water. Spring constants for each cantilever (∼30 pN/nm) were determined before each experiment in buffer solution using the equipartition theorem (75, 76) and varied by ∼10%. To compensate for uncertainties in cantilever calibration, β2AR was unfolded using at least five different cantilevers for each pulling velocity. All SMFS experiments were carried out using AFM cantilevers from the same wafer. Using Hook’s Law, the forces were calculated from the cantilever deflection.
DOPC/CHS (or DOPC) proteoliposomes containing β2AR were adsorbed over night at 4 °C onto freshly cleaved mica in SMFS buffer (300 mM NaCl, 25 mM Tris, 25 mM MgCl2, pH 7.0). After adsorption, the sample was rinsed several times with SMFS buffer to remove weakly attached membrane patches. SMFS buffer was prepared using nanopure water (≥18 MOhm/cm, PURE-LAB Ultra; ELGA LabWater) and purity grade (≥98.5%) reagents from Sigma or Merck. All SMFS experiments were performed in SMFS buffer at 24 °C.
To attach a single β2AR by unspecific interactions, the AFM stylus was pushed onto proteoliposomes, applying a constant force of 700 pN for 0.5 s. The unspecific attachment between the AFM stylus and the terminal end of the β2AR polypeptide chain is strong enough to withstand pulling forces of ∼2 nN (77, 78). Separation of stylus and membrane stretched the tethered polypeptide and exerted a force on the protein. At sufficiently high pulling force, β2AR unfolded stepwise. The F-D curves showed a characteristic force pattern, which was assigned to unfolding β2AR from its N-terminal end (SI Text).
Selection of F-D Curves and Data Analysis.
F-D curves recorded during unfolding of β2AR showed a sequence of force peaks. Each peak denoted the rupture of an unfolding barrier, which stabilized a structural segment of β2AR. The last force peak of an F-D curve described the unfolding of the last stable structural segment that remained anchored in the membrane (79). Overcoming the stability of this last segment completed the unfolding and extraction of β2AR from the membrane. For analysis, we selected F-D curves that corresponded to N-terminal unfolding of β2AR (SI Text). Only F-D curves with an overall length between 70 and 90 nm were taken into account, because they represented the complete unfolding of a β2AR. Peaks of an F-D curve were fitted using the WLC model (80) (Eq. 1):
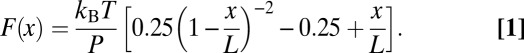 |
For WLC fitting, we assumed a persistence length P of 0.4 nm and a monomer length x of 0.36 nm (81). Contour lengths L representing the number of extended amino acids of the unfolded β2AR polypeptide stretches were obtained from fitting unfolding force peaks with the WLC model. Each of these contour lengths stretched by the AFM stylus assigned a stable structural segment of the partially folded protein that remained anchored in the membrane. The amplitude of a force peak gives the force that is required to mechanically unfold a stable structural segment. Every force peak of every F-D curve was analyzed using built-in and custom procedures of IgorPro 6 (WaveMetrics). F-D curves were superimposed and aligned to the characteristic force peak detected at the contour length of 121 aa.
Assignment of Stable Structural Segments.
The contour length determined by a WLC fit describes the length of the unfolded and stretched β2AR polypeptide that tethers AFM stylus and unfolding intermediate. Thus, each force peak assigns the end of the previous and the beginning of the following structural segment that stabilized β2AR against unfolding (31). Some stable structural segments had to be assumed to begin at the cytoplasmic β2AR surface at the opposite side of the pulling AFM stylus. In these cases, the so-called membrane compensation procedure was applied to locate the beginning of a stable structural segment (31, 79). Therefore, the thickness of the lipid membrane (4 nm ∼ 11 aa × 0.36 nm/aa) was added to the contour length of the unfolded polypeptide. If the beginning of a stable structural segment was within the membrane, fewer amino acids were added to the contour length.
Calculation of xu and k0.
The Bell–Evans model (47) describes the most probable unfolding force F* as a function of loading rate rf*. The relationship between both parameters was used to reveal insight into the free-energy barrier that stabilized a structural segment against mechanical unfolding (46) (Eq. 2):
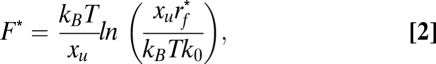 |
where kB is the Boltzmann constant, T is the absolute temperature, rf* is the most probable loading rate, xu is the distance between the free-energy minimum and the transition state, and k0 is the unfolding rate at zero applied force. The parameters xu and k0 were obtained by fitting Eq. 2 to a DFS plot using a nonlinear least squares algorithm. The loading rate was obtained using rf = kspacerv, where kspacer is the spring constant of the stretched polypeptide and v is the pulling velocity. kspacer corresponds to the slope of a force peak before rupture. Unfolding force and loading rate histograms (Figs. S8, S9, S10, and S11) were fitted using Gaussian distributions.
Calculation of Free-Energy Barrier Height and Mechanical Rigidity.
The height of the free-energy barrier ΔG‡ separating the folded state from the unfolded state was calculated using the Arrhenius equation (Eq. 3):
The diffuse relaxation time τD typically ranges from 10−7 to 10−9 s (82, 83). To calculate ΔG‡ for each stable structural segment, we used τD = 10−8 s (84). Errors of ΔG‡ were calculated propagating the errors of k0. Without having information on the energy potential shape, we assumed a simple parabolic potential from which the spring constant κ of a structural segment can be calculated using ΔG‡ and xu (57) (Eq. 4):
 |
To estimate errors of κ, errors of ΔG‡ and xu were propagated.
Supplementary Material
Acknowledgments
We thank C. Bippes, P. Bosshart, J. Fung, and J. Thoma for assistance. This research was supported by European Community’s Seventh Framework Programme FP7/2007-2013 Grant 211800, the Deutsche Forschungsgemeinschaft, and the Lundbeck Foundation.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
See Author Summary on page 20186 (volume 109, number 50).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1210373109/-/DCSupplemental.
References
- 1.Milligan G, Svoboda P, Brown CM. Why are there so many adrenoceptor subtypes? Biochem Pharmacol. 1994;48(6):1059–1071. doi: 10.1016/0006-2952(94)90141-4. [DOI] [PubMed] [Google Scholar]
- 2.Takeda S, Kadowaki S, Haga T, Takaesu H, Mitaku S. Identification of G protein-coupled receptor genes from the human genome sequence. FEBS Lett. 2002;520(1-3):97–101. doi: 10.1016/s0014-5793(02)02775-8. [DOI] [PubMed] [Google Scholar]
- 3.Bai TR. Beta 2 adrenergic receptors in asthma: A current perspective. Lung. 1992;170(3):125–141. doi: 10.1007/BF00174316. [DOI] [PubMed] [Google Scholar]
- 4.Barnes PJ. Beta-adrenoceptors on smooth muscle, nerves and inflammatory cells. Life Sci. 1993;52(26):2101–2109. doi: 10.1016/0024-3205(93)90725-i. [DOI] [PubMed] [Google Scholar]
- 5.Smiley RM, Finster M. Do receptors get pregnant too? Adrenergic receptor alterations in human pregnancy. J Matern Fetal Med. 1996;5(3):106–114. doi: 10.1002/(SICI)1520-6661(199605/06)5:3<106::AID-MFM2>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 6.Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nat Rev Drug Discov. 2006;5(12):993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- 7.Wishart DS, et al. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006;34(Database issue):D668–D672. doi: 10.1093/nar/gkj067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rasmussen SG, et al. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature. 2007;450(7168):383–387. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 9.Cherezov V, et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318(5854):1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosenbaum DM, et al. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science. 2007;318(5854):1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- 11.Rasmussen SG, et al. Structure of a nanobody-stabilized active state of the β(2) adrenoceptor. Nature. 2011;469(7329):175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rosenbaum DM, et al. Structure and function of an irreversible agonist-β(2) adrenoceptor complex. Nature. 2011;469(7329):236–240. doi: 10.1038/nature09665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Watts A, Volotovski ID, Marsh D. Rhodopsin-lipid associations in bovine rod outer segment membranes. Identification of immobilized lipid by spin-labels. Biochemistry. 1979;18(22):5006–5013. doi: 10.1021/bi00589a031. [DOI] [PubMed] [Google Scholar]
- 14.Fretten P, Morris SJ, Watts A, Marsh D. Lipid-lipid and lipid-protein interactions in chromaffin granule membranes. A spin label ESR study. Biochim Biophys Acta. 1980;598(2):247–259. doi: 10.1016/0005-2736(80)90003-6. [DOI] [PubMed] [Google Scholar]
- 15.Marsh D, et al. ESR spin-label studies of lipid-protein interactions in membranes. Biophys J. 1982;37(1):265–274. doi: 10.1016/S0006-3495(82)84675-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Engelman DM. Membranes are more mosaic than fluid. Nature. 2005;438(7068):578–580. doi: 10.1038/nature04394. [DOI] [PubMed] [Google Scholar]
- 17.Phillips R, Ursell T, Wiggins P, Sens P. Emerging roles for lipids in shaping membrane-protein function. Nature. 2009;459(7245):379–385. doi: 10.1038/nature08147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simons K, Gerl MJ. Revitalizing membrane rafts: New tools and insights. Nat Rev Mol Cell Biol. 2010;11(10):688–699. doi: 10.1038/nrm2977. [DOI] [PubMed] [Google Scholar]
- 19.Lingwood D, Simons K. Lipid rafts as a membrane-organizing principle. Science. 2010;327(5961):46–50. doi: 10.1126/science.1174621. [DOI] [PubMed] [Google Scholar]
- 20.Oates J, Watts A. Uncovering the intimate relationship between lipids, cholesterol and GPCR activation. Curr Opin Struct Biol. 2011;21(6):802–807. doi: 10.1016/j.sbi.2011.09.007. [DOI] [PubMed] [Google Scholar]
- 21.Lee AG. How lipids affect the activities of integral membrane proteins. Biochim Biophys Acta. 2004;1666(1–2):62–87. doi: 10.1016/j.bbamem.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 22.Gimpl G, Burger K, Fahrenholz F. Cholesterol as modulator of receptor function. Biochemistry. 1997;36(36):10959–10974. doi: 10.1021/bi963138w. [DOI] [PubMed] [Google Scholar]
- 23.Pang L, Graziano M, Wang S. Membrane cholesterol modulates galanin-GalR2 interaction. Biochemistry. 1999;38(37):12003–12011. doi: 10.1021/bi990227a. [DOI] [PubMed] [Google Scholar]
- 24.Burger K, Gimpl G, Fahrenholz F. Regulation of receptor function by cholesterol. Cell Mol Life Sci. 2000;57(11):1577–1592. doi: 10.1007/PL00000643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pucadyil TJ, Chattopadhyay A. Cholesterol modulates ligand binding and G-protein coupling to serotonin(1A) receptors from bovine hippocampus. Biochim Biophys Acta. 2004;1663(1-2):188–200. doi: 10.1016/j.bbamem.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 26.Pucadyil TJ, Chattopadhyay A. Role of cholesterol in the function and organization of G-protein coupled receptors. Prog Lipid Res. 2006;45(4):295–333. doi: 10.1016/j.plipres.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 27.Rajendran L, Knölker HJ, Simons K. Subcellular targeting strategies for drug design and delivery. Nat Rev Drug Discov. 2010;9(1):29–42. doi: 10.1038/nrd2897. [DOI] [PubMed] [Google Scholar]
- 28.Xiang Y, Rybin VO, Steinberg SF, Kobilka B. Caveolar localization dictates physiologic signaling of beta 2-adrenoceptors in neonatal cardiac myocytes. J Biol Chem. 2002;277(37):34280–34286. doi: 10.1074/jbc.M201644200. [DOI] [PubMed] [Google Scholar]
- 29.Yao Z, Kobilka B. Using synthetic lipids to stabilize purified beta2 adrenoceptor in detergent micelles. Anal Biochem. 2005;343(2):344–346. doi: 10.1016/j.ab.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 30.Hanson MA, et al. A specific cholesterol binding site is established by the 2.8 A structure of the human beta2-adrenergic receptor. Structure. 2008;16(6):897–905. doi: 10.1016/j.str.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kedrov A, Janovjak H, Sapra KT, Müller DJ. Deciphering molecular interactions of native membrane proteins by single-molecule force spectroscopy. Annu Rev Biophys Biomol Struct. 2007;36:233–260. doi: 10.1146/annurev.biophys.36.040306.132640. [DOI] [PubMed] [Google Scholar]
- 32.Engel A, Gaub HE. Structure and mechanics of membrane proteins. Annu Rev Biochem. 2008;77:127–148. doi: 10.1146/annurev.biochem.77.062706.154450. [DOI] [PubMed] [Google Scholar]
- 33.Janovjak H, Sapra KT, Kedrov A, Müller DJ. From valleys to ridges: Exploring the dynamic energy landscape of single membrane proteins. ChemPhysChem. 2008;9(7):954–966. doi: 10.1002/cphc.200700662. [DOI] [PubMed] [Google Scholar]
- 34.Park PS, et al. Stabilizing effect of Zn2+ in native bovine rhodopsin. J Biol Chem. 2007;282(15):11377–11385. doi: 10.1074/jbc.M610341200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kedrov A, Appel M, Baumann H, Ziegler C, Muller DJ. Examining the dynamic energy landscape of an antiporter upon inhibitor binding. J Mol Biol. 2008;375(5):1258–1266. doi: 10.1016/j.jmb.2007.11.032. [DOI] [PubMed] [Google Scholar]
- 36.Bippes CA, et al. Substrate binding tunes conformational flexibility and kinetic stability of an amino acid antiporter. J Biol Chem. 2009;284(28):18651–18663. doi: 10.1074/jbc.M109.004267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kedrov A, et al. Probing the interactions of carboxy-atractyloside and atractyloside with the yeast mitochondrial ADP/ATP carrier. Structure. 2010;18(1):39–46. doi: 10.1016/j.str.2009.11.009. [DOI] [PubMed] [Google Scholar]
- 38.Damaghi M, et al. Dual energy landscape: The functional state of the β-barrel outer membrane protein G molds its unfolding energy landscape. Proteomics. 2010;10(23):4151–4162. doi: 10.1002/pmic.201000241. [DOI] [PubMed] [Google Scholar]
- 39.Ge L, Perez C, Waclawska I, Ziegler C, Muller DJ. Locating an extracellular K+-dependent interaction site that modulates betaine-binding of the Na+-coupled betaine symporter BetP. Proc Natl Acad Sci USA. 2011;108(43):E890–E898. doi: 10.1073/pnas.1109597108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fung JJ, et al. Ligand-regulated oligomerization of beta(2)-adrenoceptors in a model lipid bilayer. EMBO J. 2009;28(21):3315–3328. doi: 10.1038/emboj.2009.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oates J, et al. The role of cholesterol on the activity and stability of neurotensin receptor 1. Biochim Biophys Acta. 2012;1818(9):2228–2233. doi: 10.1016/j.bbamem.2012.04.010. [DOI] [PubMed] [Google Scholar]
- 42.Gimpl G, Fahrenholz F. Cholesterol as stabilizer of the oxytocin receptor. Biochim Biophys Acta. 2002;1564(2):384–392. doi: 10.1016/s0005-2736(02)00475-3. [DOI] [PubMed] [Google Scholar]
- 43.Chae PS, et al. Maltose-neopentyl glycol (MNG) amphiphiles for solubilization, stabilization and crystallization of membrane proteins. Nat Methods. 2010;7(12):1003–1008. doi: 10.1038/nmeth.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saxena R, Chattopadhyay A. Membrane cholesterol stabilizes the human serotonin(1A) receptor. Biochim Biophys Acta. 2012;1818(12):2936–2942. doi: 10.1016/j.bbamem.2012.07.032. [DOI] [PubMed] [Google Scholar]
- 45.Evans E. Probing the relation between force—lifetime—and chemistry in single molecular bonds. Annu Rev Biophys Biomol Struct. 2001;30:105–128. doi: 10.1146/annurev.biophys.30.1.105. [DOI] [PubMed] [Google Scholar]
- 46.Evans E, Ritchie K. Dynamic strength of molecular adhesion bonds. Biophys J. 1997;72(4):1541–1555. doi: 10.1016/S0006-3495(97)78802-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Evans E. Energy landscapes of biomolecular adhesion and receptor anchoring at interfaces explored with dynamic force spectroscopy. Faraday Discuss. 1998;111(111):1–16. doi: 10.1039/a809884k. [DOI] [PubMed] [Google Scholar]
- 48.Janovjak H, et al. Probing the energy landscape of the membrane protein bacteriorhodopsin. Structure. 2004;12(5):871–879. doi: 10.1016/j.str.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 49.Sapra KT, Balasubramanian GP, Labudde D, Bowie JU, Muller DJ. Point mutations in membrane proteins reshape energy landscape and populate different unfolding pathways. J Mol Biol. 2008;376(4):1076–1090. doi: 10.1016/j.jmb.2007.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sapra KT, Doehner J, Renugopalakrishnan V, Padrós E, Muller DJ. Role of extracellular glutamic acids in the stability and energy landscape of bacteriorhodopsin. Biophys J. 2008;95(7):3407–3418. doi: 10.1529/biophysj.108.131904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kawamura S, Colozo AT, Müller DJ, Park PS. Conservation of molecular interactions stabilizing bovine and mouse rhodopsin. Biochemistry. 2010;49(49):10412–10420. doi: 10.1021/bi101345x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wells JW. Analysis and interpretation of binding at equilibrium. In: Hulme EC, editor. Receptor–Ligand Interactions: A Practical Approach. Oxford: Oxford Univ Press; 1992. [Google Scholar]
- 53.Motulsky HJ, Christopoulos A. Fitting Models to Biological Data Using Linear and Nonlinear Regression: A Practical Guide to Curve Fitting. New York: Oxford Univ Press; 2004. [Google Scholar]
- 54.Janovjak H, Kessler M, Oesterhelt D, Gaub H, Müller DJ. Unfolding pathways of native bacteriorhodopsin depend on temperature. EMBO J. 2003;22(19):5220–5229. doi: 10.1093/emboj/cdg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kumar S, Ma B, Tsai CJ, Sinha N, Nussinov R. Folding and binding cascades: Dynamic landscapes and population shifts. Protein Sci. 2000;9(1):10–19. doi: 10.1110/ps.9.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oliveberg M, Wolynes PG. The experimental survey of protein-folding energy landscapes. Q Rev Biophys. 2005;38(3):245–288. doi: 10.1017/S0033583506004185. [DOI] [PubMed] [Google Scholar]
- 57.Dietz H, Berkemeier F, Bertz M, Rief M. Anisotropic deformation response of single protein molecules. Proc Natl Acad Sci USA. 2006;103(34):12724–12728. doi: 10.1073/pnas.0602995103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Evans E, Needham D. Giant vesicle bilayers composed of mixtures of lipids, cholesterol and polypeptides. Thermomechanical and (mutual) adherence properties. Faraday Discuss Chem Soc. 1986;81:267–280. doi: 10.1039/dc9868100267. [DOI] [PubMed] [Google Scholar]
- 59.Needham D, Nunn RS. Elastic deformation and failure of lipid bilayer membranes containing cholesterol. Biophys J. 1990;58(4):997–1009. doi: 10.1016/S0006-3495(90)82444-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dudko OK, Hummer G, Szabo A. Theory, analysis, and interpretation of single-molecule force spectroscopy experiments. Proc Natl Acad Sci USA. 2008;105(41):15755–15760. doi: 10.1073/pnas.0806085105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hebert TE, et al. A peptide derived from a beta2-adrenergic receptor transmembrane domain inhibits both receptor dimerization and activation. J Biol Chem. 1996;271(27):16384–16392. doi: 10.1074/jbc.271.27.16384. [DOI] [PubMed] [Google Scholar]
- 62.Terrillon S, Bouvier M. Roles of G-protein-coupled receptor dimerization. EMBO Rep. 2004;5(1):30–34. doi: 10.1038/sj.embor.7400052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Milligan G. G protein-coupled receptor dimerization: Function and ligand pharmacology. Mol Pharmacol. 2004;66(1):1–7. doi: 10.1124/mol.104.000497.. [DOI] [PubMed] [Google Scholar]
- 64.Liang Y, et al. Organization of the G protein-coupled receptors rhodopsin and opsin in native membranes. J Biol Chem. 2003;278(24):21655–21662. doi: 10.1074/jbc.M302536200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Guo W, et al. Dopamine D2 receptors form higher order oligomers at physiological expression levels. EMBO J. 2008;27(17):2293–2304. doi: 10.1038/emboj.2008.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zocher M, Fung JJ, Kobilka BK, Müller DJ. Ligand-specific interactions modulate kinetic, energetic, and mechanical properties of the human β2 adrenergic receptor. Structure. 2012;20(8):1391–1402. doi: 10.1016/j.str.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kirilovsky J, Schramm M. Delipidation of a beta-adrenergic receptor preparation and reconstitution by specific lipids. J Biol Chem. 1983;258(11):6841–6849. [PubMed] [Google Scholar]
- 68.Kobilka BK, et al. Chimeric alpha 2-,beta 2-adrenergic receptors: Delineation of domains involved in effector coupling and ligand binding specificity. Science. 1988;240(4857):1310–1316. doi: 10.1126/science.2836950. [DOI] [PubMed] [Google Scholar]
- 69.Gether U, Kobilka BK. G protein-coupled receptors. II. Mechanism of agonist activation. J Biol Chem. 1998;273(29):17979–17982. doi: 10.1074/jbc.273.29.17979. [DOI] [PubMed] [Google Scholar]
- 70.Ben-Arie N, Gileadi C, Schramm M. Interaction of the beta-adrenergic receptor with Gs following delipidation. Specific lipid requirements for Gs activation and GTPase function. Eur J Biochem. 1988;176(3):649–654. doi: 10.1111/j.1432-1033.1988.tb14326.x. [DOI] [PubMed] [Google Scholar]
- 71.Reiter E, Lefkowitz RJ. GRKs and beta-arrestins: Roles in receptor silencing, trafficking and signaling. Trends Endocrinol Metab. 2006;17(4):159–165. doi: 10.1016/j.tem.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 72.Xiang Y, Kobilka B. The PDZ-binding motif of the beta2-adrenoceptor is essential for physiologic signaling and trafficking in cardiac myocytes. Proc Natl Acad Sci USA. 2003;100(19):10776–10781. doi: 10.1073/pnas.1831718100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kobilka BK, Deupi X. Conformational complexity of G-protein-coupled receptors. Trends Pharmacol Sci. 2007;28(8):397–406. doi: 10.1016/j.tips.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 74.Struckmeier J, et al. Fully automated single-molecule force spectroscopy for screening applications. Nanotechnology. 2008;19(38):384020. doi: 10.1088/0957-4484/19/38/384020. [DOI] [PubMed] [Google Scholar]
- 75.Butt HJ, Jaschke M. Calculation of thermal noise in atomic-force microscopy. Nanotechnology. 1995;6:1–7. [Google Scholar]
- 76.Florin EL, et al. Sensing specific molecular-interactions with the atomic-force microscope. Biosens Bioelectron. 1995;10:895–901. [Google Scholar]
- 77.Müller DJ, Engel A. Atomic force microscopy and spectroscopy of native membrane proteins. Nat Protoc. 2007;2(9):2191–2197. doi: 10.1038/nprot.2007.309. [DOI] [PubMed] [Google Scholar]
- 78.Muller DJ. AFM: A nanotool in membrane biology. Biochemistry. 2008;47(31):7986–7998. doi: 10.1021/bi800753x. [DOI] [PubMed] [Google Scholar]
- 79.Müller DJ, et al. Stability of bacteriorhodopsin alpha-helices and loops analyzed by single-molecule force spectroscopy. Biophys J. 2002;83(6):3578–3588. doi: 10.1016/S0006-3495(02)75358-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bustamante C, Marko JF, Siggia ED, Smith S. Entropic elasticity of lambda-phage DNA. Science. 1994;265(5178):1599–1600. doi: 10.1126/science.8079175. [DOI] [PubMed] [Google Scholar]
- 81.Rief M, Gautel M, Oesterhelt F, Fernandez JM, Gaub HE. Reversible unfolding of individual titin immunoglobulin domains by AFM. Science. 1997;276(5315):1109–1112. doi: 10.1126/science.276.5315.1109. [DOI] [PubMed] [Google Scholar]
- 82.Krieger F, Fierz B, Bieri O, Drewello M, Kiefhaber T. Dynamics of unfolded polypeptide chains as model for the earliest steps in protein folding. J Mol Biol. 2003;332(1):265–274. doi: 10.1016/s0022-2836(03)00892-1. [DOI] [PubMed] [Google Scholar]
- 83.Dietz H, Rief M. Exploring the energy landscape of GFP by single-molecule mechanical experiments. Proc Natl Acad Sci USA. 2004;101(46):16192–16197. doi: 10.1073/pnas.0404549101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gräter F, Grubmüller H. Fluctuations of primary ubiquitin folding intermediates in a force clamp. J Struct Biol. 2007;157(3):557–569. doi: 10.1016/j.jsb.2006.11.005. [DOI] [PubMed] [Google Scholar]
