

Abstract
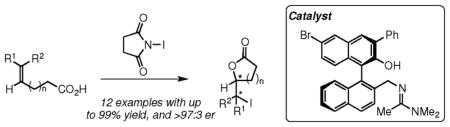
The enantioselective iodolactonizations of a series of diversely-substituted olefinic carboxylic acids are promoted by a BINOL-derived, bifunctional catalyst. Reactions involving 5-alkyl- and 5-aryl-4(Z)-pentenoic acids and 6-alkyl- and 6-aryl-5(Z)-hexenoic acids provide the corresponding γ- and δ-lactones having stereogenic C–I bonds in excellent yields and >97:3 er. Significantly, this represents the first organocatalyst that promotes both bromo- and iodolactonization with high enantioselectivities. The potential of this catalyst to induce kinetic resolutions of racemic unsaturated acids is also demonstrated.
Electrophilic halocyclizations of α,ω-functionalized alkenes represent an important class of reactions in organic chemistry.1 In particular, halolactonizations of unsaturated acids have been widely used in organic chemistry to prepare intermediates in the synthesis of biologically active natural products and other compounds having potential utility.2 Given the importance of these cyclizations, there has been considerable interest in developing enantioselective variants, and a number of notable advances in developing enantioselective bromolactonizations of unsaturated acids 1 to give lactones 2 (X = Br, Figure 1) have been recently reported.3 However, the corresponding catalytic, enantioselective chloro-3f,4 and iodolactonizations5 to give 2 (X = Cl and I, Figure 1) are less common and much more restrictive in substrate scope.
Figure 1.

Enantioselective Halolactonizations
The first catalytic, enantioselective iodolactonization was reported in 2004 by Gao, who used quaternary ammonium salts derived from cinchonidine to promote cyclizations of 5-aryl-4(E)-pentenoic acids. However, these reactions proceeded with only moderate enantioselectivity, and mixtures of γ- and δ-lactones were often obtained.5a Gao and coworkers later found that a salen-Co(II) complex catalyzed enantioselective iodolactonizations of several 4-aryl-4-pentenoic acids to give γ-lactones with up to 92:8 er.5b Veitch and Jacobsen recently discovered that 5-aryl-5-hexenoic acids undergo enantioselective iodolactonizations, typically with >95:5 er, in the presence of a tertiary aminourea catalyst.5c Most recently Dobish and Johnston utilized a chiral Brønsted acid catalyst to promote enantioselective iodolactonizations of an extensive series of 5-aryl-5-hexenoic acids, usually with >98:2 er.5d Despite these achievements, there are a number of significant limitations to existing methods. In particular, there are few examples of iodolactonizations of olefinic acids bearing alkyl groups on the double bond,5b–d and these cyclizations invariably have occurred with lower enantioselectivity than their aryl counterparts. Moreover, there are no examples of iodolactonizations that generate new C–I bonds at stereogenic centers. Because the products of such cyclizations may bear two new stereocenters, existing iodolactonization methods are not applicable to the direct synthesis of compounds containing multiple stereocenters.
We envisioned that these and other gaps in prevailing methods might be addressed by adapting our recently reported protocol for enantioselective bromolactonizations that are catalyzed by 3.3i In particular, we queried whether 4, which is formed quantitatively in situ by bromination of 3 under conditions used for bromolactonization, might promote highly enantioselective iodolactonizations. We thus discovered that 4 catalyzed the iodolactonizations of various disubstituted olefinic acids with N-iodosuccinimide (NIS) to deliver iodolactones in excellent yields and enantioselectivities.6 To our knowledge, this is the first instance where the same organocatalyst is highly effective for both bromo- and iodolactonizations. Herein we report the details of some of our findings.
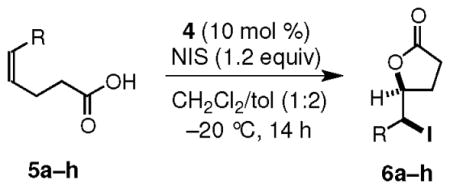
We found that both 5-alkyl-4(Z)-pentenoic acids 5a–d and 5-aryl-4(Z)-pentenoic acids 5e–h undergo iodolactonization in the presence of 4 (10 mol %) and NIS (1.2 eq) at −20 °C to furnish the γ-lactones 6a–h as the only isolable products in >97:3 er and in excellent yield (Table 1, entries a–h). The reaction with 5a was also ran on 0.5 mmol scale to give the iodolactone product 6a in 95% yield and 97.5:2.5 er (Table 1, entry a). These cyclizations proceed via an exo mode of ring closure with high regioselectivity to generate products with two contiguous stereogenic centers. Diastereomeric products were observed only when the starting Z-alkene contained detectable amounts of the inseparable E-isomer. The presence of electron withdrawing groups on the aromatic ring is well tolerated, but the products obtained from substrates having electron rich aromatic rings, such as p-methoxyphenyl, are unstable and suffer extensive decomposition during isolation and purification. In the reactions of cis-aryl olefinic acids 5e–h, no products arising from a 6-endo cyclization pathway were observed. We tentatively attribute this observation to the difficulty of achieving the planar arrangement in the transition state necessary for maximum stabilization of the developing positive charge at the benzylic position.7
Table 1.
Iodolactonization of 5-Substituted-4(Z)-pentenoic Acidsa
| ||||
|---|---|---|---|---|
| entry | R | product | yieldb (%) | erc,d |
| a | i-Pr | 6a | 93 (95)e | 97.5:2.5 |
| b | i-Bu | 6b | 94 | 98:2 |
| c | t-Bu | 6c | 99 | 97:3 |
| d | c-Hex | 6d | 97 | 98:2 |
| ef | Ph | 6e | 93 | 98.5:1.5 |
| f | p-NC-C6H4 | 6f | 95 | 99:1 |
| gg | p-Cl-C6H4 | 6g | 89 | 98:2 |
| hh | 2-Np | 6h | 94 | 98:2 |
Reactions run on 0.1 mmol scale.
Isolated yield after column chromatography.
er determined by chiral HPLC.
Absolute stereochemistry of 6c and 6e established by X-ray analysis; 6a–b, 6d, 6f–h were based on analogy.
Yield in parentheses obtained on 0.5 mmol scale.
Z/E ratio of 5e, 20:1.
Z/E ratio of 5g, 11:1.
Z/E ratio of 5h, 17:1.
The utility of organocatalyst 4 is further exemplified by the enantioselective iodolactonizations of 6-aryl- and 6-alkyl-5(Z)-hexenoic acids such as 7a–d to provide the corresponding δ-lactones 8a–d in high yields and up to 99:1 er (Table 2). No regioisomeric products were observed, and diastereomeric products were detected only if the starting Z-alkene contained discernable amounts of E-isomer. Although an electron-withdrawing group slowed the iodolactonization reaction, the selectivity was not adversely affected (Table 2, entry b).
Table 2.
Iodolactonization of 6-Substituted-5(Z)-hexenoic Acidsa
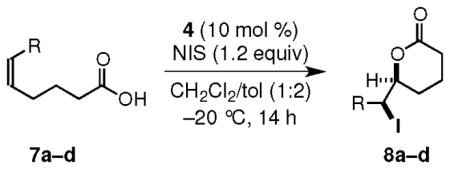
| ||||
|---|---|---|---|---|
| entry | R | product | yieldb (%) | erc,d |
| ae | Ph | 8a | 89 | 99:1 |
| bf,g | p-NC-C6H4 | 8b | 88 | 99:1 |
| cg | 2-Np | 8c | 93 | 98.5:1.5 |
| d | t-Bu | 8d | 98 | 98:2 |
Reactions run on 0.1 mmol scale.
Isolated yield after column chromatography.
er determined by chiral HPLC.
Absolute stereochemistry of 8a–d are based on analogy with 6c and 6e.
Z/E ratio of 7a, 14:1.
Z/E ratio of 7b, 20:1.
Results obtained after 38 h at −20 °C.
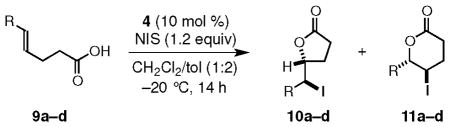
In contrast to the high enantioselectivities observed for 5-substituted-4(Z)-pentenoic acids 5a–h, iodolactonizations of 5-alkyl-4(E)-pentenoic acids are somewhat less selective, as exemplified by the cyclization of 9a to give 10a (Table 3, entry a). The divergent levels of enantioselectivity in the iodolactonizations of 5-alkyl-4(E)- and 5-alkyl-4(Z)-pentenoic acids mirrors the related bromolactonizations,3i and it may reflect a preferential side-on approach of the olefin during formation of the iodonium ion in analogy with known enantioselective epoxidations of olefins.8 The iodolactonization of 9b was not regioselective, giving an inseparable mixture (1.3:1.0) of the γ- and δ-lactones 10b and 11b via 5-exo and 6-endo cyclization pathways, respectively (Table 3, entry b). For other 5-aryl-4(E)-pentenoic acids, we find, like Gao,5a that the electronic nature of substituents on the aromatic ring has a marked effect upon the regioselectivity of the iodolactonization (Table 3, entries c,d). For example, 9c undergoes both regio- and enantioselective iodolactonization preferentially via an endo mode of ring closure (6-endo:5-exo >20:1) to give 11c with 86.5:13.5 er. The presence of an electron-withdrawing group favors iodolactonization by an exo mode of closure that is not very enantioselective (Table 3, entry d).
Table 3.
Regiochemistry in Iodolactonization of 5-Substituted-4(E)-pentenoic Acidsa
| ||||
|---|---|---|---|---|
| entry | R | product | yieldb (%) | erc,d |
| a | i-Pr | 10a | 78 | 67:33 |
| b | Ph | 10b+11b | 89e | 52:48(95:5)f |
| c | p-MeO-C6H4 | 11c | 89 | 86.5:13.5 |
| dg | p-NC-C6H4 | 10d | 94 | 58:42 |
Reactions run on 0.1 mmol scale.
Isolated yield after column chromatography.
er determined by chiral HPLC.
Absolute stereochemistry of iodolactones was assigned based on analogy with corresponding bromolactone analogs.3i
Combined yield; ratio of 1.3:1.0 based upon NMR analysis.
er shown in parentheses is for 11b,
Reaction performed for 14 h at −20 °C and 48 h at −10 °C in CH2Cl2/tol (1:1).
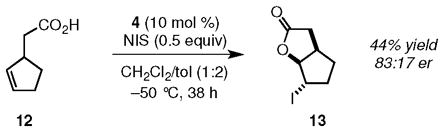
Catalytic enantioselective transformations may be applied to kinetic resolutions, which can provide an enantiomerically pure product from a racemic starting material. There are, however, fewer examples of such processes with organic catalysts than with metal or enzyme catalysts. Although there is a recent report of a kinetic resolution of a racemic, unsaturated acid by bromolactonization,9 we are aware of no example of a kinetic resolution by an iodolactonization. It is thus notable that 4 promotes the kinetic resolution of commercially available 2-cyclopentene-1-acetic acid (12) to give bicyclic iodolactone (+)–13 in 44% yield (88% of theoretical) in 83:17 er (eq 1).10 Compound (+)–13 and its enantiomer are important intermediates in the syntheses of a number of targets.11
 |
(1) |
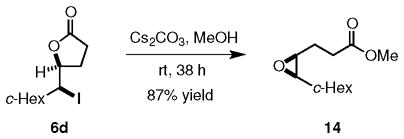
As mentioned previously, halolactones are useful intermediates for the synthesis of a number of other compounds. One important application of their utility is exemplified by their conversion into epoxides. Although there are some excellent methods for the enantioselective synthesis of epoxides,12 there remain some gaps in methods for preparing epoxides having certain substitution patterns. For example, enantioselective epoxidation of unconjugated, cis-1,2-dialkyl alkenes lacking suitable directing groups is not well-documented. 13 It is thus significant that treatment of 6d with Cs2CO3 in MeOH afforded the corresponding epoxy ester 14 in 87% yield with essentially complete retention of enantiopurity (eq 2). Application of this iodolactonization-epoxidation sequence therefore provides a practical approach to the enantioselective synthesis of cis-epoxides that were heretofore not accessible.
 |
(2) |
In summary, we have discovered that the BINOL-derived catalyst 4 is a useful bifunctional catalyst for promoting the highly enantioselective iodolactonizations of a wide array of olefinic acids. Moreover, to our knowledge 4 is the first organocatalyst that is capable of promoting both highly enantioselective bromo- and iodolactonizations. The iodolactonizations of 5-substituted-4(Z)-pentenoic and 6-substituted-5(Z)-hexenoic acids typically proceed with >97:3 er to give the corresponding γ- and δ-lactones having two contiguous stereogenic centers. Indeed, these reactions represent the first examples where new C–I bonds are formed with high enantioselectivity by iodolactonizations. The potential of 4 and analogs thereof for effecting enantioselective syntheses of iodolactones by kinetic resolution is also demonstrated. Moreover, the product lactones can be converted into epoxides, thereby enabling the highly enantioselective preparation of certain substituted epoxides that were heretofore inaccessible. The utility of 4 and related catalysts to promote highly enantioselective reactions as key steps in natural product synthesis is under active investigation as are studies to elucidate the mechanism and the origin of stereoinduction in these cyclizations.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health (GM31077) and the Robert A. Welch Foundation (F-0652) for generous support of this research. DHP thanks the NIH for a postdoctoral fellowship (GM 96557). We are also grateful to Dr. Vincent Lynch (The University of Texas) for X-ray crystallography.
Footnotes
Supporting Information Available: Experimental procedures, characterization of new compounds, and Xray crystal data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For recent reviews, see: Chen G, Ma S. Angew Chem Int Ed. 2010;49:8306–8308. doi: 10.1002/anie.201003114.Tan CK, Zhou L, Yeung Y. Synlett. 2011:1335–1339.Snyder SA, Treitler DS, Brucks AP. Aldrichimica Acta. 2011;44:27–40.Denmark SE, Kuester WE, Burk MT. Angew Chem Int Ed. 2012;51:10938–10953. doi: 10.1002/anie.201204347.
- 2.(a) Dowle MD, Davies DI. Chem Soc Rev. 1979;8:171–197. [Google Scholar]; (b) Ranganathan S, Muraleedharan KM, Vaish NK, Jayaraman N. Tetrahedron. 2004;60:5273–5308. [Google Scholar]; (c) Lava MS, Banerjee AK, Cabrera EV. Curr Org Chem. 2009;13:720–730. [Google Scholar]; (d) Rodriguez F, Fananas FJ. In: Handbook of Cyclization Reactions. Ma S, editor. Vol. 4. Wiley-VCH; New York: 2010. pp. 951–990. [Google Scholar]
- 3.(a) Zhang W, Zheng S, Liu N, Werness JB, Guzei IA, Tang W. J Am Chem Soc. 2010;132:3664–3665. doi: 10.1021/ja100173w. [DOI] [PubMed] [Google Scholar]; (b) Murai K, Matsushita T, Nakamura A, Fukushima S, Shimura M, Fujioka H. Angew Chem, Int Ed. 2010;49:9174–9177. doi: 10.1002/anie.201005409. [DOI] [PubMed] [Google Scholar]; (c) Zhou L, Tan CK, Jiang X, Chen F, Yeung Y. J Am Chem Soc. 2010;132:15474–15476. doi: 10.1021/ja1048972. [DOI] [PubMed] [Google Scholar]; (d) Tan CK, Zhou L, Yeung Y. Org Lett. 2011;13:2738–2741. doi: 10.1021/ol200840e. [DOI] [PubMed] [Google Scholar]; (e) Tan CT, Le C, Yeung Y. Chem Commun. 2012;48:5793–5795. doi: 10.1039/c2cc31148h. [DOI] [PubMed] [Google Scholar]; (f) Zhang W, Liu N, Schienebeck CM, Decloux K, Zheng S, Werness JB, Tang W. Chem–Eur J. 2012;18:7296–7305. doi: 10.1002/chem.201103809. [DOI] [PubMed] [Google Scholar]; (g) Murai K, Nakamura A, Matsushita T, Shimura M, Fujioka H. Chem– Eur J. 2012;18:8448–8453. doi: 10.1002/chem.201200647. [DOI] [PubMed] [Google Scholar]; (h) Jiang X, Tan CK, Zhou L, Yueng Y. Angew Chem, Int Ed. 2012;51:7771–7775. doi: 10.1002/anie.201202079. [DOI] [PubMed] [Google Scholar]; (i) Paull DH, Fang C, Donald JR, Pansick AD, Martin SF. J Am Chem Soc. 2012;134:11128–11131. doi: 10.1021/ja305117m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Whitehead DC, Yousefi R, Jaganathan A, Borhan B. J Am Chem Soc. 2010;132:3298–3300. doi: 10.1021/ja100502f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Wang M, Gao LX, Mai WP, Xia AX, Wang F, Zhang SB. J Org Chem. 2004;69:2874–2876. doi: 10.1021/jo035719e. [DOI] [PubMed] [Google Scholar]; (b) Ning Z, Jin R, Ding J, Gao L. Synlett. 2009:2291–2294. [Google Scholar]; (c) Veitch GE, Jacobsen EN. Angew Chem Int Ed. 2010;49:7332–7335. doi: 10.1002/anie.201003681. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Dobish MC, Johnston JN. J Am Chem Soc. 2012;134:6068–6071. doi: 10.1021/ja301858r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (a) Tungen JE, Nolsøe JMJ, Hansen TV. Org Lett. doi: 10.1021/ol302798g. [DOI] [Google Scholar]
- 6.In preliminary experiments, we found that iodolactonizations catalyzed by 4 were slightly faster and/or higher yielding than those promoted by 3; both catalysts can be recovered, unmodified, from iodolactonization reactions.
- 7.(a) Nicolaou KC, Prasad CVC, Somers PK, Hwang C-K. J Am Chem Soc. 1989;111:5330–5334. [Google Scholar]; (b) Nicolaou KC, Prasad CVC, Somers PK, Hwang CK. J Am Chem Soc. 1989;111:5335–5340. [Google Scholar]
- 8.(a) Brandes BD, Jacobsen EN. J Org Chem. 1994;59:4378–4380. [Google Scholar]; (b) Fukuda T, Irie R, Katsuki T. Synlett. 1995:197–198. [Google Scholar]
- 9.Ikeuchi K, Ido S, Yoshimura S, Asakawa T, Inai M, Hamashima Y, Kan T. Org Lett. doi: 10.1021/ol302908a. [DOI] [PubMed] [Google Scholar]
- 10.Seeman M, Schöller M, Kudis S, Helmchen G. Eur J Org Chem. 2003:2122–2127. [Google Scholar]
- 11.For recent examples, see Röhrig S, Hennig L, Findeisen M, Welzel P. Tetrahedron. 1998;54:3439–3456.Canham SM, France DJ, Overman LE. J Am Chem Soc. 2010;132:7876–7877. doi: 10.1021/ja103666n.
- 12.For reviews and leading references, see Johnson RA, Sharpless KB. In: Catalytic Asymmetric Synthesis. 2. Ojima I, editor. Wiley-CVH; New York: 2000. pp. 231–280.Jacobsen EN, Wu MH. In: Comprehensive Asymmetric Catalysis. 1. Jacobsen EN, Pfaltz A, Yamamoto H, editors. Vol. 2. Springer; New York: 1999. pp. 649–677.Katsuki T. Adv Synth Catal. 2002;344:131–147.Shi Y. Acc Chem Res. 2004;37:488–496. doi: 10.1021/ar030063x.Yang D. Acc Chem Res. 2004;37:497–505. doi: 10.1021/ar030065h.Denmark SE, Wu Z. Synlett. 1999:847–859.Shibasaki M, Kanai M, Matsunaga S. Aldrichachim Acta. 2006;39:31–39.Zhang FY, Corey EJ. Org Lett. 1999;1:1287–1290. doi: 10.1021/ol990964z.Page PCB, Buckley NR, Blacker AJ. Org Lett. 2004;6:1543–1546. doi: 10.1021/ol049782h.Marigo M, Franzen J, Poulsen TB, Zhuang W, Jørgensen KA. J Am Chem Soc. 2005;127:6964–6965. doi: 10.1021/ja051808s.Lee S, MacMillan DWC. Tetrahedron. 2006;62:11413–11424.
- 13.Recently Shi reported a modified fructose-derived ketone to promote enantioselective epoxidation of limited scope of cis-olefins with good enantioselectivity: Burke CP, Shi Y. Org Lett. 2009;11:5150–5153. doi: 10.1021/ol901724v.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.